

Abstract
Tumor molecular datasets are becoming increasingly complex, making it nearly impossible for humans alone to effectively analyze them. Here, we demonstrate the power of using machine learning (ML) to analyze a single-cell, spatial, and highly multiplexed proteomic dataset from human pancreatic cancer and reveal underlying biological mechanisms that may contribute to clinical outcome. We designed a multiplex immunohistochemistry antibody panel to compare T-cell functionality and spatial localization in resected tumors from treatment-naive patients with localized pancreatic ductal adenocarcinoma (PDAC) with resected tumors from a second cohort of patients treated with neoadjuvant agonistic CD40 (anti-CD40) monoclonal antibody therapy. In total, nearly 2.5 million cells from 306 tissue regions collected from 29 patients across both cohorts were assayed, and over 1,000 tumor microenvironment (TME) features were quantified. We then trained ML models to accurately predict anti-CD40 treatment status and disease-free survival (DFS) following anti-CD40 therapy based upon TME features. Through downstream interpretation of the ML models’ predictions, we found anti-CD40 therapy reduced canonical aspects of T-cell exhaustion within the TME, as compared to treatment-naive TMEs. Using automated clustering approaches, we found improved DFS following anti-CD40 therapy correlated with an increased presence of CD44+CD4+ Th1 cells located specifically within cellular neighborhoods characterized by increased T-cell proliferation, antigen-experience, and cytotoxicity in immune aggregates. Overall, our results demonstrate the utility of ML in molecular cancer immunology applications, highlight the impact of anti-CD40 therapy on T cells within the TME, and identify potential candidate biomarkers of DFS for anti-CD40-treated patients with PDAC.
Keywords: tumor microenvironment, machine learning, T cell, pancreatic ductal adenocarcinoma, spatial proteomics
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive treatment-refractory cancers with a median overall survival rate of just months (1). Thus, there is a critical need for improved understanding of the immunobiology of PDAC to inform future treatment strategies for this disease. Recent reports reveal immunological responses in PDAC are induced via approaches that promote priming of T-cell responses against PDAC, such as occurs following agonistic CD40 monoclonal antibodies (anti-CD40) (2) and mRNA vaccination (3) strategies. We and others have previously reported that anti-CD40 binds to CD40 on dendritic cells (DCs), thereby licensing DCs to subsequently enhance T-cell activation and bolster antitumor immunity (4). In addition, we previously described global immune contexture of the PDAC tumor microenvironment (TME) at baseline and after anti-CD40 therapy and found anti-CD40-treated TMEs contained reduced densities of M2-like tumor-associated macrophages, increased DC maturation, and increased T-cell enrichment (2, 5). However, little is known regarding selective impact of anti-CD40 therapy on granular T-cell states within the TME, and studies interrogating how anti-CD40 stimulation sculpts the T-cell response specifically (6, 7) are needed to optimize anti-CD40 use in the clinical setting.
Upon antigen stimulation, T cells exist along a spectrum of diverse differentiation states with varying functionalities (8). On one end, T cells possess stem-cell-like plasticity, accompanied by memory, proliferative, and cytotoxic capabilities, and are identified by expression of T-BET, and/or TCF-1 (9, 10). On the other end, T cells are exhausted and/or dysfunctional and express TOX1 and/or EOMES (11). Along the spectrum, expressed in varying combinations, T cells express immune checkpoint molecules such as PD-1, LAG-3, and TIM3, with increased expression of immune checkpoint molecules correlating with more exhausted T cells (12). These partially exhausted T cells are susceptible to reinvigoration by immune checkpoint blockade (ICB) and regain the ability to proliferate and produce effector cytokines (13). However, terminally differentiated T cells expressing TOX1 or EOMES are resistant to rescue by ICB, and fail to proliferate or exert cytotoxic activity (14). Flow cytometric analyses of T cells in preclinical tumor models or tissues following viral infection have elucidated notable T-cell states; however, characterizations of effector versus exhausted T-cell phenotypes from tumors in patients are scant. Moreover, with the advent of single-cell sequencing approaches, the diversity of T-cell subsets within tumors is seemingly endless (15, 16). We sought to clarify T-cell characteristics within the PDAC TME and identify subsets associated with therapeutic anti-CD40 responses. Recognizing that both the cellular composition and spatial organization of cells is a critical metric associated with therapeutic response and clinical outcome (17–22), we investigated the impact of anti-CD40 therapy on the complex spatial contexture of T cells within the PDAC TME and associated survival.
We previously developed a multiplex immunohistochemistry (mIHC) single-cell spatial proteomics imaging platform to interrogate leukocyte heterogeneity and spatial landscape within various TMEs (5, 23, 24). Following a cyclical staining protocol, the mIHC platform iteratively deploys up to 30 antibodies on a single tissue specimen, thus preserving TME spatial context. Resulting data provide single-cell resolution maps quantifiable by a number of metrics that can be correlated with therapeutic response or clinical outcome. However, this reductionist approach does not combine or weight TME features together to capture biological complexities of the TME. Machine learning (ML), a form of artificial intelligence, can address TME complexity by creating computational models that weigh and combine data features to predict a given output. ML models can be used to make accurate predictions for new data and analyzed to identify the feature combinations most important in the predictions. As a result of this capability, ML is becoming widely utilized in precision oncology to decipher patterns in large datasets resulting from deep interrogations (21, 25–27).
Here, we leveraged ML to elucidate the frequency of various T-cell states in PDAC and investigated the impact of anti-CD40 therapy on those states. We first designed an mIHC antibody panel to deeply audit T-cell functionality and spatial organization in patients from either treatment-naive or neoadjuvant anti-CD40-treated PDAC cohorts. Using this mIHC panel, we generated a dataset of nearly 2.5 million cells with spatially-resolved single-cell phenotypic and functional measurements. Interrogation of this dataset presented a unique opportunity to elucidate: first, the types of T cells present at baseline in a treatment-refractory disease, and second, to what degree anti-CD40 therapy sustains T-cell functionality in situ, or instead promotes T-cell dysfunction that may limit potential use of anti-CD40 or other T-cell priming therapies in the clinical setting. Given the vast amount of spatially-resolved data and complexity of T-cell function in the TME, we leveraged ML approaches to discern new biological insights regarding T cells in the pancreatic TME and their association with clinical outcome for pancreatic cancer patients.
MATERIALS AND METHODS
Tissue Acquisition
Human PDAC tissue specimens from cohort 1 were obtained from patients with approval from the Oregon Pancreas Tissue Registry under Oregon Health & Science University (OHSU) IRB protocol #3609 and Dana Farber Harvard Cancer Center protocols #03–189 and #12–013. Cohort 1 consisted of 18 treatment-naive tumors, selected as a representative subset of a larger PDAC cohort (n=104 tumors) from a prior study evaluating global immune contexture (5). These 18 samples were selected based on cellular subsets not statistically differing from the full cohort in terms of leukocyte densities or patient survival durations (Supplementary Table S1). Specimens from cohort 2 were obtained from patients treated with neoadjuvant selicrelumab with approval under the IRBs of four sites across the United States involved in an open-label phase I clinical trial (Cancer Immunotherapy Trials Network CITN11-01; NCT02588443), including 8 patients with neoadjuvant selicrelumab alone and 3 patients with neoadjuvant selicrelumab combined with gemcitabine and nab-paclitaxel. Patients from this trial were combined into a single cohort for the present study, as the anti-CD40-treated cohort was previously analyzed in two separate cohorts for total immune contexture (2). Additionally, healthy tonsil and spleen samples were from the Knight Tissue Bank, collected under the OHSU IRB protocol #4918. All studies were conducted in accordance with the Declaration of Helsinki and written informed consent was obtained.
Multiplex Immunohistochemistry Image Acquisition and Analysis
Formalin-fixed, paraffin-embedded (FFPE) surgical tissue samples were sectioned and assessed using hematoxylin and eosin (H&E), as well as chromogen-based mIHC. Briefly, slides with 5 µm tissue sections were rehydrated as described (5), followed by 12 minutes in hematoxylin, 2 minutes in tap water, 10 seconds in eosin, and 2 minutes in tap water. Slides were then dehydrated and coverslipped prior to scanning and annotation by a pathologist. Next, using the pathologist annotations overlaid from the H&E-stained slides, serial sections were assessed with regard to histopathologic regions of interest annotated as tumor (T), immune aggregate (IA), tumor-adjacent stroma (TAS), or normal-adjacent pancreas (NAP), as defined by the pathologist (5). Regions were selected with the aim of maximizing both diversity of regions selected from each patient and the tissue area captured for each histopathologic region. Across all patients, a mean of 15.2% of tissue was analyzed per entire resection, ranging from 4.4% to 35.6%, and across the entire dataset, 14.6% of available tissue was analyzed (Supplementary Fig. S1A, Supplementary Table S2). Of note, our prior investigation of PDAC immune contexture found NAP regions to contain increased leukocyte density as compared to true healthy normal pancreas tissue collected from organ donors (5). Multiplex staining was performed on 5 µm sections, as previously described, and each stained image was scanned at 20x magnification on an Aperio AT2 scanner (Leica Biosystems) (5). The antibody panel used in the present study delineated 18 T-cell subpopulations and contained 10 functionality biomarkers to assess differentiation/exhaustion status on all T cells (TOX1, TIM3, TCF-1, CD38, PD-1, EOMES, CD39, CD44, LAG-3, and T-BET), as well as antibodies for proliferation (Ki-67) and cytotoxicity (granzyme B, GrzB) (Supplementary Fig. S1B, Supplementary Table S3). Additionally, the panel only broadly delineated epithelial cells, mesenchymal cells, B cells, and myeloid cells, given our previous efforts at describing those subsets in these same patient cohorts (2, 5, 19). Human tonsil and spleen were included in all rounds of mIHC as staining controls. Image processing was performed using previously described methods (5). Each region was registered to the final hematoxylin using Matlab Computer Vision Toolbox (The Mathworks, Inc., Natick, MA), color deconvolution and watershed-based nuclei segmentation was performed using ImageJ, and single cell mean intensity for each stain was quantified using Cell Profiler (28). Single biomarker positivity thresholds were set using FCS Express Image Cytometry RUO (De Novo Software, Glendale, CA) to visually validate protein biomarker expression overlaid on signal extracted images. Single cell classification was performed using R Statistical Software based on filtering exclusive populations in a defined hierarchy.
T-cell Phenotyping
423,317 T cells were identified by CD3 expression and then subsequently stratified by CD8α expression. CD8+ T cells were further classified as one of six cell states (TNAIVE, TEFF, TEM, TEMRA, TEX, or TTEX). Due to biomarker selection and positional restrictions within the cyclic multiplex panel, a CD4 antibody was not included. However, the majority (72%) of CD3+CD8– T cells were CD4+ as determined in a testing panel using a subset of the data (Supplementary Fig. S1C); therefore CD3+CD8– T cells are referred to as CD4+ T cells herein, although it is possible other minor lineages may be represented (15). Based on this schema, CD3+CD8– T cells were further evaluated; CD4+ Th1 cells were defined by T-BET+ expression, and further classified as one of three cell states (TEFF, TEM, or TEMRA). Only 6% of CD8+ T or CD4+ Th1 cells were phenotyped as one of these six T-cell states (TNAIVE, TEFF, TEM, TEMRA, TEX, or TTEX)(Supplementary Table S4). The other 94% of the T cells were labeled as TOTHER and were stratified based on expression of CD44, a canonical biomarker of prior cognate antigen experience (29). A population of T-BET–CD4+ Th cells (non-Th1-specific T helper cells) were also stratified into two cell states based upon CD44 expression. The remaining CD3+CD8– T cells were FOXP3+ regulatory T (TREG) cells and classified into three cell states (Naive TREG, mTREG, and T-BET+ TREG). All 18 T-cell states were included in downstream analyses given calculations from previous single-cell studies (30), and the fact that these populations were manually gated and thus represent real phenotypes of T cells present in the PDAC TME despite low numbers of certain T-cell states (Supplementary Table S4).
TME Feature Quantifications
Three approaches were used to quantify treatment-naive and anti-CD40-treated PDAC TMEs, resulting in a total of 1,252 TME features quantified per tissue region:
Cell State Densities
To identify the types and amounts of cell states present in the PDAC TME, densities of each of the 23 cell states for each tissue region assayed were quantified by dividing raw counts of cells (Supplementary Table S4) by tissue area of the region.
T-cell Functionality Barcodes
To investigate T-cell functionality, we assigned all T-cell states a “Functionality Barcode,” as defined by binary positive or negative expression of unique combinations of 10 T-cell functionality biomarkers (TOX1, TIM3, TCF-1, CD38, PD-1, EOMES, CD39, CD44, LAG-3, and T-BET). Among the 423,317 T cells present in the dataset, we identified expression of 961 unique barcodes (Supplementary Table S5).
Cell–Cell Spatial Interactions
To address spatial organization of cells in the PDAC TME, we leveraged the fact that mIHC preserves spatial context and quantified frequency of two cell states interacting, based on their cell centers being within 20 µm from each other, as previously reported (31). Total cell–cell interactions were normalized by dividing summed densities of cell states involved in the interactions to avoid skewing by cell states present in high abundances. We identified 268 unique pairs of cell–cell interactions present in the dataset (Supplementary Table S6).
Machine Learning Classifiers and Feature Importance Analyses
Elastic net (EN) classifier models were built using scikit-learn’s (32) LogisticRegression function to predict: 1) treatment status and 2) disease-free survival (DFS), from 1,252 TME features calculated herein. EN models perform well on datasets as generated in this present study where there are more data features than examples for learning (33). EN models use mathematical regularization approaches to identify and upweight the most informative subset of features to make model predictions while accounting for feature collinearity (34). Using regularization reduces model overfitting, which is important when there are a large number of data features and a limited number of examples. Further, this approach is unbiased, as no prior feature selection is performed. Instead, all 1,252 TME features were provided to the models, leveraging the EN algorithm’s ability to perform aggressive feature selection within model training.
Predictions were made on an individual region basis, rather than a patient basis, to maximize sample size and model robustness, as well as to mitigate tissue region selection biases. As such, tissue regions were not averaged across patients and were instead evaluated independently, thus: 1) reducing the impact of each region on overall model performance and 2) providing the models with more examples to extract biologically-meaningful information from the dataset. Separate models were created for regions of each histopathologic site to: 1) compare performance of models derived from different histopathologic sites; 2) identify where therapy was exerting greatest impact; and 3) mitigate broad variation in average tissue area from each histopathologic site. A leave-one-patient-out cross-validation approach was used to split the train and test sets. Thus, within each cross-validation loop, a new EN model was created and trained on all regions except those from one patient, and testing was then performed on regions from the patient withheld from training. This process was repeated until all patients were cycled through the test set. This approach prevents data leakage by ensuring regions from the same patient were not in both the train and test sets for one model, thus preventing the EN models from learning patient-specific features, which often artificially increases model accuracy.
Test set predictions were aggregated across all cross-validation loops to construct one final confusion matrix, from which performance of the models was assessed by calculating accuracy, F1 score, and area under the receiver operating characteristic curve (AUC). These metrics address both precision and recall (F1 score), in addition to the true positive rate and false positive rate (AUC) – these are often used to assess performance of classifier models. Model overfitting was mitigated by using the same model hyperparameters across cross-validation loops. The penalty term was set to “elasticnet,” and the “l1_ratio” hyperparameter was set to 0.5, representing an equal balance of the lasso model and ridge model effects. All features were log10+1 normalized and scaled using a minmax [0,1] scaler to equally compare features spanning different orders of magnitude and improve model interpretability. To further prevent data leakage, in each cross-validation loop, the scaler was fit to the train set and then applied to the train set and subsequently the test set. Test feature outliers were clipped to [0,1] following this normalization. The train set was balanced within each cross-validation loop using Synthetic Minority Over-sampling Technique (SMOTE) to up-sample the minority class to equal the majority class (35). Feature importance analyses were conducted by computing Shapley Additive exPlanations (SHAP) values for each model (36). SHAP values enable the interpretation of which combinations of features contribute to the overall model predictions, as SHAP values denote the relative importance of a given feature in driving a model’s prediction. SHAP values have been used to explain ML predictions in prior cancer studies (25, 26).
Recurrent Cellular Neighborhood Analysis
Recurrent cellular neighborhoods were quantified to assess spatial organization of tissues. A neighborhood was created for every cell by counting all cells within a 60 µm radius of each seed cell’s center, as inferred from previous studies (37, 38). Using proportions of cells comprising the neighborhoods as features, neighborhoods were grouped using K-means clustering. The elbow method was used to determine the number of clusters, resulting in groupings of spatial neighborhoods that were similar in cellular composition that could be found across all regions of interest in the analysis.
Statistics
Mann-Whitney U tests were used to determine statistically significant differences in top TME features between treatment cohorts or DFS groups. The Benjamini-Hochberg correction was used to account for multiple hypothesis testing for each analysis. P-values less than 0.05 were considered statistically significant. Statistical calculations were performed with the Scipy and statsmodels packages using Python software (39, 40).
RESULTS
Deep phenotyping of T cells within the PDAC TME using mIHC
29 PDAC tumors were surgically resected from patients across two treatment cohorts (Fig. 1A). Tumors from 18 treatment-naive patients comprised cohort 1 and were previously evaluated for immune contexture in a larger study (5). Specimens from cohort 1 served as a representative baseline comparison to the 11 specimens from cohort 2, which reflected patients who had received neoadjuvant anti-CD40 therapy alone (n=8) or in combination with gemcitabine and nab-paclitaxel (n=3) prior to resection (2), and were combined here to a single cohort to evaluate impact of CD40 stimulation on T cells given our prior study using this cohort (2). Three to 26 tissue regions per PDAC resection were selected by a pathologist and quantitatively assayed by mIHC, with each region annotated as one of four histopathologic sites within the resected samples: tumor (T), immune aggregate (IA), tumor-adjacent stroma (TAS), or normal-adjacent pancreas (NAP) (Fig. 1B) (5). Breakdown of region types assayed per patient are shown (Fig. 1C).
Figure 1. Deep phenotyping of T cells within the PDAC TME using mIHC.
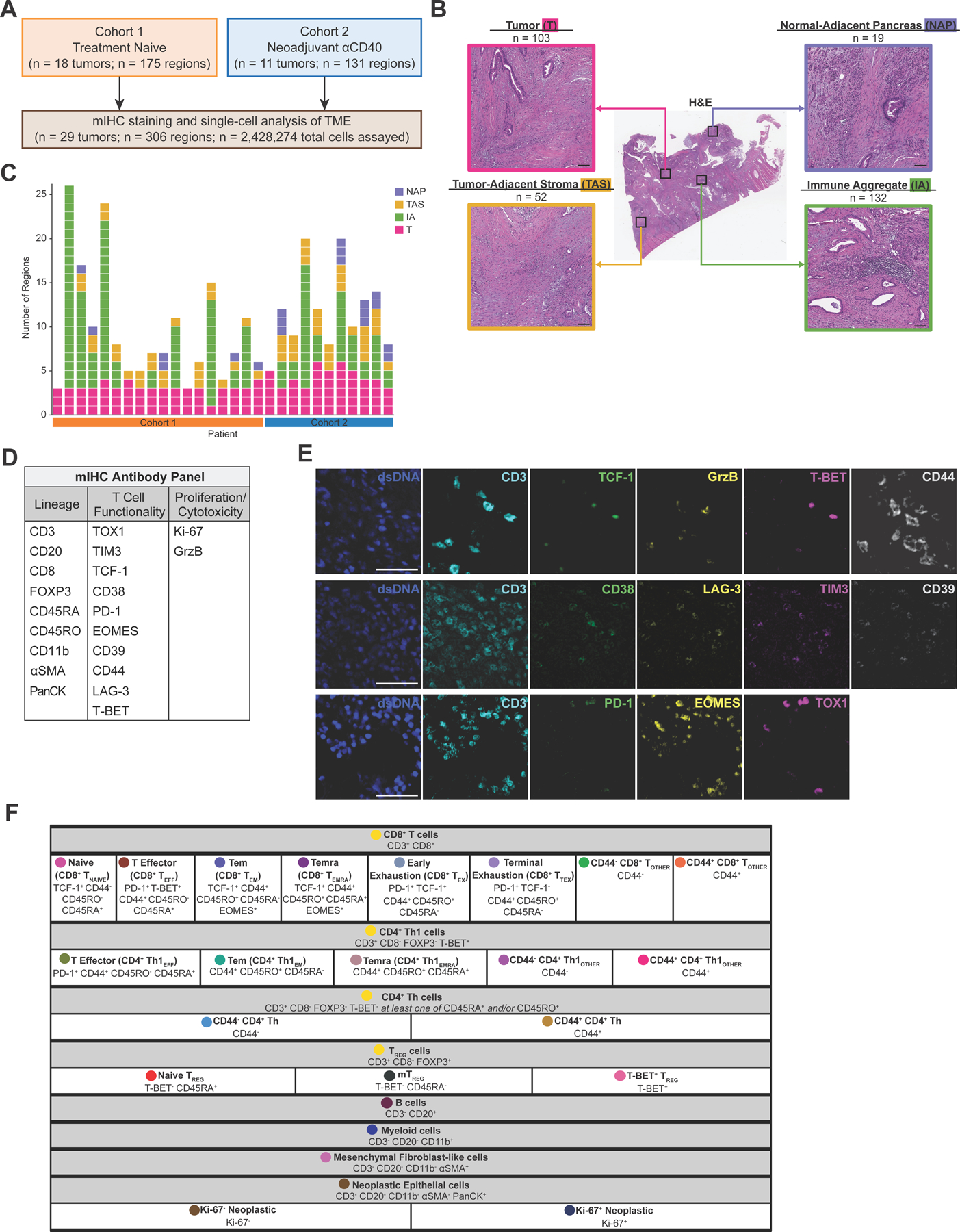
A. Overview of two PDAC cohorts assayed via mIHC.
B. Representative PDAC tissue resection stained with H&E (middle) showing four histopathologic sites annotated. Total number of regions assayed per histopathologic site are listed. Scale bars = 100 µm.
C. Number of regions assayed per patient. Each box represents one tissue region and is colored according to its histopathologic site.
D. 21-antibody mIHC panel used to assay tissue regions.
E. Representative pseudo-colored mIHC images showing T cell functionality biomarkers with CD3 expression. Scale bars = 50 µm.
F. Cell phenotyping strategy by hierarchical gating of lineage and functional biomarkers. Circles indicate colors associated with each cell state in the following figures.
In total, nearly 2.5 million cells were assayed across 306 tissue regions by our 21-antibody mIHC panel (Fig. 1D and E, Supplementary Table S3, Supplementary Fig. S1B). 423,317 T cells were identified by CD3 and CD8 expression (Supplementary Fig. S1C) and subsequently classified into 18 distinct T-cell states (Fig. 1F), including eight CD8+ T-cell states, five CD4+ Th1-cell states, two CD4+ Th-cell states (non-Th1-specific T helper cells), and three TREG states. In addition, T-cell functionality was further assessed by 10 biomarkers characterizing differentiation/exhaustion, and by biomarkers of proliferation and cytotoxicity (Fig. 1D, E). The remaining CD3– (non-T) cells were defined by a hierarchical gating strategy and classified as B cells, myeloid cells, mesenchymal fibroblast-like cells (also referred to as mesenchymal cells), or neoplastic epithelial cells (Fig. 1F, Supplementary Fig. S1D). Altogether, cells were phenotyped as one of 23 different cell lineages and states (Fig. 1F).
Interrogating cell states and spatial interactions within the PDAC TME
Using single-cell spatial data collected from the mIHC assay, we calculated three types of TME features to create a granular map of leukocyte infiltration, T-cell functionality status, and cellular spatial orientation in the PDAC TME.
Varying densities of leukocytes, mesenchymal fibroblast-like cells, and neoplastic epithelial cells were present in annotated histopathologic regions across treatment cohorts (Fig. 2A, top). T regions were dominated by neoplastic epithelial cells; IA regions were dominated by T and B cells, and distal NAP regions were dominated by myeloid cells. TAS regions, which encompassed tumor borders, comprised a mix of neoplastic cells, T cells, myeloid cells, and mesenchymal cells. On average, CD4+ T cells were present at a density that was nearly two-fold that of CD8+ T cells (Fig. 2A, middle, bottom rows) across regions and cohorts. However, average densities of the CD8+ and CD4+ T-cell states often differed by histopathologic site and treatment cohort, demonstrating the importance of identifying spatial and histopathological information for interpreting how—and where—anti-CD40 therapy alters T cells in the PDAC TME, important information that could not be captured by flow cytometric methodologies.
Figure 2. Interrogating cell states and spatial interactions within the PDAC TME.

A. Stacked bar charts showing the average cell state densities for each treatment cohort and histopathologic site. Top row: lineage defining cells including neoplastic epithelial cells, mesenchymal fibroblast-like cells, myeloid cells, B cells, and CD3+ T cells; Middle row: CD8+ T cell states; Bottom row: CD4+ T cell states. Columns denote histopathologic site, and each plot is further broken into treatment cohort.
B. Bar charts showing average densities of barcoded T cells for each treatment cohort and histopathologic site. Only the 15 most abundant barcodes are shown as measured by average density. Rows denote histopathologic site, and columns denote treatment cohort. Brown bars denote barcoded T cells that are in the top 15 most abundant barcodes in both cohorts. Orange bars denote barcoded T cells that are in the top 15 most abundant barcodes in the treatment-naive cohort only. Blue bars denote barcoded T cells that are in the top 15 most abundant barcodes in the anti-CD40-treated cohort only.
C. Heatmaps showing average number of spatial interactions between two cell states for each treatment cohort and histopathologic site. Cell states are denoted by colors shown in Figure 1F. Interactions were normalized first by density of cells participating in the interaction and were then log10+1 transformed. Rows denote treatment cohort and columns denote histopathologic site.
D. Overview schematic of analyses performed in this study. TME features were calculated for each tissue region. Two ML classifier models were built for each histopathologic site to predict treatment status and DFS. Feature importance analyses were performed to interpret biological meaning.
T-cell functionality was then assessed through quantification of a T cell “Functionality Barcode,” for each T cell present in the dataset. The top 15 most common barcodes by average density are shown for each histopathologic type and treatment cohort (Fig. 2B). Over half of the most common barcoded T cells were present in both treatment cohorts, regardless of histopathologic site, as indicated by brown bars (Fig. 2B). The majority of barcodes present in the treatment-naive cohort (orange bars) contained two functionality biomarkers while the most abundant barcodes present in the anti-CD40 cohort only (blue bars, Fig. 2B) contained three or more functionality biomarkers. This result supports the hypothesis that anti-CD40 therapy shifts T-cell differentiation/functionality within the PDAC TME, as represented by an increase in the number of functionality biomarkers expressed.
Finally, cellular spatial organization was assessed by calculating cell–cell spatial interactions. On average, there were increased interactions between CD4+ T cells with other CD4+ T cells in the anti-CD40 cohort, regardless of histopathologic site (Fig. 2C, white boxes). Altogether, these results support the hypothesis that anti-CD40 drives an increase in CD4+ T-cell density, functional capacity, and spatial proximity in the PDAC TME, as compared to treatment-naive PDAC TMEs.
Following these single-cell quantifications, all 2,428,274 cells present were phenotyped as one of 23 cell states; all 423,317 T cells were assigned one of 961 T-cell functionality barcodes, and the immediate spatial neighbors of each cell were computed and binned into one of 268 types of pairwise cell-cell interactions. This quantification resulted in 1,252 TME features computed for each of the 306 regions, each annotated as one of four histopathologic sites. Given the complexity and large amount of data, we leveraged ML and feature importance analyses to identify: 1) impact of anti-CD40 therapy on these TME metrics, and 2) the likely mechanism(s) we hypothesized underlying improved clinical outcome following anti-CD40 therapy. To do this, we trained EN classifier models to predict treatment status and DFS within the anti-CD40-treated cohort from the 1,252 TME features quantified above (Fig. 2D). Finally, SHAP values were used to identify which combinations of TME features drove model predictions and thus interpret the cellular biology underpinning model predictions (36).
ML models classify anti-CD40-treated TMEs as having reduced T-cell exhaustion phenotypes
To reveal the impact of anti-CD40 therapy on T cell–exhaustion phenotype, we trained four EN classifier models—one per histopathologic annotation—to predict the treatment status of the tissue. All models performed well, as measured by the accuracy, F1 score, and AUC for test sets of each of the models (Fig. 3A and B). Across the four models, accuracy ranged from 0.83 to 0.85, F1 score ranged from 0.73 to 0.89, and AUC ranged from 0.87 to 0.90. As models were trained to differentiate treatment-naive from anti-CD40-treated PDAC, high performance of all four models indicates that anti-CD40 modulates all types of histopathologic regions across the TME evaluated herein.
Figure 3. ML models classify anti-CD40-treated TMEs as having reduced T cell exhaustion phenotypes.
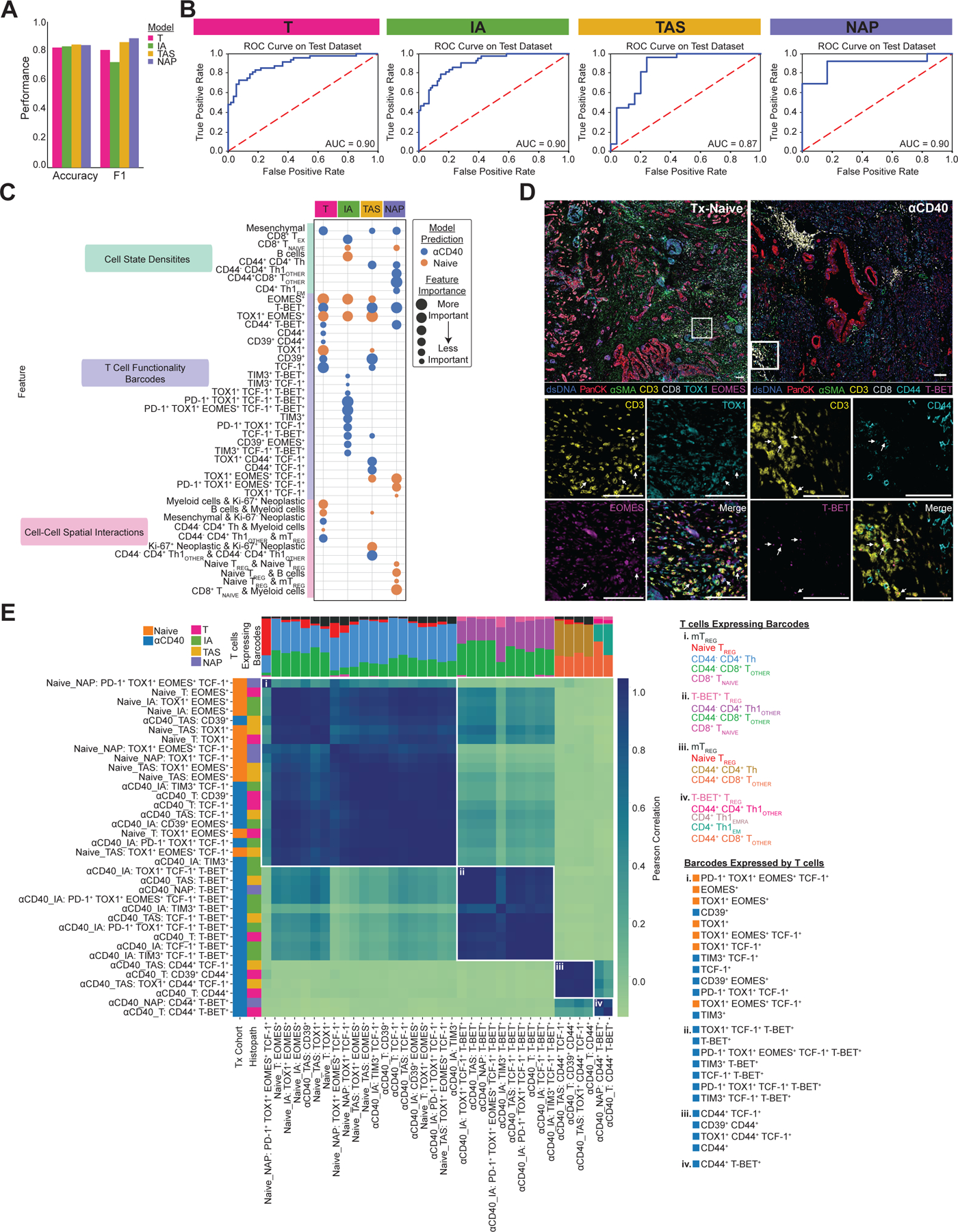
A. Bar chart showing accuracy and F1 score for each histopathologic model that predicts treatment status.
B. ROC curve with corresponding AUC for each histopathologic model.
C. Bubble chart showing top 15 features whose increased presence drove each histopathologic model to predict treatment-naive (orange) or anti-CD40-treated (blue). Features are grouped by TME feature type (density, barcode, interaction). Bubble size denotes relative importance of the feature for a given histopathologic model. Bubbles appearing multiple times in the same row indicate TME feature is a top feature across histopathologic models.
D. Representative pseudo-colored mIHC images showing TOX1+ and/or EOMES+ CD3+ T cells in treatment-naive tissue (left) and CD44+ and/or T-BET+ CD3+ T cells in anti-CD40-treated tissue (right).
E. Matrix showing correlations between top barcodes from the models with each other based on types and proportions of T cell states expressing the barcodes. Stacked bars at the top of correlation matrixes show proportions of T cell states expressing barcodes, with T cells color coded and listed for each group to the right of the heatmap, along with corresponding barcodes in each group. Leftmost columns are color coded according to which treatment group the presence of the barcode was predicted by the model, followed by the histopathologic site the model was derived from.
To identify the features driving model predictions, and thus reveal how anti-CD40 therapy impacted T cells in the PDAC TME, we calculated SHAP feature importance values for each of the four models (Supplementary Fig. S2A). The top 30 most important features out of 1,252 total features accounted for the majority of importance according to SHAP values (T model: 84%; IA model: 74%; TAS model: 87%; NAP model: 92%). Comparison of the top 15 features driving model predictions for each of the four histopathologic models revealed 13 of the same features were top contributors across multiple models (Fig. 3C), indicating shared T-cell densities, differentiation states, and spatial organizations across histopathologic sites within a given treatment cohort. We then compared the normalized values for each of the top features quantified by SHAP analysis between treatment-naive samples versus anti-CD40-treated samples within each histopathologic site (Supplementary Fig. S2B, C, D, and E). All 15 features derived from the T model were significantly different between treatment cohorts, 14 of 15 features derived from the IA and TAS models were significantly different between treatment cohorts, and 9 of 15 features derived from the NAP models were significantly different between treatment cohorts, likely because NAP had the fewest number of regions present in the data.
Overall, the models identified anti-CD40-treated TMEs as containing increased densities of mesenchymal fibroblast–like cells and several T-cell states, including three CD4+ T-helper populations and two antigen-experienced CD8+ T-cell populations (TEX and CD44+ TOTHER) (Fig. 3C, Supplementary Fig. S2B, C, D, and E), as compared to treatment-naive TMEs, which contained increased densities of naive CD8+ T cells and B cells. In addition to cell-state densities, analysis of T-cell functionality barcodes revealed that anti-CD40-treated TMEs contained increased densities of T cells expressing combinations of T-BET, CD44, CD39, TIM3, and TCF-1 (Fig. 3C, Supplementary Fig. S2B, C, D, and E). On the other hand, treatment-naive TMEs contained increased densities of T cells expressing combinations of TOX1 and EOMES, concordant with mIHC stained tissue images (Fig. 3D). Finally, spatial interactions involving CD4+ Th1 cells were associated with the αCD40-treated cohort, whereas interactions involving myeloid cells, naive TREGs, and Ki-67+ neoplastic epithelial cells were associated with treatment-naive tissue (Fig. 3C, Supplementary Fig. S2B, C, D, and E). Altogether, these results indicate anti-CD40 TMEs contained increased presence of T cells in close spatial proximity to one another—in particular, CD4+ T helper cells—with reduced exhaustion profiles, as compared to treatment-naive TMEs.
Of the top features across all four models, the majority of features were densities of T cell–functionality barcodes. As all barcodes present on any T cell were provided to the ML models, we sought to determine whether the top barcodes identified by the models were expressed by similar or different T-cell states. Thus, barcodes were correlated based on the types and proportions of T-cell states expressing each barcode (Fig. 3E). This analysis resulted in four clusters (i. – iv.) of barcodes, each with distinct compositions of T cells and not influenced by histopathologic site. Barcodes belonging to cluster (i) were expressed by antigen-inexperienced (as defined by lack of CD44 expression) CD8+ and CD4+ T-helper cells, naive TREGs, and mTREGs. However, barcodes containing TOX1+ and EOMES+ dominated these cluster (i) T cells when predictive of treatment-naive samples, while T cells predictive of anti-CD40-treated samples expressed barcodes containing CD39+ TIM3+, and TCF-1+. This result supports the notion that, while the same T-cell types were present regardless of therapy exposure, their functional capacity differed following anti-CD40 treatment. Higher densities of barcodes on multiple antigen-experienced CD4+ Th1 cell states and T-BET+ TREGs—reported to be similar to CD4+ Th1 cells in their function (41)—in clusters (ii), (iii), and (iv) included combinations of T-BET+, TIM3+, TCF-1+, and CD44+ and were predictive of tissue samples from patients treated with anti-CD40 therapy.
Long disease-free survivors have enhanced T-cell effector functionality following anti-CD40 therapy
The clinical trial from which the anti-CD40-treated specimens were derived was not designed to assess correlates with survival. However, despite the small size of our cohort, we hypothesized that we could train ML models to accurately predict DFS for these patients, with the goal of identifying the combinations of TME features associated with long versus short DFS within the anti-CD40-treated cohort. The median DFS timepoint (9.8 months) across all patients in the anti-CD40-treated cohort was used to segregate long and short disease-free survivors. Separate models were built for each histopathologic site, although NAP region presence was insufficient to build a model. Only the model trained from IA regions performed well in predicting both long and short DFS, with an accuracy of 0.81, F1 score of 0.88, and AUC of 0.77 (Fig. 4A and B).
Figure 4. Long disease-free survivors have enhanced effector T-cell functionality following anti-CD40 therapy.

A. Bar chart showing accuracy and F1 score for each histopathologic model predicting anti-CD40 DFS.
B. ROC curve with corresponding AUC for the IA histopathologic model.
C. Bubble chart showing the top 15 features whose increased presence drove the IA model to predict short DFS (purple) or long DFS (green). Features are grouped by TME feature type (density, barcode, interaction). Bubble size denotes relative importance of the feature.
D. Representative pseudo-colored mIHC images showing TOX1+ and/or PD-1+ CD3+ T cells in short DFS tissue (left) and CD44+ and/or CD39+ CD3+ T cells in long DFS tissue (right). Scale bars = 100 µm.
E. Matrix showing correlations between the top barcodes from the IA model with each other based on types and proportions of T cell states expressing the barcodes. Stacked bars at the top of the correlation matrix show proportions of T cell states expressing the barcodes, and T cells are color coded and listed for each group to the right of the heatmap, along with the corresponding barcodes in each group. Leftmost column is color coded according to which DFS group the presence of the barcode predicted by the model.
To identify TME features driving predictions for the IA-derived model, we followed a similar model interpretation analysis using SHAP values (Supplementary Fig. S3A, Fig. 4C). The top 30 features out of 1,252 features available to the model accounted for 78% of feature importance. All of the top 15 ranked features identified by the SHAP analysis were significantly different between DFS groups (Supplementary Fig. S3B), demonstrating how ML can be used to reveal potential combinations of candidate biomarkers of DFS in the PDAC TME.
Of note, we observed an increased density of CD44+CD4+ Th1 cells and increased spatial interactions between CD4+ Th1 TEM or CD8+ TEMRA cells and T-BET+ TREGs in IA regions from patients with long versus short DFS (Fig. 4C, Supplementary Fig. S3B). However, increased densities of T cell–functionality barcodes were the most common feature predictive of DFS, accounting for 12 of the top 15 features. Of these, eight barcode densities were associated with short DFS and four were associated with long DFS (Fig. 4C, Supplementary Fig. S3B). TOX1 was expressed on all eight barcodes associated with short DFS, while PD-1 expression was found exclusively in five of the eight barcodes associated with short DFS. In contrast, expression of CD44, CD38, CD39, TIM3, and LAG-3 were unique to the four barcodes associated with long DFS (Fig. 4C, D).
To determine if the T-cell states expressing each of the predictive barcodes were similar, we again correlated the barcodes by T cell–state expression (Fig. 4E). We found four distinct clusters of barcodes, and within each cluster, the barcodes were expressed by similar types of T-cell states and in similar proportions. All but one of the barcodes in clusters (i) and (ii) were among the features whose increased densities were associated with short DFS and were expressed by antigen-inexperienced CD8+ and CD4+ T helper cells and TREGs subsets. In contrast, increased densities of all barcodes in clusters (iii) and (iv) were associated with long DFS. Cluster (iii) consisted of one barcode that was uniquely expressed by CD8+ TEM and TEMRA cells, CD44+CD4+ T helper cells, as well as naive TREG cells. Finally, cluster (iv) barcodes were expressed by CD44+CD8+ T cells, CD4+ Th1 TEM and TEMRA cells, CD44+CD4+ Th1 cells, and T-BET+ TREGs. In summary, these findings indicate the following TME changes in IAs – an antitumor T-cell response, characterized by presence of CD44+ T cells and in particular CD4+ Th1 cells – are associated with prolonged DFS in our patient cohort.
T-cell spatial organization correlates of DFS after anti-CD40 therapy
The majority of top TME feature types driving the anti-CD40 DFS IA model predictions were densities of specifically barcoded T cells. However, the spatial organization of these cells was not clear from our model predictions. TME spatial architecture is associated with clinical outcomes across cancer types (17–22); thus, we aimed to identify the spatial neighbors of the top barcoded T cells whose densities were associated with DFS following anti-CD40 therapy within IA regions.
To quantify the spatial organization of the tissue, we performed a recurrent cellular neighborhood (RCN) analysis across all IA regions within the anti-CD40-treated cohort (18, 22) (Fig. 5A). This resulted in seven RCNs (Supplementary Fig. S4A) – each representing spatial neighborhood of cells present across multiple IAs that were distinct in proportions and types of cell states located within the neighborhood. The average cellular composition of each RCN is shown (Fig. 5B). We confirmed that no single RCN dominated the IA regions analyzed (Supplementary Fig. S4B and C) and that no RCN was exclusively derived from any single IA region or patient (Supplementary Fig. S4D and E). Upon viewing the scatterplot reconstructions of regions, clearly defined spatial patterns within IAs were revealed (Fig. 5C, Supplementary Fig. S4F). For example, cells in RCN1, whose neighborhood consisted mostly of B cells, were often found to be spatially clustered together, potentially representing “germinal center”-like pockets within IA regions.
Figure 5. T-cell spatial organization correlates with DFS following anti-CD40 therapy.

A. Schematic depicting RCN analysis. Cellular neighborhoods were defined by identifying all cells within a 60 µm radius of a given cell. Neighborhoods were calculated for all cells in anti-CD40-treated IA regions. Neighborhoods were then grouped using K-means clustering to identify RCNs. Created with BioRender.com.
B. Stacked bar chart showing average cellular composition of each of seven RCNs from the anti-CD40-treated IA regions. Bars are colored by cell state and represent average proportions (out of 1.0) of each cell state present in neighborhoods assigned to each RCN.
C. Representative IA tissue region as depicted by scatterplot reconstructions. Each dot represents a cell present in the IA, and each cell is colored by its original cell state phenotype (left scatterplot) or RCN assignment (right scatterplot).
D. Heatmap showing top T cell barcodes from the anti-CD40 IA DFS model clustered by proportion of RCNs the T cell barcodes were assigned to. Rows are barcoded T cells from IA regions from patients associated with short DFS or long DFS ordered by hierarchical clustering of their RCN assignment. Columns are RCNs used as clustering features. Proportion of RCNs was normalized using a log10+1 transformation prior to clustering. Leftmost columns are color coded by DFS group followed by barcode.
E. Stacked bar chart showing average fraction of RCNs barcoded T cells were assigned to for each of two hierarchically clustered groups (C1 or C2).
F. Bar chart showing percentage of T cells expressing Ki-67 residing in each of seven RCNs for anti-CD40 IA regions. Horizontal dashed line represents percentage of Ki-67+ T cells across all anti-CD40 IA regions, regardless of RCN assignment.
G. Bar chart showing percentage of T cells expressing GrzB residing in each of seven RCNs for anti-CD40 IA regions. Horizontal dashed line represents percentage of GrzB+ T cells across all anti-CD40 IA regions, regardless of RCN assignment. TREG populations were excluded from this analysis.
H. Representative IA region from a patient with long DFS with cells colored by RCN assignment in the upper left scatterplot. Remaining images show mIHC staining of GrzB+ CD44+ CD3+ T cells localized within RCN5. Scale bars = 100 µm.
Given our goal of identifying neighbors surrounding the top barcoded T cells from the anti-CD40 DFS model, we first identified which of the seven RCNs the barcoded T cells were assigned to. We then performed unsupervised clustering of barcoded T cells from long and short DFS patients together based on proportions of the seven RCNs the barcoded T cells resided in (Fig. 5D). This resulted in two distinct clusters of barcoded T cells, which also segregated according to DFS. Cluster 1 (C1) included all barcoded T cells from patients with long DFS, as well as CD44+T-BET+ barcoded T cells from patients with short DFS. Cluster 2 (C2) consisted of all remaining barcoded T cells from patients with short DFS.
The most striking difference between the two clusters was proportions of barcoded T cells residing in RCN5 (Fig. 5E). Of all RCNs, RCN5 contained the greatest proportion of CD44+ T cells, spanning both CD8+ T cells and CD4+ T helper lineages, and with only 28% of cells representing non-T cell lineages, including mesenchymal cells, myeloid cells, a small subset of neoplastic cells, and B cells (Fig. 5B). 45% of T cells in C1 (long DFS) resided in RCN5, whereas only 3% of T cells from C2 (short DFS) resided in RCN5 (Fig. 5E). Thus, T cells correlated with long DFS were frequently found to be surrounded by CD44+ T cells, supporting the hypothesis that anti-CD40 therapy prolongs survival through the promotion of T-cell priming and/or recruitment of primed T cells to PDAC TMEs.
Finally, to further elucidate potential cellular mechanisms active within various RCNs, we calculated proportions of T cells expressing Ki-67 or GrzB in each RCN and compared values to the overall proportion of Ki-67+ or GrzB+ T cells across all anti-CD40 IA regions. Given our prior findings that the majority of T-cell barcodes whose increased density correlated with long DFS were assigned to RCN5 (Fig. 5E), we hypothesized that T cells assigned to RCN5 would possess increased proliferative and/or cytotoxic capabilities. We found a larger proportion of T cells expressing Ki-67 residing in RCN1 and RCN5 as compared to the overall T-cell population (dashed line) (Fig. 5F). In addition, we found a larger proportion of T cells (excluding TREGs) expressing GrzB residing in RCN5, RCN6, and RCN7, as compared to the overall non-TREG T-cell population (dashed line) (Fig. 5G). However, raw counts of GrzB+ T cells in RCN6 and RCN7 were low (RCN6: n=8 cells; RCN7: n=11 cells), whereas RCN5 contained the highest level of GrzB+ T cells across all RCNs (n=168). Visualization of a representative mIHC image depicts presence of GrzB+ T cells localized to RCN5 in an IA region from a patient with long DFS (Fig. 5H). Collectively, these results provide further support that T cells within RCN5 are likely activated and possess an effector phenotype capable of an antitumor cytotoxic response.
DISCUSSION
In this study, we integrated spatial proteomic imaging technology with ML approaches to understand the role of T-cell phenotypes and spatial organization in the complex TME of human pancreatic cancer. In contrast to previous single-cell spatial proteomic studies, which often group T cells together as CD8+ T cells, CD4+ T cells, or TREGs (17–19, 21), our mIHC panel was curated to phenotype T cells as one of 18 distinct states along with functionality status from 10 different biomarkers, all while preserving the spatial orientation of each cell in the TME. In considering the full spectrum of T-cell states in addition to their spatial organization, ML approaches were necessary due to the complexity of data. ML was used to identify combinations of TME features most associated with anti-CD40 therapy exposure or prolonged DFS (Fig. 6). This study demonstrates the value of merging single-cell spatial proteomic assays with ML analyses to interrogate how immunotherapy modulates the PDAC TME and potentially drives improved survival.
Figure 6. Spatial features of T cells associated with anti-CD40 therapy and prolonged DFS in the PDAC TME.

A. T cell subsets that best define resected tumor samples from treatment-naive (left) or anti-CD40-treated (right) patients. In the absence of therapy, T cells appear in an exhausted state, while T cells present with activated and effector phenotypes after CD40 agonism. Created with BioRender.com.
B. T cell phenotypes in IAs from anti-CD40-treated patients associated with long (top) or short (bottom) DFS. IAs from patients with long DFS are characterized by the presence of spatial neighborhoods of effector T cells capable of proliferating and cytotoxicity, while IAs from patients with short DFS have a preponderance of exhausted T cell states. Created with BioRender.com.
Despite the multitude of unique T-cell states identified herein, our ML models identified T-cell subsets associated with antitumor characteristics and prolonged DFS. Consistent with our preclinical studies revealing that CD4+ T cells are a major contributor to PDAC immunity following anti-CD40 therapy (42), our ML models revealed that anti-CD40-treated patient tumors contain increased densities of effector memory cells specifically within the CD4+ Th1 lineage, while no increase was observed in CD8+ memory T-cell populations. Our models also identified antigen-experienced CD4+ Th1 cells as the main cell type whose density associated with prolonged DFS following anti-CD40 therapy. This result is concordant with two independent studies, including characterization of immune cells in biopsied PDAC liver metastases following CD40 agonism (6), and the second investigating primary resected PDAC TMEs after treatment with a granulocyte-macrophage colony-stimulating factor-secreting allogenic PDAC vaccine (GVAX) (43). Both studies reported that presence of CD4+ T helper cells contribute to improved survival following immunotherapy in PDAC (6, 43). Here, we further characterized expression features of the CD4+ T helper cells as CD44+ Th1 cells, which correlated with improved outcomes. Reports have also highlighted the direct role of CD4+ T cells in mediating antitumor immunity, including via cytotoxicity (44) and production of effector cytokines (45). As such, we hypothesize that future therapies designed to harness effector and memory functions of CD4+ T helper cells following administration of anti-CD40 therapy may be clinically beneficial.
In addition to investigating presence of various T-cell states, our ML analyses show that localization and spatial organization of T cells within PDAC TMEs are associated with prolonged survival following anti-CD40 treatment. High performance of the IA-derived DFS prediction model indicates that IAs are a major site of anti-CD40-induced immune response contributing to prolonged DFS. We previously reported increased IAs following anti-CD40 treatment in PDAC-bearing mice (7), and in the aforementioned GVAX study, survival was linked to increased CD4+ T helper pathway genes specifically within IAs (and not tumor regions) (43). Moreover, a recent study found enrichment of gene signatures representing mature tertiary lymphoid structures in pre-treatment PDACs associated with improved survival in patients following treatment with varying chemoimmunotherapies (46). Our RCN analyses further revealed that the key T cells associated with prolonged DFS were often surrounded by antigen-experienced CD8+ and CD4+ T helper cells, as well as a higher proportion of proliferating and cytotoxic T cells, as compared to all T cells regardless of spatial neighborhood. While our mIHC panel in this study focused on T cells, we observed a minor presence of B cells (compared to other RCNs) as well as a subset of myeloid cells and mesenchymal cells in RCN5 that could also contribute locally to T-cell activation or function. Collectively, the results indicate IAs may function as sites of T-cell priming or second signal, promoting T-cell activation and function in PDAC TMEs, contributing to prolonged DFS following multiple types of immunotherapies.
Our interrogation of IAs revealed T-cell states concordant with dysfunctional tumor-infiltrating T-cell phenotypes correlated with short DFS following anti-CD40 therapy. TOX1+ T cells are at the far end of the exhausted T–cell spectrum (11), and TOX1 expression correlates with PD-1 on T cells and impaired immunotherapy response in hepatocellular carcinoma (47). Correspondingly, we observed expression of TOX1 and/or PD-1 on CD8+ and CD4+ T helper cells associated with shorter DFS following anti-CD40 therapy. T cells linked to short DFS expressing TOX1 and/or PD-1 were largely CD44–, which may represent a population of antigen-naive T cells that aberrantly upregulated these proteins, or T cells that are terminally exhausted due to repeated T cell–receptor stimulation (48). We also found an increased presence of TOX1+ T cells within treatment-naive TMEs, indicating a baseline terminally exhausted T–cell phenotype in the PDAC TME. Despite this, and in agreement with our prior study (5), we did not identify PD-1+ T cells as a major subset, in contrast to tumors such as melanoma (49), where PD-1+ T cells are abundant. The paucity of PD-1+ T cells may contribute to the failure of ICBs targeting PD-1 or PD-L1 in the majority of patients with PDAC (50). Together, our data support the conclusion that TOX1, but not PD-1, is a dominant feature of exhausted T cells in PDAC. Thus, therapies that modulate TOX1+ T cells in the TME—such as anti-CD40 agonism—may improve clinical outcomes for patients with PDAC.
Notably, the dataset we used to conduct this study was unique in several aspects—including therapy administered, mIHC panel deployed, and histopathologic sites assayed—making validation of our results challenging, highlighting the need for additional studies on larger cohorts to build models capable of more generalized predictions, and cautioning integration of our results in a clinical setting prior to additional validation. Patient samples were collected from multiple institutions per treatment cohort, yet our ML models still accurately classified samples according to therapy. This indicates that any institutional or technical differences in tissue processing were not driving features in model predictions. Importantly, the ML models we trained performed comparably to or better than models derived from similar studies (21, 25–27). Additionally, our biological conclusions on impacts of anti-CD40 are concordant with several prior studies (6, 7, 11, 42, 43, 47, 50), providing further support for methods herein and findings despite small sample sizes. Future antibody panels may incorporate additional biomarkers, such as chemokine receptors, to further characterize key T-cell subsets, or additional lineage biomarkers that denote myeloid or B-cell subsets (20), to further phenotype cell–cell interactions. It should be noted that in the present study, treatment with anti-CD40 did not prolong DFS as compared to the treatment-naive cohort, and future data analyses from clinical trials with larger cohorts powered for survival analyses will be useful to validate our findings that associate TME features with longer DFS. Finally, tumors were resected 12 days after anti-CD40 administration; thus, it is possible that T cells involved in prolonged DFS had insufficient time to transit beyond IAs and into surrounding TMEs following priming. Further analyses investigating timing of T-cell trafficking throughout PDAC TMEs are necessary to determine if analysis of T or TAS regions sampled at later timepoints following treatment could be used to assess clinical outcome for these patients.
This study provides proof-of-principle for leveraging ML approaches to evaluate highly multiplexed cancer datasets and supports the use of similar analytics in future studies to identify important, and otherwise inconspicuous alterations in TMEs correlating with patient treatment or response. Future studies could utilize these findings to target pathways identified via this approach to improve treatment strategies for cancer patients.
DATA AVAILABILITY
mIHC data used for this study is available for download on Zenodo at https://zenodo.org/records/10476868. All other data are available in the article and its supplementary files or from the corresponding author upon reasonable request.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Drs. David L. Bajor, E. Gabriela Chiorean, Daniel A. Laheru, and Mark H. O’Hara for efforts as site investigators for the neoadjuvant selicrelumab (CD40 agonist) clinical trial. We also thank John Wherry for helpful discussion related to panel design and biomarker selection. This research was supported by the National Cancer Institute (NCI) of the National Institutes of Health grants T32CA254888 (K.E.B.), P50 CA127003 (J.A.N.), R01 CA248857 (J.A.N.), R01 CA205406 (J.A.N.), R01 CA169141 (J.A.N.), R35 CA197735 (J.A.N.), U01 CA250549 (J.A.N.), U01 CA224012 (LMC), U24CA231877 (J.G.), and U2CCA233280 (J.G.), the DFCI Hale Family Center for Pancreatic Cancer Research (J.A.N. and B.M.W.), the Lustgarten Foundation Dedicated Laboratory program (J.A.N., B.M.W.), the Parker Institute for Cancer Immunotherapy (R.H.V. and K.T.B.), the Brenden-Colson Center for Pancreatic Care (L.M.C., S.S., C.B.B., K.T.B., K.B., N.K.), the Robert L. Fine Cancer Research Foundation (K.T.B.), funding from the Prospect Creek Foundation to the OHSU SMMART (Serial Measurement of Molecular and Architectural Responses to Therapy) Program (J.G. and L.M.C.), and a generous startup package from the Knight Cancer Institute and the Brenden-Colson Center for Pancreatic Care (K.T.B.).
Footnotes
Conflict of Interest: R.H.V. is an inventor on licensed patents relating to cancer cellular immunotherapy and cancer vaccines, and mutant Kras specific T cell receptors; has received consulting fees from BMS; and receives royalties from Children’s Hospital Boston for a licensed research-only monoclonal antibody and from the University of Pennsylvania for licensed research cell lines. J.A.N. receives consulting fees from Leica Biosystems and research support from Natera. B.M.W. receives research funding from AstraZeneca, Celgene/BMS, Eli Lilly, Novartis, and Revolution Medicines, and consulting for Celgene, GRAIL, Ipsen, Mirati, Third Rock Ventures unrelated to the current work. C.B.B. is an employee of, and holds equity in, Akoya Biosciences, Inc. K.T.B. receives royalties from the University of Pennsylvania for licensed research cell lines and has received consulting fees from Guidepoint. L.M.C. has received reagent support from Cell Signaling Technologies, Syndax Pharmaceuticals, Inc., ZielBio, Inc., and Hibercell, Inc.; holds sponsored research agreements with Syndax Pharmaceuticals, Hibercell, Inc., Prospect Creek Foundation, Lustgarten Foundation for Pancreatic Cancer Research, Susan G. Komen Foundation, and the National Foundation for Cancer Research; is on the Advisory Board for Carisma Therapeutics, Inc., CytomX Therapeutics, Inc., Kineta, Inc., Hibercell, Inc., Cell Signaling Technologies, Inc., Alkermes, Inc., Raska Pharma, Inc., NextCure, Guardian Bio, AstraZeneca Partner of Choice Network (OHSU Site Leader), Genenta Sciences, Pio Therapeutics Pty Ltd., and Lustgarten Foundation for Pancreatic Cancer Research Therapeutics Working Group, Inc.
CODE AVAILABILITY
The code used to generate all computational results of this research was created using Python version 3.9.4 and is available at https://github.com/kblise/PDAC_mIHC_paper.
REFERENCES
- 1.Pishvaian MJ, Blais EM, Brody JR, Lyons E, DeArbeloa P, Hendifar A, et al. Overall survival in patients with pancreatic cancer receiving matched therapies following molecular profiling: a retrospective analysis of the Know Your Tumor registry trial. Lancet Oncol 2020;21(4):508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byrne KT, Betts CB, Mick R, Sivagnanam S, Bajor DL, Laheru DA, et al. Neoadjuvant Selicrelumab, an Agonist CD40 Antibody, Induces Changes in the Tumor Microenvironment in Patients with Resectable Pancreatic Cancer. Clin Cancer Res 2021;27(16):4574–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023;618(7963):144–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vonderheide RH. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu Rev Med 2020;71:47–58. [DOI] [PubMed] [Google Scholar]
- 5.Liudahl SM, Betts CB, Sivagnanam S, Morales-Oyarvide V, da Silva A, Yuan C, et al. Leukocyte Heterogeneity in Pancreatic Ductal Adenocarcinoma: Phenotypic and Spatial Features Associated with Clinical Outcome. Cancer Discov 2021;11(8):2014–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Padron LJ, Maurer DM, O’Hara MH, O’Reilly EM, Wolff RA, Wainberg ZA, et al. Sotigalimab and/or nivolumab with chemotherapy in first-line metastatic pancreatic cancer: clinical and immunologic analyses from the randomized phase 2 PRINCE trial. Nat Med 2022;28(6):1167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Byrne KT, Vonderheide RH. CD40 Stimulation Obviates Innate Sensors and Drives T Cell Immunity in Cancer. Cell Rep 2016;15(12):2719–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oliveira G, Wu CJ. Dynamics and specificities of T cells in cancer immunotherapy. Nat Rev Cancer 2023;23(5):295–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat Immunol 2011;12(7):663–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Hu J, Li Y, Xiao M, Wang H, Tian Q, et al. The Transcription Factor TCF1 Preserves the Effector Function of Exhausted CD8 T Cells During Chronic Viral Infection. Front Immunol 2019;10:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature 2019;571(7764):211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wherry EJ. T cell exhaustion. Nat Immunol 2011;12(6):492–9. [DOI] [PubMed] [Google Scholar]
- 13.Huang AC, Orlowski RJ, Xu X, Mick R, George SM, Yan PK, et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat Med 2019;25(3):454–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity 2016;45(2):389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chu Y, Dai E, Li Y, Han G, Pei G, Ingram DR, et al. Pan-cancer T cell atlas links a cellular stress response state to immunotherapy resistance. Nat Med 2023;29(6):1550–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng L, Qin S, Si W, Wang A, Xing B, Gao R, et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 2021;374(6574):abe6474. [DOI] [PubMed] [Google Scholar]
- 17.Jackson HW, Fischer JR, Zanotelli VRT, Ali HR, Mechera R, Soysal SD, et al. The single-cell pathology landscape of breast cancer. Nature 2020;578(7796):615–20. [DOI] [PubMed] [Google Scholar]
- 18.Blise KE, Sivagnanam S, Banik GL, Coussens LM, Goecks J. Single-cell spatial architectures associated with clinical outcome in head and neck squamous cell carcinoma. NPJ Precis Oncol 2022;6(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mi H, Sivagnanam S, Betts CB, Liudahl SM, Jaffee EM, Coussens LM, et al. Quantitative Spatial Profiling of Immune Populations in Pancreatic Ductal Adenocarcinoma Reveals Tumor Microenvironment Heterogeneity and Prognostic Biomarkers. Cancer Res 2022;82(23):4359–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dias Costa A, Vayrynen SA, Chawla A, Zhang J, Vayrynen JP, Lau MC, et al. Neoadjuvant Chemotherapy Is Associated with Altered Immune Cell Infiltration and an Anti-Tumorigenic Microenvironment in Resected Pancreatic Cancer. Clin Cancer Res 2022;28(23):5167–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Risom T, Glass DR, Averbukh I, Liu CC, Baranski A, Kagel A, et al. Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stroma. Cell 2022;185(2):299–310 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schurch CM, Bhate SS, Barlow GL, Phillips DJ, Noti L, Zlobec I, et al. Coordinated Cellular Neighborhoods Orchestrate Antitumoral Immunity at the Colorectal Cancer Invasive Front. Cell 2020. [DOI] [PMC free article] [PubMed]
- 23.Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep 2017;19(1):203–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banik G, Betts CB, Liudahl SM, Sivagnanam S, Kawashima R, Cotechini T, et al. High-dimensional multiplexed immunohistochemical characterization of immune contexture in human cancers. Methods Enzymol 2020;635:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buk Cardoso L, Cunha Parro V, Verzinhasse Peres S, Curado MP, Fernandes GA, Wunsch Filho V, et al. Machine learning for predicting survival of colorectal cancer patients. Sci Rep 2023;13(1):8874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang C, Xiu Y, Qiao K, Yu X, Zhang S, Huang Y. Prediction of lymph node metastasis in patients with breast invasive micropapillary carcinoma based on machine learning and SHapley Additive exPlanations framework. Front Oncol 2022;12:981059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sammut SJ, Crispin-Ortuzar M, Chin SF, Provenzano E, Bardwell HA, Ma W, et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature 2022;601(7894):623–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 2006;7(10):R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baaten BJ, Tinoco R, Chen AT, Bradley LM. Regulation of Antigen-Experienced T Cells: Lessons from the Quintessential Memory Marker CD44. Front Immunol 2012;3:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis A, Gao R, Navin NE. SCOPIT: sample size calculations for single-cell sequencing experiments. BMC Bioinformatics 2019;20(1):566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carstens JL, Correa de Sampaio P, Yang D, Barua S, Wang H, Rao A, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun 2017;8:15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: Machine Learning in Python. JMLR 2011;12:2825–30. [Google Scholar]
- 33.Jang IS, Neto EC, Guinney J, Friend SH, Margolin AA. Systematic assessment of analytical methods for drug sensitivity prediction from cancer cell line data. Pac Symp Biocomput 2014:63–74. [PMC free article] [PubMed]
- 34.Zou H, Hastie T. Regularization and variable selection via the elastic net. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 2005;67(2):301–20. [Google Scholar]
- 35.Chawla NV, Bowyer KW, Hall LO, Kegelmeyer WP. SMOTE: Synthetic Minority Over-sampling Technique. JAIR 2002;16:321–57. [Google Scholar]
- 36.S.M. L, S. L. A Unified Approach to Interpreting Model Predictions. NIPS 2017:4765–74.
- 37.Busse D, de la Rosa M, Hobiger K, Thurley K, Flossdorf M, Scheffold A, et al. Competing feedback loops shape IL-2 signaling between helper and regulatory T lymphocytes in cellular microenvironments. Proc Natl Acad Sci U S A 2010;107(7):3058–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oyler-Yaniv A, Oyler-Yaniv J, Whitlock BM, Liu Z, Germain RN, Huse M, et al. A Tunable Diffusion-Consumption Mechanism of Cytokine Propagation Enables Plasticity in Cell-to-Cell Communication in the Immune System. Immunity 2017;46(4):609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods 2020;17(3):261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seabold S, Perktold J. statsmodels: Econometric and statistical modeling with python. Proceedings of the 9th Python in Science Conference 2010. [Google Scholar]
- 41.Duhen T, Duhen R, Lanzavecchia A, Sallusto F, Campbell DJ. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood 2012;119(19):4430–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morrison AH, Diamond MS, Hay CA, Byrne KT, Vonderheide RH. Sufficiency of CD40 activation and immune checkpoint blockade for T cell priming and tumor immunity. Proc Natl Acad Sci U S A 2020;117(14):8022–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol Res 2014;2(7):616–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sledzinska A, Vila de Mucha M, Bergerhoff K, Hotblack A, Demane DF, Ghorani E, et al. Regulatory T Cells Restrain Interleukin-2- and Blimp-1-Dependent Acquisition of Cytotoxic Function by CD4(+) T Cells. Immunity 2020;52(1):151–66 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen D, Varanasi SK, Hara T, Traina K, Sun M, McDonald B, et al. CTLA-4 blockade induces a microglia-Th1 cell partnership that stimulates microglia phagocytosis and anti-tumor function in glioblastoma. Immunity 2023;56(9):2086–104 e8. [DOI] [PubMed] [Google Scholar]
- 46.Kinker GS, Vitiello GAF, Diniz AB, Cabral-Piccin MP, Pereira PHB, Carvalho MLR, et al. Mature tertiary lymphoid structures are key niches of tumour-specific immune responses in pancreatic ductal adenocarcinomas. Gut 2023;72(10):1927–41. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, He Q, Shen H, Xia A, Tian W, Yu W, et al. TOX promotes the exhaustion of antitumor CD8(+) T cells by preventing PD1 degradation in hepatocellular carcinoma. J Hepatol 2019;71(4):731–41. [DOI] [PubMed] [Google Scholar]
- 48.Abu Eid R, Ahmad S, Lin Y, Webb M, Berrong Z, Shrimali R, et al. Enhanced Therapeutic Efficacy and Memory of Tumor-Specific CD8 T Cells by Ex Vivo PI3K-delta Inhibition. Cancer Res 2017;77(15):4135–45. [DOI] [PubMed] [Google Scholar]
- 49.Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009;114(8):1537–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morrison AH, Byrne KT, Vonderheide RH. Immunotherapy and Prevention of Pancreatic Cancer. Trends Cancer 2018;4(6):418–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
mIHC data used for this study is available for download on Zenodo at https://zenodo.org/records/10476868. All other data are available in the article and its supplementary files or from the corresponding author upon reasonable request.