

Abstract
Cancer is a complex cellular ecosystem where malignant cells coexist and interact with immune, stromal and other cells within the tumor microenvironment (TME). Recent technological advancements in spatially resolved multiplexed imaging at single-cell resolution have led to the generation of large-scale and high-dimensional datasets from biological specimens. This underscores the necessity for automated methodologies that can effectively characterize molecular, cellular and spatial properties of TMEs for various malignancies. This study introduces SpatialCells, an open-source software package designed for region-based exploratory analysis and comprehensive characterization of TMEs using multiplexed single-cell data. The source code and tutorials are available at https://semenovlab.github.io/SpatialCells. SpatialCells efficiently streamlines the automated extraction of features from multiplexed single-cell data and can process samples containing millions of cells. Thus, SpatialCells facilitates subsequent association analyses and machine learning predictions, making it an essential tool in advancing our understanding of tumor growth, invasion and metastasis.
Keywords: spatial analysis, region-based profiling, multiplexed single-cell data, spatial omics, tumor immune infiltration, tumor microenvironment
INTRODUCTION
Cancer presents an intricate cellular ecosystem, which plays a critical role in tumor development, progression and therapeutic outcomes. The spatial organization of cells and their interactions within the tumor microenvironment (TME) contain essential insights into the course of tumor growth and progression. Characterizing molecular, cellular and spatial properties of TMEs across diverse malignancies has gained substantial attention [1, 2].
Recent technological breakthroughs in spatially resolved multiplexed imaging at single-cell resolution, such as CODEX and CyCIF, are highly effective in studying TMEs and intratumoral heterogeneity within solid tumors [3–6]. However, the lack of systematic computational methodologies to leverage the large volume of data generated by these technologies poses a significant challenge for their scalable deployment in clinical settings.
Currently, the available tools for handling multiplexed imaging data are designed to address specific aspects of multiplexed imaging data analysis. Some tools focus on converting multi-channel whole-slide images into single-cell data (e.g. MCMICRO [7]), while others prioritize preprocessing, cell type phenotyping and visualizing the obtained single-cell data (e.g. SCIMAP [8]). Spatial analysis of single-cell data represents only a small portion of the functions in HALO [9] and often demands manual annotation. Furthermore, this platform is proprietary and not freely available for public use. Another toolkit, Squidpy, specializes in neighborhood graph construction and analysis [10]. Consequently, the capability of these tools to effectively conduct comprehensive spatial analysis remains limited.
Furthermore, the considerable progress in leveraging machine learning for predictive tasks presents a critical consideration for clinical outcomes. However, these algorithms often require substantial sample sizes to achieve robust performance. Overall, the high-dimensional, heterogeneous and complicated dependency structures inherent in multiplexed single-cell data, coupled with the need for large sample sizes in machine learning algorithms, present challenges to conventional manual annotation and statistical techniques. Thus, there is an urgent need to develop computational methodologies to analyze multiplexed single-cell data in a scalable manner and enable the development of forecasting models that would inform clinical decision-making and enhance our understanding of disease progression.
In this study, we introduce SpatialCells, an open-source software package designed to perform region-based exploratory analysis and characterization of TMEs using multiplexed single-cell data. This tool is equipped to efficiently analyze tissue samples containing millions of cells and automatically extract quantitative features, enabling subsequent association analyses and machine learning predictions at scale.
IMPLEMENTATION
Overview
This study is grounded in existing literature on tumor and immune parameters associated with cancer progression and patient survival. Our primary goal is to develop automated methodologies for quantifying these parameters with spatially resolved single-cell data. By integrating clinicopathologic features extracted from electronic medical records, we will be equipped to perform comprehensive association analyses or make predictions about patient outcomes, as illustrated in Figure 1A and B. We have provided detailed tutorials on data exploration, tumor cell-centric analysis, immune cell-oriented analysis and others.
Figure 1.
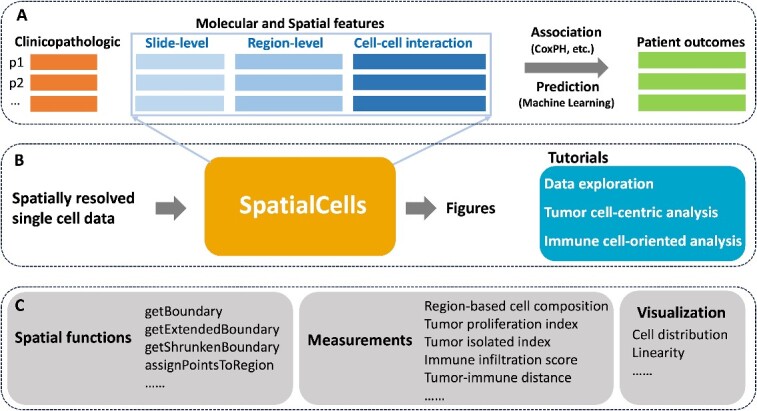
Overview of the framework. (A) The role of SpatialCells within downstream analyses. In this context, ‘p1, p2, …’ represent patient IDs, and CoxPH (Cox proportional hazards) modeling is one of the association analysis methods. (B) The input and output of SpatialCells. It takes the spatially resolved multiplexed single-cell data as input and provides quantified features along with the corresponding visualizations as output. Tutorials for data exploration, tumor cell–centric analysis, immune cell-oriented analysis and others have been provided. (C) Key modules in SpatialCells. SpatialCells incorporates several main modules to facilitate its functionality, such as tumor proliferation index, immune infiltration score and tumor-immune distance.
SpatialCells is featured by its capability to define regions of interest (ROIs) based on any group of cells and subsequently conduct region-based analyses. Figure 1C presents the main modules incorporated into SpatialCells. Our workflow starts with developing a Spatial module, including functions to establish regional boundaries and annotate each cell with the corresponding region in which it is located. The Measurements module contains functions to extract properties of tumor cells and tumor-immune cell interactions, including tumor proliferation index, immune infiltration score and tumor-immune cell distance. These properties can be assessed for the whole tissue or local regions. Importantly, our methods can efficiently process datasets containing millions of cells.
Spatially resolved single-cell data
SpatialCells takes spatially resolved multiplexed data at single-cell resolution as input (Figure 1B). Our collaborators commonly utilize cyclic immunofluorescence imaging (CyCIF) [4] to generate such data. Supplementary Figure 1 outlines the data generation process and the data format. For each tissue specimen, multi-channel images are first generated by CyCIF. This procedure is followed by cell segmentation and quantification using MCMICRO [7]. Based on relative protein expression levels of cells, most cells can be assigned to specific cell types [11]. This process produces data that include spatial coordinates, marker intensity values and cell types for up to 107 cells.
While our experiments primarily focus on CyCIF data, SpatialCells can be applied to analyzing other spatially resolved omics data, such as 10X Genomics Visium data and iterative indirect immunofluorescence imaging (4i) data [12, 13].
Data exploration
In the data exploration, we have incorporated measurement and visualization tools that can provide initial insights into samples. These include:
Whole-slide-level cell composition. This involves assessing the percentages of different cell types present in the samples, such as tumor cells and immune cells.
ROI-level cell composition. Whole-slide tissues often have a significant amount of blank background. SpatialCells computationally defines ROIs and removes background in a standardized manner across all tissues in the study cohort. This is accomplished by initially calculating the tumor boundary and then extending this boundary by a specified distance (a user-defined parameter).
Region-based cell composition. SpatialCells provides the capability for the geometric partition of tissues in various ways. For instance, it can divide the tumor region into subregions based on distance from the centroid or angle from the zero degree. Following the partitioning, SpatialCells can enumerate cell types within each tissue subregion for detailed compositional analysis.
Region-based clustering. Similarly, after partitioning, SpatialCells provides the ability to cluster cells within a specific region.
Tumor cell-centric analysis
Tumor cell-centric analysis focuses on the characterization and study of tumor cells to gain insights into their biology, heterogeneity and behavior within the TME. Specifically, the American Joint Committee on Cancer/Union for International Cancer Control (AJCC/UICC) staging system provides guidelines for classifying the extent of cancer spread, considering factors such as tumor size and depth of invasion [14–16]. Additionally, the mitotic rate of various tumors is highly relevant in predicting patient outcomes [17–19]. In this analysis, we have emphasized the following tumor characteristics that may be linked to patient prognosis:
Tumor area and tumor cell density. SpatialCells offers functions for defining regions based on user-specified markers and calculating the area of a region and the density of any cell type within the region.
Tumor multivariate proliferation index (MPI). Gaglia et al. demonstrated the effectiveness of an MPI in differentiating proliferating from non-proliferating tumor cells [20]. Building on this work, SpatialCells provides a function for calculating an MPI of a given cell type, including tumor cells. The input of this function includes two marker lists of interest: (1) mitosis/proliferation markers (e.g. Serine-10 phosphorylated histone H3, Ki67, PCNA and MCM2) and (2) cell cycle arrest markers (e.g. p21 and p27). Adapted from the definition in the work by Gaglia et al. [20], the MPI is defined as follows:
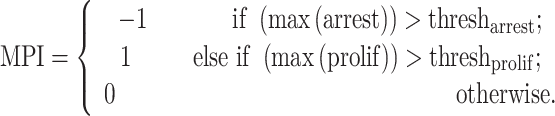 |
The threshold values for proliferation and arrest are data dependent. For normalized markers of expression levels from 0 to 1, the values are set to 0.5 by default. The 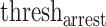 can be tuned based on a commonly used proliferation marker, such as Ki67. Instead of computing a single MPI for the whole tissue, SpatialCells enables calculating MPI for subregions of interest within the tissue sample. We demonstrate this method by applying a sliding window of a user-defined size over the tissue and computing the MPI for each subregion.
can be tuned based on a commonly used proliferation marker, such as Ki67. Instead of computing a single MPI for the whole tissue, SpatialCells enables calculating MPI for subregions of interest within the tissue sample. We demonstrate this method by applying a sliding window of a user-defined size over the tissue and computing the MPI for each subregion.
Tumor isolation index. Categorizing tumors into immune hot, immune suppressed and immune cold/isolated groups has considerable prognostic value in various malignancies [21, 22]. Here, we focus on defining a tumor isolation index, which is the fraction of tumor cells in the region without immune cells over all tumor cells. This index is accomplished by dividing the overall ROI (whole-slide tissue with background removed) into two subregions: immune-rich region and region with almost no tumor-infiltrating immune cells.
Immune cell-oriented analysis
The TME is a spatially organized landscape characterized by the presence of lymphocytes, macrophages and other cell types located both within the central region and at the invasive margin of the tumor. Understanding the TME composition is essential for developing effective cancer therapies and predicting patient prognosis. Here, we have conducted macro-region and micro-region analyses, along with cell-to-cell interactions:
Macro-region analysis. The overall ROI, which encompasses the whole-slide tissue with the background removed, is divided into four subregions: tumor, tumor border, stroma border and stroma, as illustrated in Figure 2A. We subsequently evaluate the cell compositions within each region, with a particular focus on immune cells.
Micro-region analysis. This analysis focuses on small regions, defined as micro-regions, within the tumor area, as illustrated in Figure 2B. By identifying these micro-regions, we can perform detailed characterizations of cells within them, enabling a finer-grained understanding of the cellular landscape.
Cell-to-cell interactions. An important aspect of our analysis involves investigating the interactions between various cell types within the TME. For instance, SpatialCells can be applied to quantify the interactions between tumor cells and immune cells. As shown in Figure 2C, we assess the degree of tumor immune infiltration by measuring the distance between a tumor cell and its nearest immune cell. This information provides valuable insights into the interplay between different cell populations within the TME.
Figure 2.
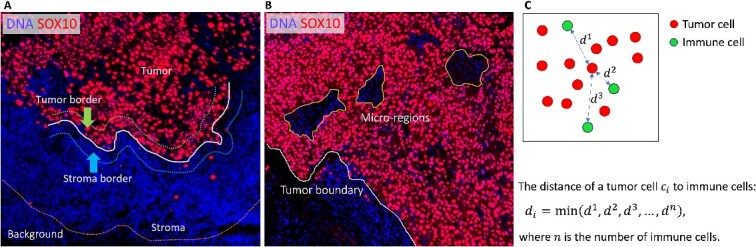
Macro-regions, micro-regions and cell-to-cell distance. (A) The background defined based on a user-specified distance from the tumor boundary is removed from the whole-slide image. The remaining ROI is partitioned into tumor region, tumor border, stroma border and stroma region (macro-regions). (B) Within the tumor region, there are small regions of interest, defined as micro-regions, which allow fine-grained analyses. (C) The distance of a cell (e.g. a tumor cell) to another cell type (e.g. immune cells) is defined as the distance between the cell and its nearest neighbor of another cell type.
Compatibility with other software
SpatialCells is built on top of Anndata, Shapely and Scanpy frameworks, making it easy to integrate with other commonly used toolkits [23–25]. In tutorials, we have provided examples of integrating SpatialCells with SCIMAP, Squidpy and Scanpy [8, 10, 25].
RESULTS
In this section, we demonstrate the functionality of SpatialCells by presenting results in data exploration, tumor cell–centric analysis and immune cell-oriented analysis. Additional analyses can be found in tutorials, providing researchers with a comprehensive toolkit for in-depth investigations.
Our analyses involve publicly available multiplexed imaging data of a cutaneous melanoma sample (MEL1) consisting of 1 110 585 cells [26]. MEL1 was imaged using CyCIF [4] with 30 antibody markers (e.g. SOX10, Keratin, CD31, CD3D, CD8A, CD11C, MITF and Ki67) and preprocessed using MCMICRO [7], which transforms multi-channel whole-slide images into single-cell data. The output of MCMICRO is the input of SpatialCells. Additional details of this sample can be found in the provided reference [26].
Data exploration
SpatialCells is a versatile tool for region-based data exploration, particularly for assessing cell composition or clustering within whole-slide images or specific regions of interest. Figure 3 illustrates how SpatialCells can be applied to compute cell compositions for three different types of regions within the MEL1 sample.
Figure 3.

Cell composition in regions of interest. (A) Gradient of MITF on CyCIF imaging data. (B) Keratin, SOX10 and CD3D expressions on CyCIF imaging data. (C) Boundary of the main tumor area. (D) Cell type composition in subregions based on distance from the centroid. (E) Cell type composition in subregions based on the angle from the zero-degree reference. (F) Cell type composition in subregions based on distance from the epidermis.
The principal tumor (T) category for invasive melanoma is defined as the maximum tumor thickness measured with a calibrated ocular micrometer at a right angle to the adjacent normal skin to the deepest point of tumor invasion [16, 27]. In this case, the MEL1 section shows fragmentation artifacts at the deep aspect of the invasive front. To ensure accurate measurements of the distance from the centroid or the epidermis to the tumor boundary, we excluded the fragmented area with SOX10+ cells and focused on the main tumor area. If we included these separate fragments of SOX10+ cells along with the intervening empty spaces, the measurements would have been artificially increased.
Figure 3A and B presents the expression levels of related markers, including keratin, SOX10, MITF and CD3D, within the imaging data. SOX10 expression is a sensitive and specific marker for melanoma tumor cells. MITF is an important melanoma oncogene and plays a role in determining therapeutic resistance [28, 29]. CD3D is a marker commonly associated with immune cells, particularly T lymphocytes (or T cells). Figure 3C shows the boundary of the main tumor area as identified by SpatialCells. In Figure 3D, the subregions are defined based on the distance from the centroid of the main tumor area. In Figure 3E, the subregions are based on their angles from the zero-degree reference. The percentage of MITF+SOX10+ cells within each subregion is computed. The two barplots in Figure 3D and E demonstrate the gradient of MITF+ tumor cells. Figure 3F provides cell composition within subregions, which are defined based on their distance from the epidermis. The centroid (start point) and the epidermis (start line) are computationally determined. It is worth noting that SpatialCells offers support for user-defined start points and lines, allowing users to tailor analyses that align with specific requirements.
Tumor isolation index and MPI
SpatialCells also facilitates tumor-cell-centric analyses. Figure 4 showcases the tumor isolation index and the MPI. These analyses are part of our dedicated tumor-cell-centric tutorial.
Figure 4.
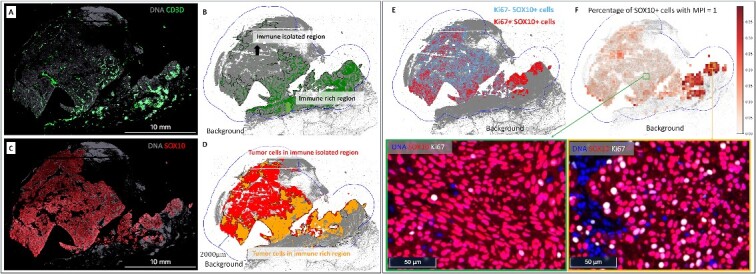
Tumor isolation index (A–D) and tumor MPI (E, F). (A) Expression level of CD3D+ cells in the MEL1 sample. (B) Division of the overall ROI into two subregions based on CD3D+ cells: (1) immune-isolated region, which has almost no immune cells; (2) immune-rich region. (C) Expression level of tumor (SOX10+) cells in the MEL1 sample. (D) Visualization of tumor cells in the immune-isolated region and immune-rich region, respectively. The tumor isolation index for this sample is 46.9%, which quantifies the percentage of tumor cells in the immune-isolated region. (E) Display of Ki67+ and Ki67− tumor cells. Ki67 is a proliferative marker. The overall percentage of proliferative tumor cells (MPI = 1) in the MEL1 sample is 7.4%. (F) Percentage of tumor cells with MPI = 1 in subregions and the zoomed-in CyCIF images. A sliding window of 300  300 microns is applied over the ROI to compute the percentage of tumor cells with MPI = 1. In this example, the MPI is computed using Ki67 due to the availability of markers. Additionally, zoomed-in CyCIF images are provided to visualize areas with varying levels of Ki67+ tumor cells.
300 microns is applied over the ROI to compute the percentage of tumor cells with MPI = 1. In this example, the MPI is computed using Ki67 due to the availability of markers. Additionally, zoomed-in CyCIF images are provided to visualize areas with varying levels of Ki67+ tumor cells.
Figure 4A shows the expression of CD3D+ cells in the MEL1 sample. In Figure 4B, the overall ROI is divided into two distinct subregions based on the presence of CD3D+ cells: (1) immune-isolated region, characterized by minimal immune cell presence, and (2) immune-rich region, characterized by a higher presence of immune cells. Figure 4C shows the expression of SOX10+ cells in the MEL1 sample. Figure 4D visually contrasts tumor cells in the immune-isolated and immune-rich regions. The tumor isolation index for this sample is quantified as 46.9%, which is the percentage of tumor cells in the immune-isolated region.
Figure 4E displays Ki67+ and Ki67- tumor cells. Ki67 is a marker of cell proliferation. The overall percentage of proliferating tumor cells (MPI = 1) in the MEL1 sample is 7.4%. Figure 4F shows the percentage of tumor cells with MPI = 1 in a sliding window of 300 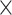 300 microns. In this example, the MPI is computed using Ki67 due to the availability of markers. Zoomed-in CyCIF images are provided to visualize areas with varying levels of Ki67+ tumor cells. In Figure 4E and F, a greater proportion of Ki67+/SOX10+ cells are along the periphery of the main tumor area and, most prominently, at the advancing edge of the invasive front. The region with the greatest proportion of SOX10+ cells with MPI = 1 is within an immune-rich region, as seen in Figure 4D.
300 microns. In this example, the MPI is computed using Ki67 due to the availability of markers. Zoomed-in CyCIF images are provided to visualize areas with varying levels of Ki67+ tumor cells. In Figure 4E and F, a greater proportion of Ki67+/SOX10+ cells are along the periphery of the main tumor area and, most prominently, at the advancing edge of the invasive front. The region with the greatest proportion of SOX10+ cells with MPI = 1 is within an immune-rich region, as seen in Figure 4D.
Macro-region and micro-region analyses
The immune context within the TME plays an important role in cancer prognosis and therapeutic efficacy [22]. Figure 5 provides an overview of macro-region (A, B and C) and micro-region (D, E, F, G and H) analyses, empowering users to comprehensively characterize the immune and cellular landscape within the TME.
Figure 5.

Macro-region analysis (A–C) and micro-region analysis (D–H). (A) The overall ROI is divided into four subregions: Tumor (T), Tumor border (Tb), Stroma border (Sb) and Stroma (S). The tumor region boundary is 100 microns away from the tumor boundary; the stroma border region boundary is 200 microns away from the tumor boundary; the boundary for the overall ROI is 800 microns away from the tumor boundary. Users can specify these distances. (B) The corresponding counts of tumor cells, T cells and other cells in the four regions. (C) The cell fraction among all cells within each region. (D) The micro-regions within the tumor area. (E) The zoomed-in micro-regions. Each micro-region is indexed, allowing users to pick the ones of interest. (F) Each micro-region boundary is extended and shrunk (offset = 30 microns) to get the ‘In’, ‘Border inside’ and ‘Border outside’ subregions, as shown in (H). (G) Cell compositions within the three subregions for each micro-region.
Tumor margins are exposed to cell infiltration, physical contact and diffusible chemotactic gradients from the stromal regions and vice versa. Therefore, defining tumor and stromal boundaries allows us to more specifically assess the complex interplay between invasive melanoma tumor cells and the microenvironment and to identify novel markers [26, 30]. In Figure 5A, the overall ROI is divided into four distinct subregions: Tumor (T), which represents the core tumor region; Tumor border (Tb), which is the transitional area immediately adjacent to the tumor; Stroma border (Sb), marking the border between tumor and stroma; and Stroma (S), situated farther from the tumor. These subregions are defined based on specific distances from the tumor boundary, with the tumor region boundary set at 100 microns, the stroma border region boundary at 200 microns and the boundary for the overall ROI at 800 microns away from the tumor boundary. Importantly, users have the flexibility to customize these distances. In Figure 5B, the corresponding counts of tumor cells, T cells and other cells are presented for each of the four defined regions. Figure 5C further provides insights by illustrating the cell fraction among all cells within each defined region. Figure 5D displays micro-regions within the tumor area. Each micro-region is indexed, enabling users to pinpoint specific micro-regions of interest. Figure 5E offers a closer look at these micro-regions. In Figure 5F, every micro-region boundary is extended and contracted (offset = 30 microns) to define ‘In’, ‘Border inside’ and ‘Border outside’ subregions, as visually demonstrated in Figure 5H. The cell compositions within these three subregions for each micro-region are presented in Figure 5G.
The Stroma region in Figure 5A includes some SOX10+ tumor cells on the right side of the image. This is due to the presence of fragmentation artifacts in this sample [26]. In practice, users may need to analyze the data separately for each area, as was the case in the provided reference [26]. In our tutorials, we include detailed instructions on how to perform these analyses.
Experimental results on spatial omics data
To demonstrate the broad utility of SpatialCells for analyzing other spatial omics data modalities, we further conducted experiments on a public Visium dataset of a human lymph node sample [31]. The data were deconvoluted following the standard Cell2location protocol and analyzed with SpatialCells [31]. The details are available in our tutorials, with results presented in Supplementary Figure 2. SpatialCells can automate the identification of all germinal centers and delineate their region boundaries that are consistent with the authors’ manual annotation, eliminating the necessity of manual annotation that could introduce significant bias.
DISCUSSION AND FUTURE DIRECTIONS
SpatialCells represents a significant advance in the field of spatial analysis for multiplexed single-cell image data, enabling the computational quantification and standardized analyses of critical features within the TME. Its functionalities encompass various aspects of TME analysis, such as region-based cell composition, tumor proliferation index, tumor isolation index, immune cell infiltration and tumor-immune distance. By providing these analytical tools, SpatialCells empowers researchers to gain deeper insights into the intricate spatial relationships and characteristics of cells within the TME.
One of the limitations of the SpatialCells software is that the cell segmentation provided by upstream imaging data preprocessing tools, such as MCMICRO [7], may introduce errors in the cell counts. However, SpatialCells is a customizable software that can analyze multiple samples within a study in a consistent and standardized manner. This capability involves applying similar settings and parameters across all samples, thus mitigating some of the variability introduced by cell segmentation. Another limitation of the SpatialCells software is the reliance on the quality of gating and calling cell types, which involve converting continuous marker expression levels into binary variables. This binary conversion introduces subjectivity, leading to variability in cell count. Finally, the adaptability of SpatialCells, with its support for user-defined parameters, affords users the freedom to configure settings based on their specific needs. However, this flexibility can also be a source of errors, as suboptimal parameter choices may impact the accuracy of the analysis.
In the future, we will continue to improve this software and provide clear guidelines to users on best practices for parameter selection. We will include more functions as well as tutorials, particularly for multimodal analyses within the field of spatial biology. For example, we will provide functions for migrating ROIs from other modalities, such as hematoxylin and eosin stain imaging and spatial transcriptomics, to multiplexed imaging, allowing integrative analyses across different modality data.
CONCLUSIONS
In summary, SpatialCells is a novel software solution for spatially analyzing the TME using multiplexed imaging data in a streamlined fashion with the capacity to process samples containing millions of cells. This software is of critical importance in the analysis of the TME and in furthering our understanding of the factors leading to tumor progression as it facilitates subsequent association analyses and machine learning predictions.
Key Points
SpatialCells is a software package for spatially analyzing multiplexed single-cell imaging data, which is featured by its capability to define regions of interest based on any group of cells and subsequently conduct region-based analyses.
Functionalities of SpatialCells encompass various aspects of tumor microenvironment analyses, such as region-based cell composition, tumor proliferation index, tumor isolation index, immune cell infiltration and tumor-immune distance.
SpatialCells allows preprocessing and analyzing data in a standardized manner with user-defined parameters and can process samples containing millions of cells.
Detailed tutorials on data exploration, tumor cell-centric analysis and immune cell-oriented analysis and documentation of SpatialCells have been provided.
Supplementary Material
Author Biographies
Guihong Wan, PhD, is a computer scientist and an Instructor of Dermatology at Massachusetts General Hospital and Harvard Medical School. Her research focuses on the development of computational methodologies for multimodal analyses and explainable machine learning models of biomedical data.
Zoltan Maliga, PhD, is a cell biologist and immunologist who co-leads the tissue imaging platform at the Laboratory of Systems Pharmacology at Harvard Medical School. His research interests are in the mechanisms of melanoma progression and the adverse reactions to immunotherapy.
Boshen Yan has completed his master’s degree in Biomedical Informatics at Harvard Medical School and his bachelor’s degree in Computational Biology at the National University of Singapore. His research interests include the development of machine learning models and multimodal integration methods to analyze disease progression.
Tuulia Vallius, MD, PhD, is a postdoctoral fellow in the Laboratory of Systems Pharmacology at the Department of Systems Biology, Harvard Medical School. Her research focuses on early melanoma progression and cell-to-cell interactions in the melanoma tumor microenvironment using cyclic immunofluorescence Imaging and spatial transcriptomics.
Yingxiao Shi (TK) is a PhD candidate studying in the Biological and Biomedical Sciences program at Harvard University, co-mentored by Dr David Liu and Dr Eliezer Van Allen. His PhD research, in collaboration with Dr Peter Sorger’s lab, mainly focuses on the biological mechanism of cancer progression in melanoma patients via multimodal analysis.
Sara Khattab is receiving her doctorate in medicine from the Chobanian and Avedisian Boston University School of Medicine and is currently a research fellow at Massachusetts General Hospital, working with Dr Yevgeniy Semenov studying melanoma and immune-related adverse events to immunotherapy.
Crystal Chang is receiving her doctorate in medicine from the Kaiser Permanente School of Medicine and is currently completing a master’s degree at Stanford University in the clinical informatics and management program. Her research interests include genetic predictors of melanoma treatment response.
Ajit J. Nirmal, PhD, is an assistant professor in the Department of Dermatology at Brigham and Women’s Hospital, Harvard Medical School, and the Director of Next Generation Tissue Analysis and Imaging Core. His research focuses on advancing cancer treatments by developing innovative methods and analyzing complex datasets.
Kun-Hsing Yu, MD, PhD, is an assistant professor in the Department of Biomedical Informatics at Harvard Medical School. His lab develops machine learning methods for integrating digital pathology imaging and multi-omics profiles to predict patients’ clinical phenotypes.
David Liu, MD, is a medical oncologist at Dana-Farber Cancer Institute and Brigham and Women’s Hospital and an Assistant Professor of Medicine at Harvard Medical School. His research interests include the leveraging of computational biology to explore treatment resistance and response to chemotherapeutic agents and immunotherapy.
Christine G. Lian, MD, is a practicing dermatopathologist at Mass General Brigham and an Associate Professor of Pathology at Harvard Medical School. Her laboratory focuses on understanding the epigenetic regulation of melanoma and other skin disorders.
Mia S. DeSimone, MD, is a dermatopathologist at Brigham and Women’s Hospital and an Instructor in Pathology at Harvard Medical School. She also serves as the Medical Director of Quality and Safety in Pathology. Her broad research interests span from clinical translational research investigating melanoma and other rare cutaneous tumors to quality and safety.
Peter K. Sorger, PhD, is a systems and cancer biologist and an Otto Krayer Professor of Systems Pharmacology at the Department of Systems Biology at Harvard Medical School. He is the founding head of the Harvard Program in Therapeutic Science and the director of its Laboratory of Systems Pharmacology. His research focuses on understanding the signal transduction and cell survival networks whose mutation causes cancer and determines responsiveness to therapy.
Yevgeniy R. Semenov, MD, MA, is a practicing dermatologist and an Assistant Professor of Dermatology at Massachusetts General Hospital and Harvard Medical School. He is also the Co-Director of the Oncodermatology Program. His primary areas of clinical and research interest are in oncodermatology and cutaneous oncology. He received his MD degree from Johns Hopkins University School of Medicine and completed residency training in Dermatology at the Washington University in Saint Louis. He has additional training in Applied Mathematics & Statistics and Applied Economics.
Contributor Information
Guihong Wan, Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA; Laboratory of Systems Pharmacology, Program in Therapeutic Science, Harvard Medical School, Boston, MA, USA.
Zoltan Maliga, Laboratory of Systems Pharmacology, Program in Therapeutic Science, Harvard Medical School, Boston, MA, USA.
Boshen Yan, Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA; Department of Biomedical Informatics, Harvard Medical School, Boston, MA, USA.
Tuulia Vallius, Laboratory of Systems Pharmacology, Program in Therapeutic Science, Harvard Medical School, Boston, MA, USA; Ludwig Center for Cancer Research at Harvard, Harvard Medical School, Boston, MA.
Yingxiao Shi, Department of Medicine, Dana-Farber Cancer Institute, Boston, MA, USA.
Sara Khattab, Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA.
Crystal Chang, Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA.
Ajit J Nirmal, Laboratory of Systems Pharmacology, Program in Therapeutic Science, Harvard Medical School, Boston, MA, USA; Department of Dermatology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA.
Kun-Hsing Yu, Department of Biomedical Informatics, Harvard Medical School, Boston, MA, USA; Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA.
David Liu, Department of Medicine, Dana-Farber Cancer Institute, Boston, MA, USA.
Christine G Lian, Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA.
Mia S DeSimone, Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, USA.
Peter K Sorger, Laboratory of Systems Pharmacology, Program in Therapeutic Science, Harvard Medical School, Boston, MA, USA.
Yevgeniy R Semenov, Department of Dermatology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, USA; Laboratory of Systems Pharmacology, Program in Therapeutic Science, Harvard Medical School, Boston, MA, USA.
AUTHOR CONTRIBUTIONS
Guihong Wan (conceived and designed the study, wrote the scripts, implemented the SpatialCells package and wrote the tutorials, generated the figures and wrote the manuscript), Zoltan Maliga (conceived and designed the study and wrote the manuscript), Boshen Yan (conceived and designed the study, wrote the scripts, implemented the SpatialCells package and wrote the tutorials and generated the figures), Tuulia Vallius (conceived and designed the study), Yingxiao Shi (conceived and designed the study), Sara Khattab (wrote the manuscript), Peter K. Sorger (conceived and designed the study, conducted under the supervision), Yevgeniy R. Semenov (conceived and designed the study, conducted under the supervision). All authors reviewed and approved the manuscript. All co-authors provided an intellectual contribution to this study.
FUNDING
G.W. is supported by the National Cancer Institute of the National Institutes of Health under Award Number K99CA286966. Y.R.S. is supported in part by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number K23AR080791, the Department of Defense under Award Number W81XWH2110819 and the Melanoma Research Alliance Young Investigator Award. K.-H.Y. is partly supported by the National Institute of General Medical Sciences grant R35GM142879, the Department of Defense Peer Reviewed Cancer Research Program Career Development Award HT9425-23-1-0523, Google Research Scholar Award and the Blavat Nik Center for Computational Biomedicine Award.
DATA AVAILABILITY
The code and data are available at https://semenovlab.github.io/SpatialCells
References
- 1. Srivastava S, Ghosh S, Kagan J, et al. The making of a PreCancer atlas: promises, challenges, and opportunities. Trends Cancer 2018;4(8):523–36. [DOI] [PubMed] [Google Scholar]
- 2. Consortium H. The human body at cellular resolution: the NIH human biomolecular atlas program. Nature 2019;574(7777):187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Black S, Phillips D, Hickey JW, et al. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat Protoc 2021;16(8):3802–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lin JR, Izar B, Wang S, et al. Highly multiplexed immunofluorescence imaging of human tissues and Tumors using T-CyCIF and conventional optical microscopes. Elife 2018;7:e31657 https://www.ncbi.nlm.nih.gov/pubmed/29993362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tirosh I, Izar B, Prakadan SM, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016;352(6282):189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin JR, Wang S, Coy S, et al. Multiplexed 3D atlas of state transitions and immune interaction in colorectal cancer. Cell 2023;186(2):363–381.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schapiro D, Sokolov A, Yapp C, et al. MCMICRO: a scalable, modular image-processing pipeline for multiplexed tissue imaging. Nat Methods 2022;19(3):311–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nirmal AJ. SCIMAP—SCIMAP Accessed October 3, 2023. https://scimap.xyz/.
- 9. Spatial analysis . Indica labs Published April 18, 2023. Accessed October 3, 2023. https://indicalab.com/products/spatial-analysis-2/.
- 10. Palla G, Spitzer H, Klein M, et al. Squidpy: a scalable framework for spatial omics analysis. Nat Methods 2022;19(2):171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Overton JA, Vita R, Dunn P, et al. Reporting and connecting cell type names and gating definitions through ontologies. BMC Bioinformatics 2019;20(S5):182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rao N, Clark S, Habern O. Bridging genomics and tissue pathology: 10x genomics explores new frontiers with the Visium spatial gene expression solution. Genetic Engineering & Biotechnology News 2020;40(2):50–1. [Google Scholar]
- 13. Gut G, Markus D, Herrmann L. Multiplexed protein maps link subcellular organization to cellular states. Science Published online 2018;361(6401):eaar7042. [DOI] [PubMed] [Google Scholar]
- 14. Locker GY, Hamilton S, Harris J, et al. ASCO 2006 update of recommendations for the use of tumor markers in gastrointestinal cancer. J Clin Oncol 2006;24(33):5313–27. [DOI] [PubMed] [Google Scholar]
- 15. Brierley JD, Gospodarowicz MK, Wittekind C, et al. TNM Classification of Malignant Tumours, Eighth Edition. Oxford, UK; Hoboken, NJ: John Wiley & Sons, Inc., 2017. [Google Scholar]
- 16. Keung EZ, Gershenwald JE. The eighth edition American joint committee on cancer (AJCC) melanoma staging system: implications for melanoma treatment and care. Expert Rev Anticancer Ther 2018;18(8):775–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dematteo RP, Gold JS, Saran L, et al. Tumor mitotic rate, size, and location independently predict recurrence after resection of primary gastrointestinal stromal tumor (GIST). Cancer: Interdisciplinary International Journal of the American Cancer Society 2008;112(3):608–15. [DOI] [PubMed] [Google Scholar]
- 18. Wan G, Nguyen N, Liu F, et al. Prediction of early-stage melanoma recurrence using clinical and histopathologic features. NPJ Precis Oncol 2022;6(1):79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wan G, Leung BW, Desimone MS, et al. Development and validation of time-to-event models to predict metastatic recurrence of localized cutaneous melanoma. J Am Acad Dermatol 2024;90(2):288–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gaglia G, Kabraji S, Rammos D, et al. Temporal and spatial topography of cell proliferation in cancer. Nat Cell Biol 2022;24(3):316–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer 2020;20(11):662–80. [DOI] [PubMed] [Google Scholar]
- 22. Pagès F, Mlecnik B, Marliot F, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet 2018;391(10135):2128–39. [DOI] [PubMed] [Google Scholar]
- 23. Virshup I, Rybakov S, Theis FJ, et al. Anndata: annotated data bioRxiv 2021. 10.1101/2021.12.16.473007. [DOI]
- 24. Gillies S, van der Wel C, Van den Bossche J, et al. Shapely. Zenodo, 2022. https://zenodo.org/records/7263102. [Google Scholar]
- 25. Wolf FA, Angerer P, Theis FJ. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 2018;19(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nirmal AJ, Maliga Z, Vallius T, et al. The spatial landscape of progression and immunoediting in primary melanoma at single-cell resolution. Cancer Discov 2022;12(6):1518–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frishberg DP, Balch C, Balzer BL, et al. Protocol for the examination of specimens from patients with melanoma of the skin. Archives of Pathology & Laboratory Medicine 2009;133(10):1560–7. [DOI] [PubMed] [Google Scholar]
- 28. Garraway LA, Widlund HR, Rubin MA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005;436(7047):117–22. [DOI] [PubMed] [Google Scholar]
- 29. Bai X, Fisher DE, Flaherty KT. Cell-state dynamics and therapeutic resistance in melanoma from the perspective of MITF and IFNγ pathways. Nat Rev Clin Oncol 2019;16:549–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Centofanti E, Wang C, Iyer S, et al. The spread of interferon-γ in melanomas is highly spatially confined, driving nongenetic variability in tumor cells. Proc Natl Acad Sci U S A 2023;120(35):e2304190120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kleshchevnikov V, Shmatko A, Dann E, et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat Biotechnol 2022;40(5):661–71. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code and data are available at https://semenovlab.github.io/SpatialCells