

Significance
Diffuse large B cell lymphoma (DLBCL) is a molecularly heterogeneous disease with a poor prognosis for patients who do not respond to first-line standard therapy. We characterized genetic loss of the putative tumor suppressor gene TRAF3 in DLBCL patients. In both primary tumors harboring TRAF3 deletions and isogenic TRAF3 loss-of-function (LOF) cell line models, we observed enhanced noncanonical (NC) NF-κB signaling, driven by stabilization of the central kinase NF-κB-inducing kinase (NIK). Pharmacological blockage of NIK abolished NC NF-κB signaling and killed TRAF3 LOF lymphoma cells, indicating that a subset of NC NF-κB-driven lymphomas may be susceptible to targeted inhibition of this signaling axis.
Keywords: lymphoma, somatic mutations, functional models, targeted therapies, tumor microenvironment
Abstract
Here, we report recurrent focal deletions of the chr14q32.31-32 locus, including TRAF3, a negative regulator of NF-κB signaling, in de novo diffuse large B cell lymphoma (DLBCL) (24/324 cases). Integrative analysis revealed an association between TRAF3 copy number loss with accumulation of NIK, the central noncanonical (NC) NF-κB kinase, and increased NC NF-κB pathway activity. Accordingly, TRAF3 genetic ablation in isogenic DLBCL model systems caused upregulation of NIK and enhanced NC NF-κB downstream signaling. Knockdown or pharmacological inhibition of NIK in TRAF3-deficient cells differentially impaired their proliferation and survival, suggesting an acquired onco-addiction to NC NF-κB. TRAF3 ablation also led to exacerbated secretion of the immunosuppressive cytokine IL-10. Coculturing of TRAF3-deficient DLBCL cells with CD8+ T cells impaired the induction of Granzyme B and interferon (IFN) γ, which were restored following neutralization of IL-10. Our findings corroborate a direct relationship between TRAF3 genetic alterations and NC NF-κB activation, and highlight NIK as a potential therapeutic target in a defined subset of DLBCL.
Diffuse large B cell lymphoma (DLBCL) is the most common B cell non-Hodgkin lymphoma, accounting for more than 30% of newly diagnosed lymphoma cases worldwide (1). Despite the introduction of first-line treatment regimens incorporating rituximab, targeting the pan-B cell antigen CD20, over a third of patients suffer from relapsed or refractory disease with a median overall survival (OS) of approximately 6 mo (2–5).
Transcriptional profiling studies have identified “cell-of-origin” (COO) germinal center B cell-like (GCB) and activated B cell-like (ABC) subtypes of DLBCL, which helped uncover some of the biological heterogeneity impacting treatment response, such as worse 3-y progression-free survival (PFS) in the ABC (40 to 50%) vs. GCB (75%) subtype (5–7). Among the good prognosis GCB subgroup, 21% express a germinal center dark-zone gene expression signature (DZsig) associated with poor prognosis (2-y OS of 57% DZsigpos vs. 89% DZsigneg) (8, 9). Recently proposed DLBCL genetic and microenvironment subtypes revealed additional features reflective of tumor intrinsic (e.g., deregulated cellular signaling) and extrinsic (e.g., tumor microenvironment composition) biology (10–14). In aggregate, these studies provided clinical rationales for, and have led to the development of, targeted therapies including signaling pathway inhibitors (e.g., proteasome inhibitors) and immunotherapies (e.g., PD1 inhibitors). However, several of these therapies have been met with limited success in clinical trials, suggesting a heterogeneous background requiring a more directed approach (15–17).
Mutations in the canonical NF-κB signaling pathway (12, 18–23) are well-established drivers of DLBCL proliferation and survival, particularly in the ABC subtype. A second NF-κB pathway, known as the “noncanonical” (NC) branch, is required for normal B cell survival and differentiation (24). In nontransformed B cells, activation of this pathway can be triggered by engagement of TNF-family receptors, such as CD40/TNFRSF5 and BAFF-R/TNFRSF13C, and is tightly regulated by the TRAF3/TRAF2/BIRC2/BIRC3 ubiquitin E3 ligase complex (25, 26). Microenvironment-derived signals trigger downstream posttranslational processing events, ultimately leading to the degradation of TRAF3 and stabilization of the NC NF-κB master regulator, the NIK kinase (26). Mutations in components of this alternative pathway (e.g., TRAF3, MAP3K14, BIRC3, RELB, NFKB2) contribute to the pathogenesis of several lymphoid malignancies (27–36). Emerging works are now focused on addressing the therapeutic targetability of cells harboring recurrent mutations in the NC NF-κB pathway in DLBCL (28, 29, 37).
We and others previously reported recurrent focal deletions of chromosome 14q32.31-32, containing RCOR1 and TRAF3, in cohorts of R-CHOP-treated DLBCL patients, and showed an association of these events with inferior outcomes (11, 36, 38). These observations motivated us to further explore the functional impact of TRAF3 deletions in DLBCL, with the goal to identify druggable dependencies for this patient population in need.
Results
14q32.31-32 Deletions Correlate with Reduced TRAF3 Expression in DLBCL Tumors.
As a first step, we extended our original cohort (n = 11/139) of uniformly R-CHOP treated de novo DLBCLs from a population-based registry (38) and found additional tumors containing focal 14q32.31-32 deletions by Affymetrix SNP6.0 arrays (Fig. 1A and SI Appendix, Tables S1 and S2, total n = 24/324, 7.4%). There were no differences in routinely reported clinical and phenotypic characteristics between TRAF3-deleted vs. wild-type (WT) cases (SI Appendix, Table S3). We integrated SNP6.0 findings with RNA-sequencing (RNA-seq) data from the same cohort and identified a significant association between TRAF3 mRNA levels and copy number status (Fig. 1B), suggesting TRAF3 deletions cause haploinsufficiency (39). In agreement with previous observations for 14q32.31-32 deletions (38), TRAF3 expression was associated with 5-y disease-specific survival (DSS) in multivariable Cox regression analyses containing the International Prognostic Index score and COO classification as covariables (P = 0.03, HR 95% CI 0.6393-0.9935, SI Appendix, Fig. S1A and Table S4). We next assessed whether TRAF3 deletions were associated with previously established molecular or genetic subtypes of DLBCL. We observed no significant differences in the enrichment of TRAF3 deletions with COO, DZsig (9) or LymphGen (12) compared to TRAF3 WT tumors (Fig. 1 C–E). These findings were generally corroborated in an independent DLBCL cohort (11), except differences in GCB-associated subtypes were observed given the greater enrichment of ABC cases in the Chapuy et al. cohort (SI Appendix, Fig. S1B and Table S5).
Fig. 1.
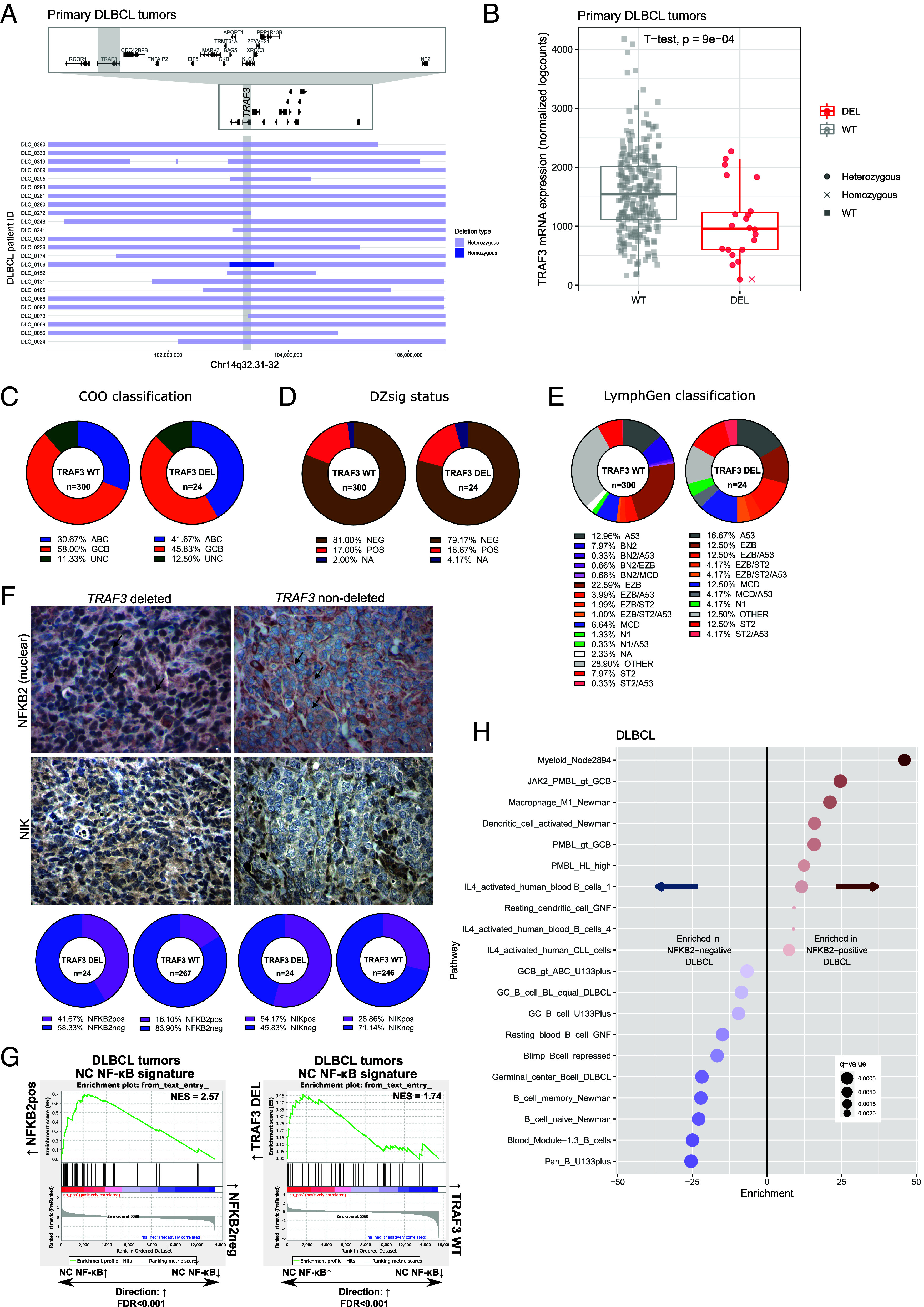
Genetic characterization of TRAF3 deletions in DLBCL. (A) Horizontal bars correspond to focal copy number loss along chromosome 14q32.31-32 (n = 24) centered on the TRAF3 gene (gray vertical bar). Light blue represents hemizygous deleted regions, and dark blue represents homozygous deleted regions in each case. Individual gene bodies and exons in the visualized region are depicted above including a zoomed view. (B) Copy number-to-mRNA expression correlation plot on 313 DLBCL tumors. Box-and-whisker plot shows the median centered around the upper and lower quartiles with each dot representing an individual patient sample. (C–E) Pie graphs illustrating the proportion of subtypes within TRAF3 WT or DEL cases according to the following classification algorithms: COO (C), DZsig (D), and LymphGen (E). (F) Representative IHC images for NFKB2 and NIK stained DLBCL samples. Magnification at 40×, and a 50 μm scale is provided. In the upper images, the black arrows indicate the nuclear compartment of tumor cells stained with NFKB2. The number of stained samples is provided in SI Appendix, Table S6. (G) GSEA using the NC NF-κB expression signature in NFKB2pos vs. NFKB2neg tumors (Left) and TRAF3-deleted vs. TRAF3 WT tumors (Right). (H) Top pathways from LymphoChip enriched in NFKB2pos (Right) or NFKB2neg cases (Left) in the DLBCL cohort. Red and blue color gradients indicate the fold enrichment of pathways.
TRAF3 Loss Is Associated with NC NF-κB Activation in Primary DLBCL.
To assess whether TRAF3 loss correlates with increased NC NF-κB signaling cascade activity, we first assessed cytoplasmic staining of NIK and nuclear staining of NFKB2 in malignant cells by immunohistochemistry (IHC) on DLBCL tissue microarrays. NIK positive (NIKpos, n = 88/292, 30.1%, SI Appendix, Table S6) stained cases were significantly associated with TRAF3 deletions (P = 0.0188, Fig. 1F). Following the stabilization of NIK in the absence of TRAF3, the transcriptional cofactor NFKB2 is processed by the proteasome, leading to the conversion of the p100 precursor to the mature p52 protein, which translocates to the nucleus in a complex with the transcription factor RelB (40). Consistent with this mechanism, we found 57/303 cases (18.8%) were positive for nuclear NFKB2 (NFKB2pos, Fig. 1F) and strongly associated with NIKpos status (P = 0.0001) and TRAF3 deletions (P = 0.0045). The percentage of tumor cells positive for nuclear NFKB2 was significantly correlated with NFKB2 transcript expression (SI Appendix, Fig. S1C). By contrast, we also reanalyzed previously published IHC data from Xia et al. (19) and observed no significant association of NIK staining nor TRAF3 deletions with nuclear p65 (RelA) status, a marker of canonical NF-κB activation.
To determine the downstream transcriptional consequences associated with NC NF-κB signaling, we next conducted a supervised differential gene expression and pathway analysis in primary DLBCL specimens, stratified by nuclear NFKB2 status (i.e., NFKB2pos vs. neg, SI Appendix, Tables S7 and S8) or TRAF3 genetic status (i.e., TRAF3-deleted vs. WT, SI Appendix, Tables S9 and S10). Using previously established NC NF-κB gene expression signatures in lymphoid malignancies (22, 41, 42), we curated a NC NF-κB gene signature (selection criteria detailed in SI Appendix, Table S11) and interrogated our RNA-seq dataset using gene set enrichment analysis (GSEA). We observed enrichment of this signature in both NFKB2pos vs. NFKB2neg and TRAF3-deleted vs. TRAF3 WT comparisons (Fig. 1G and SI Appendix, Table S12), strongly suggesting a functional role of TRAF3 deletions in NC NF-κB signaling in DLBCL. In addition to NF-κB-associated signaling pathways, we also found marked downregulation of naive and germinal center B cell expression signatures in NFKB2pos cases (Fig. 1H). Previous reports have shown enforced Nik expression in mice negatively impacts the GC reaction and induces plasma cell differentiation (27), whereas deletion of Nfkb2 in murine GC B cells impairs the development of antigen-specific plasma cells (24), indicating a central role of NC NF-κB signaling in regulating B cell differentiation. Taken together, these findings establish recurrent TRAF3 deletions as a candidate driver of dysregulated NC NF-κB transcriptional rewiring in DLBCL.
TRAF3 Loss-of-Function (LOF) Enhances NC NF-κB Signaling in DLBCL.
To delineate the specific role of TRAF3 LOF in DLBCL, we generated isogenic TRAF3 knockout DLBCL cell lines (SI Appendix, Fig. S2A) using CRISPR-Cas9. First, we investigated the activity of the canonical and NC NF-κB pathways by immunoblots, as both branches contribute to the pathogenesis of lymphomas (43, 44). Consistent with NC NF-κB pathway activation, we observed increased nuclear localization of subunits RelB and p52 in TRAF3 LOF cells, as compared to gRNA control clones with WT sequences, termed “WT” (Fig. 2 A and B and SI Appendix, Fig. S2 B and E). Compared to GCB-subtype cells, ABC-subtype cells showed higher baseline nuclear enrichment of NC NF-κB transcription factors (Fig. 2 A and B), which may explain the lower fold change of NC NF-κB activation following TRAF3 knockout. Accordingly, we saw increased total NF-κB driven transcriptional activity using a luciferase reporter system (45) only in GCB models (Fig. 2C). On the other hand, nuclear localization of canonical NF-κB transcription factor complex subunits RelA (Fig. 2B and SI Appendix, Fig. S2 C and F) and c-Rel (SI Appendix, Fig. S2G) was not affected by TRAF3 LOF. Nuclear expression of the cofactor p50, however, was enriched in TRAF3 LOF DLBCL cells, suggesting potential crosstalk between the two NF-κB branches. To elucidate the underlying transcriptional effects specifically triggered by TRAF3 LOF, and ensuing NC NF-κB activation, we next performed RNA-seq on our cell line model systems and observed upregulation of NC NF-κB targets such as IL10 and BIRC3 in TRAF3-deficient cells (SI Appendix, Fig. S3 A–D and Tables S13 and S14). In line with our differential gene expression analysis (DGEA) of primary tumor samples (Fig. 1G), we observed enrichment of the curated NC NF-κB gene signature in TRAF3 LOF cells (SI Appendix, Table S12 and Fig. 2D), suggesting that TRAF3 LOF is a critical driver of enhanced NC NF-κB signaling and ensuing transcriptional rewiring in DLBCL.
Fig. 2.
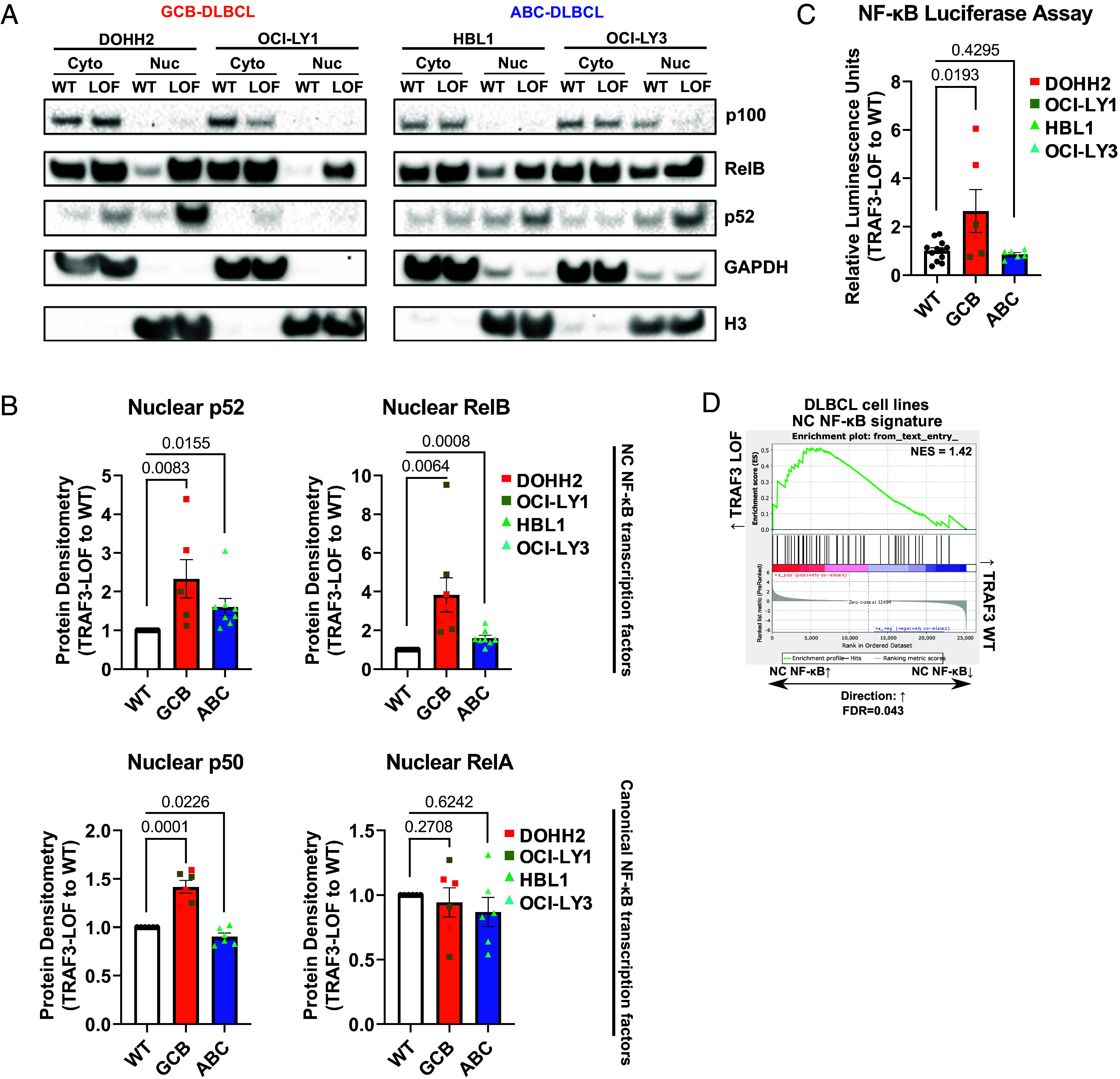
TRAF3 LOF enhances the NC NF-κB pathway in DLBCL. (A) Representative immunoblotting images of p100/p52 and RelB from TRAF3 WT and LOF DLBCL cell line models (DOHH2, OCI-LY1, HBL1, OCI-LY3). (B) Summaries of nuclear protein expression of p52, RelB, p50, and RelA. Band densities of TRAF3 LOF are compared to WT and normalized to Histone H3 (nuclear loading control) expression. (C) NF-κB luciferase reporter assays summarized as relative luminescence units of TRAF3 LOF to WT cells. (D) GSEA using the NC NF-κB expression signature in TRAF3 LOF vs. WT cell lines. For (B and C), each bar graph shows the mean values ± SEM, and each dot represents an independent replicate.
TRAF3-Deficient DLBCL Cells Require NIK for Growth and Survival.
We wondered whether this increase in NC NF-κB signaling caused by TRAF3 LOF would create a targetable dependency that could be exploited clinically. Activation of both NF-κB branches is tightly regulated by proteasome activity (46). To determine whether bortezomib, an FDA-approved proteasome inhibitor recently shown to improve outcomes in nonGCB-DLBCL (47), could kill TRAF3 LOF DLBCL cells more effectively than WT control cells, we performed dose–response studies. We did not observe differential killing between TRAF3 WT and LOF cells (SI Appendix, Fig. S4A). At the molecular level, we observed that proteasome inhibition with a similar drug (MG132) effectively reduced nuclear p50 levels but not nuclear p52 levels (SI Appendix, Fig. S4 B and C). These results suggest canonical NF-κB signaling, but not NC NF-κB, can be inhibited by proteasome blockade in DLBCL cells. Under steady-state conditions, the central NC NF-κB kinase NIK is actively degraded by the proteasome in a TRAF3-dependent manner (26). We observed increased accumulation of NIK in TRAF3 LOF cells compared to WT cells, at basal level (Fig. 3A and SI Appendix, Fig. S2 A and D). NIK upregulation was dramatically exacerbated, particularly in TRAF3 LOF cells, upon treatment with MG132 (SI Appendix, Fig. S4D), suggesting proteasome inhibition may be inadequate at inhibiting TRAF3 LOF-dependent NC NF-κB signaling.
Fig. 3.
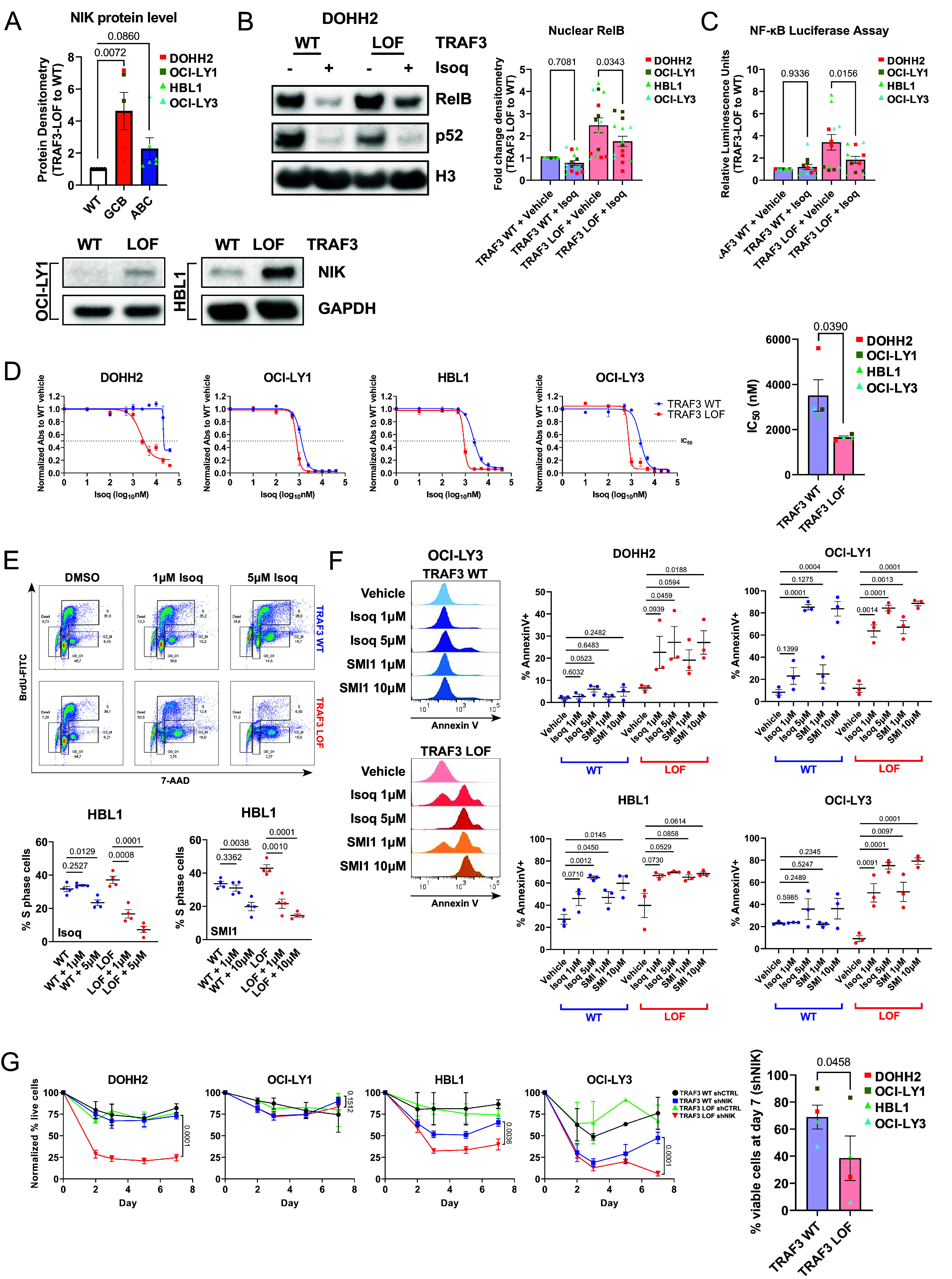
NIK is a critical player in NC NF-κB signaling in DLBCL. (A) NIK protein densitometry summary of TRAF3 WT and LOF whole-cell lysates. Band densities of each treatment condition are compared to TRAF3 WT and normalized to GAPDH (loading control) expression. Each dot represents an independent replicate. (B) Immunoblotting analysis of nuclear RelB and p52 expression in NIK inhibitor Isoq or vehicle control treated DOHH2 cells. Protein densitometry for RelB was normalized to GAPDH, prior to comparing each treatment condition to “TRAF3 WT + vehicle.” The full blot for DOHH2 is included in SI Appendix, Fig. S5A (excerpt from Top Left). (C) NF-κB luciferase reporter assays summarized as relative luminescence units of TRAF3 LOF to WT cells. (D) Dose–response analysis of the NIK inhibitor Isoq was performed using the WST-1 assay. Raw absorbance values were normalized to the “TRAF3 WT + vehicle” treatment. Each dot represents the average of independent replicates of each cell line. (E) BrdU/7-AAD flow cytometry staining and summary of percent gated S-phase events following Isoq or SMI1 treatment for HBL1. Shown above is the cell cycle gating strategy for G0_G1, S, G2_M, and dead cells. (F) AnnexinV flow cytometry staining was performed on DLBCL cells. Shown on the Left are representative histograms of Isoq or SMI1 treated OCI-LY3 TRAF3 WT and LOF cells, and summaries of DOHH2, OCI-LY1, HBL1, and OCI-LY3 are shown on the Right. (G) MAP3K14 (shNIK) knockdown in DLBCL cells. For (A–C and E–G), each dot represents the average of independent replicates of each cell line. For all panels, each graph shows the mean values ± SEM.
These findings prompted us to assess direct NIK inhibition as a therapeutic strategy. We employed a small molecule inhibitor of NIK, isoquinoline-1,3(2H,4H)-dione (Isoq), which has been reported to repress NC NF-κB pathway activation in NIK-overexpressing Hodgkin-Reed Sternberg cells (32). We observed a reduction of nuclear RelB localization (Fig. 3B and SI Appendix, Fig. S5A) and NF-κB transactivation capacity (Fig. 3C) following Isoq treatment. Canonical NF-κB activation (nuclear RelA, p50) was not strongly affected by Isoq, confirming the specificity of NIK inhibition on NC NF-κB signaling (SI Appendix, Fig. S5 A and B).
We next performed dose–response studies on DLBCL cells using Isoq to assess the impact on cell survival. Three of four TRAF3 LOF cell lines were more sensitive to Isoq compared to WT cells (IC50 LOF = 1.6 to 1.8 µM vs. IC50 WT = 2.4 to 5.6 µM) (Fig. 3D and SI Appendix, Fig. S5C). Similar findings were observed with a second inhibitor NIK-SMI1 (SMI1), which has been reported to suppress NC NF-κB activation in primary B cells and TRAF3 mutant multiple myeloma cells (39, 48) (SI Appendix, Fig. S5D). Treatment with NIK inhibitors resulted in a dose-dependent overall reduction in proliferation based on progressive dilution of CellTrace Violet (CTV) staining (SI Appendix, Fig. S6A) and in the fraction of cycling S phase cells, based on BrdU incorporation (Fig. 3E and SI Appendix, Fig. S6B), independent of basal proliferation rates. In addition, we saw an increase of AnnexinV+ cells after treatment (Fig. 3F and SI Appendix, Fig. S6C), suggesting that NIK inhibition further compromises the survival of TRAF3-deficient cells. Isoq and SMI1 induced cell cycle arrest and killed TRAF3 LOF cells at concentrations as low as 1 µM, a dose which had no significant effect on TRAF3 WT cells. To validate these findings through an orthogonal approach, we performed MAP3K14 (NIK) shRNA knockdown and observed a significant reduction in NC NF-κB activation and cell viability in three of four TRAF3 LOF cell lines (Fig. 3G and SI Appendix, Fig. S6 D and E), in contrast to TRAF3 WT controls. In summary, our results suggest TRAF3 LOF cells acquire dependence on NIK for growth and survival and highlight a subset of lymphomas which may be therapeutically targetable by direct NIK inhibition.
DLBCL-Derived IL-10 Suppresses Granzyme B and IFNγ Expression in CD8+ T Cells.
NC NF-κB activation is strongly linked to the expression of both pro- and anti-inflammatory cytokines and chemokines in normal and disease contexts (40, 49–51). Therefore, to gain further insight into the potential effects of NC NF-κB activation in DLBCL cells, we performed a Luminex-based assessment of secreted factors in conditioned media from our TRAF3-deficient models (Fig. 4A). One of the cytokines that appeared consistently upregulated in these cell models was IL-10. This cytokine has been previously shown to exert immunosuppressive effects on T cells (52–55) and to promote ABC-DLBCL growth and survival (56, 57). To validate our observations through an orthogonal approach, we measured IL-10 secretion via ELISA. Basal IL-10 levels were higher in WT-treated HBL1 and OCI-LY3, than in DOHH2 and OCI-LY1, which showed almost undetectable IL-10 levels (Fig. 4B). In line with our Luminex-based findings, IL-10 secretion was further enhanced in TRAF3 LOF cells (except in DOHH2 where it was not detectable), strongly suggesting a direct role of TRAF3 LOF in the regulation of this cytokine, in agreement with previous reports on Traf3 KO or haploinsufficient murine B cells (58, 59). IL10 mRNA was also found consistently up-regulated in our RNA-seq datasets (SI Appendix, Fig. S3 B–D). Importantly, Isoq specifically reduced IL10 mRNA levels (Fig. 4C and SI Appendix, Fig. S7A) without impeding the expression of another secreted factor, LTA (SI Appendix, Fig. S7B). These results indicate that TRAF3 LOF exerts its regulatory effects on IL-10 expression at the transcriptional level in DLBCL. More broadly, previous studies have established IL-10 expression as a general feature in ABC-DLBCLs, driven by oncogenic canonical BCR/NF-κB signaling (21, 60). Indeed, IL10 transcript levels were highly enriched in human DLBCL specimens of the ABC-associated MYD88/CD79B (MCD) genetic subtype in our study cohort and external cohorts (Fig. 4D).
Fig. 4.
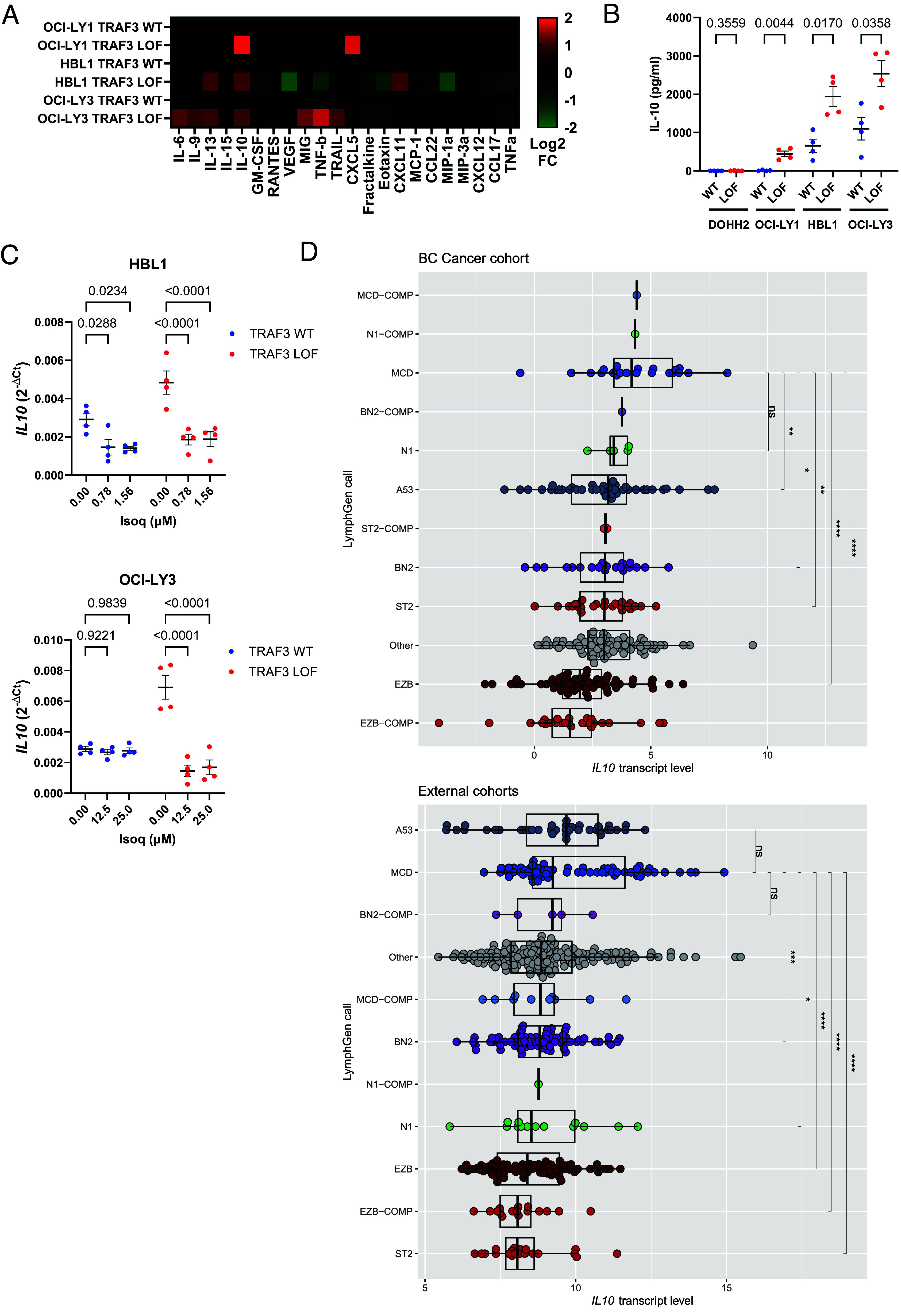
IL-10 expression induced by TRAF3 LOF is dependent on NIK in DLBCL. (A) Luminex profiling of cell line supernatants; a heatmap of the log2 fold change is shown comparing TRAF3 LOF to WT. (B) IL-10 ELISA validation of cell line supernatants. (C) RT-qPCR analysis of IL10 in TRAF3 WT and LOF HBL1 (Top) and OCI-LY3 (Bottom) cells following 6-h treatment at the indicated doses of Isoq. GAPDH was used as the internal control and the reference gene for ΔCt calculations. (D) Normalized log2-transformed counts-per-million scaling of IL10 mRNA transcript levels from RNA-seq in DLBCL cases grouped by LymphGen genetic subtypes (12) in the study cohort (Top) and external cohorts (Bottom). Additional details for “External cohorts” are described in SI Appendix. For panels B and C, each dot represents an independent replicate, and each graph shows the mean values ± SEM. For panel D, each dot represents an individual tumor sample, with the box plots and whiskers showing the 95% CI, and for pairwise comparisons ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05, ns = not significant.
As previous reports have proposed a role for IL-10 in suppressing CD8+ T cell effector function (52–55), we decided to explore whether TRAF3 inactivation in DLBCL cells could similarly mediate immune evasive effects. To this end, we performed coculture assays between in vitro activated CD8+ T cells from healthy donors, and our TRAF3-deficient lymphoma models. Expression of IL-10 was primarily derived from lymphoma cells (SI Appendix, Fig. S8A). We assessed a panel of effector (GZMB, IFNγ) and exhaustion markers (TIM3, PD1, CTLA4, LAG3, by which expression of the latter two was not detectable) on cocultured CD8+ T cells by flow cytometry (gating strategy shown in SI Appendix, Fig. S8 B and C). CD69 expression was induced to a comparable extent in coculture with TRAF3 WT or LOF cells, indicating that broad T cell activation was not impaired by TRAF3 LOF (SI Appendix, Fig. S8D). On the other hand, we observed a reduced percentage of GZMB+/IFNγ+ among CD8+ T cells cocultured with TRAF3 LOF HBL1 and OCI-LY3 cells, as compared to TRAF3 WT cells (Fig. 5A). We also observed increased percentages of TIM3+ and PD1+ CD8+ T cells cocultured with TRAF3 LOF cells, as compared to control cells (Fig. 5 B and C), while retaining similar proliferative capacities (SI Appendix, Fig. S8 E and F). In contrast, TRAF3 LOF DOHH2 and OCI-LY1 failed to elicit detectable changes in the percentages of GZMB+/IFNγ+, TIM3+ or PD1+ CD8+ T cells (SI Appendix, Fig. S8 G and H).
Fig. 5.
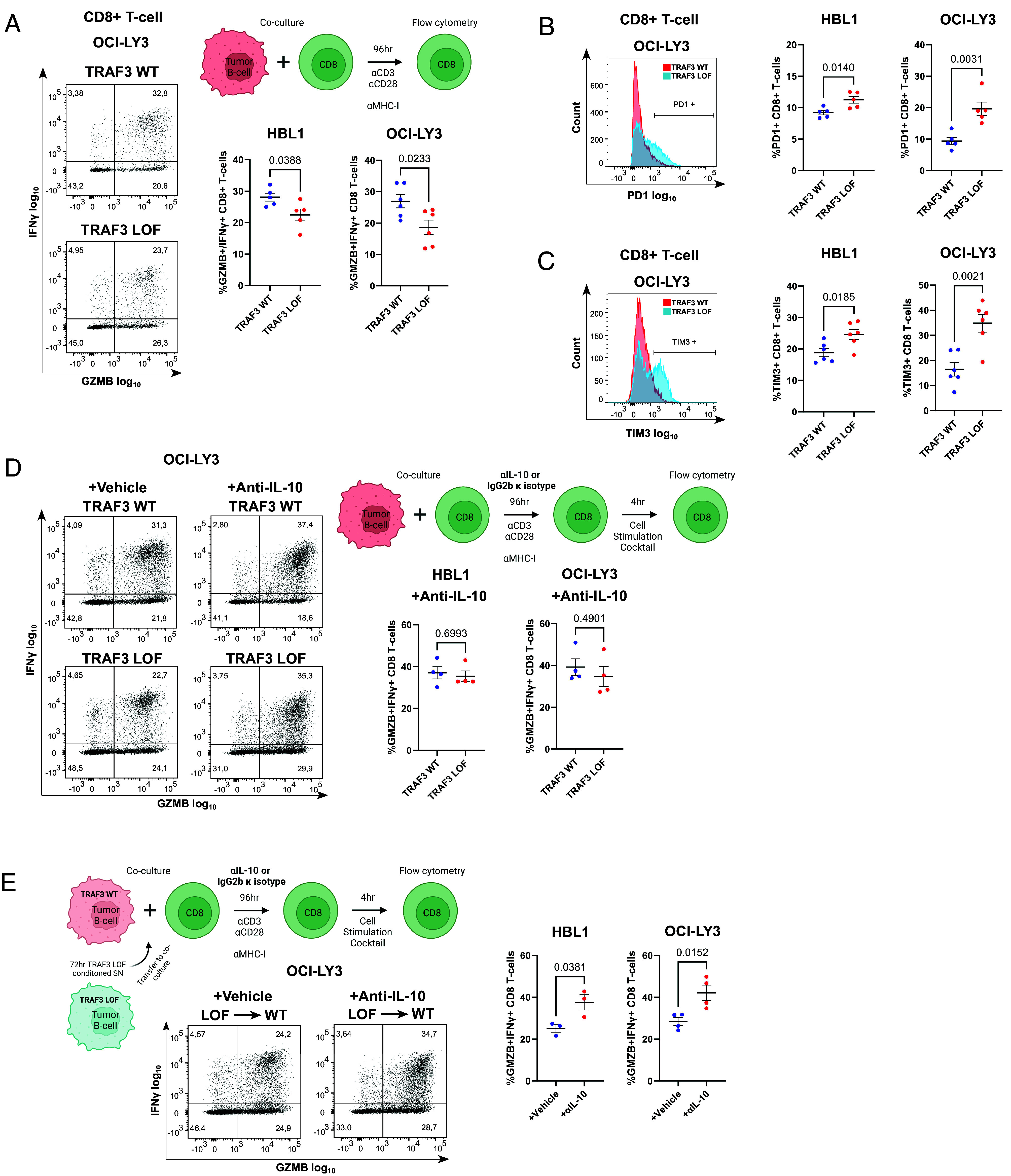
TRAF3 LOF in DLBCL impairs CD8+ T cell effector capacity. (A–C) GZMB/IFNγ (A), PD1 (B), and TIM3 (C) expression of CD8+ T cells cocultured with HBL1 or OCI-LY3 TRAF3 WT (red) or LOF (blue) DLBCL cells. A diagram of the experimental setup is included in panel C. (D) GZMB/IFNγ expression on CD8+ T cells following coculture with HBL1 or OCI-LY3 TRAF3 WT or LOF cells treated with anti-IL-10 or vehicle control IgG2b κ isotype. (E) GZMB/IFNγ expression on CD8+ T cells cocultured with HBL1 or OCI-LY3 TRAF3 WT cells after incubation with TRAF3 LOF conditioned supernatant (SN). A diagram of the experimental setup is included at the Top. For all panels, each dot represents an independent replicate, and each graph shows the mean values ± SEM.
To determine whether IL-10 was functionally contributing to the observed T cell phenotypes, we next blocked IL-10 signaling in our coculture system, using an anti-IL-10 antibody. Notably, IL-10 blockage rescued the reduction in GZMB+/IFNγ+ CD8+ T cells introduced by TRAF3 LOF to similar levels as TRAF3 WT (Fig. 5D). Next, to further assess whether the NC NF-κB-driven lymphoma secretome would be sufficient to mediate the observed effects, we cocultured CD8+ T cells and TRAF3 WT HBL1 and OCI-LY3 cells, in the presence of conditioned supernatant from TRAF3 LOF HBL1 and OCI-LY3, respectively. Indeed, the addition of conditioned media was sufficient to repress GZMB and IFNγ expression in CD8+ T cells, and this effect could again be rescued by adding anti-IL-10 treatment, indicating a predominantly interleukin-driven suppressive effect on GZMB and IFNγ expression, mediated by IL-10 (Fig. 5E). The lack of suppressive effects on CD8+ T cells observed in TRAF3 LOF DOHH2 and OCI-LY1 coculture models is consistent with the lower absolute quantity of IL-10 secreted by these cells, as compared to TRAF3 WT HBL1 and OCI-LY3 (Fig. 4B). Interestingly, TIM3 and PD1 expression were not rescued by anti-IL-10 treatment (SI Appendix, Fig. S8I), suggesting independent control mechanisms (61, 62). Taken together, our results demonstrate a critical extrinsic role of NC NF-κB in DLBCL, whereby IL-10 secretion attenuates the effector capacity of CD8+ T cells.
Discussion
Studies in GC-derived B cell lymphomas have implicated somatic mutation-driven aberrant NC NF-κB activation as a prevalent mechanism of pathogenesis (27–36). In this study, we performed comprehensive molecular and genetic characterization of the NC NF-κB regulator TRAF3 in a large DLBCL cohort. We identified a subgroup of TRAF3-deleted tumors with active NC NF-κB signaling, which were more commonly found in SGK1/TET2 (ST2) and MCD genetic subtypes containing other NF-κB altering lesions in genes such as NFKBIA, MYD88 and CD79B (12). Lacy et al. (63) identified somatic TRAF3 variants which were exclusively nonsense, frameshift, and essential splice site mutations (n = 7/928 tumors, ~0.8%, summarized in SI Appendix, Table S15). Those mutations occurred upstream of the region encoding the TRAF-C domain of TRAF3 mediating interaction with NIK (26), which together with somatic TRAF3 deletions strongly suggest a convergence on a LOF phenotype in DLBCL. Interestingly, TRAF3 is one of the most frequently mutated genes in canine BCLs, which histologically recapitulate many aspects of human DLBCLs (36, 64). A recent report correlated TRAF3 monogenic germline variants with reduced protein expression in B cells of immunodeficient and autoimmune patients (39), suggesting that TRAF3 haploinsufficiency likely mirrors the phenotype of somatically acquired TRAF3 deletions.
Unlike other immune microenvironment-rich lymphomas, such as classic Hodgkin lymphoma, which respond well to immune checkpoint blockade, clinical trials in DLBCL have shown only marginal improvements in response rates (65). We found that lymphoma-derived IL-10, up-regulated in part by TRAF3 LOF, constitutes one of the mechanisms leading to downregulation of GZMB and IFNγ expression in CD8+ T cells, further expanding on IL-10’s pleiotropic roles in antitumor immunity and chronic infection (57, 66, 67). This suppressive effect may depend on the local concentration of IL-10 in the microenvironment milieu, as reduced T cell expression of GZMB and IFNγ was detected only in coculture systems with high IL-10-expressing TRAF3 LOF HBL1 and OCI-LY3 cells. This is further evidenced by the lack of a suppressive phenotype conferred by TRAF3 LOF OCI-LY1 cells. Low baseline IL-10 expression in the WT cells likely accentuates the magnitude of change in the TRAF3 LOF model. Production of IL-10 by NC NF-κB activation complements canonical NF-κB in ABC and MCD tumors, which is reported to be another main oncogenic signaling pathway contributing to elevated IL-10 baseline expression (21, 56, 60, 68–70). Our findings suggest that the complex relationship between IL-10-expressors and TME constituents requires further exploration in the context of DLBCL.
We provide evidence that constitutive NC NF-κB activation is not restricted to the ABC subtype, reconciling previous findings showing enforced p100 or p52 expression can reprogram GCB cells into an NF-κB phenotype characteristic of ABC cells (71). NFKB2 is a target gene of the canonical NF-κB pathway (71, 72), and NFKB2 overexpression can directly lead to increased nuclear accumulation of p52 and RelB in experimental models (71). These prior findings suggest signaling crosstalk in the two NF-κB pathways might be one of the explanations for why a fraction of DLBCL cases exhibit nuclear NFKB2 staining without concomitant NIK accumulation or TRAF3 aberration. Interestingly, we observed that the canonical NF-κB cofactor p50 was differentially enriched in the nuclear fraction of TRAF3 WT and LOF cells, suggesting other roles for p50, perhaps as part of promiscuous heterocomplexes (e.g., with BCL3) that may influence NF-κB transactivation (73).
Proteasome inhibition promotes the accumulation of NIK protein in lymphoma cells, more markedly in TRAF3 LOF cells, while p52 levels are unchanged, suggesting additional mechanisms such as IKKα-dependent NIK degradation may play an important role in regulating NIK protein levels (74). Based on our in vitro data, NIK inhibition serves as proof-of-concept for the induction of apoptosis in DLBCLs with heightened NC NF-κB signaling. Although some heterogeneity in the magnitude of response in each cell line is observed, TRAF3 LOF cells were consistently more sensitive to NIK inhibition compared to WT cells. Our data complement previous work demonstrating reduced proliferation of DLBCL cells (75, 76) and increased sensitivity to chemotherapeutics (28, 29, 37) following RELB shRNA knockdown or genetic ablation. Taken together, these findings warrant further investigation into direct lymphoma cell killing using NC NF-κB inhibitors (e.g., NIK inhibitors) in combination with chemotherapeutics or immunotherapeutics (e.g., anti-IL-10).
Materials and Methods
Study Cohort Description.
A total of 324 de novo DLBCL patients uniformly treated with rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP) from the BC Cancer population-based registry were included in this study. Patient characteristics are summarized in SI Appendix, Table S3. Then, 313 samples with matched RNA-seq, COO assignment (Lymph2Cx), and copy number data were used for integrative genomics analysis. In addition, 293 samples with matched clinical annotations were included for outcome correlation. This study was reviewed and approved by the University of British Columbia—BC Cancer Research Ethics Board, in accordance with the Helsinki Declaration. All patients provided written informed consent with the exception of patients where waiver of consent was granted by the Research Ethics Board.
Copy Number Analysis.
OncoSNP (v1.3, 36) was used to define “gene-centric” copy number alterations and segment copy number data from Affymetrix Human SNP6.0 Arrays of 324 DLBCL samples as previously described (14). Tumor states associated with tumor copy number <2 were defined as “deletion events” in processed samples (SI Appendix, Table S1).
Isogenic TRAF3 LOF Generation in DLBCL.
Alt-R CRISPR-Cas9 genome editing (Integrated DNA Technologies, Coralsville, IA) was used to generate TRAF3 knockout cell lines using the following guide RNA sequences: 5′-TCTTGACACGCTGTACATTT-3′ and 5′-GCCCACACTCGGTCTGCTTC-3′ targeting exons 4 and 3 of TRAF3, respectively. For each of the four DLBCL isogenic cell line model systems, three single-cell expanded knockout clones were generated. TRAF3 genotype and TRAF3 protein expression were validated using Sanger sequencing and immunoblotting, respectively (SI Appendix, Fig. S2A and Table S16).
DGEA.
RNA-seq was performed on three biological replicates for each cell line (DOHH-2, OCI-LY1, HBL-1, OCI-LY3), according to the manufacturer’s instructions, on a NextSeq550 instrument (Illumina, San Diego, CA). Data processing and DGEA (DESeq2 v1.26.0) for primary lymphoma and cell line samples were performed as previously described (14). Preranked gene-set enrichment analysis (GSEA v4.1.0, https://www.gsea-msigdb.org/gsea/index.jsp) was performed using the log2FC between TRAF3_del vs. TRAF3_WT, or TRAF3 LOF vs. TRAF3 WT for gene ordering, and interrogating a collection of MSigDB (Broad Institute) annotated gene-sets [Hallmark, BioCarta, KEGG, Reactome, and Gene Ontology (GO) terms, https://www.gsea-msigdb.org/gsea/msigdb] as well as a curated list of NC NF-κB pathway genes (selection criteria described in SI Appendix, Table S11). DGEA between NFKB2-positive and NFKB2-negative DLBCL samples was performed using limma. GO analysis using the LymphoChip database (77) was applied to identify enriched gene expression signatures and biological pathways among the most significantly differentially expressed genes.
T Cell Coculture and Characterization.
Peripheral blood mononuclear cells were isolated from whole blood of healthy donors after which CD8+ cell fractions were selected using magnetic activated cell sorting (MACS, Miltenyi Biotec, Gaithersburg, MD). CellTrace Violet (Invitrogen, Waltham, MA) labeling at a final concentration of 5 μM was performed on 1 × 107 cells/ml CD8+ T cells in 5 mL polystyrene tubes and incubated at 37 °C for 15 min with gentle vortexing every 5 min. Cells were seeded at a 50:1 ratio (T:B cell) in RPMI + FBS10% + MHC-I blocking antibodies at 10 μg/mL to reduce alloreactivity (HLA-ABC antibody clone W6/32, Invitrogen, Waltham, MA). Human IL-10 antibody (MAB217, R&D Systems, Minneapolis, MN) or mouse IgG2b κ isotype ctrl antibody (BioLegend, San Diego, CA) was added at a final concentration of 2 μg/mL. After 96 h, samples were prepared for flow cytometry analysis using the following markers (SI Appendix, Table S17): LIVE/DEAD Fixable Yellow (Invitrogen, Waltham, MA), CD4, CD8, PD-1, CD69, TIM3, LAG3, CTLA4. For intracellular marker detection [Granzyme B (GZMB) and IFNγ], samples were pretreated 4 h with Cell Stimulation Cocktail and GolgiStop/GolgiPlug (Invitrogen, Waltham, MA). Please refer to SI Appendix for additional details.
Drug Dose–Response and Cell Cycle Assays.
DLBCL cells (DOHH2, OCI-LY1, HBL1, OCI-LY3) were seeded in 96-well plates at 2 × 104 cells per well and incubated with serial dilutions of bortezomib (MilliporeSigma, Burlington, MA), isoquinoline-1,3(2H,4H)-dione (Isoq, SantaCruz, Dallas, TX), NIK-SMI1 (SMI1, MedChemExpress, Princeton, NJ) or vehicle control (DMSO) for 48 h at 37 °C in complete media. WST-1 reagent (Roche, Basel, Switzerland) was added, and the cells were incubated for 3 h. IC50 was determined for each drug using the GraphPad Prism ver 9.4.1 (GraphPad Software Inc, https://www.graphpad.com). Cell cycle state was determined using bromodeoxyuridine/7-aminoactinomycin D staining (BrdU/7-AAD, FITC BrdU Flow Kit, BD Biosciences, Franklin Lakes, NJ). Then, 2 × 105 cells were cultured for 48 h with Isoq (1 μM, 5 μM), SMI1 (1 μM, 10 μM) or vehicle DMSO treatment and incubated with 10 μM BrdU in complete culture medium for 30 min at 37 °C. For assessment of IL10 and LTA mRNA expression, Isoq was serially diluted starting from 25 μM and added to cells for 6 h. Real-time qPCR was performed using the TaqMan gene expression probe system (Invitrogen, Waltham, MA, cat #4331182).
Statistical Analysis.
Statistical testing between groups was performed using one-sample t test, paired or unpaired two-sample t test, one-way ANOVA, or two-way ANOVA, where appropriate (R ver 4.2.0 & GraphPad Prism ver 9.4.1). Fisher’s exact tests were performed on categorical data. The Benjamini–Hochberg procedure was carried out to control the false discovery rate for multiple comparisons, and an adjusted P-value < 0.05 was considered statistically significant. For survival analysis, univariable and multivariable Cox proportional hazard regression models were used to evaluate the risk of the proposed factors for DSS, time-to-progression, PFS, and OS (SI Appendix, Table S4). Normalized counts for TRAF3 expression were included as a continuous covariate in these analyses.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank the British Columbia Cancer Foundation (BCCF) for their support. We also greatly thank Elizabeth A. Chavez for next-generation sequencing experiments and excellent technical support. Some figures were created using BioRender (https://biorender.com). This work was supported by a project grant from the Terry Fox Research Institute (Grants #1061 and #1108), the Leukemia and Lymphoma Society, and the Leukemia and Lymphoma Society of Canada Translational Research Program (Grant #6663-23) (C.S.). M.Y.L. is a PhD candidate at The University of British Columbia, Canada and is supported by an Elizabeth C. Watters Research Fellowship. E.V. is the recipient of a Michael Smith Foundation for Health Research Fellowship award. I.E. and G.K. were funded by the Lotte & John Hecht Memorial Foundation (#4340). L.V. is a Michael Smith Health Research BC Scholar and is supported by the BCCF and a Canadian Institutes of Health Research Project Grant (#180613). C.S. has received research funding from Epizyme and Trillium Therapeutics. D.W.S. has received research funding Roche/Genentech and Janssen.
Author contributions
M.Y.L., W.N., E.V., T.A., S.H., G.K., L.V., D.W.S., and C.S. designed research; M.Y.L., L.C.C., G.D., A.L., B.W., A.J., A.T., S.B.-N., G.W.S., and I.E. performed research; M.Y.L., L.C.C., G.D., A.L., B.W., A.J., S.B.-N., and G.W.S. analyzed data; and M.Y.L., L.V., and C.S. wrote the paper.
Competing interests
C.S. has performed consultancy for Bayer. D.W.S. has served as a consultant for Abbvie, AstraZenenca, Incyte, and Janssen. C.S. and D.W.S. are named inventors on a patent filed by the National Cancer Institute “Methods for determining lymphoma type.” The remaining authors declare no competing financial interests.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
Cell line RNA-seq data (78) are deposited at Gene Expression Omnibus (accession GSE218750). Study cohort raw RNA-seq and SNP6.0 data can be found at European Genome-phenome Archive (accession EGAS00001002657 https://ega-archive.org/studies/EGAS00001002657) (79). All other data are included in the manuscript and/or SI Appendix.
Supporting Information
References
- 1.Horvat M., et al. , Diffuse large B-cell lymphoma: 10 years’ real-world clinical experience with rituximab plus cyclophosphamide, doxorubicin, vincristine and prednisolone. Oncol. Lett. 15, 3602–3609 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Coiffier B., et al. , CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 346, 235–242 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Cunningham D., et al. , Rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisolone in patients with newly diagnosed diffuse large B-cell non-Hodgkin lymphoma: A phase 3 comparison of dose intensification with 14-day versus 21-day cycles. Lancet 381, 1817–1826 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Johnson N. A., et al. , Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J. Clin. Oncol. 30, 3452–3459 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sehn L. H., Salles G., Diffuse large B-cell lymphoma. N. Engl. J. Med. 384, 842–858 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alizadeh A. A., et al. , Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Scott D. W., et al. , Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood 123, 1214–1217 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ennishi D., et al. , Double-hit gene expression signature defines a distinct subgroup of germinal center B-cell-like diffuse large B-cell lymphoma. J. Clin. Oncol. 37, 190–201 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alduaij W., et al. , Molecular determinants of clinical outcomes in a real-world diffuse large B-cell lymphoma population. Blood 141, 2493–2507 (2022), 10.1182/blood.2022018248. [DOI] [PubMed] [Google Scholar]
- 10.Schmitz R., et al. , Genetics and pathogenesis of diffuse large B-cell lymphoma. N. Engl. J. Med. 378, 1396–1407 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chapuy B., et al. , Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 24, 679–690 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wright G. W., et al. , A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell 37, 551–568.e14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kotlov N., et al. , Clinical and biological subtypes of B-cell lymphoma revealed by microenvironmental signatures. Cancer Discov. 11, 1468–1489 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duns G., et al. , Characterization of DLBCL with a PMBL gene expression signature. Blood 138, 136–148 (2021). [DOI] [PubMed] [Google Scholar]
- 15.Dunleavy K., et al. , Differential efficacy of bortezomib plus chemotherapy within molecular subtypes of diffuse large B-cell lymphoma. Blood 113, 6069–6076 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davies A., et al. , Gene-expression profiling of bortezomib added to standard chemoimmunotherapy for diffuse large B-cell lymphoma (REMoDL-B): An open-label, randomised, phase 3 trial. Lancet Oncol. 20, 649–662 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Younes A., et al. , Randomized phase III trial of ibrutinib and rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in non-germinal center B-cell diffuse large B-cell lymphoma. J. Clin. Oncol. 37, 1285–1295 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis R. E., et al. , Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 463, 88–92 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia M., et al. , BCL10 mutations define distinct dependencies guiding precision therapy for DLBCL. Cancer Discov. 12, 1922–1941 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lenz G., et al. , Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 319, 1676–1679 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Ngo V. N., et al. , Oncogenically active MYD88 mutations in human lymphoma. Nature 470, 115–119 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Compagno M., et al. , Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 459, 717–721 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kato M., et al. , Frequent inactivation of A20 in B-cell lymphomas. Nature 459, 712–716 (2009). [DOI] [PubMed] [Google Scholar]
- 24.De Silva N. S., et al. , Transcription factors of the alternative NF-κB pathway are required for germinal center B-cell development. Proc. Natl. Acad. Sci. U.S.A. 113, 9063–9068 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerondakis S., Siebenlist U., Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol. 2, a000182 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun S. C., Non-canonical NF-κB signaling pathway. Cell Res. 21, 71–85 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang B., et al. , An oncogenic role for alternative NF-κB signaling in DLBCL revealed upon deregulated BCL6 expression. Cell Rep. 11, 715–726 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eluard B., et al. , The alternative RelB NF-κB subunit is a novel critical player in diffuse large B-cell lymphoma. Blood 139, 384–398 (2022). [DOI] [PubMed] [Google Scholar]
- 29.Nuan-Aliman S., Bordereaux D., Thieblemont C., Baud V., The alternative RelB NF-kB subunit exerts a critical survival function upon metabolic stress in diffuse large B-cell lymphoma-derived cells. Biomedicines 10, 348 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mottok A., et al. , Integrative genomic analysis identifies key pathogenic mechanisms in primary mediastinal large B-cell lymphoma. Blood 134, 802–813 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Rahal R., et al. , Pharmacological and genomic profiling identifies NF-κB-targeted treatment strategies for mantle cell lymphoma. Nat. Med. 20, 87–92 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Ranuncolo S. M., Pittaluga S., Evbuomwan M. O., Jaffe E. S., Lewis B. A., Hodgkin lymphoma requires stabilized NIK and constitutive RelB expression for survival. Blood 120, 3756–3763 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demchenko Y. N., et al. , Classical and/or alternative NF-kappaB pathway activation in multiple myeloma. Blood 115, 3541–3552 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pasqualucci L., Klein U., NF-κB mutations in germinal center B-cell lymphomas: Relation to NF-κB function in normal B cells. Biomedicines 10, 2450 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pasqualucci L., et al. , Analysis of the coding genome of diffuse large B-cell lymphoma. Nat. Genet. 43, 830–837 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bushell K. R., et al. , Genetic inactivation of TRAF3 in canine and human B-cell lymphoma. Blood 125, 999–1005 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Lim S. K., et al. , Sustained activation of non-canonical NF-κB signalling drives glycolytic reprogramming in doxorubicin-resistant DLBCL. Leukemia 37, 441–452 (2023). [DOI] [PubMed] [Google Scholar]
- 38.Chan F. C., et al. , An RCOR1 loss-associated gene expression signature identifies a prognostically significant DLBCL subgroup. Blood 125, 959–966 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Rae W., et al. , Immunodeficiency, autoimmunity, and increased risk of B cell malignancy in humans with TRAF3 mutations. Sci. Immunol. 7, eabn3800 (2022). [DOI] [PubMed] [Google Scholar]
- 40.Sun S. C., The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 17, 545–558 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Annunziata C. M., et al. , Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 12, 115–130 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saba N. S., et al. , Pathogenic role of B-cell receptor signaling and canonical NF-κB activation in mantle cell lymphoma. Blood 128, 82–92 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bista P., et al. , TRAF3 controls activation of the canonical and alternative NFkappaB by the lymphotoxin beta receptor. J. Biol. Chem. 285, 12971–12978 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pham L. V., et al. , Constitutive BR3 receptor signaling in diffuse, large B-cell lymphomas stabilizes nuclear factor-κB-inducing kinase while activating both canonical and alternative nuclear factor-κB pathways. Blood 117, 200–210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou W., et al. , MicroRNA-223 suppresses the canonical NF-κB pathway in basal keratinocytes to dampen neutrophilic inflammation. Cell Rep. 22, 1810–1823 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kanarek N., London N., Schueler-Furman O., Ben-Neriah Y., Ubiquitination and degradation of the inhibitors of NF-kappaB. Cold Spring Harb. Perspect. Biol. 2, a000166 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davies A. J., et al. , Differential efficacy from the addition of bortezomib to R-CHOP in diffuse large B-cell lymphoma according to the molecular subgroup in the REMoDL-B study with a 5-year follow-up. J. Clin. Oncol. 41, 2718–2723 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brightbill H. D., et al. , NF-κB inducing kinase is a therapeutic target for systemic lupus erythematosus. Nat. Commun. 9, 179 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu L. G., Shu H. B., TNFR-associated factor-3 is associated with BAFF-R and negatively regulates BAFF-R-mediated NF-kappa B activation and IL-10 production. J. Immunol. 169, 6883–6889 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Cao S., Zhang X., Edwards J. P., Mosser D. M., NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J. Biol. Chem. 281, 26041–26050 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu H., Lin L., Zhang Z., Zhang H., Hu H., Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 5, 209 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Inoue S., Leitner W. W., Golding B., Scott D., Inhibitory effects of B cells on antitumor immunity. Cancer Res. 66, 7741–7747 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Wei X., et al. , Regulatory B cells contribute to the impaired antitumor immunity in ovarian cancer patients. Tumour Biol. 37, 6581–6588 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Chen Z., et al. , Role of regulatory B cells in the progression of cervical cancer. Mediators Inflamm. 2019, 6519427 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang X., et al. , IL-10-producing B cells in differentiated thyroid cancer suppress the effector function of T cells but improve their survival upon activation. Exp. Cell Res. 376, 192–197 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Béguelin W., et al. , IL10 receptor is a novel therapeutic target in DLBCLs. Leukemia 29, 1684–1694 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Stirm K., et al. , Tumor cell-derived IL-10 promotes cell-autonomous growth and immune escape in diffuse large B-cell lymphoma. Oncoimmunology 10, 2003533 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xie P., et al. , Enhanced Toll-like receptor (TLR) responses of TNFR-associated factor 3 (TRAF3)-deficient B lymphocytes. J. Leukoc. Biol. 90, 1149–1157 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pérez de Diego R., et al. , Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 33, 400–411 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lam L. T., et al. , Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood 111, 3701–3713 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu C., et al. , An IL-27/NFIL3 signalling axis drives Tim-3 and IL-10 expression and T-cell dysfunction. Nat. Commun. 6, 6072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chihara N., et al. , Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 558, 454–459 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lacy S. E., et al. , Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A Haematological Malignancy Research Network report. Blood 135, 1759–1771 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Richards K. L., et al. , Gene profiling of canine B-cell lymphoma reveals germinal center and postgerminal center subtypes with different survival times, modeling human DLBCL. Cancer Res. 73, 5029–5039 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ansell S. M., et al. , Nivolumab for relapsed/refractory diffuse large B-cell lymphoma in patients ineligible for or having failed autologous transplantation: A single-arm, phase ii study. J. Clin. Oncol. 37, 481–489 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smith L. K., et al. , Interleukin-10 directly inhibits CD8(+) T cell function by enhancing N-glycan branching to decrease antigen sensitivity. Immunity 48, 299–312.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Das A., et al. , IL-10-producing regulatory B cells in the pathogenesis of chronic hepatitis B virus infection. J. Immunol. 189, 3925–3935 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rui L., et al. , Epigenetic gene regulation by Janus kinase 1 in diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 113, E7260–E7267 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu L., et al. , Gene regulation and suppression of type I interferon signaling by STAT3 in diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 115, E498–E505 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Azzaoui I., et al. , T-cell defect in diffuse large B-cell lymphomas involves expansion of myeloid-derived suppressor cells. Blood 128, 1081–1092 (2016). [DOI] [PubMed] [Google Scholar]
- 71.Guo X., et al. , Molecular impact of selective NFKB1 and NFKB2 signaling on DLBCL phenotype. Oncogene 36, 4224–4232 (2017). [DOI] [PubMed] [Google Scholar]
- 72.Lombardi L., et al. , Structural and functional characterization of the promoter regions of the NFKB2 gene. Nucleic Acids Res. 23, 2328–2336 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nolan G. P., et al. , The bcl-3 proto-oncogene encodes a nuclear I kappa B-like molecule that preferentially interacts with NF-kappa B p50 and p52 in a phosphorylation-dependent manner. Mol. Cell Biol. 13, 3557–3566 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Razani B., et al. , Negative feedback in noncanonical NF-kappaB signaling modulates NIK stability through IKKalpha-mediated phosphorylation. Sci. Signal. 3, ra41 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang M., et al. , RelA NF-κB subunit activation as a therapeutic target in diffuse large B-cell lymphoma. Aging (Albany NY) 8, 3321–3340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rovsing A. B., et al. , Resistance to vincristine in DLBCL by disruption of p53-induced cell cycle arrest and apoptosis mediated by KIF18B and USP28. Br. J. Haematol. 202, 825–839 (2023). [DOI] [PubMed] [Google Scholar]
- 77.Alizadeh A., et al. , The lymphochip: A specialized cDNA microarray for the genomic-scale analysis of gene expression in normal and malignant lymphocytes. Cold Spring Harb. Symp. Quant. Biol. 64, 71–78 (1999). [DOI] [PubMed] [Google Scholar]
- 78.Li M. Y., et al. , Data from “TRAF3 loss-of-function Reveals the Non-Canonical NF-κB Pathway as a Therapeutic Target in Diffuse Large B-cell Lymphoma.” Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE218750. Deposited 25 November 2022.
- 79.Ennishi D., et al. , Data from “Detection of clinically relevant genetic and transcriptomic landscape in DLBCL uniformly treated by R-CHOP.” European Genome-Phenome Archive. https://ega-archive.org/studies/EGAS00001002657. Deposited 11 September 2017.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
Cell line RNA-seq data (78) are deposited at Gene Expression Omnibus (accession GSE218750). Study cohort raw RNA-seq and SNP6.0 data can be found at European Genome-phenome Archive (accession EGAS00001002657 https://ega-archive.org/studies/EGAS00001002657) (79). All other data are included in the manuscript and/or SI Appendix.