

Abstract
Since 2016, A(H5Nx) high pathogenic avian influenza (HPAI) virus of clade 2.3.4.4b has become one of the most serious global threats not only to wild and domestic birds, but also to public health. In recent years, important changes in the ecology, epidemiology, and evolution of this virus have been reported, with an unprecedented global diffusion and variety of affected birds and mammalian species. After the two consecutive and devastating epidemic waves in Europe in 2020–2021 and 2021–2022, with the second one recognized as one of the largest epidemics recorded so far, this clade has begun to circulate endemically in European wild bird populations. This study used the complete genomes of 1,956 European HPAI A(H5Nx) viruses to investigate the virus evolution during this varying epidemiological outline. We investigated the spatiotemporal patterns of A(H5Nx) virus diffusion to/from and within Europe during the 2020–2021 and 2021–2022 epidemic waves, providing evidence of ongoing changes in transmission dynamics and disease epidemiology. We demonstrated the high genetic diversity of the circulating viruses, which have undergone frequent reassortment events, providing for the first time a complete overview and a proposed nomenclature of the multiple genotypes circulating in Europe in 2020–2022. We described the emergence of a new genotype with gull adapted genes, which offered the virus the opportunity to occupy new ecological niches, driving the disease endemicity in the European wild bird population. The high propensity of the virus for reassortment, its jumps to a progressively wider number of host species, including mammals, and the rapid acquisition of adaptive mutations make the trend of virus evolution and spread difficult to predict in this unfailing evolving scenario.
Keywords: high pathogenic avian influenza A(H5) viruses, phylodynamics, Europe, reassortments, spatial spread
Introduction
In 1996, high pathogenic avian influenza (HPAI) A(H5Nx) virus of the A/goose/Guangdong/1/1996 (Gs/Gd) lineage was identified for the first time in poultry in China. In late 2003 and early 2004, multiple outbreaks were reported almost simultaneously in poultry in several East Asian countries (Li et al. 2004). The situation dramatically changed in spring 2005, when, following a mass die-off of wild migratory birds infected with A(H5N1) in Qinghai Lake in central China (Chen et al. 2006), this strain spread over long distances reaching for the first time Europe and Africa during the wild bird autumn migration (Olsen et al. 2006). Since then, the HPAI A(H5Nx) virus of the Gs/GD lineage has widely spread across Asia, Europe, Africa, and North America (Fusaro et al. 2019; Lycett et al. 2020), becoming a serious global threat for poultry, endangered wild bird species, as well as representing a zoonotic risk for human health, with 962 HPAI A(H5Nx) zoonotic infections reported up to 14 July 2023 (World Health Organization 2023). The global circulation of this lineage has led to the diversification of the hemagglutinin (HA) gene segment into ten first-order clades and multiple second to fifth-order subclades (Smith and Donis 2015). Since 2014, subclade 2.3.4.4, which originated in China in 2008, has dominated the global scene and has been responsible for all the Gs/Gd epidemic waves reported in Europe. According to the World Health Organization (WHO), this clade has evolved into eight subclades (a–h) (World Health Organization 2021), with 2.3.4.4b being responsible for the largest HPAI epidemic waves recorded in Europe in 2016–2017, 2020–2021, and 2021–2022, as well as for the most recent one in 2022–2023. The 2016–2017 epidemic was predominantly caused by HPAI A(H5N8) viruses, which affected most European countries with a significant impact on both wild (1,563 cases) and domestic birds (1,218 outbreaks) (Adlhoch et al. 2023). Over the following years, 2017–2018 and 2019–2020, two minor incursions were reported, with a limited number of countries affected during the winter seasons. Since October 2020, consecutive epidemic waves have been reported in Europe (Floyd et al. 2021; Bordes et al. 2023; Tammiranta et al. 2023; Vreman et al. 2023), which heavily affected poultry and wild and captive birds, with occasional spill over into mammalian species. The first epidemic wave lasted from October 2020 to September 2021, with 3,792 HPAI A(H5Nx) detections (2,407 in wild birds and 1,385 in domestic birds) across thirty-one European countries. This was followed by an even larger second wave, which lasted from October 2021 to September 2022, counting a total of 6,707 HPAI A(H5Nx) detections (3,936 in wild and 2,771 in domestic birds) reported across thirty-seven countries. In 2022, for the first time the virus caused several mass mortality events in wild birds during the summer months, likely due to the virus spreading to seabird breeding colonies, a category of wild birds, which had very rarely been affected before by HPAI A(H5Nx) (Adlhoch et al. 2022). The 2020–2021 and 2021–2022 waves were characterized by the circulation of different subtypes and genotypes, as a result of reassortment events with low pathogenicity avian influenza (LPAI) viruses (Lewis et al. 2021; Briand et al. 2022; Engelsma et al. 2022; Grant et al. 2022; Nagy, Černíková, and Stará 2022; Pohlmann et al. 2022). While the A(H5N8) subtype mainly drove the first (2020–2021) epidemic wave, A(H5N1) was responsible for the majority of the 2021–2022 and 2022–2023 outbreaks (Adlhoch et al. 2023).
These epidemics did not affect only Europe, but had global impact. Since January 2021, an A(H5N1) related to the 2020–2021 European viruses had been circulating in West Africa, and soon reached South African countries (Abolnik et al. 2022; Letsholo et al. 2022; Lo et al. 2022; Makalo et al. 2022; Ouoba et al. 2022; Sanogo et al. 2022). From late 2021, A(H5N1) viruses of clade 2.3.4.4b have been detected in South and East Asia (Isoda et al. 2022; Sagong et al. 2022; Ye et al. 2022). In December 2021, the A(H5N1) clade 2.3.4.4b closely related to the A(H5N1) identified in Northern Europe during the 2020–2021 epidemic season was introduced into North America (Caliendo et al. 2022). At the beginning of 2022, a new introduction into North America via the Pacific flyway of an A(H5N1) related to the viruses circulating in Japan was identified (Alkie et al. 2022). Since then, the virus has spread throughout North America, undergoing reassortment events with LPAI viruses of the American lineages (Alkie et al. 2022). In October 2022, A(H5N1) viruses spread for the first time to Mexico and soon after to South America, with mass mortality events reported in wild birds (Adlhoch et al. 2023), wild mammals, and poultry farms.
In this study, we characterized the spatiotemporal patterns of the A(H5Nx) virus spread to/from and within Europe during the 2020–2021 and 2021–2022 epidemic waves, providing important insights on the regions at high risk of virus incursions. We demonstrated the high genetic diversity of the circulating viruses, which underwent frequent reassortment events, and suggested a nomenclature to describe the circulating genotypes. Importantly, we described the relevant changes between the two epidemic waves in terms of spatiotemporal dynamics of virus spread and host range, which have dramatically modified the disease epidemiology in Europe with serious consequences for the poultry industry, wild bird populations (such as sea birds), and farmed carnivores (e.g. minks and foxes), as well as concerns around the increased zoonotic risk for public health.
Materials and methods
Dataset
During the 2020–2021 and 2021–2022 European epidemic waves, sequencing analyses of the complete genome of HPAI A(H5Nx) viruses were performed by the European Union Reference Laboratory (EURL—IZSVe), by National Reference Laboratories (NRLs), and by some other academic partners, for a total of 1,956 complete viral genomes. The samples originated from thirty-two European countries with 875 being collected during the 2020–2021 wave and 1,081 during the 2021–2022 wave—(up to August 2022). All the sequences were deposited in the GISAID EpiFlu database (http://www.gisaid.org, Supplementary Table S1).
Among avian hosts, we identified five different host categories: domestic birds (Galliformes, Anseriformes, and others), waterfowls (mainly Anseriformes and Charadriformes), raptors, colony breeding seabirds (mainly Laridae and Sulidae), and others (Supplementary Table S1).
Phylogenetic analysis and genotype identification
Sequences of the eight gene segments were aligned in MAFFT v7 (Katoh and Standley 2013) and compared to the most related sequences determined using the Nucleotide Basic Local Alignment Search Tool (BLAST) search available in the GISAID EpiFlu™ database (accessed on 7 September 2022). Maximum likelihood (ML) phylogenetic trees of all gene segments were generated in IQTREE v1.6.6 (Nguyen et al. 2015; Thi Hoang et al. 2017), performing an ultrafast bootstrap resampling analysis (1,000 replications). ModelFinder, available in IQTREE, was used to select the best-fit model of sequence evolution. FigTree v1.4.4 was used to visualize the phylogenetic tree of each gene segment (http://tree.bio.ed.ac.uk/software/figtree/).
The identification of the genotype was based on the phylogenetic tree topology. Monophyletic clusters for genotype assignment were identified for all the trees, using the following criteria: (1) each HPAI A(H5Nx) cluster had to be supported by ultrafast bootstrap values >90, and (2) they had to show a higher identity with low or (rarely) high pathogenic influenza viruses (the possible donors of the gene segment) than the one observed with the other 2020–2022 HPAI A(H5Nx) clusters. These donors, located at the base of each recognized cluster, were identified from the BLAST search of the 2020–2022 HPAI A(H5Nx) viruses under study. A reference sequence was selected among the donors for each gene segment and each cluster, to represent the potential progenitor virus of that cluster. An identification (ID) number (cluster ID) was assigned to each cluster and the combination of the eight IDs (one for each gene segment) defined a genotype (Fig. 1, Supplementary Figs. S1–S13).
Figure 1.

Graphical representation of the A(H5Nx) genotypes identified in Europe during the first (2020–2021) and second (2021–2022) epidemic wave. A number and associated colour code is assigned to each gene based on its genetic clustering in the phylogenetic trees. The reference sequences represent the possible progenitor virus for each specific gene.
HA datasets’ refinement
To investigate the spatial spread of the 2.3.4.4b HPAI A(H5Nx) viruses during the 2020–2021 and 2021–2022 epidemic waves, we generated three different HA datasets: a global dataset and two European datasets.
For the global analysis, we downloaded from the GISAID EpiFlu™ database (accessed on 18 January 2024) all the available A(H5Nx) HA sequences at least 1,600 nucleotides long belonging to clade 2.3.4.4b from Africa, Asia, and North America, sampled in a time frame ranging from January 2018 to August 2022. Sequences were phylogenetically analysed together with the HA gene sequences of the European viruses; all the HA sequences that shared a common progenitor with the European viruses were retained for the following analyses (about 3,700 sequences). Six discrete geographic regions—Africa, North America, East Asia, Central Asia, Europe, and the Middle East—were defined. To mitigate sampling bias and ensure a balanced dataset, a ML phylogenetic tree using IQTREE v1.6.6 (Nguyen et al. 2015; Thi Hoang et al. 2017) was generated for each geographic trait (Africa, North America, East Asia, Central Asia, and Europe), except for the Middle East, in which case all the forty-seven available sequences were retained. The Phylogenetic Diversity Analyzer (PDA) tool (Chernomor et al. 2014) was then used to select about 150 sequences from each tree based on their phylogenetic diversity, making sure to include the first detected viruses from each geographic region. For each trait (geographic area), the number of sequences ranged from 47 (the Middle East) to 165 (Europe), for a total of 806 sequences.
To explore the dynamics of virus spread within Europe, we generated two datasets, one for each epidemic wave. Seven discrete regions were defined: North-Central Europe (the Czech Republic, Denmark, Germany, Norway, Poland, and Sweden), North-Eastern Europe (Estonia, Finland, Latvia, and Lithuania), North-Western Europe (Belgium, the Republic of Ireland, Luxembourg, the Netherlands, and the UK), Central Asia (Russia and Kazakhstan), South-Central Europe (Albania, Austria, Croatia, Greece, Hungary, Italy, Kosovo, Slovakia, Slovenia, and Switzerland), South-Eastern Europe (Bulgaria, Moldova, and Romania), and South-Western Europe (France, Portugal, and Spain). To reduce possible bias associated with different sequences availability from each country, we calculated the percentage of sequenced outbreaks for each epidemic wave in each geographic area: North-Central Europe (16 per cent W1, 11 per cent W2), North-Eastern Europe (17 per cent W1, 17 per cent W2), North-Western Europe (71 per cent W1, 15 per cent W2), South-Central Europe (47 per cent W1, 51 per cent W2), South-Eastern Europe (100 per cent W1, 42 per cent W2), and South-Western Europe (2 per cent W1, 7 per cent W2). For the regions with <20 per cent of characterized outbreaks or with a number of sequences ≤10, all the genetic data were retained. For the remaining geographic areas, a number of sequences corresponding to 20 per cent of the reported outbreaks were selected using the PDA tool, starting from ML phylogenetic trees generated for each geographic area. The European dataset of the first epidemic wave (2020–2021) included 617 sequences, whereas the one generated for the second epidemic wave (2021–2022) included 838 sequences collected up to August 2022.
Evolutionary and phylogeographic analyses
Markov chain Monte Carlo (MCMC) analyses of the HA gene segment were performed using BEAST v1.10.4 in combination with the BEAGLE library (Suchard et al. 2018). We employed an uncorrelated lognormal relaxed molecular clock, the coalescent constant population size model, and the SRD06 substitution model (HKY85 + Γ4 with two partitions—first + second positions vs third position—base frequencies and Γ-rate heterogeneity unlinked across all codon positions) (Shapiro, Rambaut, and Drummond 2006). MCMC chains were run for 400–700 million iterations and convergence was assessed using Tracer v1.7.1 (Shapiro, Rambaut, and Drummond 2006; Drummond and Rambaut 2007; Suchard and Rambaut 2009; Shapiro et al. 2011). The maximum clade credibility (MCC) trees were summarized using TreeAnnotator v1.10.4 and visualized in FigTree v1.4.4 (http://beast.bio.ed.ac.uk/TreeAnnotator/, http://tree.bio.ed.ac.uk/software/figtree/).
To explore the pattern of spatial diffusion among the geographic regions, discrete phylogeographic analyses using location as a trait were performed (Lemey et al. 2009). We assumed an asymmetric non-reversible transition model and incorporated Bayesian stochastic search variable selection (Lemey et al. 2009). Spread D3 v0.9.6 (Bielejec et al. 2016) was used to visualize the spatial dispersal of the viral strain through time and to assess Bayes factor (BF) supports for individual transitions between discrete locations. We interpreted the strength of statistical support as positive for 5 < BF ≤ 20, strong for 20 < BF ≤ 150, and very strong for BF >150.
We employed the Markov jump counts to measure the number of viral movements along the branches of the phylogeny and estimated the Markov rewards to quantify the time the virus spent in each geographical region (Minin and Suchard 2008). We also counted all jumps ‘from’ and all jumps ‘to’ each geographic region, to evaluate which were the main donor and recipient sites of the virus movements.
Results
Genetic variability of the European viruses and shift of the circulating genotypes between the two waves
ML phylogenetic analysis of the eight gene segments was used to assess the number and variety of co-circulating genotypes, each defined by a unique gene composition inferred from tree topologies and inconsistencies among the genetic clustering of the eight gene segments (Supplementary Figs. S1–S13). The first identified A(H5Nx) viral genome showing a particular gene constellation was used as a reference strain for a specific genotype (e.g. H5N1-A/Eurasian_Wigeon/Netherlands/1/2020-like). In order to make communication easier and to distinguish the genotypes identified during each epidemic wave, we named them with the indication of the lineage (e.g. EA, Eurasian), the year of identification and with letters—a single letter (i.e. A, B, C) for the genotypes which had originated during the 2020–2021 wave and two letters (i.e. AA, AB, ac) for the genotypes of the 2021–2022 wave (e.g. EA-2020-C, EA-2021-AB).
Overall, we defined fifty different A(H5Nx) genotypes from multiple inter- and intra-subtype reassortment events (Fig. 1). The highest genetic diversity in terms of number of co-circulating genotypes (from 13 to 25) was observed during the winter months in both waves: January–March 2021 and November 2021–February 2022 (Fig. 2). In most cases (73 per cent) gene segments had been acquired by reassortments with LPAI viruses circulating in wild birds. Of note, the frequency of reassortment was not homogenous among the eight gene segments. While the HA and matrix (M) gene segments were stable, multiple switches were observed for the polymerase basic (PB) 2, PB1, polymerase acidic (PA), and nucleoprotein (NP) segments that were classified into 14, 12, 11, and 16 clusters, respectively.
Figure 2.
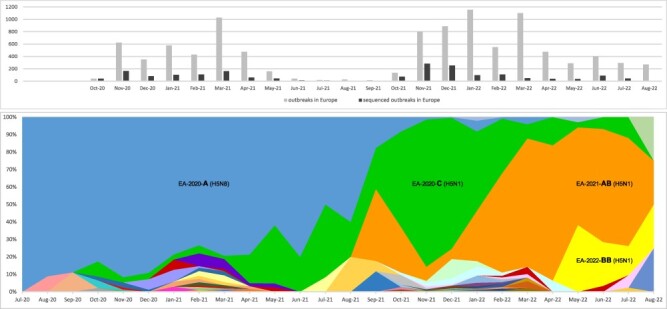
Upper panel: distribution of total number of notified (grey) and genetically characterized (black) HPAI A(H5Nx) viruses in Europe by month of detection. Lower panel: monthly distribution of the A(H5Nx) genotypes identified in Europe between July 2020 and August 2022. Each colour represents a different genotype. The four main genotypes EA-2020-A, EA-2020-C, EA-2021-AB and EA-2022-BB are marked in the graph.
During the 2020–2021 epidemic wave, we identified nineteen genotypes belonging to the A(H5N8) (N = 5), A(H5N5) (N = 8), A(H5N1) (N = 3), A(H5N4) (N = 2), and A(H5N3) (N = 1) subtypes. The great majority of the viruses (80.7 per cent) belonged to genotype EA-2020-A (H5N8-A/duck/Chelyabinsk/1207-1/2020-like), identified for the first time in July 2020 in central Russia, close to the Kazakh border, followed by the EA-2020-C genotype (H5N1-A/Eurasian_Wigeon/Netherlands/1/2020-like) (7 per cent). EA-2020-A (H5N8), together with EA-2020-C (H5N1), were the first two genotypes identified in Europe (The Netherlands) in mid-October 2020. As genotype EA-2020-C (H5N1) had never been identified before October 2020 in other areas outside Europe, it was assumed to have emerged in Europe from the A(H5N8) subtype through a single reassortment event involving six gene segments (PB2, PB1, PA, NP, neuraminidase (NA), and non-structural (NS)), although it is not possible to discount that this occurred in a step-wise manner involving un-sampled intermediates including outside Europe. The EA-2020-A genotype appeared to dominate over all the other genotypes until August 2021. With the beginning of the second wave in Europe in October 2021 and the rise in the number of A(H5Nx) virus detections, we witnessed a dramatic shift in the genetic composition of circulating subtypes and genotypes (Fig. 2). While in 2020–2021, A(H5N8) had been the most prevalent subtype, the 2021–2022 wave was dominated almost exclusively by A(H5N1). Only three of the genotypes identified during the first wave, namely EA-2020-A (H5N8), EA-2020-C (H5N1), and EA-2021-I (H5N5), continued to be detected through the summer months, while all the other genotypes that were detected in the 2021–2022 wave had either emerged from reassortment events with LPAI viruses circulating in wild birds in Europe or were the result of novel virus introductions, such as genotype EA-2021-AB (H5N1-A/duck/Saratov/29-02V/2021-like). This genotype was identified for the first time in September 2021 in the Russian Federation (Saratov oblast) and during October 2021 it was also detected in Germany, The Netherlands, and Denmark (Supplementary Table S1). Unfortunately, the scarcity of genetic data from countries outside Europe makes it impossible to assess for certain the geographic source area of the different genotypes. Overall, thirty-four different genotypes, thirty belonging to the A(H5N1) subtype, were characterized during the 2021–2022 wave, indicating that the diversity of genotypes detected was even greater than in the previous wave. With a frequency of 52.9 per cent and 29.5 per cent, the A(H5N1) genotypes EA-2020-C and EA-2021-AB were the most prevalent in 2021–2022. At the beginning of this epidemic wave, a rise of genotype EA-2020-C was observed in Europe, likely as a result of a combination between an increase in the number of local cases and novel virus incursions (probably from Russia) of the same genotype. Indeed, the topology of the phylogenetic trees revealed that the 2021–2022 European viruses of the EA-2020-C genotype split into two sub-clusters, one comprising the A(H5N1) viruses collected in European countries since October 2020, the other including A(H5N1) collected in Russia since the beginning of October 2021 and subsequently (end of October) in Europe (Supplementary Figs. S1–S13). This genotype remained the most prevalent (frequency ranging from 46 per cent to 84 per cent) from October 2021 to January 2022. In February 2022, it was overtaken by genotype EA-2021-AB, which in February–August 2022 reached a frequency of 33 per cent to 78 per cent, thus becoming the most frequent and widespread (No. of countries = 19) genotype in Europe (Fig. 2), based on the available data.
In May 2022, a new A(H5N1) genotype likely emerged from reassortment with A(H13) LPAI viruses, a subtype primarily found in gulls, from which it acquired the PA, NP, and NS gene segments. This new genotype, named EA-2022-BB (H5N1-A/Herring_gull/France/22P015977/2022-like), was identified for the first time in a herring gull in France and, subsequently, in the Netherlands and Belgium. It circulated extensively during the summer months, being the second most frequently detected genotype (22 per cent of the viruses sequenced between June and August 2022), mainly in gulls (86 per cent), with the European herring gull as the most commonly affected species (70 per cent).
During the 2020–2021 and 2021–2022 epidemic waves, multiple spillover events from birds to ten different mammalian species were reported (Adlhoch et al. 2023). Sequences were generated for a total of forty-nine viruses collected during the 2020–2021 (N = 10) and 2021–2022 (N = 39) waves, in eleven countries from nine different species: red fox (Vulpes vulpes) (N = 34), harbour seal (Phoca vitulina) (N = 5), grey seal (Halichoerus grypus) (N = 1), Eurasian otter (Lutra lutra) (N = 2), ferret (Mustela furo) (N = 1), European polecat (Mustela putorius) (N = 4), lynx (Lynx lynx) (N = 1), European badger (Meles meles) (N = 1), and porpoise (Phocoena phocoena) (N = 1). The analysed sequences belonged to seven different A(H5N8) and A(H5N1) genotypes—A, C, Q, AF, AB, AH, BB—with 52 per cent and 31 per cent of the viruses belonging, respectively, to the A(H5N1) genotypes EA-2020-C and EA-2021-AB, the two most widespread in Europe during the 2021–2022 wave, when most of the cases in mammals were reported (Fig. 3A).
Figure 3.
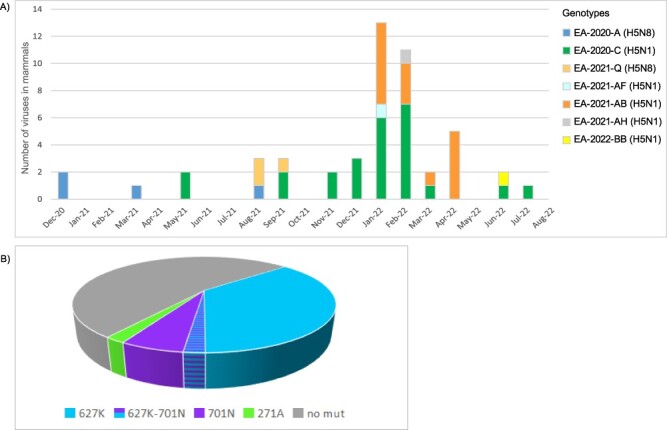
Panel A—bars represent the distribution of the genetically characterized (complete genome) A(H5Nx) viruses collected from mammals in Europe from December 2020 to August 2022, by month of detection, coloured according to the genotype. Panel B—frequency of the molecular markers of mammalian adaptation in the PB2 protein (271A, 627K, 701N) identified in viruses collected from mammalian species in Europe between October 2020 and August 2022.
Geographic and host distribution of the different genotypes
The fifty genotypes identified during the 2020–2021 and 2021–2022 waves are likely the result of novel virus incursions during the fall migration of wild birds, and/or may have emerged de novo from reassortment events with viruses circulating in wild birds in Europe. Three genotypes, specifically genotypes EA-2020-C (H5N1), EA-2020-A (H5N8), and EA-2021-I (H5N5), persistently circulated during both epidemic waves. With respect to the first wave (2020–2021), in 2021–2022, the EA-2020-A and EA-2021-I genotypes reduced their geographic spread, which was limited to three countries (Kosovo, Denmark, and Albania) for EA-2020-A and to one country (Norway) for EA-2021-I. Conversely, during the second wave, genotype EA-2020-C showed a remarkable geographical expansion (twenty-one countries in 2021–2022 vs eleven countries in 2020–2021) (Fig. 1, Fig. 4, Supplementary Tables S2 and S3).
Figure 4.
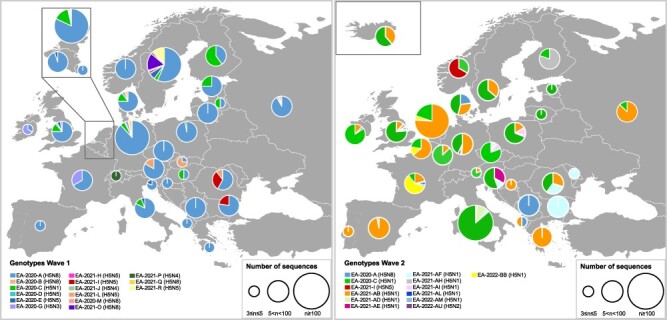
Geographic distribution of the major A(H5Nx) genotypes (represented by at least three viruses) in Europe during the first (2020–2021, left panel) and second (2021–2022, right panel) epidemic wave. The pie charts display the frequency of the genotype in each country. The size of the circle is proportional to the number of sequences analysed.
The highest genotype diversity was observed in the Netherlands (fifteen genotypes), France (twelve genotypes), Sweden (eleven genotypes), and Germany (ten genotypes), followed by Belgium, Croatia, Czech Republic, Denmark, Ireland, Italy, Poland, Romania, Slovenia, and the UK, with five to ten circulating genotypes (Fig. 4, Supplementary Tables S2 and S3). However, bias due to the number of available sequences from each country cannot be excluded, as surveillance activities and genetic characterization efforts widely differ (the proportion of sequenced virus samples in each country ranges from 0 per cent to 100 per cent). Specifically, during the 2020–2021 wave, the highest genotype diversity (5–9 genotypes) was detected in Northern Europe (Denmark, Germany, the Netherlands, Sweden, and the UK), while during the 2021–2022 wave, a more widespread genotype diversity was observed, with the Czech Republic, France, Germany, Italy, the Netherlands, Poland, Romania, and the UK having four to nine genotypes each.
About 43 per cent of the genotypes were identified only in wild birds, 20 per cent only in domestic birds, and 37 per cent being detected in both wild and domestic birds. Domestic birds (both Galliformes and Anseriformes) and waterfowl (Anseriformes) are the host categories harbouring the highest genetic diversity among the detected viruses, while the lowest was observed in seabirds (Fig. 5). In this latter group, the frequency of the detected genotypes also differed considerably from what was noted in the other host categories (domestic, waterfowl, raptors), showing a higher level of circulation of genotype EA-2020-C during the 2020–2021 wave and of genotype EA-2022-BB during the 2021–2022 wave compared to other host categories (Fig. 5).
Figure 5.
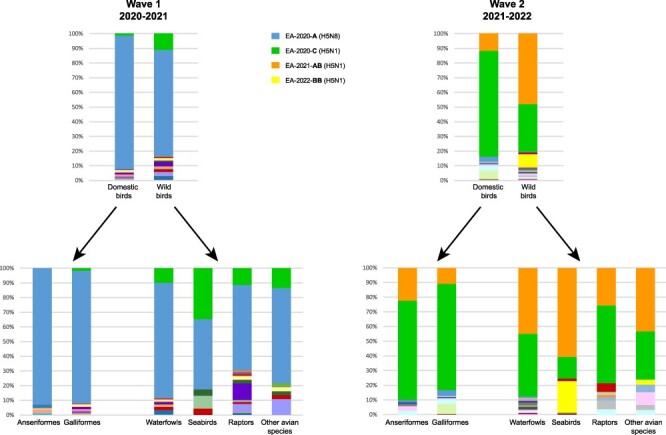
Distribution of the different genotypes by bird host categories during the first (2020–2021, left panel) and second (2021–2022, right panel) epidemic wave. The colours assigned to the four major genotypes are reported in the figure legend.
Global analysis—the origin and global spread of clade 2.3.4.4b HPAI H5 viruses affecting Europe in 2020–2022
To attempt to define the origins of the HPAI A(H5Nx) viruses of clade 2.3.4.4b introduced in Europe in October 2020, and in order to reconstruct their spatial spread, a discrete phylogeographic analysis was performed on the HA gene of 806 representative HPAI A(H5Nx) viruses from six distinct geographical regions, namely East Asia, Africa, North America, Europe, the Middle East, and Central Asia (Russia/Kazakhstan/Georgia) (Fig. 6). According to the most probable location at the root of the MCC tree of the HA gene, the Middle East seems to be the most likely geographical source of this virus, which, between the second half of 2019 and the beginning of 2020, reached Central Asia (Supplementary Fig. S1), which agrees with earlier studies (Lewis et al. 2021). Our analysis indicates that Central Asia, which represents one of the most important breeding sites for multiple wild bird species, was a key source of the virus for most of the other geographic locations (52.42 per cent Markov jumps), including East Asia, the Middle East, and Europe (Fig. 7). Of note, the estimated tMRCAs (Supplementary Fig. S14) indicated that the viruses had been introduced from Central Asia to the different geographic areas over the same time period (summer–autumn) during both epidemic waves. Europe proved to be the major source of the virus for Africa and North America. In Africa, two different virus incursions from Europe of the A(H5N8) and A(H5N1) subtypes belonging to European genotypes EA-2020-C (H5N1) and EA-2020-A (H5N8) (Lo et al. 2022; Meseko et al. 2023) had occurred at the end of 2020. In North America, a first virus introduction of an A(H5N1) closely related to the viruses detected in Europe (genotype EA-2020-C, corresponding to genotype A in Youk et al. (2023) had been identified at the end of 2021 along the eastern coast, while a second transpacific virus incursion of A(H5N1) viruses closely related to the ones circulating in East Asia had occurred in early 2022 along the western coast, confirming the results from previous studies (Alkie et al. 2022). Markov rewards, which estimate the proportion of time the A(H5Nx) virus spent in each geographical area, revealed that at a global level the virus spent most of the time in East Asia (52.5 per cent) and Europe (48.7 per cent) and similar amounts of time in Central Asia (35.6 per cent), Africa (35.1 per cent), and North America (32.8 per cent) (Fig. 7), although bias due to the number of available sequences from each geographic area cannot be excluded.
Figure 6.
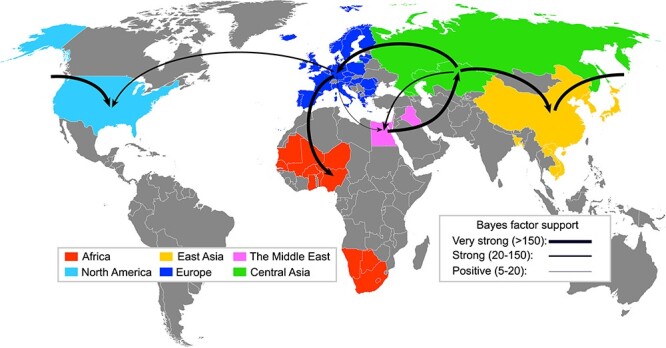
Global migration rates among the geographic regions of clade 2.3.4.4b (2019–2022). The thickness of the lines representing the rates is proportional to the relative strength by which rates are supported: very strong (BF > 150, thick lines) and positive (5 < BF < 20, thin lines).
Figure 7.
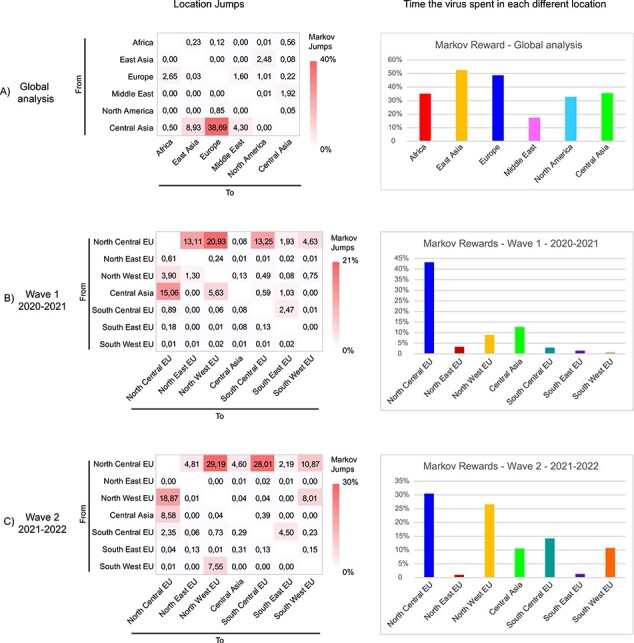
Pattern of geographic jumps and hotspots obtained from the analyses of (A) global dataset, (B) European dataset—first wave (2020–2021), and (C) European dataset—second wave (2021–2022). The three heatmaps on the left indicate the frequency of transitions between locations estimated using a discrete trait phylogenetic model. The number of location transitions was determined using Markov jumps. The bar charts on the right indicate the proportion of time the virus spent in each location.
European analysis—Clade 2.3.4.4b HPAI A(H5Nx) virus spread within Europe, 2020–2022
To explore the pattern of virus spread within Europe during the 2020–2021 and 2021–2022 epidemic waves, a discrete phylogeographic analysis of the HA gene was undertaken by dividing the dataset into seven geographic regions: North-Central Europe, North-Eastern Europe, North-Western Europe, South-Central Europe, South-Eastern Europe, South-Western Europe, and Central Asia. Analyses confirmed Central Asia as being the major source of the virus for the whole of Europe during both epidemic waves (Fig. 8, Supplementary Fig. S15). North-Central Europe likely acted as the main point of entry (Markov Jumps from Central Asia to North-Central Europe: 15.06 per cent—first wave, 8.58 per cent—second wave) (Fig. 7) and played a key role in the virus spread to the rest of Europe during both epidemic waves (first wave—53.85 per cent Markov jumps; second wave—75.07 per cent Markov jumps), with the largest number of Markov jumps (>10 per cent) estimated for viral flow from North-Central Europe to North-Western Europe (20.93 per cent and 29.19 per cent Markov Jumps for the first and the second epidemic waves, respectively), South-Central Europe (first wave—13.25 per cent Markov jumps; second wave—28.01 per cent Markov Jumps), North-Eastern Europe (first wave—13.11 per cent Markov jumps), and South-Western Europe (second wave—10.87 per cent Markov Jumps). North-Western and South-Western Europe as well as South-Central Europe and South-Eastern Europe were also connected in one or both waves (Fig. 7), indicating multiple directions of virus spread within Europe: among the northern regions, from northern to southern regions, and—to a lesser extent—also among the southern regions (Fig. 7). Estimation of the time the A(H5Nx) had remained in each region using Markov rewards demonstrated that the virus had spent most of the time in North-Central Europe (43.3 per cent) in 2020–2021, while in 2021–2022 a comparable amount of time across North-Central Europe (30.5 per cent) and North-Western Europe (26.6 per cent) (Fig. 7). A(H5Nx) spent the least amount of time in the South-Eastern and North-Eastern regions of Europe, although bias due to different intensity of genomic surveillance in each geographic region cannot be excluded.
Figure 8.
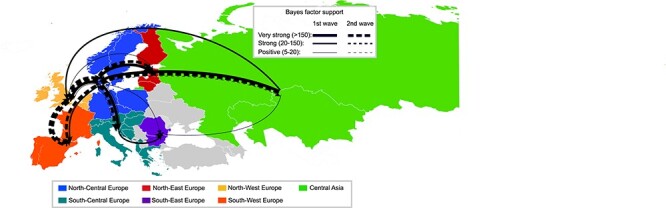
Migration rates between European regions of clade 2.3.4.4b during the first (continuous lines) and second (dashed lines) epidemic wave. The thickness of the lines representing the rates is proportional to the relative strength by which rates are supported: very strong (BF > 150, thick lines), strong (20 < BF < 150, medium lines), and positive (5 < BF < 20, thin lines).
Molecular markers affecting the biological characteristics of the European HPAI A(H5Nx) viruses
Molecular analyses of the European A(H5Nx) viruses investigated in this study (October 2020–August 2022) indicate that they retained the avian-type-receptor-binding signature (222Q and 224G (H5 numbering)) in the HA receptor binging site (Stevens et al. 2006). However, we identified several mutations with a varying frequency, which have been previously demonstrated to affect the biological characteristics of avian influenza viruses (Suttie et al. 2019) (Table 1). These include mutations associated with (1) enhanced polymerase activity and replication in mammals or mammalian cells, (2) increased virulence in mammals or avian species, (3) antiviral drug resistance, and (4) increased in vitro binding to human-type receptors alpha-2,6-linked sialic acids. The phenotypic effect of most of these mutations on the biological characteristics of the viruses is still unknown and further studies are needed to improve existing knowledge. Among the mutations in the HA protein that have proved to increase in vitro binding to human-type receptors, some (i.e. S133A, S154N, T156A) have been identified in the majority of the A(H5N1) viruses circulating in Europe in the period under study (October 2020–August 2022), while others (i.e. D94N, S155N, T188I, V210I, E251K, and S123P-496K) have only been sporadically detected. Moreover, we identified 84 viruses carrying mutations in the second sialic acid-binding site contact residues (2SBS) of the NA, which can affect the binding and cleavage of receptors and virus replication. Of note, all the viruses belonging to the BB genotype contained mutation S369I (N1 numbering) in the NA. A single mutation in this position of the N1 protein has been demonstrated to disrupt 2SBS and negatively affect N1 activity (Du et al. 2018). This mutation has been observed in all N1 sequences from human (seasonal) and H1N1pdm09 pandemic viruses and in several other viruses adapted to mammalian host species. Similarly, mutation K432E in the NA, which is also proposed to have an impact on 2SBS, has been detected only among viruses of the EA-2021-AB genotype (12 per cent of the EA-2021-AB viruses).
Table 1.
Mutations with a described biological effect identified in the European HPAI A(H5Nx) viruses, 2020–2022.
| Effect | Protein | Mutation | N. viruses | N. viruses from mammals | References |
|---|---|---|---|---|---|
| Enhance polymerase activity and replication in mammals or mammalian cells | PB2 | D253N | 1 | – | (Zhang et al. 2018) |
| T271A | 1 | 1 | (Bussey et al. 2010) | ||
| I292V | 798 | – | (Gao et al. 2019) | ||
| K389R | All/almost all viruses (N = 1,637) | (Hu et al. 2017b) | |||
| K482R | 19 | 3 | (Yamayoshi et al. 2018, Yamayoshi et al. 2014) | ||
| K526R | 4 | – | (Song et al. 2014) | ||
| A588V | 4 | – | (Xiao et al. 2016) | ||
| V598T/I | All/almost all viruses (N = 1,945) | (Hu et al. 2017b) | |||
| E627K | 32 | 18 | (Bortz et al. 2011; Fornek et al. 2009; Hatta et al. 2001, Hatta et al. 2007; Herfst et al. 2012; Kim et al. 2010; Manzoor et al. 2009; Richard et al. 2017; Shinya et al. 2004) | ||
| D701N | 6 | 4 | (Gao et al. 2009; Le et al. 2009; Li et al. 2005; Steel et al. 2009; Taft et al. 2015) | ||
| PA | V63I | 16 | 1 | (Hu et al. 2017a, Hu et al. 2016) | |
| T97I | 2 | – | (Taft et al. 2015) | ||
| K158R | 7 | – | (Elgendy et al. 2017) | ||
| K356R | 3 | – | (Xu et al. 2016) | ||
| K497R | 118 | – | (Yamayoshi et al. 2018) | ||
| NP | I41V | 29 | 3 | (Zhu et al. 2015) | |
| N319K | 36 | 5 | (Gabriel, Herwig, and Klenk 2008, Gabriel et al. 2005) | ||
| NS1 | I106M | All/almost all viruses (N = 1,954) | (Ayllon et al. 2014) | ||
| C138F | All/almost all viruses (N = 1,943) | (Li et al. 2018) | |||
| In vitro increase binding to human-type receptors alpha2,6-SA | HAa | D94N (D101N) | 6 | – | (Su et al. 2008) |
| S133A (S137A) | All/almost all viruses (N = 1,952) | (Yang et al. 2007) | |||
| S154N (S158N) | All/almost all viruses (N = 1,931) | (Wang et al. 2010) | |||
| S155N (S159N) | 5 | – | (Wang et al. 2010) | ||
| T156A (T160A) | All/almost all viruses (N = 1,939) | (Gao et al. 2009; Wang et al. 2010) | |||
| T188I (T192I) | 1 | – | (Yang et al. 2007) | ||
| V210I (V214I) | 3 | – | (Watanabe et al. 2011) | ||
| E251K (E255K) | 7 | – | (Chen et al. 2012) | ||
| S123P-496K (S128P-R496K (HA2-R167K)) | 4 | – | (Yamada et al. 2006) | ||
| Increase virulence in mice | PB2 | D9N | 2 | – | (Kim et al. 2010) |
| NS1 | P42S | All/almost all viruses (N = 1,922) | (Jiao et al. 2008) | ||
| L103F, I106M | All/almost all viruses (N = 1,919) | (Kuo and Krug 2009; Spesock et al. 2011) | |||
| 227ESEV230 (PDZ domain) | 1,115 | – | (Jackson et al. 2008; Soubies et al. 2010) | ||
| Enhance replication in mammalian cells, decrease IF response | NS1 | K55E, K66E, C138F | 1,087 | – | (Li et al. 2018) |
| Resistance toward antiviral drugs: $ Oseltamivir and/or Zanamivir § Laninamivir # Baloxavir ^ Amantidine-Rimantidine |
NAb | I117T$ | 1 | – | (Kode et al. 2019) |
| I117V$ | 2 | – | (Adams et al. 2019) | ||
| R152K§ | 1 | – | (Gubareva et al. 2017) | ||
| I223T$ | 1 | – | (Samson et al. 2014) | ||
| S247R$ | 1 | – | (Gubareva et al. 2017) | ||
| S247N$ | 5 | – | (Hurt et al. 2011) | ||
| PA | 34R# | 1 | – | (Govorkova et al. 2022) | |
| 199G# | 5 | – | (Govorkova et al. 2022; Kiso et al. 2020) | ||
| M2 | V27A^ | 4 | – | (Abed, Goyette, and Boivin 2005; Cheung et al. 2006; Ilyushina, Govorkova, and Webster 2005; Lan et al. 2010) | |
| V27I^ | 1 | – | (Chizhmakov et al. 2003) | ||
| A30S^ | 1 | – | (Cheung et al. 2006; Ilyushina, Govorkova, and Webster 2005) | ||
| S31N^ | 7 | – | (Buranathai et al. 2007; Cheung et al. 2006; He et al. 2008; Ilyushina, Govorkova, and Webster 2005; Lan et al. 2010; Puthavathana et al. 2005) | ||
| Decrease antiviral response in ferrets | NS1 | N205S (with NS2: T47A) | 1,112 | – | (Imai et al. 2010) |
| Disruption of the second sialic acid binding site (2SBS) | NAb | S369I | 44 | – | (Du et al. 2021, Du et al. 2018) |
| S369C/N/R | 3 | – | (Du et al. 2021, Du et al. 2018) | ||
| K432E/N | 38 | – | (Du et al. 2021, Du et al. 2018) | ||
| Evasion human BTN3A3 | NP | Y52N | 43 | 1 | (Pinto et al. 2023, Pinto et al. 2022) |
| Adaptation to domestic birds/increased virulence | NA | 22-AA deletion in the NA stalk region | 7 | – | (Hoffmann et al. 2012; Munier et al. 2010; Sorrell et al. 2010; Zhou et al. 2009) |
H5 numbering (H3 numbering).
N1 numbering.
Molecular markers associated with increased replication and/or virulence in mammals were rarely detected in birds. Specifically, the well-known molecular markers of mammalian adaptation E627K and D701N (Subbarao, London, and Murphy 1993; Hatta et al. 2001, 2007; Shinya et al. 2004; Salomon et al. 2006; Labadie et al. 2007; Mehle and Doudna 2008; Fornek et al. 2009; Li et al. 2009; Rameix-Welti et al. 2009; Steel et al. 2009) were observed in sixteen A(H5Nx) viruses collected from birds (N = 14 E627K, N = 2 PB2-D701N, all domestic/captive) in four European countries, which represented 0.8 per cent of the analysed sequences from avian species. Despite these markers being only sporadically detected in birds, it is worth noting that eleven out of the seventeen of the sequences showing the PB2-627K were detected in viruses collected from a cluster of outbreaks in poultry in Germany, as previously described (King et al. 2022). This suggests that viruses with increased zoonotic potential may retain the capability to infect and be transmitted among poultry farms The same markers (E627K and D701N) as well as mutation PB2-T271A were detected in about half of the viruses characterized from mammalian species (21/45), with the majority of them (17/21) bearing mutation PB2-627K (Fig. 3B).
Mutations associated with antiviral resistance were identified occasionally. Specifically, thirteen viruses contained mutations associated with reduced sensitivity to amantadine and rimantadine, eleven viruses showed mutations causing reduced susceptibility to neuraminidase inhibitors (oseltamivir/zanamivir), and six viruses displayed mutations able to reduce the virus sensitivity to baloxavir (Table 1).
Moreover, almost all the A(H5N1) viruses belonging to the A(H5N1) genotypes EA-2022-BB and EA-2020-C contained the mutation NP-Y52N/H, which allows the evasion of human butyrophilin subfamily 3 member A3 (BTN3A3) protein. BTN3A3 is a major histocompatibility complex associated protein constitutively expressed in human airways and is a potent inhibitor of avian but not human influenza A viruses (Pinto et al. 2022).
Discussion
Since October 2020, Europe has witnessed an important change in the ecology, epidemiology, and evolution of the HPAI A(H5Nx) viruses of clade 2.3.4.4b, exemplified by the following: (1) the geographic expansion of this virus that for the first time reached countries (i.e. Norway, Iceland, and Svalbard Islands) (Madslien et al. 2021) that had never before been affected by the Gs/GD lineage; (2) the variety of affected wild bird species (Adlhoch et al. 2023); (3) the high number of mammalian infections with sporadic episodes where field data suggested a potential mammal-to-mammal transmission, as reported in Spain (Agüero et al. 2023), but also in other countries outside Europe (Leguia et al. 2023); (4) the unprecedented number of reassortment events; (5) the virus persistence throughout summer 2022, with a severe impact on birds breeding in colonies in northern European countries (Adlhoch et al. 2022); and (6) the first reports of clade 2.3.4.4b A(H5Nx) in humans in countries (i.e. Russia, the UK, the USA, Chile, and Ecuador) where the HPAI the Gs/GD A(H5Nx) virus is not endemically circulating in poultry (Bruno et al. 2023; Human Infection caused by Avian Influenza A (H5N1) - Chile, n.d.; Threat Assessment Brief: First identification of human cases of avian influenza A(H5N8) infection, n.d.; U.S. Case of Human Avian Influenza A(H5) Virus Reported| CDC Online Newsroom| CDC, n.d.; Oliver et al. 2022).
In this study, we explored the changing pattern of the HPAI virus evolution between 2020 and 2022 in Europe, one of the most affected continents in terms of geographical spread, number of poultry outbreaks, and number of affected wild bird species. The complete data from the 2022–2023 wave were not available at the time of our analyses; therefore, they have not been included in the present paper. In agreement with previous studies (Lewis et al. 2021; Hagag et al. 2022; Xie et al. 2023), our spatial analyses identified the Middle East as the place where most likely the A(H5N8) virus responsible for the 2020–2021 epidemic wave in Europe originated. In Egypt, the A(H5N8) virus has been endemically circulating in poultry since 2016 (Hagag et al. 2022; Salaheldin et al. 2022). In spring 2020, viruses closely related to those collected in poultry in the Middle East were first identified in wild birds in Central Asia and then in Europe, which suggests that the virus may have jumped from the domestic to the wild bird population, although unsampled ancestry makes definitive assessment challenging. However, where this jump occurred—in the Middle East, Russia, or elsewhere—cannot be ascertained based on the available data. A similar scenario was observed in 2019, when one A(H5N8) reassortant virus of clade 2.3.4.4b, containing six gene segments of sub-Saharan African origin and two genes from LPAI viruses identified in wild birds in Western Siberia, was detected in eastern Europe (Swieton et al. 2020). Since the first transcontinental spread of the Gs/GD in 2005–2006, Eastern Asia has been recognized as the primary source location of A(H5Nx) clades for Europe (Sonnberg, Webby, and Webster 2013; Lycett et al. 2016; Verhagen, Fouchier, and Lewis 2021; Zhang et al. 2023). These two recent incursions in Europe of viruses originating from areas south of the Mediterranean Sea highlight a possible shift in the diffusion dynamics of the Gs/GD lineage and the emergence of new pathways for virus introductions in Europe. Despite data indicating that new hotspots (i.e. the Middle East and North-Central Europe) for virus evolution and subsequent spread may exist, this study confirms the main route of virus introduction into Europe through wild bird migrations from breeding sites in Central Asia, as previously reported (Lee et al. 2017; More et al. 2017; Fusaro et al. 2019; Lycett et al. 2020).
At the European level, North-Central Europe emerged as the major source of A(H5Nx) viruses for the rest of Europe and the area where the virus was detected for the longest period of time. The coastal area of North-Central Europe (i.e. the Wadden Sea) is of particular importance for migratory birds, which use this region for wintering or as a stop-over site on their annual migrations between southern wintering and northern breeding area, with about 10–12 million waterbirds spending at least part of their annual life cycle on these coasts (Scheiffarth 2003). A caveat of this study is that phylodynamic analyses rely on the availability of sequences; big efforts were made to increase wild bird surveillance and to generate sequencing data. The structure, design, and implementation of surveillance for wild birds across Europe are highly variable (i.e. target species, numbers examined, and number of geolocations within country). Furthermore, additional bias cannot be excluded given the different resources and sequencing capabilities of the different countries.
Clade 2.3.4.4b viruses have a high propensity to undergo multiple reassortment events, as observed during the European 2016–2017 epidemic wave (Verhagen, Fouchier, and Lewis 2021). However, compared to the previously described epidemic waves in Europe (Bragstad et al. 2007; Lycett et al. 2020), we detected an unprecedented genetic diversity, with the identification of nineteen and thirty-four distinct genotypes during the 2020–2021 and 2021–2022 epidemic waves, respectively. The greater genetic diversity in the second wave may be explained by the unprecedented number of cases and geographical extent (thirty-seven European affected countries) observed in 2021–2022, which represents the most devastating HPAI A(H5N1) epidemic affecting Europe to date. The highest number of reassortment events were observed for the gene segments encoding for the polymerase complex (PB1, PB1, and PA) and the nucleoprotein gene, while the HA and M genes—during the first wave—and the HA, NA, and M genes—during the second wave—were very stable. Previous studies demonstrated the crucial role of the M gene in virus morphology, transmission, and HA/NA balance (Bourmakina and García-Sastre 2003; Elleman and Barclay 2004; Campbell et al. 2014). It has recently been suggested that the M gene of clade 2.3.4.4b is associated with a more filamentous morphology, which may provide a selective advantage in vivo (Leyson et al. 2021). An M-HA effective cooperation has been demonstrated to be necessary for efficient virus replication (Webster, Kawaoka, and Bean 1989; Scholtissek et al. 2002; Leyson et al. 2021). Moreover, Campbell et al. (2014) showed that co-adapted M, NA, and HA segments are required to provide optimal transmissibility to the A(H1N1) 2009 pandemic virus and revealed the significant impact of the M gene on the increase of NA activity and contact transmission in the 2009 A(H1N1) pandemic virus (Campbell et al. 2014).
Of note, reassortments of the NA gene, which were frequently detected during the first wave and resulted in the co-circulation of five different NA subtypes clustering into seven distinct genetic groups, were extremely rare during the second wave, in which the A(H5N1) subtype was the most prevalent one (97 per cent of the sequences). The A(H5N1) subtype (genotype EA-2020-C) emerged in autumn 2020, most likely within Europe, through reassortment events, preserving only the HA and M segment from the A(H5N8) subtype. This novel A(H5N1) subtype was sporadically detected in Europe during the A(H5N8)-dominated 2020–2021 wave. However, it spread globally and, in the subsequent epidemic wave, it became clearly dominant in Europe, which suggests that it may have had a higher fitness advantage than the other subtypes.
Among the characterized genotypes, EA-2022-BB A(H5N1)—which had emerged in spring 2022 from reassortment events with H13 LPAI viruses, a subtype circulating primarily in gulls, with unique species-specific gene signatures emerging for prolonged genetic isolation and co-evolution with this species (Olsen et al. 2006)—is of particular concern as it is associated with several mass mortality events in Laridae. In the summer 2022, this genotype had been detected mainly in European herring gulls in Northern Europe (France, Belgium, the Netherlands, and occasionally in the UK). However, starting from autumn 2022, a southward spread of this genotype to Spain and then Italy, Switzerland, and Austria, followed by an eastward and northward spread to Poland, the Czech Republic, Croatia, Sweden, Norway, and Denmark, was reported (Adlhoch et al., 2023a, 2023b). The ongoing and widespread circulation of this genotype in the gull population, with black-headed gulls representing the most affected species since winter 2022–2023, highlights the serious threat posed by this variant to the sea-bird population, with mass mortality events persistently reported in several colonies of gulls and terns from different countries (Adlhoch et al., 2023a). Of note, the association of this genotype with the infection in Laridae may indicate it is well adapted to the members of this family. This genotype possesses the PA, NP, and NS gene segments of A(H13). Gull-specific clades have previously been demonstrated for the PB2, NP, and NS gene segments, regardless of the geographical region. This suggests an intra-host evolution and adaptation of these virus genes that may favour virus replication in and/or transmission between gulls (Fouchier et al. 2005; Wille et al. 2011) and related host taxa, as well as species in shared habitats with gulls. Moreover, the NP and PA genes have been shown to share a common evolutionary history in different host species, which suggests they tend not to reassort independently (Webster et al. 1992). Viruses of the A(H13) subtype do not readily infect ducks when they are inoculated experimentally (Fouchier et al. 2005), which may explain why in the viruses under study the EA-2022-BB genotype was not identified in Anseriformes and was detected only once in Galliformes (domestic chicken) during the 2021–2022 epidemic wave.
Of note, this genotype possesses mutations in the NP and NA proteins that can contribute to increase its zoonotic potential (Du et al. 2018; Pinto et al. 2023). The EA-2022-BB genotype has also been associated with infections in mammals, as in the outbreak reported in a mink farm in Spain in October 2022—where mink-to-mink transmission was suggested (Agüero et al. 2023)—in the outbreaks in fur farms in Finland in July 2023 (Lindh et al. 2023), as well as is some asymptomatic human detections in the UK (‘Investigation into the risk to human health of avian influenza (influenza A H5N1) in England: technical briefing 4 - GOV.UK’, n.d.). Whether the zoonotic potential of this genotype is higher than that of the other genotypes is unknown. Based on the available data, the majority of the infections in mammalian species have been caused by genotypes circulating with the highest frequency in birds at the time of the infection, suggesting direct and separate spillover events from birds to scavenging mammals in particular. Of note, our data indicate that mutations in the polymerase proteins associated with adaptation to mammalian species can be rapidly acquired by the virus during replication in a mammalian host, which seems to indicate that mammals may represent an important source of viruses with an increased zoonotic potential. However, recent serological evidence of A(H5Nx) infections in wild and domestic mammals, including pets reared or living in farms where poultry outbreaks were reported, clearly suggests that we have likely uncovered only the tip of the iceberg (Chestakova et al. 2023; Moreno et al. 2023; Rosone et al. 2023) and further studies are needed to increase our knowledge on the extent of virus circulation in mammals. Furthermore, this study, jointly with the recent infection cases reported in cats in Poland (Domańska-Blicharz et al. 2023) and in fur farms in Finland (Lindh et al. 2023), highlights the compelling need to adapt surveillance and to include mammals among the species posing a considerable risk for zoonotic spread of HPAI. This would provide health authorities with adequate information for a proper management of outbreaks in species belonging to this class.
For the first time, this study presents a complete overview of the multiple genotypes that had been circulating in Europe from October 2020 to August 2022, providing information on their geographic and host distribution. However, such data should always be interpreted taking into account the sampling bias within wild bird surveillance towards sick or dead birds, rather than resilient or asymptomatic birds. We assigned each genotype a name to simplify and encourage discussion between European laboratories. Although this work focuses on the 2020–2021 and 2021–2022 epidemic waves, it is worth mentioning that genotypes EA-2021-AB and EA-2022-BB (Adlhoch et al., 2023b)—that emerged in September 2021 and May 2022, respectively—were the main genotypes that had been circulating in Europe throughout the latest 2022/2023 epidemic.
We have also provided evidence of the emergence of a new genotype with gull adapted genes (EA-2022-BB), which offered the virus the opportunity to occupy new ecological niches and to persist during the summer months, causing massive mortality events in Laridae and multiple spill over events in farmed carnivores (Agüero et al. 2023; Lindh et al. 2023). This changing ecology, accompanied by viruses endemically circulating in wild birds in Europe in a progressively wider number of hosts, may change both the hot spots and the target species for virus surveillance, making trends of virus evolution and spread difficult to predict.
Supplementary Material
Acknowledgements
The authors wish to thank Francesca Ellero, Joe James, and Caroline Bröjer for their careful revision of the manuscript. We gratefully acknowledge also the authors, originating and submitting laboratories of the sequences from GISAID’s EpiFlu™ Database on which this research is based in part (Supplementary Table S4).
The author Francesca Baldinelli is employed with the European Food Safety Authority (EFSA) in the Unit BIOHAW that provides scientific and administrative support to the Panel on Animal Health and Welfare in the area of animal health. However, the present article is published under the sole responsibility of the author Francesca Baldinelli and may not be considered as an EFSA scientific output. The positions and opinions presented in this article are those of the authors alone and do not necessarily represent the views/any official position or scientific works of EFSA. To know about the views or scientific outputs of EFSA, please consult its website under http://www.efsa.europa.eu.
Contributor Information
Alice Fusaro, European Reference Laboratory (EURL) for Avian Influenza and Newcastle Disease, Istituto Zooprofilattico Sperimentale delle Venezie, viale dell'universita 10, Legnaro, Padua 35020, Italy.
Bianca Zecchin, European Reference Laboratory (EURL) for Avian Influenza and Newcastle Disease, Istituto Zooprofilattico Sperimentale delle Venezie, viale dell'universita 10, Legnaro, Padua 35020, Italy.
Edoardo Giussani, European Reference Laboratory (EURL) for Avian Influenza and Newcastle Disease, Istituto Zooprofilattico Sperimentale delle Venezie, viale dell'universita 10, Legnaro, Padua 35020, Italy.
Elisa Palumbo, European Reference Laboratory (EURL) for Avian Influenza and Newcastle Disease, Istituto Zooprofilattico Sperimentale delle Venezie, viale dell'universita 10, Legnaro, Padua 35020, Italy.
Montserrat Agüero-García, Ministry of Agriculture, Fisheries and Food, Laboratorio Central de Veterinaria (LCV), Ctra. M-106, Km 1,4 Algete, Madrid 28110, Spain.
Claudia Bachofen, Federal Department of Home Affairs FDHA Institute of Virology and Immunology IVI, Sensemattstrasse 293, Mittelhäusern 3147, Switzerland.
Ádám Bálint, Veterinary Diagnostic Directorate (NEBIH), Laboratory of Virology, National Food Chain Safety Office, Tábornok utca 2, Budapest 1143, Hungary.
Fereshteh Banihashem, Department of Microbiology, National Veterinary Institute (SVA), Travvägen 20, Uppsala 75189, Sweden.
Ashley C Banyard, WOAH/FAO international reference laboratory for Avian Influenza and Newcastle Disease, Virology Department, Animal and Plant Health Agency-Weybridge, Woodham Lane, New Haw, Addlestone KT15 3NB, United Kingdom.
Nancy Beerens, Department of Virology Wageningen Bioveterinary Research, Houtribweg 39, Lelystad 8221 RA, The Netherlands.
Manon Bourg, Luxembourgish Veterinary and Food Administration (ALVA), State Veterinary Laboratory, 1 Rue Louis Rech, Dudelange 3555, Luxembourg.
Francois-Xavier Briand, Agence Nationale de Sécurité Sanitaire, de l’Alimentation, de l’Environnement et du Travail, Laboratoire de Ploufragan-Plouzané-Niort, Unité de Virologie, Immunologie, Parasitologie Avaires et Cunicoles, 41 Rue de Beaucemaine – BP 53, Ploufragan 22440, France.
Caroline Bröjer, Department of Pathology and Wildlife Disease, National Veterinary Institute (SVA), Travvägen 20, Uppsala 75189, Sweden.
Ian H Brown, WOAH/FAO international reference laboratory for Avian Influenza and Newcastle Disease, Virology Department, Animal and Plant Health Agency-Weybridge, Woodham Lane, New Haw, Addlestone KT15 3NB, United Kingdom.
Brigitte Brugger, Icelandic Food and Veterinary Authority, Austurvegur 64, Selfoss 800, Iceland.
Alexander M P Byrne, WOAH/FAO international reference laboratory for Avian Influenza and Newcastle Disease, Virology Department, Animal and Plant Health Agency-Weybridge, Woodham Lane, New Haw, Addlestone KT15 3NB, United Kingdom.
Armend Cana, Kosovo Food and Veterinary Agency, Sector of Serology and Molecular Diagnostics, Kosovo Food and Veterinary Laboratory, Str Lidhja e Pejes, Prishtina 10000, Kosovo.
Vasiliki Christodoulou, Laboratory for Animal Health Virology Section Veterinary Services (1417), 79, Athalassa Avenue Aglantzia, Nicosia 2109, Cyprus.
Zuzana Dirbakova, Department of Animal Health, State Veterinary Institute, Pod Dráhami 918, Zvolen 96086, Slovakia.
Teresa Fagulha, I.P. (INIAV, I.P.), Avenida da República, Instituto Nacional de Investigação Agrária e Veterinária, Quinta do Marquês, Oeiras 2780 – 157, Portugal.
Ron A M Fouchier, Department of Viroscience, Erasmus MC, Dr. Molewaterplein 40, Rotterdam 3015 GD, The Netherlands.
Laura Garza-Cuartero, Department of Agriculture, Food and the Marine, Central Veterinary Research Laboratory (CVRL), Backweston Campus, Stacumny Lane, Celbridge, Co. Kildare W23 X3PH, Ireland.
George Georgiades, Thessaloniki Veterinary Centre (TVC), Department of Avian Diseases, 26th October Street 80, Thessaloniki 54627, Greece.
Britt Gjerset, Immunology & Virology department, Norwegian Veterinary Institute, Arboretveien 57, Oslo Pb 64, N-1431 Ås, Norway.
Beatrice Grasland, Agence Nationale de Sécurité Sanitaire, de l’Alimentation, de l’Environnement et du Travail, Laboratoire de Ploufragan-Plouzané-Niort, Unité de Virologie, Immunologie, Parasitologie Avaires et Cunicoles, 41 Rue de Beaucemaine – BP 53, Ploufragan 22440, France.
Oxana Groza, Republican Center for Veterinary Diagnostics (NRL), 3 street Murelor, Chisinau 2051, Republic of Moldova.
Timm Harder, Institute of Diagnostic Virology, Friedrich-Loeffler-Institut, Südufer 10, Greifswald-Insel Riems 17493, Germany.
Ana Margarida Henriques, I.P. (INIAV, I.P.), Avenida da República, Instituto Nacional de Investigação Agrária e Veterinária, Quinta do Marquês, Oeiras 2780 – 157, Portugal.
Charlotte Kristiane Hjulsager, Department for Virus and Microbiological Special Diagnostics, Statens Serum Institut, 5 Artillerivej, Copenhagen DK-2300, Denmark.
Emiliya Ivanova, National Reference Laboratory for Avian Influenza and Newcastle Disease, National Diagnostic and Research Veterinary Medical Institute (NDRVMI), 190 Lomsko Shose Blvd., Sofia 1231, Bulgaria.
Zygimantas Janeliunas, National Food and Veterinary Risk Assessment Institute (NFVRAI), Kairiukscio str. 10, Vilnius 08409, Lithuania.
Laura Krivko, Institute of Food Safety, Animal Health and Environment (BIOR), Laboratory of Microbilogy and Pathology, 3 Lejupes Street, Riga 1076, Latvia.
Ken Lemon, Virological Molecular Diagnostic Laboratory, Veterinary Sciences Division, Department of Virology, Agri-Food and Bioscience Institute (AFBI), Stoney Road, Belfast BT4 3SD, Northern Ireland.
Yuan Liang, Department of Veterinary and Animal Sciences, University of Copenhagen, Grønnegårdsvej 15, Frederiksberg 1870, Denmark.
Aldin Lika, Animal Health Department, Food Safety and Veterinary Institute, Rruga Aleksandër Moisiu 10, Tirana 1001, Albania.
Péter Malik, Veterinary Diagnostic Directorate (NEBIH), Laboratory of Virology, National Food Chain Safety Office, Tábornok utca 2, Budapest 1143, Hungary.
Michael J McMenamy, Virological Molecular Diagnostic Laboratory, Veterinary Sciences Division, Department of Virology, Agri-Food and Bioscience Institute (AFBI), Stoney Road, Belfast BT4 3SD, Northern Ireland.
Alexander Nagy, Department of Molecular Biology, State Veterinary Institute Prague, Sídlištní 136/24, Praha 6-Lysolaje 16503, Czech Republic.
Imbi Nurmoja, National Centre for Laboratory Research and Risk Assessment (LABRIS), Kreutzwaldi 30, Tartu 51006, Estonia.
Iuliana Onita, Institute for Diagnosis and Animal Health (IDAH), Str. Dr. Staicovici 63, Bucharest 050557, Romania.
Anne Pohlmann, Institute of Diagnostic Virology, Friedrich-Loeffler-Institut, Südufer 10, Greifswald-Insel Riems 17493, Germany.
Sandra Revilla-Fernández, Austrian Agency for Health and Food Safety (AGES), Institute for Veterinary Disease Control, Robert Koch Gasse 17, Mödling 2340, Austria.
Azucena Sánchez-Sánchez, Ministry of Agriculture, Fisheries and Food, Laboratorio Central de Veterinaria (LCV), Ctra. M-106, Km 1,4 Algete, Madrid 28110, Spain.
Vladimir Savic, Croatian Veterinary Institute, Poultry Centre, Heinzelova 55, Zagreb 10000, Croatia.
Brigita Slavec, University of Ljubljana – Veterinary Faculty/National Veterinary Institute, Gerbičeva 60, Ljubljana 1000, Slovenia.
Krzysztof Smietanka, Department of Poultry Diseases, National Veterinary Research Institute, Al. Partyzantow 57, Puławy 24-100, Poland.
Chantal J Snoeck, Luxembourg Institute of Health (LIH), Department of Infection and Immunity, 29 Rue Henri Koch, Esch-sur-Alzette 4354, Luxembourg.
Mieke Steensels, Avian Virology and Immunology, Sciensano, Rue Groeselenberg 99, Ukkel 1180, Ukkel, Belgium.
Vilhjálmur Svansson, Biomedical Center, Institute for Experimental Pathology, University of Iceland, Keldnavegi 3 112 Reykjavík Ssn. 650269 4549, Keldur 851, Iceland.
Edyta Swieton, Department of Poultry Diseases, National Veterinary Research Institute, Al. Partyzantow 57, Puławy 24-100, Poland.
Niina Tammiranta, Finnish Food Authority, Animal Health Diagnostic Unit, Veterinary Virology, Mustialankatu 3, Helsinki FI-00790, Finland.
Martin Tinak, Department of Animal Health, State Veterinary Institute, Pod Dráhami 918, Zvolen 96086, Slovakia.
Steven Van Borm, Avian Virology and Immunology, Sciensano, Rue Groeselenberg 99, Ukkel 1180, Ukkel, Belgium.
Siamak Zohari, Department of Microbiology, National Veterinary Institute (SVA), Travvägen 20, Uppsala 75189, Sweden.
Cornelia Adlhoch, European Centre for Disease Prevention and Control, Gustav III:s boulevard 40, Solna 169 73, Sweden.
Francesca Baldinelli, European Food Safety Authority (EFSA), Via Carlo Magno 1A, Parma 43126, Italy.
Calogero Terregino, European Reference Laboratory (EURL) for Avian Influenza and Newcastle Disease, Istituto Zooprofilattico Sperimentale delle Venezie, viale dell'universita 10, Legnaro, Padua 35020, Italy.
Isabella Monne, European Reference Laboratory (EURL) for Avian Influenza and Newcastle Disease, Istituto Zooprofilattico Sperimentale delle Venezie, viale dell'universita 10, Legnaro, Padua 35020, Italy.
Supplementary data
Supplementary data is available at VEVOLU Journal online.
Funding
Support for this work was provided by the European Commission within the framework of the activities foreseen by the European Union Reference Laboratory for Avian Influenza and Newcastle Disease under grant agreement SI2.870510. This work was also partially supported by KAPPA-FLU HORIZON-CL6-2022-FARM2FORK-02-03 (grant agreement No 101084171) and FLU-SWITCH Era-Net ICRAD (grant agreement No 862605). ACB, AMPB, and IHB were supported by the Biotechnology and Biological Sciences Research Council (BBSRC) and Department for Environment, Food and Rural Affairs (Defra, UK) research initiative ‘FluMAP’ [grant number BB/X006204/1]. Funding was also provided by Defra and the devolved administrations of Scotland and Wales through SE2213, SV3032, and SV3400.
Conflict of interest:
None declared.
Data Availability
All sequences are published in the GISAID EpiFlu database and sequence accession numbers are available in supplementary material (Supplementary Table S1); xml, log, and trees files generated from the BEAST analyses are available on request.
References
- Abed Y., Goyette N., and Boivin G. (2005) ‘Generation and Characterization of Recombinant Influenza A (H1N1) Viruses Harboring Amantadine Resistance Mutations’, Antimicrobial Agents And Chemotherapy, 49: 556–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abolnik C. et al. (2022) ‘Wild Bird Surveillance in the Gauteng Province of South Africa during the High-Risk Period for Highly Pathogenic Avian Influenza Virus Introduction’, Viruses, 14: 2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams S. E. et al. (2019) ‘Effect of Influenza H1N1 Neuraminidase V116A and I117V Mutations on NA Activity and Sensitivity to NA Inhibitors’, Antiviral Research, 169: 104539. [DOI] [PubMed] [Google Scholar]
- Adlhoch C. et al. (2022) ‘Avian Influenza Overview June–September 2022’, EFSA Journal, 20: 7597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adlhoch C. et al. (2023a) ‘Avian Influenza Overview April–June 2023’, EFSA Journal. European Food Safety Authority, 21: e08191. 10.2903/J.EFSA.2023.8191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adlhoch C. et al. (2023b) ‘Avian Influenza Overview March–April 2023’, EFSA Journal. European Food Safety Authority, 21: e08039. 10.2903/j.efsa.2023.8039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agüero M. et al. (2023) ‘Highly Pathogenic Avian Influenza A(H5N1) Virus Infection in Farmed Minks, Spain, October 2022’, Eurosurveillance, 28: 2300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkie T. N. et al. (2022) ‘A Threat from Both Sides: Multiple Introductions of Genetically Distinct H5 HPAI Viruses into Canada via Both East Asia-Australasia/Pacific and Atlantic Flyways’, Virus Evolution, 8: veac077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayllon J. et al. (2014) ‘A Single Amino Acid Substitution in the Novel H7N9 Influenza A Virus NS1 Protein Increases CPSF30 Binding and Virulence’, Journal of Virology, 88: 12146–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielejec F. et al. (2016) ‘SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes’, Molecular Biology and Evolution, 33: 2167–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordes L. et al. (2023) ‘Highly Pathogenic Avian Influenza H5N1 Virus Infections in Wild Red Foxes (Vulpes Vulpes) Show Neurotropism and Adaptive Virus Mutations’, Microbiology Spectrum, 11: e02867–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bortz E. et al. (2011) ‘Host- and Strain-specific Regulation of Influenza Virus Polymerase Activity by Interacting Cellular Proteins’, MBio, 2: 10–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourmakina S. V., and García-Sastre A. (2003) ‘Reverse Genetics Studies on the Filamentous Morphology of Influenza A Virus’, Journal of General Virology, 84: 517–27. [DOI] [PubMed] [Google Scholar]
- Bragstad K. et al. (2007) ‘First Introduction of Highly Pathogenic H5N1 Avian Influenza A Viruses in Wild and Domestic Birds in Denmark, Northern Europe’, Virology Journal, 4: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briand F. X. et al. (2022) ‘Multiple Independent Introductions of Highly Pathogenic Avian Influenza H5 Viruses during the 2020-2021 Epizootic in France’, Transboundary and Emerging Diseases, 69: 4028–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno A. et al. (2023) ‘First Case of Human Infection with Highly Pathogenic H5 Avian Influenza a Virus in South America: A New Zoonotic Pandemic Threat for 2023?’, Journal of Travel Medicine, 30: taad032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buranathai C. et al. (2007) ‘Surveillance Activities and Molecular Analysis of H5N1 Highly Pathogenic Avian Influenza Viruses from Thailand, 2004–2005’, Avian Diseases, 51: 194–200. [DOI] [PubMed] [Google Scholar]
- Bussey K. A. et al. (2010) ‘PB2 Residue 271 Plays A Key Role in Enhanced Polymerase Activity of Influenza A Viruses in Mammalian Host Cells’, Journal of Virology, 84: 4395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caliendo V. et al. (2022) ‘Transatlantic Spread of Highly Pathogenic Avian Influenza H5N1 by Wild Birds from Europe to North America in 2021’, Scientific Reports, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell P. J. et al. (2014) ‘The M Segment of the 2009 Pandemic Influenza Virus Confers Increased Neuraminidase Activity, Filamentous Morphology, and Efficient Contact Transmissibility to A/Puerto Rico/8/1934-Based Reassortant Viruses’, Journal of Virology, 88: 3802–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. et al. (2006) ‘Properties and Dissemination of H5N1 Viruses Isolated during an Influenza Outbreak in Migratory Waterfowl in Western China’, Journal of Virology, 80: 5976–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. M. et al. (2012) ‘In Vitro Evolution of H5N1 Avian Influenza Virus toward Human-type Receptor Specificity’, Virology, 422: 105–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomor O. et al. (2014) ‘Split Diversity in Constrained Conservation Prioritization Using Integer Linear Programming’, Methods in Ecology and Evolution, 6: 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chestakova I. V. et al. (2023) ‘High Number of HPAI H5 Virus Infections and Antibodies in Wild Carnivores in the Netherlands, 2020-2022’, Emerg Microbes Infect. 12: 2270068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C. L. et al. (2006) ‘Distribution of Amantadine-resistant H5N1 Avian Influenza Variants in Asia’, Journal of Infectious Diseases, 193: 1626–9. [DOI] [PubMed] [Google Scholar]
- Chizhmakov I. V. et al. (2003) ‘Differences in Conductance of M2 Proton Channels of Two Influenza Viruses at Low and High pH’, The Journal of Physiology, 546: 427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domańska-Blicharz K. et al. (2023) ‘Outbreak of Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4b Virus in Cats, Poland, June to July 2023’, Eurosurveillance, 28: 2300366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J., and Rambaut A. (2007) ‘BEAST: Bayesian Evolutionary Analysis by Sampling Trees’, BMC Evolutionary Biology, 7: 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W. et al. (2018) ‘Substrate Binding by the Second Sialic Acid-Binding Site of Influenza A Virus N1 Neuraminidase Contributes to Enzymatic Activity’, Journal of Virology, 92: 10–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W. et al. (2021) ‘Second Sialic Acid-binding Site of Influenza A Virus Neuraminidase: Binding Receptors for Efficient Release’, The FEBS Journal, 288: 5598–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgendy E. M. et al. (2017) ‘Identification of Polymerase Gene Mutations that Affect Viral Replication in H5N1 Influenza Viruses Isolated from Pigeons’, Journal of General Virology, 98: 6–17. [DOI] [PubMed] [Google Scholar]
- Elleman C. J., and Barclay W. S. (2004) ‘The M1 Matrix Protein Controls the Filamentous Phenotype of Influenza A Virus’, Virology, 321: 144–53. [DOI] [PubMed] [Google Scholar]
- Engelsma M. et al. (2022) ‘Multiple Introductions of Reassorted Highly Pathogenic Avian Influenza H5Nx Viruses Clade 2.3.4.4b Causing Outbreaks in Wild Birds and Poultry in the Netherlands, 2020-2021’, Microbiology Spectrum, 10: e02499–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd T. et al. (2021) ‘Encephalitis and Death in Wild Mammals at a Rehabilitation Center after Infection with Highly Pathogenic Avian Influenza A(H5N8) Virus’, Emerging Infectious Diseases, 27: 2856–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Food Safety Authority, E., Adlhoch C. et al. (2023) ‘Avian Influenza Overview September – December 2022’, EFSA Journal, 21: e07786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornek J. L. et al. (2009) ‘A Single-amino-acid Substitution in A Polymerase Protein of an H5N1 Influenza Virus Is Associated with Systemic Infection and Impaired T-cell Activation in Mice’, Journal of Virology, 83: 11102–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouchier R. A. M. et al. (2005) ‘Characterization of A Novel Influenza A Virus Hemagglutinin Subtype (H16) Obtained from Black-headed Gulls’, Journal of Virology, 79: 2814–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusaro A. et al. (2019)Disentangling the Role of Africa in the Global Spread of H5 Highly Pathogenic Avian Influenza, Nature Communications, 10: 5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel G. et al. (2005) ‘The Viral Polymerase Mediates Adaptation of an Avian Influenza Virus to a Mammalian Host’, The Proceedings of the National Academy of Sciences of the United States of America, 102: 18590–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel G., Herwig A., and Klenk H. D. (2008) ‘Interaction of Polymerase Subunit PB2 and NP with Importin Alpha1 Is A Determinant of Host Range of Influenza A Virus’, PLoS Pathogens, 4: e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y. et al. (2009) ‘Identification of Amino Acids in HA and PB2 Critical for the Transmission of H5N1 Avian Influenza Viruses in a Mammalian Host’, PLoS Pathogens, 5: e1000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W. et al. (2019) ‘Prevailing I292V PB2 Mutation in Avian Influenza H9N2 Virus Increases Viral Polymerase Function and Attenuates IFN-β Induction in Human Cells’, Journal of General Virology, 100: 1273–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govorkova E. A. et al. (2022) ‘Global Update on the Susceptibilities of Human Influenza Viruses to Neuraminidase Inhibitors and the Cap-dependent Endonuclease Inhibitor Baloxavir, 2018-2020’, Antiviral Research, 200: 105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant M. et al. (2022) ‘Highly Pathogenic Avian Influenza (HPAI H5Nx, Clade 2.3.4.4.b) In Poultry and Wild Birds in Sweden: Synopsis of the 2020–2021 Season’, Veterinary Sciences, 9: 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubareva L. V. et al. (2017) ‘Drug Susceptibility Evaluation of an Influenza A(H7N9) Virus by Analyzing Recombinant Neuraminidase Proteins’, The Journal of Infectious Diseases, 216: S566–S574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagag N. M. et al. (2022) ‘Molecular Epidemiology and Evolutionary Analysis of Avian Influenza A(H5) Viruses Circulating in Egypt, 2019-2021’, Viruses, 14: 1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatta H. et al. (2001) ‘Molecular Basis for High Virulence of Hong Kong H5N1 Influenza A Viruses’, Science, 293: 1840–2. [DOI] [PubMed] [Google Scholar]
- Hatta M. et al. (2007) ‘Growth of H5N1 Influenza A Viruses in the Upper Respiratory Tracts of Mice’, PLoS Pathogens, 3: 1374–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He G. et al. (2008) ‘Amantadine-resistance among H5N1 Avian Influenza Viruses Isolated in Northern China’, Antiviral Research, 77: 72–6. [DOI] [PubMed] [Google Scholar]
- Herfst S. et al. (2012) ‘Airborne Transmission of Influenza A/H5N1 Virus between Ferrets’, Science, 336: 1534–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann T. W. et al. (2012) ‘Length Variations in the NA Stalk of an H7N1 Influenza Virus Have Opposite Effects on Viral Excretion in Chickens and Ducks’, Journal of Virology, 86: 584–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M. et al. (2016) ‘Amino Acid Substitutions V63I or A37S/I61T/V63I/V100A in the PA N-terminal Domain Increase the Virulence of H7N7 Influenza A Virus’, Scientific Reports, 6: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M. et al. (2017a) ‘PA Nsubstitutions A37S, A37S/I61T and A37S/V63I Attenuate the Replication of H7N7 Influenza A Virus by Impairing the Polymerase and Endonuclease Activities’, Journal of General Virology, 98: 364–73. [DOI] [PubMed] [Google Scholar]
- Hu M.SS et al. (2017b) ‘PB2 Substitutions V598T/I Increase the Virulence of H7N9 Influenza A Virus in Mammals’, Virology, 501: 92–101. [DOI] [PubMed] [Google Scholar]
- Human Infection Caused by Avian Influenza A (H5N1) - Chile [WWW Document], (n.d.) <https://www.who.int/emergencies/disease-outbreak-news/item/2023-DON461> Accessed 8 Aug 2023.
- Hurt A. C. et al. (2011) ‘Increased Detection in Australia and Singapore of a Novel Influenza a(H1N1)2009 Variant with Reduced Oseltamivir and Zanamivir Sensitivity Due to a S247N Neuraminidase Mutation’, Eurosurveillance, 16: 19884. [PubMed] [Google Scholar]
- Ilyushina N. A., Govorkova E. A., and Webster R. G. (2005) ‘Detection of Amantadine-resistant Variants among Avian Influenza Viruses Isolated in North America and Asia’, Virology, 341: 102–6. [DOI] [PubMed] [Google Scholar]
- Imai H. et al. (2010) ‘The HA and NS Genes of Human H5N1 Influenza A Virus Contribute to High Virulence in Ferrets’, PLoS Pathogens, 6: e1001106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Investigation into the Risk to Human Health of Avian Influenza (influenza A H5N1) in England: Technical Briefing 4 - GOV.UK [WWW Document], (n.d.) <https://www.gov.uk/government/publications/avian-influenza-influenza-a-h5n1-technical-briefings/investigation-into-the-risk-to-human-health-of-avian-influenza-influenza-a-h5n1-in-england-technical-briefing-4> Accessed 21 Nov 2023.
- Isoda N. et al. (2022) ‘Detection of New H5N1 High Pathogenicity Avian Influenza Viruses in Winter 2021-2022 in the Far East, Which are Genetically Close to Those in Europe’, Viruses, 14: 2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson D. et al. (2008) ‘A New Influenza Virus Virulence Determinant: The NS1 Protein Four C-terminal Residues Modulate Pathogenicity’, The Proceedings of the National Academy of Sciences of the United States of America, 105: 4381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao P. et al. (2008) ‘A Single-amino-acid Substitution in the NS1 Protein Changes the Pathogenicity of H5N1 Avian Influenza Viruses in Mice’, Journal of Virology, 82: 1146–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., and Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. H. et al. (2010) ‘Role of Host-specific Amino Acids in the Pathogenicity of Avian H5N1 Influenza Viruses in Mice’, Journal of General Virology, 91: 1284–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King J. et al. (2022) ‘Connect to Protect: Dynamics and Genetic Connections of Highly Pathogenic Avian Influenza Outbreaks in Poultry from 2016 to 2021 in Germany’, Viruses, 14: 1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiso M. et al. (2020) ‘Baloxavir Marboxil Treatment of Nude Mice Infected with Influenza A Virus’, The Journal of Infectious Diseases, 221: 1699–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kode S. S. et al. (2019) ‘A Novel I117T Substitution in Neuraminidase of Highly Pathogenic Avian Influenza H5N1 Virus Conferring Reduced Susceptibility to Oseltamivir and Zanamivir’, Veterinary Microbiology, 235: 21–4. [DOI] [PubMed] [Google Scholar]
- Kuo R.-L., and Krug R. M. (2009) ‘Influenza a Virus Polymerase Is an Integral Component of the CPSF30-NS1A Protein Complex in Infected Cells’, Journal of Virology, 83: 1611–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labadie K. et al. (2007) ‘Host-range Determinants on the PB2 Protein of Influenza A Viruses Control the Interaction between the Viral Polymerase and Nucleoprotein in Human Cells’, Virology, 362: 271–82. [DOI] [PubMed] [Google Scholar]
- Lan Y. et al. (2010) ‘A Comprehensive Surveillance of Adamantane Resistance among Human Influenza A Virus Isolated from Mainland China between 1956 and 2009’, Antiviral Therapy, 15: 853–9. [DOI] [PubMed] [Google Scholar]
- Le Q. M. et al. (2009) ‘Selection of H5N1 Influenza Virus PB2 during Replication in Humans’, Journal of Virology, 83: 5278–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D. H. et al. (2017) ‘Evolution, Global Spread, and Pathogenicity of Highly Pathogenic Avian Influenza H5Nx Clade 2.3.4.4’, Journal of Veterinary Science, 18: 269–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leguia M. et al. (2023) ‘Highly Pathogenic Avian Influenza A (H5N1) in Marine Mammals and Seabirds in Peru’, Nat Commun. 14: 5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P. et al. (2009) ‘Bayesian Phylogeography Finds Its Roots’, PLoS Computational Biology, 5: e1000520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letsholo S. L. et al. (2022) ‘Emergence of High Pathogenicity Avian Influenza Virus H5N1 Clade 2.3.4.4b In Wild Birds and Poultry in Botswana’, Viruses, 14: 2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis N. S. et al. (2021) ‘Emergence and Spread of Novel H5N8, H5N5 and H5N1 Clade 2.3.4.4 Highly Pathogenic Avian Influenza in 2020’, Emerging Microbes & Infections, 10: 148–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leyson C. M. et al. (2021) ‘Multiple Gene Segments are Associated with Enhanced Virulence of Clade 2.3.4.4 H5N8 Highly Pathogenic Avian Influenza Virus in Mallards’, Journal of Virology, 95: 10–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K. S. et al. (2004) ‘Genesis of a Highly Pathogenic and Potentially Pandemic H5N1 Influenza Virus in Eastern Asia’, Nature, 430: 209–13. [DOI] [PubMed] [Google Scholar]
- Li Z. et al. (2005) ‘Molecular Basis of Replication of Duck H5N1 Influenza Viruses in a Mammalian Mouse Model’, Journal of Virology, 79: 12058–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. et al. (2009) ‘Single Mutation at the Amino Acid Position 627 of PB2 that Leads to Increased Virulence of an H5N1 Avian Influenza Virus during Adaptation in Mice Can Be Compensated by Multiple Mutations at Other Sites of PB2’, Virus Research, 144: 123–9. [DOI] [PubMed] [Google Scholar]
- Li J. et al. (2018) ‘Three Amino Acid Substitutions in the NS1 Protein Change the Virus Replication of H5N1 Influenza Virus in Human Cells’, Virology, 519: 64–73. [DOI] [PubMed] [Google Scholar]
- Lindh E. et al. (2023) ‘Highly Pathogenic Avian Influenza A(H5N1) Virus Infection on Multiple Fur Farms in the South and Central Ostrobothnia Regions of Finland, July 2023’, Eurosurveillance, 28: 2300400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo F. T. et al. (2022) ‘Intercontinental Spread of Eurasian Highly Pathogenic Avian Influenza A(H5N1) to Senegal’, Emerging Infectious Diseases, 28: 234–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Global Consortium for H5N8 and Related Influenza, Lycett S. J. et al. (2016) ‘Role for Migratory Wild Birds in the Global Spread of Avian Influenza H5N8’, Science, 354: 213–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lycett S. J. et al. (2020) ‘Genesis and Spread of Multiple Reassortants during the 2016/2017 H5 Avian Influenza Epidemic in Eurasia’, The Proceedings of the National Academy of Sciences of the United States of America, 117: 20814–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madslien K. et al. (2021) ‘First Detection of Highly Pathogenic Avian Influenza Virus in Norway’, BMC Veterinary Research, 17: 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makalo M. R. J. et al. (2022) ‘Highly Pathogenic Avian Influenza (A/H5N1) Virus Outbreaks in Lesotho, May 2021’, Emerging Microbes & Infections, 11: 757–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoor R. et al. (2009) ‘PB2 Protein of a Highly Pathogenic Avian Influenza Virus Strain A/chicken/Yamaguchi/7/2004 (H5N1) Determines Its Replication Potential in Pigs’, Journal of Virology, 83: 1572–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehle A., and Doudna J. A. (2008) ‘Inhibitory Activity Restricts the Function of an Avian-like Influenza Polymerase in Primate Cells’, Cell Host & Microbe, 4: 111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meseko C. et al. (2023) ‘The Evolution of Highly Pathogenic Avian Influenza A (H5) in Poultry in Nigeria, 2021–2022’, Viruses, 15: 1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minin V. N., and Suchard M. A. (2008) ‘Counting Labeled Transitions in Continuous-time Markov Models of Evolution’, Journal of Mathematical Biology, 56: 391–412. [DOI] [PubMed] [Google Scholar]
- More S. et al. (2017) ‘Avian Influenza’, EFSA Journal. European Food Safety Authority, 15: e04991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno A. et al. (2023) ‘Asymptomatic Infection with Clade 2.3.4.4b Highly Pathogenic Avian Influenza A(H5N1) in Carnivore Pets, Italy, April 2023’, Eurosurveillance, 28: 2300441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munier S. et al. (2010) ‘A Genetically Engineered Waterfowl Influenza Virus with A Deletion in the Stalk of the Neuraminidase Has Increased Virulence for Chickens’, Journal of Virology, 84: 940–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A., Černíková L., and Stará M. (2022) ‘A New Clade 2.3.4.4b H5N1 Highly Pathogenic Avian Influenza Genotype Detected in Europe in 2021’, Archives of Virology, 167: 1455–9. [DOI] [PubMed] [Google Scholar]
- Nguyen L.-T. et al. (2015) ‘IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-likelihood Phylogenies’, Molecular Biology and Evolution, 32: 268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- European Food Safety Authority, European Centre for Disease Prevention and Control, European Union Reference Laboratory for Avian Influenza, Adlhoch C. et al. (2023) ‘Avian Influenza Overview December 2022–March 2023’, EFSA Journal. European Food Safety Authority, 21: e07917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver I. et al. (2022) ‘A Case of Avian Influenza A(H5N1) in England, January 2022’, Eurosurveillance, 27: 2200061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen B. et al. (2006) ‘Global Patterns of Influenza a Virus in Wild Birds’, Science, 312: 384–8. [DOI] [PubMed] [Google Scholar]
- Ouoba L. B. et al. (2022) ‘Emergence of a Reassortant 2.3.4.4b Highly Pathogenic H5N1 Avian Influenza Virus Containing H9N2 PA Gene in Burkina Faso, West Africa, in 2021’, Viruses, 14: 1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto R. M.s et al. (2022) ‘Zoonotic Avian Influenza Viruses Evade Human BTN3A3 Restriction’, Nature, 619: 338–47. [DOI] [PubMed] [Google Scholar]
- Pinto R. M. et al. (2023) ‘BTN3A3 Evasion Promotes the Zoonotic Potential of Influenza A Viruses’, Nature, 619: 338–47. [DOI] [PubMed] [Google Scholar]
- Pohlmann A. et al. (2022) ‘Has Epizootic Become Enzootic? Evidence for a Fundamental Change in the Infection Dynamics of Highly Pathogenic Avian Influenza in Europe, 2021’, MBio, 13: e00609–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puthavathana P. et al. (2005) ‘Molecular Characterization of the Complete Genome of Human Influenza H5N1 Virus Isolates from Thailand’, Journal of General Virology, 86: 423–33. [DOI] [PubMed] [Google Scholar]
- Rameix-Welti M.-A. et al. (2009) ‘Avian Influenza A Virus Polymerase Association with Nucleoprotein, but Not Polymerase Assembly, Is Impaired in Human Cells during the Course of Infection’, Journal of Virology, 83: 1320–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard M. et al. (2017) ‘Mutations Driving Airborne Transmission of A/H5N1 Virus in Mammals Cause Substantial Attenuation in Chickens Only When Combined’, Scientific Reports, 71: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosone F. et al. (2023) ‘Seroconversion of a Swine Herd in a Free-Range Rural Multi-Species Farm against HPAI H5N1 2.3.4.4b Clade Virus’, Microorganisms, 11: 1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagong M. et al. (2022) ‘Emergence of Clade 2.3.4.4b Novel Reassortant H5N1 High Pathogenicity Avian Influenza Virus in South Korea during Late 2021’, Transboundary and Emerging Diseases, 69: e3255–60. [DOI] [PubMed] [Google Scholar]
- Salaheldin A. H. et al. (2022) ‘Isolation of Genetically Diverse H5N8 Avian Influenza Viruses in Poultry in Egypt, 2019-2021’, Viruses, 14: 1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomon R. et al. (2006) ‘The Polymerase Complex Genes Contribute to the High Virulence of the Human H5N1 Influenza Virus Isolate A/Vietnam/1203/04’, The Journal of Experimental Medicine, 203: 689–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson M. et al. (2014) ‘Characterization of Drug-Resistant Influenza Virus A(H1N1) and A(H3N2) Variants Selected in Vitro with Laninamivir’, Antimicrobial Agents and Chemotherapy, 58: 5220–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanogo I. N. et al. (2022) ‘Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4b Virus in Poultry, Benin, 2021’, Emerging Infectious Diseases, 28: 2534–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffarth G. (2003), Born to fly-Migratory Strategies and Stopover Ecology in the European Wadden Sea of a Long-distance Migrant, the Bar-tailed Godwit (Limosa Lapponica). Faculty of Mathematics and Science, Universität Oldenburg. <http://oops.uni-oldenburg.de/198/> [Google Scholar]
- Scholtissek C. et al. (2002) ‘Cooperation between the Hemagglutinin of Avian Viruses and the Matrix Protein of Human Influenza A Viruses’, Journal of Virology, 76: 1781–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro B. et al. (2011) ‘A Bayesian Phylogenetic Method to Estimate Unknown Sequence Ages’, Molecular Biology and Evolution, 28: 879–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro B., Rambaut A., and Drummond A. J. (2006) ‘Choosing Appropriate Substitution Models for the Phylogenetic Analysis of Protein-Coding Sequences’, Molecular Biology and Evolution, 23: 7–9. [DOI] [PubMed] [Google Scholar]
- Shinya K. et al. (2004) ‘PB2 Amino Acid at Position 627 Affects Replicative Efficiency, but Not Cell Tropism, of Hong Kong H5N1 Influenza A Viruses in Mice’, Virology, 320: 258–66. [DOI] [PubMed] [Google Scholar]
- Smith G. J. D., and Donis R. O. (2015) ‘Nomenclature Updates Resulting from the Evolution of Avian Influenza A(H5) Virus Clades 2.1.3.2a, 2.2.1, And 2.3.4 During 2013–2014’, Influenza and Other Respiratory Viruses, 9: 271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W. et al. (2014) ‘The K526R Substitution in Viral Protein PB2 Enhances the Effects of E627K on Influenza Virus Replication’, Nature Communications, 5: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnberg S., Webby R. J., and Webster R. G. (2013) ‘Natural History of Highly Pathogenic Avian Influenza H5N1’, Virus Research, 178: 63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrell E. M. et al. (2010) ‘A 27-Amino-Acid Deletion in the Neuraminidase Stalk Supports Replication of an Avian H2N2 Influenza A Virus in the Respiratory Tract of Chickens’, Journal of Virology, 84: 11831–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubies S. M. et al. (2010) ‘Species-specific Contribution of the Four C-terminal Amino Acids of Influenza A Virus NS1 Protein to Virulence’, Journal of Virology, 84: 6733–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spesock A. et al. (2011) ‘The Virulence of 1997 H5N1 Influenza Viruses in the Mouse Model Is Increased by Correcting a Defect in Their NS1 Proteins’, Journal of Virology, 85: 7048–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel J. et al. (2009) ‘Transmission of Influenza Virus in a Mammalian Host Is Increased by PB2 Amino Acids 627K or 627E/701N’, PLoS Pathogens, 5: e1000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens J. et al. (2006) ‘Structure and Receptor Specificity of the Hemagglutinin from an H5N1 Influenza Virus’, Science, 312: 404–10. [DOI] [PubMed] [Google Scholar]
- Su Y. et al. (2008) ‘Analysis of a Point Mutation in H5N1 Avian Influenza Virus Hemagglutinin in Relation to Virus Entry into Live Mammalian Cells’, Archives of Virology, 153: 2253–61. [DOI] [PubMed] [Google Scholar]
- Subbarao E. K., London W., and Murphy B. R. (1993) ‘A Single Amino Acid in the PB2 Gene of Influenza A Virus Is A Determinant of Host Range’, Journal of Virology, 67: 1761–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard M. A. et al. (2018) ‘Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10’, Virus Evolution, 4: vey016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchard M. A., and Rambaut A. (2009) ‘Many-core Algorithms for Statistical Phylogenetics’, Bioinformatics, 25: 1370–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suttie A. et al. (2019) ‘Inventory of Molecular Markers Affecting Biological Characteristics of Avian Influenza A Viruses’, Virus Genes, 55: 739–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swieton E. et al. (2020) ‘Sub-Saharan Africa and Eurasia Ancestry of Reassortant Highly Pathogenic Avian Influenza A(H5N8) Virus, Europe, December 2019 - Volume 26, Number 7—July 2020 - Emerging Infectious Diseases Journal - CDC’, Emerging Infectious Diseases, 26: 1557–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taft A. S. et al. (2015) ‘Identification of Mammalian-adapting Mutations in the Polymerase Complex of an Avian H5N1 Influenza Virus’, Nature Communications, 6: 7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammiranta N. et al. (2023) ‘Highly Pathogenic Avian Influenza A (H5N1) Virus Infections in Wild Carnivores Connected to Mass Mortalities of Pheasants in Finland’, Infection Genetics & Evolution, 111: 105423. [DOI] [PubMed] [Google Scholar]
- Thi Hoang D. et al. (2017) ‘UFBoot2: Improving the Ultrafast Bootstrap Approximation’, Molecular Biology and Evolution, 35: 518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Threat Assessment Brief: First Identification of Human Cases of Avian Influenza A(H5N8) Infection [WWW Document], (n.d.), <https://www.ecdc.europa.eu/en/publications-data/threat-assessment-first-human-cases-avian-influenza-h5n8> Accessed Aug 8 2023.
- U.S. Case of Human Avian Influenza A(H5) Virus Reported | CDC Online Newsroom | CDC [WWW Document], (n.d.), <https://www.cdc.gov/media/releases/2022/s0428-avian-flu.html> Accessed Aug 8 2023.
- Verhagen J. H., Fouchier R. A. M., and Lewis N. (2021) ‘Highly Pathogenic Avian Influenza Viruses at the Wild–Domestic Bird Interface in Europe: Future Directions for Research and Surveillance’, Viruses, 13: 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreman S. et al. (2023) ‘Zoonotic Mutation of Highly Pathogenic Avian Influenza H5N1 Virus Identified in the Brain of Multiple Wild Carnivore Species’, Pathogens, 12: 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. et al. (2010) ‘Glycosylation at 158N of the Hemagglutinin Protein and Receptor Binding Specificity Synergistically Affect the Antigenicity and Immunogenicity of a Live Attenuated H5N1 A/Vietnam/1203/2004 Vaccine Virus in Ferrets’, Journal of Virology, 84: 6570–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y. et al. (2011) ‘Acquisition of Human-Type Receptor Binding Specificity by New H5N1 Influenza Virus Sublineages during Their Emergence in Birds in Egypt’, PLoS Pathogens, 7: e1002068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster R. G. et al. (1992) ‘Evolution and Ecology of Influenza A Viruses’, Microbiological Reviews, 56: 152–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster R. G., Kawaoka Y., and Bean W. J. (1989) ‘What Is the Potential of Avirulent Influenza Viruses to Complement a Cleavable Hemagglutinin and Generate Virulent Strains?’, Virology, 171: 484–92. [DOI] [PubMed] [Google Scholar]
- Wille M. et al. (2011) ‘Extensive Geographic Mosaicism in Avian Influenza Viruses from Gulls in the Northern Hemisphere’, PLoS One, 6: e20664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization (2021) ‘Antigenic and Genetic Characteristics of Zoonotic Influenza A Viruses and Development of Candidate Vaccine Viruses for Pandemic Preparedness March 2021’.
- World Health Organization (2023) ‘Avian Influenza Weekly Update Number 895 - 12 May 2023 - Human Infection with Avian Influenza A(H5) Viruses’.
- Xiao C. et al. (2016) ‘B2–588 V Promotes the Mammalian Adaptation of H10N8, H7N9 and H9N2 Avian Influenza Viruses’, Scientific Reports, 6: 19474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie R. et al. (2023) ‘The Episodic Resurgence of Highly Pathogenic Avian Influenza H5 Virus’, Nature, 622: 810–7. [DOI] [PubMed] [Google Scholar]
- Xu G. et al. (2016) ‘Prevailing PA Mutation K356R in Avian Influenza H9N2 Virus Increases Mammalian Replication and Pathogenicity’, Journal of Virology, 90: 8105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S. et al. (2006) ‘Haemagglutinin Mutations Responsible for the Binding of H5N1 Influenza A Viruses to Human-type Receptors’, Nature, 444: 378–82. [DOI] [PubMed] [Google Scholar]
- Yamayoshi S. et al. (2014) ‘Virulence-Affecting Amino Acid Changes in the PA Protein of H7N9 Influenza A Viruses’, Journal of Virology, 88: 3127–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamayoshi S. et al. (2018) ‘Enhanced Replication of Highly Pathogenic Influenza A(H7N9) Virus in Humans’, Emerging Infectious Diseases, 24: 746–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Y. et al. (2007) ‘Immunization by Avian H5 Influenza Hemagglutinin Mutants with Altered Receptor Binding Specificity’, Science, 317: 825–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H. et al. (2022) ‘Divergent Reassortment and Transmission Dynamics of Highly Pathogenic Avian Influenza A(H5N8) Virus in Birds of China during 2021’, Frontiers in Microbiology, 13: 913551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youk S. et al. (2023) ‘H5N1 Highly Pathogenic Avian Influenza Clade 2.3.4.4b In Wild and Domestic Birds: Introductions into the United States and Reassortments, December 2021-April 2022’, Virology, 587: 109860. [DOI] [PubMed] [Google Scholar]
- Zhang J. et al. (2018) ‘The D253N Mutation in the Polymerase Basic 2 Gene in Avian Influenza (H9N2) Virus Contributes to the Pathogenesis of the Virus in Mammalian Hosts’, Virologica Sinica, 33: 531–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G. et al. (2023) ‘Bidirectional Movement of Emerging H5N8 Avian Influenza Viruses between Europe and Asia via Migratory Birds since Early 2020’, Molecular Biology and Evolution, 40: msad019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H. et al. (2009) ‘The Special Neuraminidase Stalk-motif Responsible for Increased Virulence and Pathogenesis of H5N1 Influenza A Virus’, PLoS One, 4: e6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W. et al. (2015) ‘Residues 41V And/or 210D in the NP Protein Enhance Polymerase Activities and Potential Replication of Novel Influenza (H7N9) Viruses at Low Temperature’, Virology Journal, 12: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequences are published in the GISAID EpiFlu database and sequence accession numbers are available in supplementary material (Supplementary Table S1); xml, log, and trees files generated from the BEAST analyses are available on request.