

Abstract
Tools for genome-wide rapid identification of peptide–major histocompatibility complex targets of T-cell receptors (TCRs) are not yet universally available. We present a new antigen screening method, the T-synapse (Tsyn) reporter system, which includes antigen-presenting cells (APCs) with a Fas-inducible NF-κB reporter and T cells with a nuclear factor of activated T cells (NFAT) reporter. To functionally screen for target antigens from a cDNA library, productively interacting T cell–APC aggregates were detected by dual reporter activity and enriched by flow sorting followed by antigen identification quantified by deep sequencing (Tsyn-seq). When applied to a previously characterized TCR specific for the E7 antigen derived from human papillomavirus type 16 (HPV16), Tsyn-seq successfully enriched the correct cognate antigen from a cDNA library derived from an HPV16-positive cervical cancer cell line. Tsyn-seq provides a method for rapidly identifying antigens recognized by TCRs of interest from a tumor cDNA library.
Keywords: immune synapse, T-cell receptor, antigen, epitope, screen
INTRODUCTION
T cells play important roles in a variety of immune responses, including some that can be beneficial and others that can be harmful[1]. Examples of the former include protection against infections and cancers, while the latter can contribute to pathological inflammation in settings such as autoimmunity or impaired tolerance to environmental or transplanted antigens[2]. T-cell receptors (TCRs) are cell surface molecules that each endow T cells with unique antigen specificities; together, they confer on the immune system the potential to recognize a vast pool of antigens that can be foreign or host-derived, including intracellular and extracellular proteins[3, 4]. These antigens are presented as peptides by major histocompatibility complexes (pMHC) on the cell surface of target cells. Sequencing TCRA and TCRB genes can identify unique T-cell clones, and expressing these genes in another T cell can endow the new cell with the same antigen specificity as the original. While the advent of single-cell sequencing methodologies has increased the ease and affordability of identifying TCR sequences from samples, determining which antigens are being recognized by T cells continues to be challenging.
Several well-developed methods are available for identifying TCR–pMHC interactions[5–7]. These include T-cell functional assays and pMHC multimers, which allow for the evaluation of hypotheses generated a priori. However, these methods are not readily amenable to high-throughput antigen discovery. More recently, untargeted assays have been developed utilizing antigen libraries presented by baculovirus[8], yeast[9], and mammalian cells[10]. While powerful, these methods can be laborious, often require sophisticated and non-commercially available reagents, and may be technically difficult to perform. Identification of antigens at the mammalian genome scale is particularly challenging.
Herein, we present a sequencing-based screening method based on T cell–synapse formation (Tsyn-seq). We genetically modified cell lines to functionally report when T cells and target cells interacted closely with bidirectional signaling activity. First, we designed a chimeric receptor consisting of the extracellular domain of Fas fused with the transmembrane and cytoplasmic domains of tumor necrosis factor receptor 2 (TNFR2). When coupled with an NFκB response element, this resulted in a Fas-inducible NFκB (Fas-iNFκB) reporter, which we stably expressed in an artificial antigen-presenting cell line (APCs). Concomitantly, TCR–pMHC interactions were also identified on the T-cell side of the immunological synapse by activation of a nuclear factor of activated T cell (NFAT) reporter element in a T-cell line[11]. With this dual-reporter system, we used flow cytometry to enrich for aggregates of T cells and APCs where bidirectional signaling reporter activity was observed. We found that this platform allowed, with relative ease, the evaluation of a genome-wide tumor cDNA library and a successful enrichment of the correct cognate antigen recognized by a known TCR. The Tsyn-seq system represents a method for rapid genome-wide identification of antigens functionally recognized by TCRs of interest.
MATERIALS AND METHODS
Cell culture
293T (#CRL-3216), Jurkat (RRID: CVCL_0367, clone E6–1, Cat#TIB-152), and CaSki cells (#CRM-CRL-1550) were purchased from ATCC in 2019. Cell lines were authenticated by ATCC and our institution. All cell lines were confirmed to be Mycoplasma-free using PCR Mycoplasma Detection Kit (ABM cat# G238). Cells were expanded for two or three passages before being frozen down as working stocks. Prior to their use in analyses, cells were passaged one to two times after thawing. 293T and CaSki cells were cultured in DMEM (Corning) with 10% (v/v) FBS (Hyclone), and 1X Penicillin-Streptomycin-Glutamine (100X) (Gibco). Jurkat T cells were cultured in RPMI1640 (Corning) with 10% (v/v) FBS (Hyclone), and 1X Penicillin-Streptomycin-Glutamine (100X) (Gibco).
Antibodies and flow cytometry
Human TruStain FcX (Fc Receptor Blocking Solution, Biolegend cat#422301) and all of the following fluorochrome-labeled antibodies (all for humans unless otherwise indicated) were obtained from BioLegend: CD178 (Fas ligand)-PE (NOK-1, RRID:AB_314603), CD95 (Fas)-PE (DX2), anti-mouse TCRβ-APC (H57–597), HLA-ABC-APC (W6/32), HLA-A2-APC (BB7. 2), TCRα/β-PE (IP26), and Cleaved PARP (Asp214)-PE (QA17A17). TCRα/β-APC (T10B9. 1A-31), HLA-B-PE (YTH 76.3 rMAb) and HLA-C-PE (DT-9) were obtained from BD Biosciences. HLA-BC-APC (B1.23.2) was obtained from eBioscience. CellTrace Violet (CTV) and CellTrace Far Red (CTFR) were obtained from Thermo Fisher Scientific, and cell staining was performed according to the manufacturer’s instructions. Cells were acquired on a BD LSRFortessa X-20 Cell Analyzer (BD Biosciences) and a BD FACSAria Cell Sorter (BD Biosciences) using BD FACSDiva software (RRID: SCR_001456, 8.0.1). The data were analyzed using FlowJo software 10.9.0 (RRID: SCR_008520).
Retroviral production
Retrovirus was produced from transfected 293T cells as described previously[12]. In brief, 293T cells were transiently transfected with retroviral expression vectors (indicated in the sections below), together with pUMVC (Addgene#8449) and pCMV-VSV-G plasmids (RRID: Addgene_8454) using Lipofectamine 2000 (Thermo Fisher Scientific) following the manufacturer’s protocol. The retroviral supernatant was harvested 2 days later and concentrated using PEG-it (SBI). For infection, the retrovirus was used at a multiplicity of infection (MOI) > 5 to reach approximately 80% transduction.
Lentiviral production
Lentivirus was produced from transfected 293T cells as described previously[13]. In brief, 293T cells were grown to approximately 80% confluence in a 10 cm dish before being transiently transfected with 10 μg lentiviral transfer vector encoding the gene of interest (described in the sections below), together with 5 μg packaging plasmid PsPAX2 (RRID: Addgene_12260), and 3 μg envelop plasmid PMD2.G (Addgene#12259) using Lipofectamine 2000 (Thermo Fisher Scientific) following the manufacturer’s protocol. The lentiviral supernatant was harvested 2 days after transfection, filtered (0.45 μm), and concentrated using PEG-it (System Biosciences). The lentivirus was used at an MOI > 5 for infection to ensure approximately 80% transduction.
Generation of NFκB-GFP reporter
The DNA sequence encoding GFP was amplified by PCR using the following primers (all primers synthesized by IDT): GFP-F: 5’-GCCCCCATGGTGAGCAAGGGCGAGGAG-3′ and GFP-R: 5’-AAGTCATATGTTACTTGTACAGCTCGTCC-3′. The luciferase and Ubc promoter regions in the pHAGE NFκB-TA-LUC-UBC-GFP-W plasmid (Addgene#49343) were replaced with eGFP using the NcoI and NdeI sites to generate the pHAGE NFκB-GFP plasmid. The resulting vector was introduced into 293T cells by lentiviral transduction. After Fas antibody treatment, NFκB-GFP reporter–positive cells were sorted by FACS.
Fas antibody treatment assay
Fas antibody treatment was performed as previously described[14]. Cells were seeded in 96-well plates, grown to 75% confluency, and then incubated with 1 μg/ml rabbit Fas antibody (Sigma, clone CH11) or PBS as untreated groups. After overnight treatment, the activation of NFκB-GFP reporter was determined by flow cytometry for GFP expression.
Knockout of endogenous FAS gene
The pLentiCRISPR V2 vector (RRID: Addgene_169885) with the CRISPR/Cas9 guide RNA (gRNA, 5’-GTGTAACATACCTGGAGGAC-3’) directed to cleave the FAS gene was introduced into NFκB-GFP 293T cells (all CRISPR/Cas9 gRNA vectors were purchased from GenScript). After transduction, Fas-knockout (FasKO) NFκB-GFP 293T cells were sorted after staining with anti-Fas-PE (BioLegend, Cat#305608).
Generation of Fas-TNFR2 chimeric receptor
The transmembrane domain (amino acids 258–280) and cytoplasmic domain (amino acids 281–461) of TNFR2 were amplified by PCR from the TNFR2-GFP plasmid (RRID: Addgene_111207) using the following primers: TNFR2-F: 5’-GCCCGGATCCTTCGCTCTTCCAGTTGGACTG-3′ and TNFR2-R: 5’-AAGTGATATCACTGGGCTTCATCCCAGCATC-3′. Amplified TNFR2 was then cloned into the Fas_PLX307 lentiviral vector (Addgene#98334) using BamHI and EcoRV sites to fuse with the C-terminus of the extracellular region of Fas (amino acids 1–170). The Fas-TNFR2_PLX307 plasmid was introduced into FasKO NFκB-GFP 293T cells by lentiviral transduction. Fas-TNFR2 receptor-expressing FasKO NFκB-GFP 293T cells (referred to here as FIR-APCs) were selected using 2 μg/ml puromycin (Santa Cruz Biotechnology). The expression of Fas-TNFR2 chimeric receptor was confirmed by detecting the C-terminal V5 tag using Western blotting (described below). Clones were grown from single cells, and then activation of the NFκB-GFP reporter was re-verified by flow cytometry for GFP expression after Fas antibody treatment as described above.
MTT assay
The cell viability of wild-type and engineered 293T cells was assessed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) cell proliferation assay (ATCC) according to the manufacturer’s protocol. The absorbance was measured at 570 nm using a BioTek Synergy HTX plate reader.
Knockout of endogenous HLA genes in FIR-APCs
CRISPR/Cas9 gRNAs directed at cleaving the HLA-A locus were amplified using the following primers and cloned into the pLenti-eCas9 vector (Addgene#140237): HLA-A CRISPR 1: CGTCCTGCCGGTACCCGCGG, CRISPR HLA class I F1: 5’-CACCGAGGTCAGTGTGATCTCCGCA-3′, CRISPR HLA class I R1: 5’-AAACTGCGGAGATCACACTGACCTC-3′, HLA-A CRISPR 2: TACCGGCAGGACGCCTACGA, CRISPR HLA class I F2: 5’-CACCGCGGCTACTACAACCAGAGCG-3′, and CRISPR HLA class I R2: 5’-AAACCGCTCTGGTTGTAGTAGCCGC-3′. The following CRISPR/Cas9 gRNAs were used to cleave the HLA-B and HLA-C loci: HLA-B CRISPR:5’-GGATGGCGAGGACCAAACTC-3′, and HLA-C CRISPR: 5’-GACACAGAAGTACAAGCGCC-3′. Vectors were introduced into FIR-APCs by lentiviral transduction. After transduction, HLA-negative (HLA-KO) FIR-APCs were sorted using anti-HLA-ABC, anti-HLA-BC, anti-HLA-B, and anti-HLA-C, and clones were grown from single cells.
Overexpression of HLA-A*02:01 in HLA-KO FIR-APCs
The gene expressing HLA-A*02:01 was amplified from the pMP71-HLA-A0201-His plasmid (RRID: Addgene_108214) using the following primers: HLA-A2-F: 5’-GCCCATCGATATGGCCGTCATGGCGCCCCGAAC-3′ and HLA-A2-R: 5’-AAGTGATATCCACTTTACAAGCTGTGAGAGAC-3′. Amplified HLA-A*02:01 was cloned into the Fas_PLX307 lentiviral vector (Addgene#98334) using ClaI and EcoRV sites to replace the Fas region. The resulting HLA-A2 _PLX307 plasmid was introduced into HLA-KO FIR-APCs via lentiviral transduction. After transduction, HLA-A*02:01 positive HLA-KO FIR-APCs were enriched using FACS after staining with anti-HLA-A2.
Generation of NFAT reporter in T cells
DNA encoding mCherry was amplified using the following primers: mCherryF: 5’-GCACAGATCTCGCCACCATGGTGAGCAAGGGCG-3′ and mCherryR: 5’-TTCACTCGAGCTACTTGTACAGCTCG-3′. Then, mCherry was cloned into the 8xNFAT-ZsGeen-hCD8 plasmid (Addgene#153417) to replace the ZsGreen region using BglII and XhoI sites to generate an 8xNFAT-mCherry-hCD8 plasmid. To remove the hCD8 region from the 8xNFAT-mCherry-hCD8 plasmid, we replaced it with the following DNA sequence (synthesized by IDT) encoding a random sequence (MVSKGGGGGS) using the AgeI and HindIII sites:5’-GCACAGATCTCGCCACCATGGTGAGCAAGGGCGGTGGTGGTGGTTCTTAGCTCGAGTGAA-3′. The resulting 8xNFAT-mCherry plasmid was introduced into Jurkat T cells via retroviral transduction. The resulting JR-T cells were enriched by FACS for mCherry expression after PMA and ionomycin (BioLegend) treatment. Clones were grown from single cells and reporter expression was verified by flow cytometry for mCherry expression after PMA and ionomycin treatment.
Overexpression of TCRs
Retroviral expression plasmids encoding the αHPV16E629–38 TCR genes (α and β) and αHPV16E711–19 TCR genes were obtained from Addgene (#122727 and #122728). A gene expressing αCMVpp65495–503 TCR genes[15, 16] was synthesized (IDT) and cloned into the MSGV1 retroviral vector (Addgene #122727). To avoid mispairing between the introduced TCR genes and endogenous TCR genes, the constant regions of each TCR chain were exchanged for their murine counterparts as previously described[17]. After transduction, JR-T cells with TCR expression (TCR-JR-T cells) were enriched by FACS after staining with an anti-mouse TCRβ.
Knockout of endogenous TCR genes
The CRISPR/Cas9 gRNA vector CRISPR_TRAC (Addgene Plasmid#164993) was used to edit the human TCRα constant (TRAC) locus. CRISPR/Cas9 gRNA directed to edit the human TCRβ constant 1 (TRBC1) locus was amplified using the following primers and cloned into the pLenti-eCas9 vector (Addgene#140237): CRISPR TCRβ F: 5’-CACCGCGTAGAACTGGACTTGACAG-3′ and CRISPR TCRβ R: 5’-AAACCTGTCAAGTCCAGTTCTACGC-3′. The resulting vectors were introduced into JR-T cells by lentiviral transduction. TCR-negative (TCR-KO) JR-T cell clones were grown from single cells. The absence of TCR was verified by flow cytometry using anti-TCRα/β, and further confirmed by Western blotting using anti-TCRα (Santa Cruz Biotechnology, #H-1) and anti-TCRβ (Cell Signaling, #77046).
Western blot
A total of 1 × 107 cells were collected. Cells were washed twice with PBS and lysed using RIPA Lysis Buffer (Santa Cruz Biotechnology) mixed with Protease Inhibitor Cocktail Tablets (Millipore Sigma) at 4°C for 20 minutes. After being sonicated with an ultrasound sonicator, whole cell lysates were centrifuged at 16,000 g for 10 minutes to remove cell debris. Total protein concentration was determined using Pierce™ BCA Protein Assay Kits (Thermo Scientific) with bovine serum albumin as a standard. For Western blot analysis, 45 μg protein/sample mixed with 4x Laemmli Sample Buffer (Bio-Rad) were electrophoresed on SDS-PAGE and then transferred onto polyvinylidene difluoride membranes. The membranes were blocked with 5% nonfat dry milk in PBS containing 0.05% Tween 20 and incubated with primary antibodies at 4°C overnight. Blots are then incubated with horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) according to the manufacturer’s instructions. The immunoblots were visualized by X-ray film with Pierce™ ECL Western Blotting Substrate.
Fas-iNFκB reporter activation assays with peptides
HPV16E6 antigen peptide (TIHDIILECV), HPV16E7 antigen peptide (YMLDLQPET), and CMVpp65 antigen peptide (NLVPMVATV) were synthesized with a purity greater than 95% by Genemed Synthesis. CTV-labeled FIR-APCs were pulsed with 1 μM of each peptide and seeded at 3×104 cells/well in round-bottom 96 well plates, with CTFR-labeled T cells in Jurkat T cell media (as described above in “Cell culture”) at 9×104 cells/well. Cells were cocultured for 2 days and harvested for flow cytometry analysis.
Fas-iNFκB reporter activation assays with endogenously expressed antigens
Full-length HPV16E6 and HPV16E7 DNA sequences were amplified from the pLXSN16E6E7 plasmid (RRID: Addgene_52394), and pp65CMV 9-mer (NLVPMVATV) was amplified from PresentER-NLVPMVATV plasmid (RRID: Addgene_102947) using the following primers: E6-F: 5’-GCCCGGATCCCACCATGGACCAAAAGAGAACTGCA-3′, E6-R: 5’-AAGTGTCGACTTATGGTTTCTGAGAACAGATGGGGC-3′, E7-F: 5’-GCCCGGATCCCACCATGGATGGAGATACACCTACA-3′, E7-R: 5’-AAGTGTCGACTTATGGTTTCTGAGAACAGATGGGGC-3′, CMV_F65: 5’-GCCCGGATCCACCATGGTGAACCTGGTGCCCATGGTGGCCACCGTGTGAGTCGACTGAA-3′, and CMV_R65: 5’-TTCAGTCGACTCACACGGTGGCCACCATGGGCACCAGGTTCACCATGGTGGATCCGGGC-3′. The amplified antigens were cloned into the pLenti-CMV-GFP-Zeo lentiviral vector (Addgene#17449) using BamHI and SalI sites. The resulting vectors were introduced into FIR-APCs by lentiviral transduction. FIR-APCs expressing each antigen (E6-FIR-APCs, E7-FIR-APCs, and NLV-FIR-APCs) were selected using 500 μg/ml zeocin (InvivoGen). FIR-APCs expressing antigens or CaSki cells were labeled with CTV and cocultured under the same conditions as above but without peptide pulsation (Fas-iNFκB reporter activation assays with peptides).
Receiver operating characteristics (ROC) analysis
The fluorescence intensity of reporters in individual cells was analyzed using the FlowCore R package[18]. An ROC curve[19] and the corresponding area under the ROC curve (AUC) value[20] were calculated for the fluorescence intensity of reporters as predictive of target antigen expression in FIR-APCs.
Construction of cDNA library
mRNA was extracted from 1×107 CaSki cells using the Magnetic mRNA Isolation Kit (NEB). A gateway system-compatible cDNA library was generated from 1μg of mRNA using the CloneMiner II cDNA Library Construction Kit (Thermo Fisher Scientific), according to the manufacturer’s protocol. The constructed cDNA libraries were cloned into the pLenti CMV Hygro DEST vector (RRID: Addgene_17454) and transduced into FIR-APCs. FIR-APCs expressing the CaSki cDNA library (CaSki-FIR-APCs) were selected using 200 μg/ml of hygromycin (Santa Cruz Biotechnology).
CaSki cDNA library screen
CTV-labeled CaSki-FIR-APCs were cocultured with CTFR-labeled TCR-JR-T cells at a ratio of 1:3 in a 25 cm2 flask for 2 days, after which CTV+CTFR+/mCherry+ (highest 10%)/GFP+ (highest 10%) aggregates were selected by FACS. Unsorted cocultured cells were used as controls.
Genomic DNA (gDNA) purification and adaptor PCR
gDNA was isolated from dual-reporter-positive aggregates obtained by FACS using the GeneJET gDNA purification kit (Thermo Fisher Scientific). cDNA inserts were amplified from the isolated gDNA by PCR using PrimeSTAR HS DNA polymerase (TaKaRa) and the following Gateway attB adaptor primers: attB1 primer: 5’-TGGTGGAATTCTGCAGATATCAACAAG-3’ and attB2 primer: 5’-CTGTGCTGGATATCAACCACTTTGT-3’. The amplified cDNA inserts were purified using a QIAquick PCR Purification Kit (QIAGEN) for subsequent qPCR and amplicon library construction.
Quantitative PCR (qPCR)
The abundance of E7 gene copies in the amplified cDNA inserts was quantified by qPCR. The amplified cDNA inserts were normalized to 125 ng/ 50 μl and then 50× diluted. 5 μl of this diluted cDNA was used as template for each sample. Three replicates were used for each reaction. Template was amplified using the KAPA SYBR FAST qPCR Master Mix (2X) Kit (Sigma-Aldrich) with the following primers and was normalized to the abundance of human ACTB: E7-qPCR-F: 5’-ATGCATGGAGATACACCTACATTGC-3’, E7-qPCR-R: 5’-GATTATGGTTTCTGAGAACAGATGGGGC-3’, ACTB-F: 5’-GCGCGGATCCGCGGACTATGACTTAGTTGCG-3’ and ACTB-R: 5’-GCGCGCGGCCGCCCACATTGTGAACTTTGGGGG-3’. Expression fold was calculated as 2^-(Ct target gene –Ct ACTB gene). qPCRs were run on a QuantStudio™ 6 Flex real-time PCR system (Thermo Fisher Scientific).
Amplicon deep sequencing
An amplicon library was prepared using the Illumina DNA Prep workflow. The quality of each library was assessed using the 4200 TapeStation system (Agilent). Pooled libraries were sequenced on the Illumina NovaSeq platform with a 2×150 base pair paired-end protocol, resulting in more than 5 Gb per sample. Raw DNA reads were filtered using VSEARCH 2.17.1[21] with a quality score below 15. The remaining reads were aligned to the human GCRh38 and HPV database (NC_001526.4) using Diamond version 0.9.24[22]. 4,819 genes were detected in all samples isolated by FACS. Genes were considered if 80% of samples had at least a relative abundance of 10-5. Differential gene abundance testing comparing sorted and unsorted samples was performed using DESeq2.
Statistical analysis
Data were checked for normality and similar variances, and the t-test was used to compare the values between the two groups. The Mann-Whitney U test was used to compare data between the two groups when the data did not follow a normal distribution. One-way analysis of variance (ANOVA) with post-hoc Tukey’s test was used to compare data between multiple groups. Analyses were performed using R software version 4.1.2 and GraphPad Prism (RRID: SCR_002798, version 9.0). Statistical significance was set at P <0.05.
Data Availability
The data generated in this study are available within the article and its supplementary data files or from the corresponding author upon reasonable request. The sequencing data generated in this study are publicly available in Sequence Read Archive (SRA) at PRJNA1034661.
RESULTS
A Fas-inducible NFκB reporter detects activated target cells
Our goal was to develop a tool that would allow unbiased identification of cognate antigens recognized by TCRs. We chose to employ amplicon-based deep sequencing of cDNA library subsets expressed by artificial APCs after enriching for the APCs that demonstrated T-cell synaptic interactions. We refer to the tool we developed as Tsyn-seq. Human embryonic kidney 293T (293T) cells have previously been used to generate artificial APCs[23]. Thus, we engineered 293T cells to be the artificial APCs in our Tsyn-seq tool.
Cytotoxic T lymphocytes (CTLs) recognize antigen-expressing targets through TCR–pMHC engagement and, in turn, mediate killing via mechanisms that include production of perforin/granzyme B and cytotoxic cytokines, including IFNγ and TNF, and signaling through the Fas ligand (FasL)/Fas axis[24]. T cell–antigen discovery methods have been developed to detect targeted APCs through detection of the intracellular enzymatic activity of granzyme B[6] or the presence of effector T-cell cytokines at the cell surface[25].
We developed a system that detects when Fas ligation activates NFκB signaling (Fas-iNFκB) (Fig. 1A, B). Fas is a cell surface receptor that belongs to the TNFR family. The interaction between FasL and Fas at the immunological synapse[26] is the predominant mechanism of CTL-induced apoptosis[27]. Fas signaling simultaneously produces NFκB activation[28] as well as induction of apoptosis[29] via interaction of the ligated conformation of Fas with Fas-Associated Via Death Domain (FADD)[30] (Fig. 1A).
Fig. 1. Generation of Fas-inducible reporter APCs (FIR-APCs).
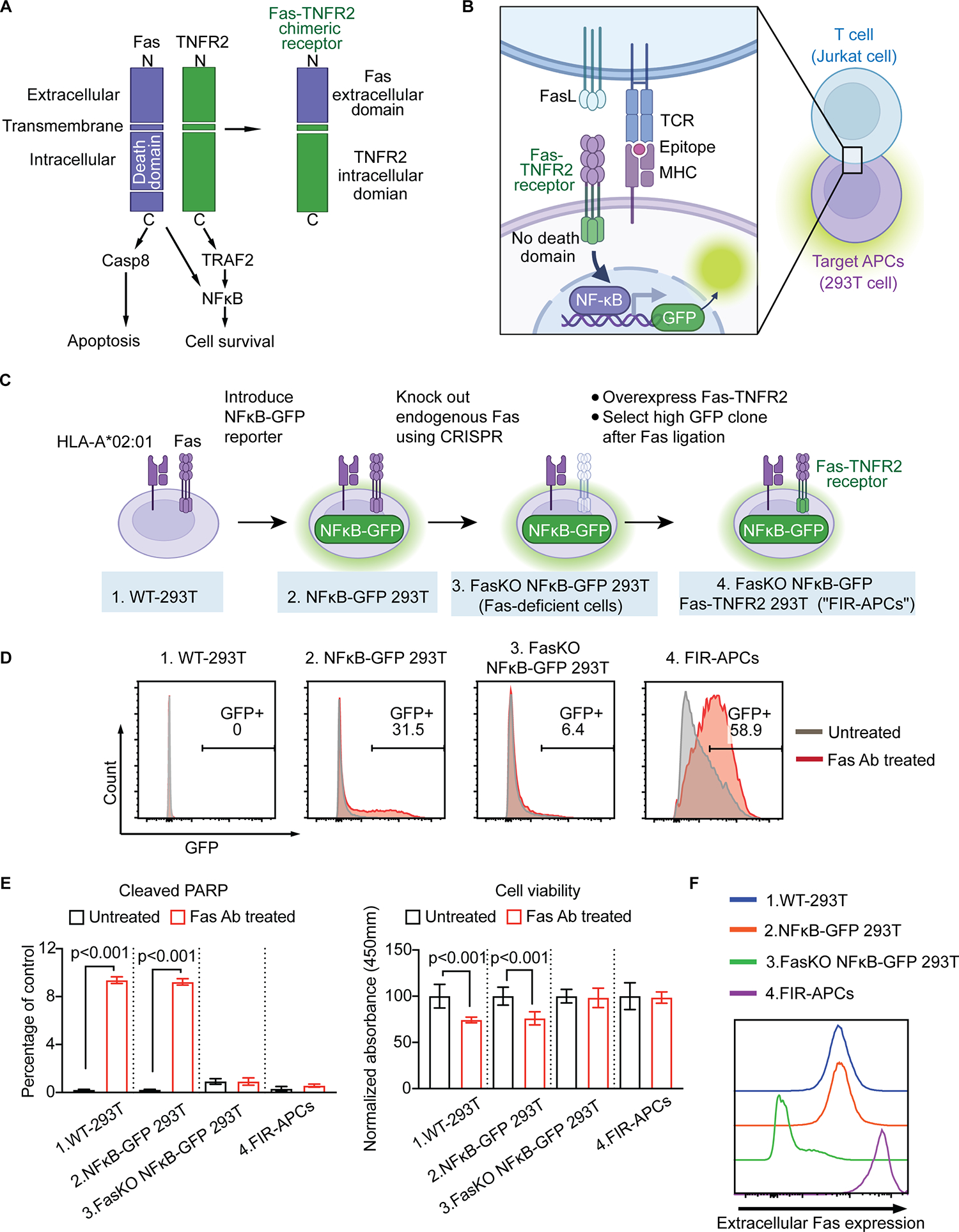
(A) Schematic of Fas-TNFR2 chimeric receptor. The death domain mediates apoptotic caspase activation, while the intracellular domain of TNFR2 recruits cytoplasmic TNF receptor-associated factor-2 (TRAF-2) resulting in initiation of NFκB signaling pathways. To prevent death domain-mediated caspase activation, we fused the extracellular domain of Fas (aa 1–170) with TNFR2 transmembrane (aa 258–280) and intracellular domains (aa 281–461). (B) T cell-target cell interactions activate Fas-TNFR2 receptor and results in NFκB-induced fluorescence. (C) Generation of Fas-TNFR2 chimeric receptor-overexpressing NFκB reporter cells. An NFκB-GFP reporter is introduced into WT-293T, followed by gene editing to remove endogenous Fas (FasKO NFκB-GFP 293T cells). A single clone of FasKO NFκB-GFP 293T cells with high GFP expression is transduced to express Fas-TNFR2 receptor (FIR-APCs). (D) Representative flow cytometry plots of GFP expression in 293T reporter cells described in C. Cells were stimulated with anti-Fas antibody (red) or PBS as untreated groups (grey). (E) Apoptosis of 293T reporter cells after anti-Fas antibody treatment compared to untreated groups. The levels of cleaved PARP in 293T NFκB reporter cells were analyzed by flow cytometry (left panel, n = 3). Cell viability is analyzed by MTT assay (right panel, n = 6). (F) Extracellular Fas expression levels in 293T reporter cells analyzed by flow cytometry. KO, knock out. Statistical significance is determined by the one-way ANOVA (ANalysis Of VAriance) with post-hoc Tukey test. Error bars indicate SD across replicates.
To generate APC reporter cells that would enable Tsyn-seq, we introduced an NFκB response element driving the expression of enhanced GFP to 293T cells (Fig. 1C). We found that ligation of Fas with an agonist antibody[31] produced GFP expression in some lentivirally transduced 293T cells at 18 hours (Fig. 1D). However, Fas ligation also induced apoptosis, quantified by the activity of cleaved poly (ADP-ribose) polymerase (PARP)[32], as well as by reduced metabolic activity, measured with an MTT assay[33] (Fig. 1E).
To reduce Fas ligation–induced apoptosis in APC reporter cells, we used CRISPR/Cas9 gene editing[34] to eliminate the expression of endogenous Fas (Fig. 1C,F). Anti-Fas treatment of Fas-deficient cells showed no changes in the activity of cleaved PARP or metabolic activity relative to untreated cells (Fig. 1E) but did not induce GFP expression (Fig. 1D). To restore NFκB signaling with Fas ligation, we overexpressed a chimeric receptor generated by fusing the extracellular domain of Fas with the transmembrane and intracellular domains of TNFR2, which we named Fas-TNFR2 (Fig. 1A). A similar strategy has previously been reported for fusing Fas with 4–1BB[26]. Fas, 4–1BB, and TNFR2 are all members of the TNFR family that form trimeric receptors and signal when interacting with their respective trimeric ligands[35]. TNFR2 lacks a death domain but maintains interactions with TRAF2[36], which can activate NFκB[37]. We found that introducing Fas-TNFR2 into Fas-deficient cells (Fig. 1C) restored NFκB signaling in response to Fas ligation (Fig. 1D) without inducing apoptosis (Fig. 1E). These cells were termed “Fas-iNFκB reporter APCs” (FIR-APCs).
FIR-APCs report when targeted by Jurkat T cells
To evaluate how FIR-APCs respond to targeting by T cells, we evaluated HPV16 E6 (E6), HPV16 E7 (E7), and CMV pp65 (NLV) as model antigens[16, 17, 38]. All three antigens contain immunogenic peptides presented by the common allele HLA-A*02:01[39]. We utilized the human Jurkat T cell line[40] and overexpressed previously-identified TCRα and TCRβ genes that recognize E6, E7, or NLV peptide antigens (αE6-, αE7- and αNLV-Jurkat T cells)[41, 42]. We pulsed FIR-APCs with E6, E7, or NLV peptide antigens and co-cultured them with each Jurkat T cell line stably overexpressing one of the three TCRs (Fig. 2A).
Fig. 2. T cell/target cell interactions activate FIR-APCs.
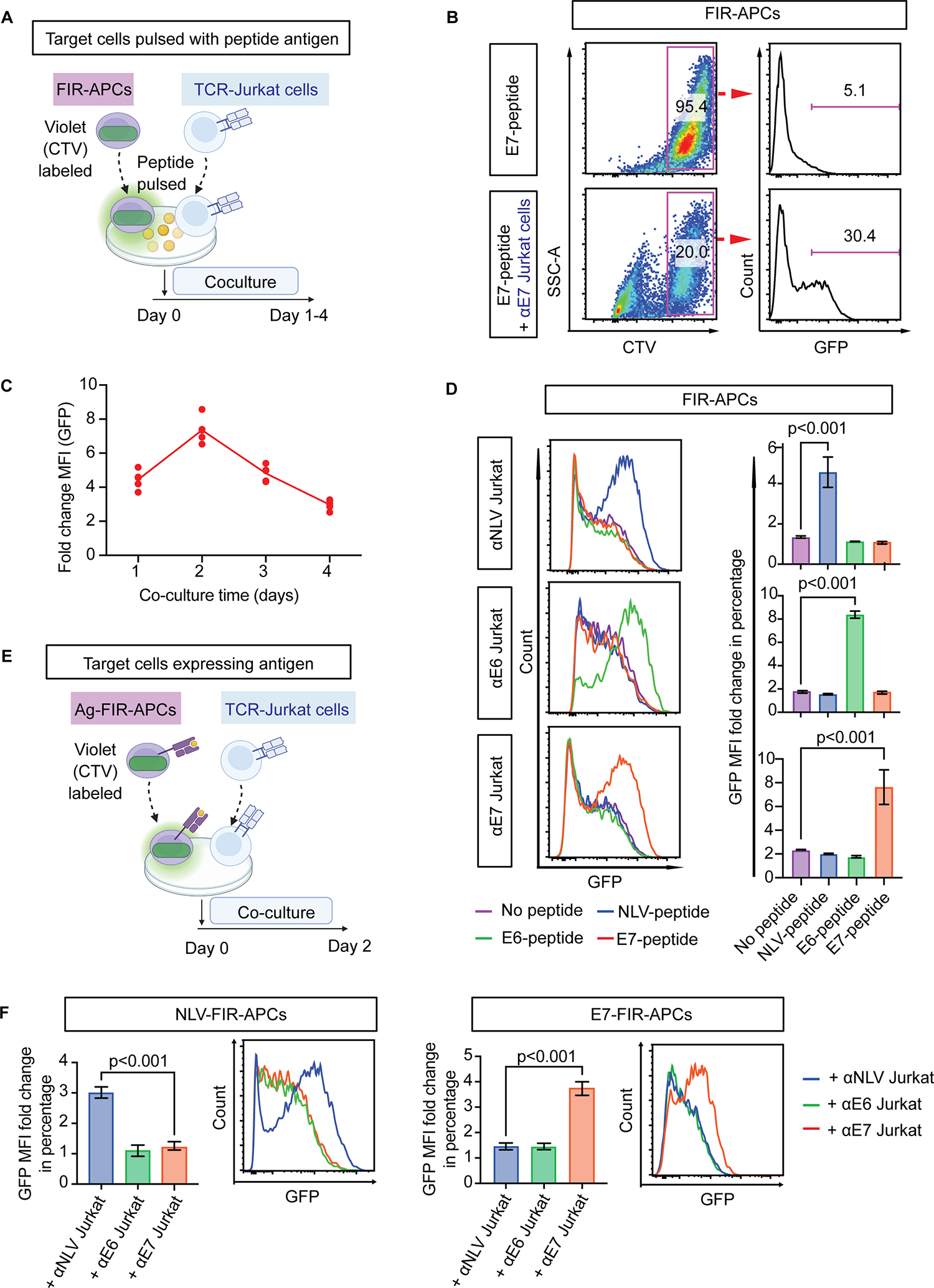
(A) Schematic of antigen peptide pulsation activation assay. FIR-APCs are labeled with CellTrace Violet (CTV) and pulsed with antigen peptides before co-culturing with αE7-Jurkat T cells. (B) GFP expression in E7-peptide-pulsed FIR-APCs quantified by flow cytometry. E7-peptide-pulsed FIR-APCs are cultured alone or with αE7-Jurkat T cells for 1 day. (C) Time course of GFP expression in E7-peptide-pulsed FIR-APCs during coculture with αE7-Jurkat T cells. The y-axis depicts fold change in mean fluorescence intensity (MFI) of GFP in FIR-APCs. (D) GFP expression in peptide-pulsed FIR-APCs after 2 days of coculture with the indicated TCR-Jurkat T cells. Representative flow cytometry plots are shown (left panel) as well as MFI fold change of GFP in FIR-APCs (right panel). Fold change is defined as the ratio of GFP MFI in the cocultured FIR-APCs relative to the mono-cultured FIR-APCs. n = 3 independent samples. (E) Schematic of antigen overexpression activation assays. (F) GFP expression in FIR-APCs with overexpressed antigens after 2 days of coculture with TCR-Jurkat T cells. n = 6 independent samples. Statistical significance is determined by the one-way ANOVA with post-hoc Tukey test. Error bars indicate SD across replicates.
We quantified the kinetics of GFP induction in FIR-APCs and found that increased GFP was detectable in E7 peptide–pulsed FIR-APCs after 1 day of co-culture with αE7-Jurkat T cells (Fig. 2B), with maximal reporter signaling observed at 2 days (Fig. 2C). Overall, we found that FIR-APCs reported when targeted by Jurkat T cells in a manner that was peptide and TCR specific, with activation observed only in the presence of the correct cognate peptide antigen and its matching TCR (Fig. 2D). To further optimize this system, we used FACS to single-cell sort FIR-APCs and selected a clone with low baseline NFκB activity as well as high upregulation of GFP upon Fas ligation (Supplementary Fig. S1A), and this clone was utilized for further experiments.
We next tested the ability of FIR-APCs to report in the setting of stable expression of antigens, in contrast to peptide pulsing in the prior experiments. To do so, we stably transduced FIR-APCs with constructs overexpressing full-length HPV16 E7 protein, HPV16 E6 protein, or the CMV pp65 NLV minigene to produce E7-, E6-, or NLV-expressing FIR-APCs (E7-FIR-APCs, E6-FIR-APCs, and NLV-FIR-APCs). We found that these cells upregulated GFP in an antigen-specific manner when cocultured with Jurkat T cells overexpressing the appropriate cognate TCRs (Fig. 2E–F). Overexpression of the antigen by FIR-APCs produced less GFP upregulation compared with peptide pulsing (Supplementary Fig. S1B), likely because of differences in the density of HLA molecules presenting cognate antigen peptides. Overall, these results indicated that FIR-APCs can report when being targeted by antigen-specific T cells both after peptide pulsing and when forced to express a protein antigen.
FIR-APCs aggregated with T cells show increased Fas signaling compared to singlet FIR-APCs
Crosstalk between T cells and APCs occurs in the setting of an immunological synapse, with an interface characterized by a central TCR–pMHC cluster surrounded by interacting costimulatory molecules[43]. It has been demonstrated that T cell–APC pairs can form aggregates detectable by flow cytometry[44]. To identify FIR-APC/Jurkat T cell aggregates, we labeled Jurkat T cells and FIR-APCs with distinct cell surface dyes, which allowed for detection of cell aggregates using fluorescence from both dyes (Fig. 3A). We found that after co-culture of labeled E7-FIR-APCs with labeled E7-specific Jurkat T cells (αE7-Jurkat T cells), the majority of the dual-labeled events detected by flow cytometry were multiparticle rather than single particles (Fig. 3B and Supplementary Fig. S2A). Furthermore, the percentage of dual-labeled events increased in the setting of matching TCRs and cognate antigens (Fig. 3C). We also compared the degree of activation of FIR-APCs that were either suspended as single particles or aggregated with the Jurkat T cells. We found that reporter activity was substantially higher in the fraction of FIR-APCs found aggregated with Jurkat T cells (Fig. 3D), and that reporter activity increased in an antigen-specific manner (Fig. 3E). These results demonstrated that a gating strategy incorporating selection of FIR-APC/Jurkat T cell aggregates, followed by selection of activated FIR-APCs within these aggregates, could allow for improved enrichment of the subset of FIR-APCs targeted by Jurkat T cells.
Fig. 3. Quantifying aggregates of interacting FIR-APCs and Jurkat T cells.
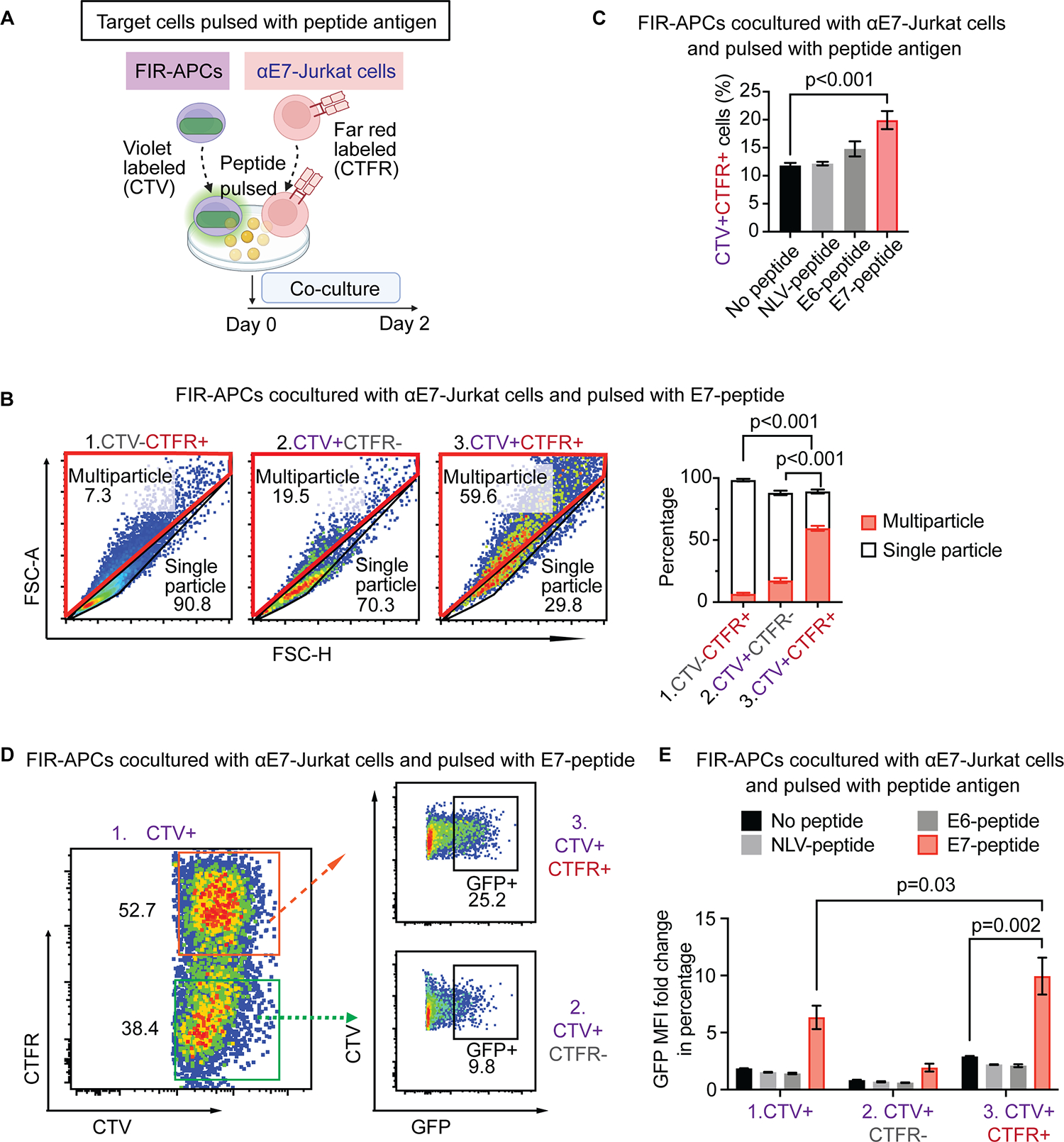
(A) Schematic: Evaluating interactions of FIR-APCs (purple) with Jurkat T cells (red). CTV-labeled FIR-APCs are pulsed with peptides and cocultured with CellTrace Far Red (CTFR) labeled αE7-Jurkat T cells for 2 days. (B) Frequencies of multiparticle and single particle in CTV and/or CTFR positive populations after co-culturing E7-peptide-pulsed FIR-APCs with Jurkat T cells. Gates for the three populations are shown in Fig. S2A. Multiparticle and single particle gates are generated after visualizing FSC-A and FSC-H. n = 6 independent samples. (C) Frequency of CTV/CTFR double-positive events after coculture following pulsing with cognate or non-cognate peptide. n = 3 independent samples. (D) GFP expression in CTV/CTFR single- or double-positive populations. Cocultured Jurkat T cells and FIR-APCs are pulsed with E7-peptide. (E) MFI fold change of GFP expression in coculture of Jurkat T cells and FIR-APCs pulsed with cognate or non-cognate peptides. n = 3 independent samples. Statistical significance of b and c is determined by the one-way ANOVA with post-hoc Tukey test, and statistical significance of e is determined by the t-test. Error bars indicate SD across replicates.
Addition of a TCR signaling reporter system to T cells further enriches for T cell–targeted FIR-APCs
At the T cell–APC immunological synapse, signaling occurs bidirectionally, with signals on the T-cell side mediated by the TCR complex, as well as by various co-stimulatory molecules[43]. We investigated whether quantifying TCR signaling could further help to identify highly interacting FIR-APC/Jurkat T cell aggregates. To evaluate this, we introduced to Jurkat T cells a construct that expressed mCherry fluorescent protein driven by an NFAT response element[11, 45], and the resulting cells were termed “Jurkat reporter T cells” (JR-T cells) (Fig. 4A). We found that treatment of JR-T cells with PMA and ionomycin, which activates T cells, led to mCherry expression (Supplementary Fig. S2B). Similarly, co-culturing E7 peptide–pulsed FIR-APCs with αE7-JR-T cells demonstrated antigen-specific reporting by JR-T cells (Supplementary Fig. S2C). When evaluating FIR-APC/JR-cell aggregates, we found that those with NFAT-activated JR-T cells demonstrated increased NFκB activation in FIR-APCs (Fig. 4B, C), indicating that highly interacting FIR-APC/JR-cell aggregates tended to have increased NFAT and NFκB activation. To identify an optimal culture duration, we quantified the kinetics of synapse formation and reporter signaling using positive and negative control FIR-APCs and JR-T cells (Supplementary Fig. S2D). We found that aggregation was higher in culture conditions where cognate TCRs and antigens were present, but was reduced when culture times were increased from 24 to 48 hours. While aggregation was reduced, reporter activity in both FIR-APCs and JR-T cells in aggregates was significantly higher in cognate culture conditions and was higher at 48 than at 24 hours. Therefore, a 48-hour co-culture was utilized for this system. We termed this system of quantifying aggregates with dual FIR-APC/JR-T cell reporter activity “T-synapse (Tsyn) reporting system”.
Fig. 4. Tsyn reporter system for detecting bidirectional signaling.
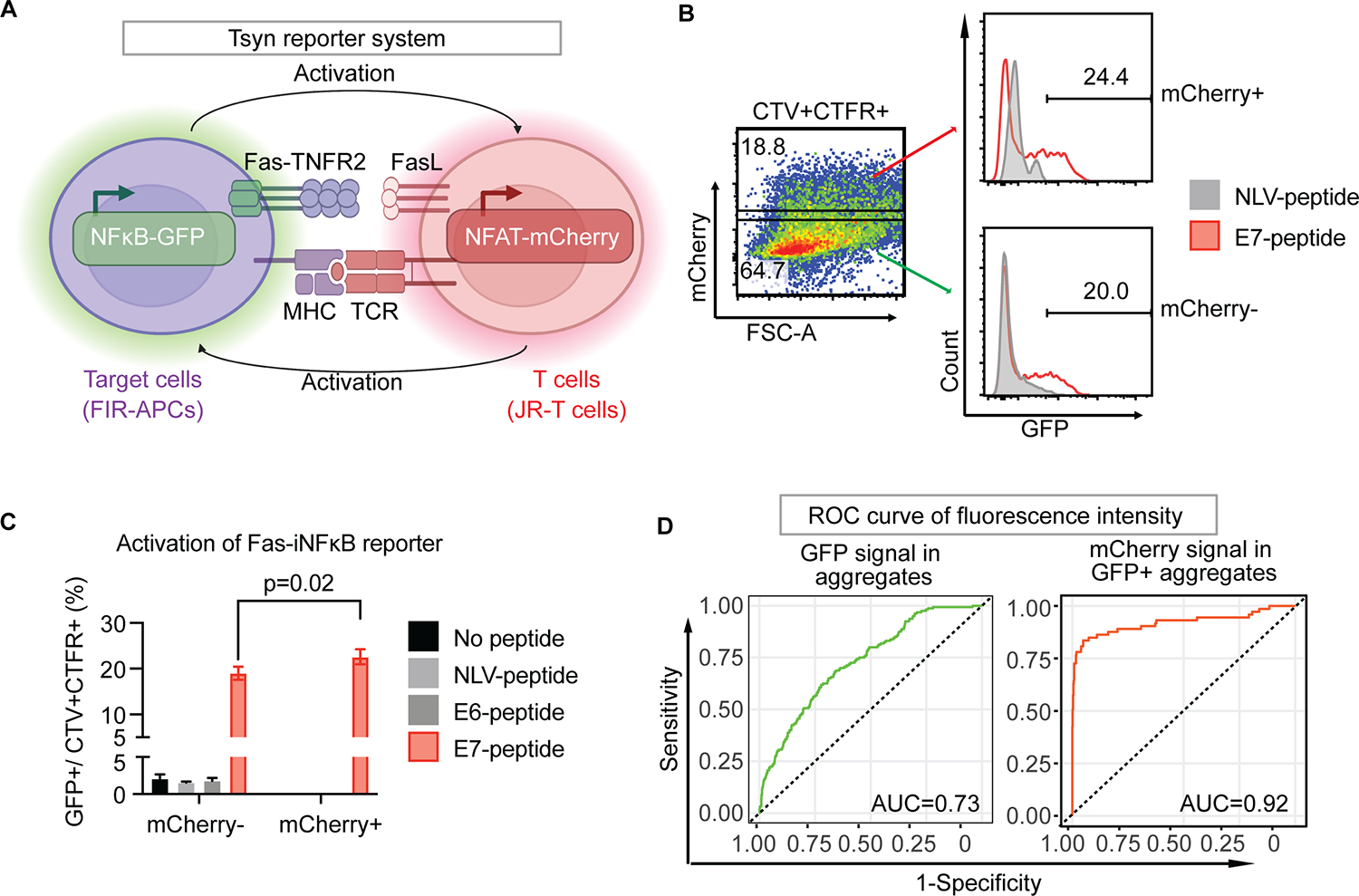
(A) Schematic of Tsyn-seq dual-reporter system. Simultaneous activation of Fas-iNFκB reporter in FIR-APCs and NFAT-mCherry reporter in JR-T cells through FasL/FasR and TCR/pMHC interactions, respectively. (B) Frequency of Fas-iNFκB reporter positive populations in NFAT-mCherry reporter positive or negative aggregates. E7-peptide-pulsed FIR-APCs are cocultured with αE7-JR-T cells (red line), while NLV-peptide-pulsed FIR-APCs are cocultured with αE7-JR-T cells as control (grey line). Gates demarcating mCherry positive (orange arrow) and negative (green arrow) populations among the CTV/CTFR double-positive aggregates are shown. (C) Coculture of JR-T cells and FIR-APCs pulsed with cognate and non-cognate peptides. Statistical significance is determined by the Mann-Whitney U test. Error bars indicate SD across 5 replicates. (D) ROC curve analysis of dual-reporter signaling in the coculture of (B). Green ROC curve represents GFP fluorescence intensity of aggregates population (left panel). Orange ROC curve represents mCherry fluorescence intensity of GFP positive population in aggregates population (right panel).
We next evaluated the ability of the Tsyn reporter system to correctly identify the small minority (1%) of FIR-APCs that expressed the correct cognate antigen (E7) from within a large population of antigen-negative FIR-APCs (Supplementary Fig. S3A). The enrichment rate was quantified by comparing the percentage of E7-FIR-APCs before and after gating for Tsyn reporter activity. We observed less heterogeneous enrichment results when cells were co-cultured in a flask compared to a 96-well U-bottom plate (Supplementary Fig. S3B); therefore, we incorporated co-culture in a flask into the standard protocol. After comparing two different FIR-cell to JR-cell ratios, 1:3 and 1:10, and observing similar results, we continued to use a 1:3 ratio (Supplementary Fig. S3B). Additionally, to further test the sensitivity of this system, we tested a smaller minority of E7-FIR-APCs (0.1%) and found that the percentage of these cells was approximately 2% in the sorted population, representing an approximately 20-fold enrichment (Supplementary Fig. S3C). Finally, to characterize the ROC of this system, we evaluated the sensitivity and specificity of the system in correctly identifying antigen-expressing FIR-APCs across all possible GFP fluorescence thresholds (Fig. 4D). This allowed us to identify an optimal GFP threshold, after which we tested all the possible mCherry thresholds. We found that the area under the ROC curve (AUC) of the Tsyn reporting system increased from 0.73 to 0.92 with the addition of NFAT-mCherry fluorescence data to NFκB-GFP fluorescence data. We established a GFP gating strategy targeting the highest 10% of the population with respect to GFP expression and observed a substantial increase in the proportion of CTV-labeled spike-in E7-FIR-APCs within this gate, as compared to the control conditions with αNLV-JR-T cells (Supplementary Fig. S3D). This demonstrated that applying a strategy of selecting the highest GFP-expressing FIR-APCs after co-culture should substantially enrich for FIR-APCs expressing a cognate antigen recognized by JR-T cells.
Finally, we performed a series of CRISPR/Cas9 gene editing procedures to remove MHC class I genes (HLA-A, B, and C) from the FIR-APCs. This was performed to eliminate the possibility of antigen presentation by more than one MHC class I molecule at a time. Similarly, we removed TCR genes (TCRα and TCRβ) from JR-T cells to eliminate the possibility of detecting TCR–pMHC interactions mediated by the endogenous TCR present in Jurkat T cells[46] rather than by the expressed TCR of interest. After removing MHC class I genes from the FIR-APCs, we re-expressed HLA-A*02:01. We termed these “cleaned-up” versions of reporter cells “Tsyn reporting system version 2” (Tsyn V.2) (Fig. 5A).
Fig. 5. Evaluating effects of non-cognate HLA and TCR genes in the Tsyn reporting system.
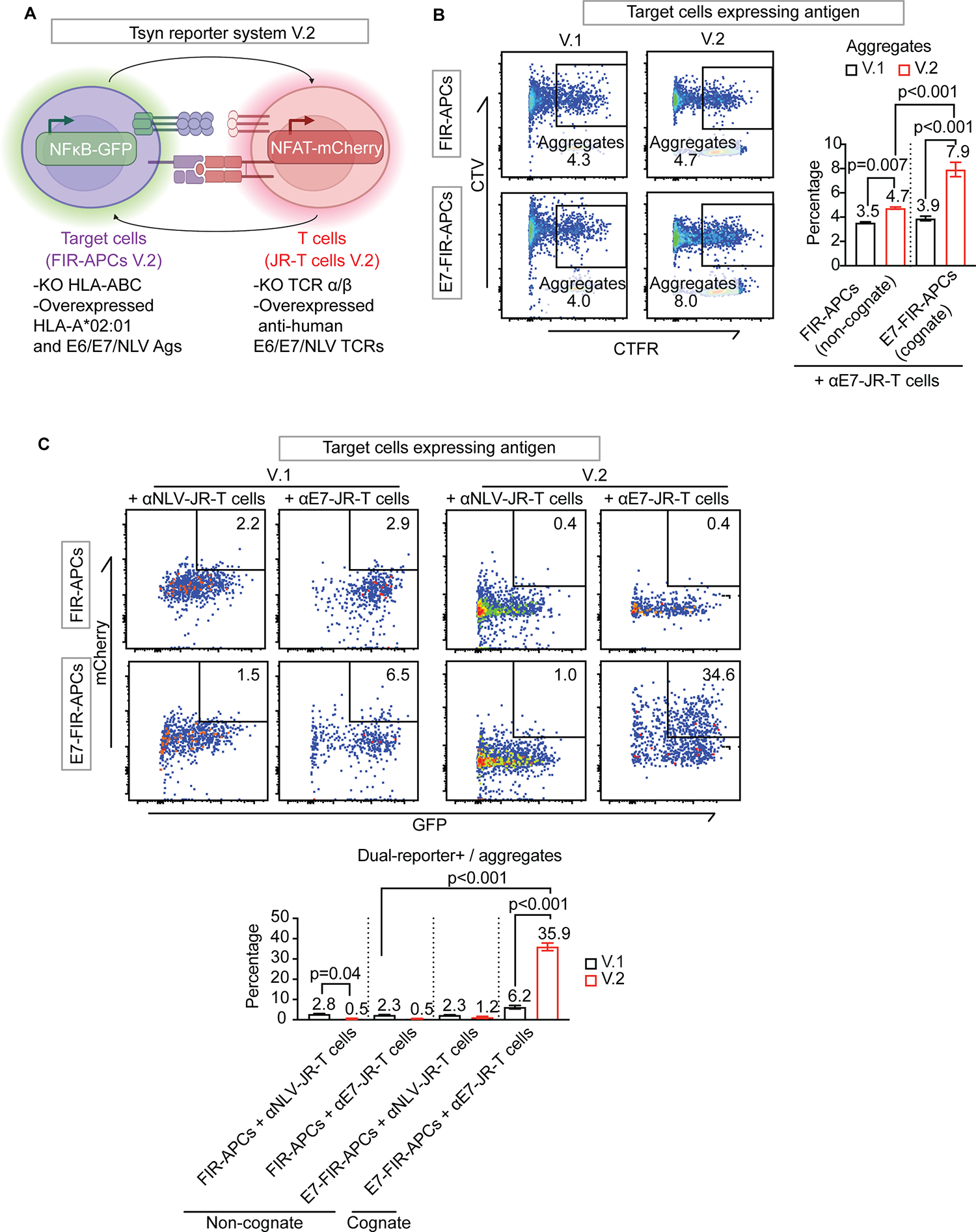
(A) Schematic: Gene editing to remove non-cognate HLA and TCR genes from the Tsyn reporting system. In FIR-APCs (green), HLA-A, B, and C genes are removed with gene editing, followed by lentiviral re-introduction of HLA-A*02:01. In JR-T cells (red), endogenous TCRα and TCRβ genes are removed with gene editing, followed by overexpression of αE6, αE7 or αNLV TCRs. V.1 refers to the Tsyn reporting system prior to gene editing; V.2 refers to the Tsyn reporting system after gene editing. (B) Representative flow cytometric plots and frequencies of aggregates in the indicated Tsyn reporting system. αE7-JR-T cells are cocultured with FIR-APCs with or without endogenous E7 antigen expression. (C) Representative flow cytometric plots and frequencies of dual-reporter positive aggregate events in the indicated Tsyn reporting system. αE7-JR or αNLV-JR-T cells are cocultured with FIR-APCs with or without endogenous E7 antigen expression. KO, knock out. Statistical significance is determined by the one-way ANOVA with post-hoc Tukey test. Error bars indicate SD across 3 replicates.
We evaluated the performance of the Tsyn V.2 system with peptide-pulsed FIR-APCs. Upon evaluation of the combined system, including aggregation and dual reporting, V.2 showed similar Tsyn reporting in non-cognate antigen settings and increased Tsyn reporting in the cognate antigen setting (3.1% versus 6.7%, Supplementary Fig. S2C). These results indicate that in the setting of peptide pulsing, which saturates pMHC complexes with the peptide antigen of interest[47], Tsyn V.2 showed slightly improved performance compared to Tsyn V.1.
We then comparatively evaluated the performance of Tsyn V.1 and V.2 using FIR-APCs overexpressing protein antigens. Co-culture of E7-expressing FIR-APCs with cognate αE7-JR-T cells demonstrated that Tsyn V.2 resulted in a higher percentage of aggregates (Fig. 5B) and an increased population of dual-reporter positive events (Fig. 5C). At the same time, with Tsyn V.2, in control conditions where cognate TCR–pMHC interactions were absent, proportions of aggregates and dual-reporter positive populations were significantly reduced (Fig. 5C). In summary, we found that gene editing to remove extraneous MHC class I and TCR genes substantially improved the performance of the Tsyn reporter system.
Genome-wide screening of antigens from a tumor cDNA library
Finally, we asked if, among the various antigens expressed by a tumor cell line, the use of the Tsyn reporter system could allow enrichment of a cognate antigen recognized by a TCR of interest. To investigate this, we examined the HPV16-positive CaSki cervical cancer cell line, which is known to harbor E7 and E6 genes[48]. We found that CaSki cells, when cultured with αE7-JR-T cells or E6-specific JR-T cells (αE6-JR-T cells), resulted in NFAT activation, indicating the expression of each of these antigens (Fig. 6A, B). We harvested mRNA from CaSki cells and performed reverse transcription to generate cDNA. Using Gateway technology[49], we constructed a CaSki cDNA–derived lentiviral expression library with extensive genome coverage (3×106 primary clones) and an average clone length of ~1. 5 Kb. We introduced this library into FIR-APCs to create a CaSki-FIR-APC library (Supplementary Fig. S4A) and confirmed by PCR the presence of the E7 gene within the amplified lentiviral inserts of the CaSki-FIR-APC library gDNA (Supplementary Fig. S4B).
Fig. 6. Antigen screening of αE7-JR-T cells against a CaSki cervical cancer cell line cDNA library using the Tsyn-seq system.
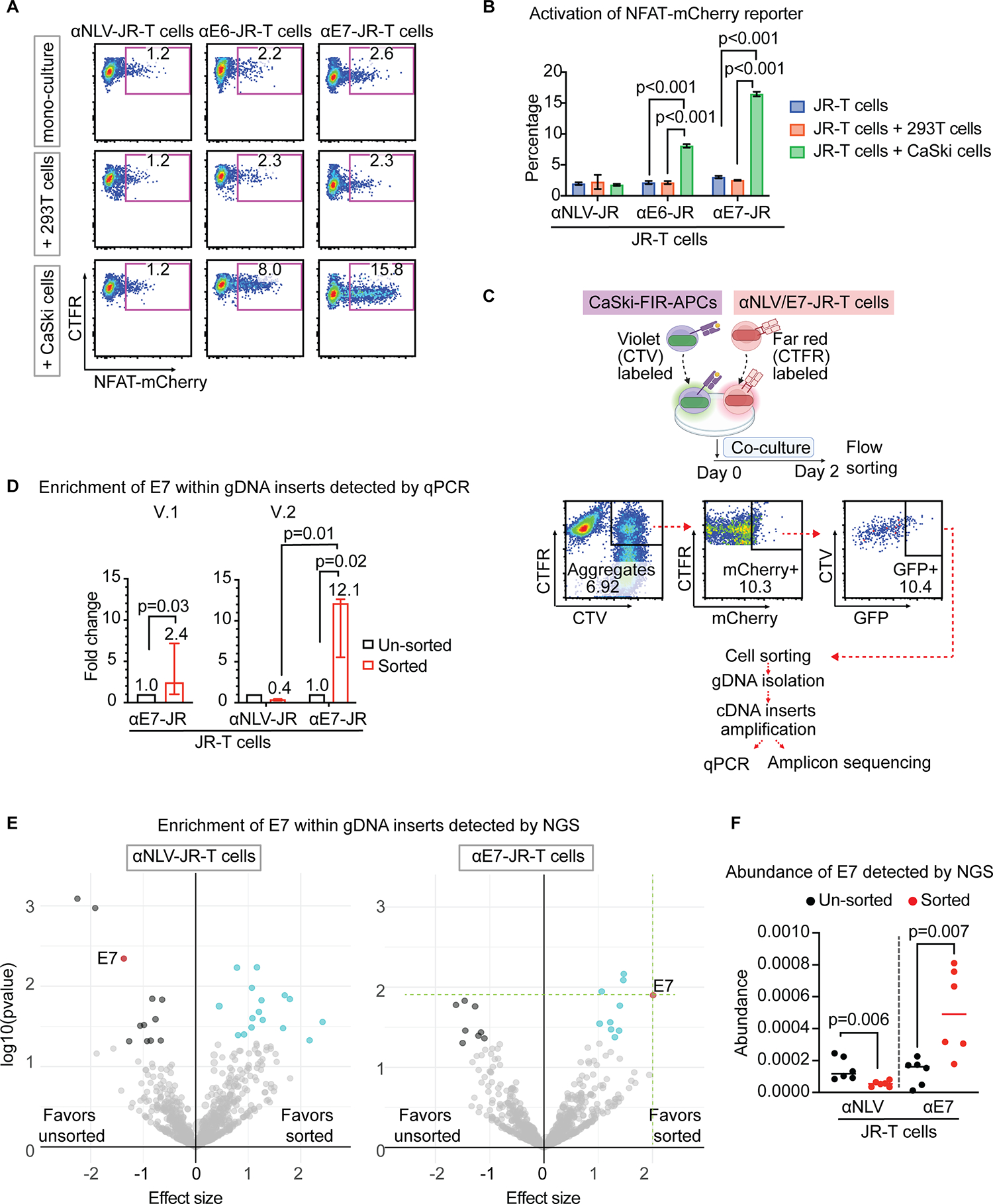
(A) Representative flow cytometric plots of NFAT-mCherry reporter activation in αE7-JR-T cells. αE7-JR-T cells are cultured alone or with control 293T cells (HLA-A*02:01-positive, HPV16-negative) or CaSki cells (HLA-A*02:01-positive, HPV16-positive). (B) Summary of frequencies in A. Error bars indicate SD across 6 replicates. Statistical significance is determined by the one-way ANOVA with post-hoc Tukey test. (C) Schematic: Antigen screening workflow of the Tsyn-seq system. CTV-labeled CaSki-FIR-APCs (purple) are cocultured with CTFR-labeled TCR-JR-T cells (red) for 2 days. Interacting CaSki-FIR-APCs and αE7-JR-T cells are sorted as dual-reporter positive CTV/CTFR double-positive aggregates, followed by gDNA isolation and cDNA insert amplification. The amplified product is then evaluated using qPCR as well as deep sequencing. (D) E7 gene quantification from cDNA inserts enriched following Tsyn-reporter testing with αE7-JR-T cells, quantified by qPCR. n = 3 independent samples. Statistical significance is determined by the t-test. Error bars indicate 95% IC across three replicates. (E) Deep sequencing analysis of cDNA inserts enriched following Tsyn-reporter testing with αE7-JR-T cells and αNLV-JR-T cells. The genes encoding the cognate E7 antigen (red) are indicated. Each dot represents one gene, with the y-axis plotting its log10 p-value, and the x-axis plotting its effect size. Both p-value and effect sizes are quantified with DESeq2 across 6 biological replicates in the sorted population relative to that in the input library prior to flow-sort enrichment. Genes with p value < 0.05 and effect size > 0 are marked cyan, and genes with p value < 0.05 and effect size < 0 are marked black. The size of the dot stands for the abundance of gene. (F) E7 gene quantification from cDNA inserts enriched following Tsyn-reporter testing with αE7-JR-T cells and control αNLV-JR-T cells. n = 6 independent samples. Statistical significance is determined using DESeq2.
We then co-cultured αE7-JR-T cells with the CaSki-FIR-APC library, followed by FACS enrichment for aggregates that were dual-reporter-positive (Fig. 6C). Lentiviral cDNA inserts were amplified from the extracted gDNA[49]. Copies of E7 genes were quantified by qPCR to allow us to determine the enrichment produced by Tsyn reporter system. We comparatively evaluated Tsyn V.1 and V.2 and found that Tsyn V.1 produced a normalized absolute abundance (sorted gene copies normalized to unsorted gene copies) of 2.4, whereas Tsyn V.2 achieved a statistically higher normalized absolute abundance of 12.1 (Fig. 6D). We also evaluated Tsyn V.2 in co-culture with non-cognate NLV-specific JR-T cells (αNLV-JR-T) as a control and found no enrichment of E7, with a significant reduction in the abundance of E7 sequences in the sorted sample.
In addition to qPCR to quantify gene copies of E7, we quantitatively profiled cDNA inserts in gDNA from Tsyn V.2 enriched CaSki-FIR-APCs library cells using amplicon deep sequencing (Tsyn-seq). Sequencing read counts of individual genes were compared between unsorted samples and sorted samples to identify genes enriched by the Tsyn reporter system. We identified the cognate E7 antigen as the most enriched gene among abundant and statistically significant genes, while there was no enrichment for E7 after sorting CaSki-FIR-APCs co-cultured with control αNLV-JR-T cells (Fig. 6E, F). These results demonstrate that Tsyn-seq can enrich cognate antigens recognized by an HPV16 E7-specific TCR from the transcriptome of a cervical cancer cDNA library.
DISCUSSION
Tsyn-seq is a high-throughput screening platform for identifying antigens recognized by TCRs of interest. It relies on the identification of functional immune synapses between T cells and target cells. This is achieved through a three-part strategy: 1) the detection of aggregated T cells and target cells, 2) quantifying T cell to target cell signaling and 3) quantifying target cell to T-cell signaling. We found that signaling reporter activity was increased in aggregates of cognate APCs and T cells, which is consistent with previous observations that physically interacting cells show increased immune activation[44].
A major strength of this system, in contrast to other TCR antigen screening methods that rely on quantifying TCR–pMHC binding[5, 6] or are purely in silico[50], is the use of T-cell functionality as a screening mechanism. Tsyn-reporting quantifies T-cell effector cytotoxicity, which requires close cell–cell interactions. The reliance on high-throughput functionality as the primary readout has potential advantages in those genes that are enriched, which have a higher likelihood of being identified as true functional targets, hence enhancing the efficiency of the workflow.
In contrast to prior functional T cell–antigen screening tools that detect granzyme B[6] or effector cytokines[51], Tsyn-seq relies on detecting Fas pathway signaling, which requires interactions between two cell-surface molecules, unlike detection of perforin/granzyme granules or cytokines, which are released by the T cell. To enable detection of Fas signaling, we developed a construct in which we replaced the apoptosis-inducing intracellular death domain of Fas with the intracellular domain of TNFR2, which also signals via NFκB but does not induce apoptosis. We also chose to simultaneously detect signaling within T cells by quantifying NFAT activity. These combined bidirectional signaling criteria markedly improved the performance of this enrichment strategy, as quantified by ROC curve analysis and enrichment indices.
To reduce potential cross-reactivity mediated by other HLA alleles expressed by the target cell line and the endogenous TCR expressed by the Jurkat T cell line, we used CRISPR/Cas9 gene editing to prevent expression of these genes. We found that this led to a reduction in the amount of background reporter activity in the setting of T cells cultured with non-cognate antigen-expressing target cells, likely resulting from reducing off-target TCR–pMHC interactions.
A potential advantage of Tsyn-seq is that it does not depend on already-generated antigen libraries[6, 52]. Instead, Tsyn-seq can screen for antigens present in a cDNA library that can be custom-generated from individual samples using commercially available kits. Hence this system, in contrast to previous APC library screening approaches, can offer increased ease of screening for antigens present in specific tissue samples, such as samples derived from genetically less characterized species (e.g., novel viruses), as well as samples for which commercially available genome libraries are not available. One application could be identifying antigens present in patient-derived tumor samples, where personal neoantigens or viral antigens could likely be of high interest. Indeed, we found that Tsyn-seq can correctly identify the cognate HPV16 E7 antigen as the most enriched antigen of those recognized by an HPV16 E7-specific TCR[38]. Had the E7 antigen not been included in a pre-existing antigen library, this finding could have been likely missed.
Another advantage of this approach, in contrast to traditional approaches to identify antigens from cDNA libraries, is the increased ease and reduced costs associated with performing the screen. Prior methods that screened genome-wide libraries have utilized individual microcultures of independent transfections to screen thousands of unique constructs[53]. Tsyn-seq is considerably less labor-intensive, requiring a co-culture performed using T cells and a mixture of target cells transduced with a cDNA library in a single flask, followed 2 days later by flow-sorting, DNA isolation, and amplicon sequencing.
While unexplored in our current study, this system could be utilized to screen for potential unexpected targeting of self-antigens by TCR-based therapies. This could provide a convenient method to quantify the therapeutic potential and safety profile of novel TCR-based candidate therapies for further development.
One limitation to our data is the observation that after sorting CaSki-FIR-APCs co-cultured with control αNLV-JR-T cells, we found that the E7 gene was de-enriched. The reasons for this are not entirely clear but suggest that this TCR, which was identified for its affinity for the NLV epitope from the pp65 CMV antigen, may also recognize other elements from the CaSki cDNA library that are enriched relative to E7. An important future direction will be demonstrating that this system can identify antigens recognized by TCRs where the cognate antigen is not yet known. Another current limitation includes the ability to only screen a single TCR at a time, in contrast to allowing the use of polyclonal primary T cells. Additionally, we have currently only established that Tsyn-seq can function for human CD8+ T cell TCRs that recognize peptides presented by HLA-A*02:01. It will be important to validate this system for other MHC-I HLA molecules, and to explore if the system could also function with murine CD8+ T cells, or MHC-II molecules presenting to human or murine CD4-derived TCRs. Finally, it is also important to consider that the reliance on 293T cell lines as APCs could produce biases in our results, due to inherent properties with respect to non-canonical transcription[54], post-translational modifications[55], or proteasomal behavior[56], all of which can have downstream impacts on peptide antigen presentation. Thus, validation of any identified antigens using other sources of APCs will be important to include in the workflow.
We anticipate that despite these current limitations, Tsyn-seq will facilitate the identification of target antigens recognized by the TCRs of interest. Improving the ease of identifying antigens recognized by T cells will be important for advancing our knowledge of T-cell antigen recognition as well as accelerating the development of T cell–based therapies for a variety of clinical indications, including infectious diseases, transplantation, autoimmune disorders, and cancer[57]. In the field of cancer immunology, this system could be applied to identify neoantigens for personalized vaccines and could also be utilized as a screening tool in the development of TCR-based therapies to evaluate potential off-target reactions that could lead to toxicity.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Synopsis:
Genome-wide identification of antigens targeted by individual TCRs can be challenging. The authors developed a new platform (Tsyn-seq) that allows for traditional cDNA library–based screening of antigen targets but is adapted for both increased ease and throughput.
ACKNOWLEDGMENTS:
We thank the MDACC Advanced Cytometry and Sorting Facility (ACSF), funded by the Cancer Center Support Grant NCI # P30CA16672. We also thank the MDACC ATGC Facility funded by the Core grant CA016672 (SMF). The results shown here are partly based on data generated by TCGA Research Network: https://www.cancer.gov/tcga.
Funding information:
This study was supported by the MD Anderson Cancer Center Advanced Cytometry and Sorting Facility and the ATGC Facility, both funded by the Cancer Center Support Grant NCI # P30CA16672.
Footnotes
Competing interests: RRJ has served as a consultant or advisory board member for MaaT Pharma, LISCure, Seres, and Prolacta, and has received patent license fees or stock options from Seres. All other authors declare no conflicts of interest.
REFERENCES
- 1.Chaplin DD, Overview of the immune response. J Allergy Clin Immunol, 2010. 125(2 Suppl 2): p. S3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shah K, et al. , T cell receptor (TCR) signaling in health and disease. Signal Transduct Target Ther, 2021. 6(1): p. 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chandran SS and Klebanoff CA, T cell receptor-based cancer immunotherapy: Emerging efficacy and pathways of resistance. Immunol Rev, 2019. 290(1): p. 127–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klebanoff CA, Rosenberg SA, and Restifo NP, Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med, 2016. 22(1): p. 26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li G, et al. , T cell antigen discovery via trogocytosis. Nat Methods, 2019. 16(2): p. 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kula T, et al. , T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes. Cell, 2019. 178(4): p. 1016–1028 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kisielow J, Obermair FJ, and Kopf M, Deciphering CD4(+) T cell specificity using novel MHC-TCR chimeric receptors. Nat Immunol, 2019. 20(5): p. 652–662. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, et al. , Using a baculovirus display library to identify MHC class I mimotopes. Proc Natl Acad Sci U S A, 2005. 102(7): p. 2476–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Birnbaum ME, et al. , Deconstructing the peptide-MHC specificity of T cell recognition. Cell, 2014. 157(5): p. 1073–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma G, Rive CM, and Holt RA, Rapid selection and identification of functional CD8(+) T cell epitopes from large peptide-coding libraries. Nat Commun, 2019. 10(1): p. 4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siewert K, et al. , Unbiased identification of target antigens of CD8+ T cells with combinatorial libraries coding for short peptides. Nat Med, 2012. 18(5): p. 824–8. [DOI] [PubMed] [Google Scholar]
- 12.Huber M, et al. , In mitosis integrins reduce adhesion to extracellular matrix and strengthen adhesion to adjacent cells. Nat Commun, 2023. 14(1): p. 2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park JS, et al. , Targeting PD-L2-RGMb overcomes microbiome-related immunotherapy resistance. Nature, 2023. 617(7960): p. 377–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hyer ML, et al. , Intracellular Fas ligand expression causes Fas-mediated apoptosis in human prostate cancer cells resistant to monoclonal antibody-induced apoptosis. Mol Ther, 2000. 2(4): p. 348–58. [DOI] [PubMed] [Google Scholar]
- 15.Knight RR, et al. , Human beta-cell killing by autoreactive preproinsulin-specific CD8 T cells is predominantly granule-mediated with the potency dependent upon T-cell receptor avidity. Diabetes, 2013. 62(1): p. 205–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyama T, et al. , Highly functional T-cell receptor repertoires are abundant in stem memory T cells and highly shared among individuals. Sci Rep, 2017. 7(1): p. 3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Draper LM, et al. , Targeting of HPV-16+ Epithelial Cancer Cells by TCR Gene Engineered T Cells Directed against E6. Clin Cancer Res, 2015. 21(19): p. 4431–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hahne F, et al. , flowCore: a Bioconductor package for high throughput flow cytometry. BMC Bioinformatics, 2009. 10: p. 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanley JA, Receiver operating characteristic (ROC) methodology: the state of the art. Crit Rev Diagn Imaging, 1989. 29(3): p. 307–35. [PubMed] [Google Scholar]
- 20.Hanley JA and McNeil BJ, A method of comparing the areas under receiver operating characteristic curves derived from the same cases. Radiology, 1983. 148(3): p. 839–43. [DOI] [PubMed] [Google Scholar]
- 21.Rognes T, et al. , VSEARCH: a versatile open source tool for metagenomics. PeerJ, 2016. 4: p. e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buchfink B, Xie C, and Huson DH, Fast and sensitive protein alignment using DIAMOND. Nat Methods, 2015. 12(1): p. 59–60. [DOI] [PubMed] [Google Scholar]
- 23.Su Q and Igyarto BZ, One-step artificial antigen presenting cell-based vaccines induce potent effector CD8 T cell responses. Sci Rep, 2019. 9(1): p. 18949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barry M and Bleackley RC, Cytotoxic T lymphocytes: all roads lead to death. Nat Rev Immunol, 2002. 2(6): p. 401–9. [DOI] [PubMed] [Google Scholar]
- 25.Lee MN and Meyerson M, Antigen identification for HLA class I- and HLA class II-restricted T cell receptors using cytokine-capturing antigen-presenting cells. Sci Immunol, 2021. 6(55). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oda SK, et al. , A Fas-4–1BB fusion protein converts a death to a pro-survival signal and enhances T cell therapy. J Exp Med, 2020. 217(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagata S, Apoptosis by death factor. Cell, 1997. 88(3): p. 355–65. [DOI] [PubMed] [Google Scholar]
- 28.LA OR, et al. , Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature, 2009. 461(7264): p. 659–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu F, et al. , NF-kappaB directly regulates Fas transcription to modulate Fas-mediated apoptosis and tumor suppression. J Biol Chem, 2012. 287(30): p. 25530–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott FL, et al. , The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature, 2009. 457(7232): p. 1019–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang DC, et al. , Activation of Fas by FasL induces apoptosis by a mechanism that cannot be blocked by Bcl-2 or Bcl-x(L). Proc Natl Acad Sci U S A, 1999. 96(26): p. 14871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boulares AH, et al. , Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem, 1999. 274(33): p. 22932–40. [DOI] [PubMed] [Google Scholar]
- 33.Frassanito MA, et al. , Fas/Fas ligand (FasL)-deregulated apoptosis and IL-6 insensitivity in highly malignant myeloma cells. Clin Exp Immunol, 1998. 114(2): p. 179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mollanoori H, et al. , CRISPR/Cas9 and CAR-T cell, collaboration of two revolutionary technologies in cancer immunotherapy, an instruction for successful cancer treatment. Hum Immunol, 2018. 79(12): p. 876–882. [DOI] [PubMed] [Google Scholar]
- 35.Ashkenazi A, Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat Rev Cancer, 2002. 2(6): p. 420–30. [DOI] [PubMed] [Google Scholar]
- 36.Park YC, et al. , Structural basis for self-association and receptor recognition of human TRAF2. Nature, 1999. 398(6727): p. 533–8. [DOI] [PubMed] [Google Scholar]
- 37.Bruggeman LA, et al. , TNFR2 interposes the proliferative and NF-kappaB-mediated inflammatory response by podocytes to TNF-alpha. Lab Invest, 2011. 91(3): p. 413–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin BY, et al. , Engineered T cells targeting E7 mediate regression of human papillomavirus cancers in a murine model. JCI Insight, 2018. 3(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ellis JM, et al. , Frequencies of HLA-A2 alleles in five U.S. population groups. Predominance Of A*02011 and identification of HLA-A*0231. Hum Immunol, 2000. 61(3): p. 334–40. [DOI] [PubMed] [Google Scholar]
- 40.Abraham RT and Weiss A, Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat Rev Immunol, 2004. 4(4): p. 301–8. [DOI] [PubMed] [Google Scholar]
- 41.Nagarsheth NB, et al. , TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat Med, 2021. 27(3): p. 419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen G, et al. , Sequence and Structural Analyses Reveal Distinct and Highly Diverse Human CD8(+) TCR Repertoires to Immunodominant Viral Antigens. Cell Rep, 2017. 19(3): p. 569–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dustin ML, The immunological synapse. Cancer Immunol Res, 2014. 2(11): p. 1023–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giladi A, et al. , Dissecting cellular crosstalk by sequencing physically interacting cells. Nat Biotechnol, 2020. 38(5): p. 629–637. [DOI] [PubMed] [Google Scholar]
- 45.Lyakh L, Ghosh P, and Rice NR, Expression of NFAT-family proteins in normal human T cells. Mol Cell Biol, 1997. 17(5): p. 2475–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heemskerk MH, et al. , Redirection of antileukemic reactivity of peripheral T lymphocytes using gene transfer of minor histocompatibility antigen HA-2-specific T-cell receptor complexes expressing a conserved alpha joining region. Blood, 2003. 102(10): p. 3530–40. [DOI] [PubMed] [Google Scholar]
- 47.Sykulev Y, et al. , Evidence that a single peptide-MHC complex on a target cell can elicit a cytolytic T cell response. Immunity, 1996. 4(6): p. 565–71. [DOI] [PubMed] [Google Scholar]
- 48.Zheng ZM, et al. , Splicing of a cap-proximal human Papillomavirus 16 E6E7 intron promotes E7 expression, but can be restrained by distance of the intron from its RNA 5’ cap. J Mol Biol, 2004. 337(5): p. 1091–108. [DOI] [PubMed] [Google Scholar]
- 49.Katzen F, Gateway((R)) recombinational cloning: a biological operating system. Expert Opin Drug Discov, 2007. 2(4): p. 571–89. [DOI] [PubMed] [Google Scholar]
- 50.Lee CH, et al. , Predicting Cross-Reactivity and Antigen Specificity of T Cell Receptors. Front Immunol, 2020. 11: p. 565096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brosterhus H, et al. , Enrichment and detection of live antigen-specific CD4(+) and CD8(+) T cells based on cytokine secretion. Eur J Immunol, 1999. 29(12): p. 4053–9. [DOI] [PubMed] [Google Scholar]
- 52.Joglekar AV, et al. , T cell antigen discovery via signaling and antigen-presenting bifunctional receptors. Nat Methods, 2019. 16(2): p. 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.van der Bruggen P, et al. , A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science, 1991. 254(5038): p. 1643–7. [DOI] [PubMed] [Google Scholar]
- 54.Lu SX, et al. , Pharmacologic modulation of RNA splicing enhances anti-tumor immunity. Cell, 2021. 184(15): p. 4032–4047 e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Srivastava AK, et al. , Post-Translational Modifications in Tumor-Associated Antigens as a Platform for Novel Immuno-Oncology Therapies. Cancers (Basel), 2022. 15(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morozov AV and Karpov VL, Proteasomes and Several Aspects of Their Heterogeneity Relevant to Cancer. Front Oncol, 2019. 9: p. 761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ott PA, et al. , An immunogenic personal neoantigen vaccine for patients with melanoma. Nature, 2017. 547(7662): p. 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files or from the corresponding author upon reasonable request. The sequencing data generated in this study are publicly available in Sequence Read Archive (SRA) at PRJNA1034661.