

Keywords: HIF-1α, PAH, PHD1, PHD2, p27
Abstract
Pulmonary arterial hypertension (PAH) is a disease characterized by increased vasoconstriction and vascular remodeling. Pulmonary artery smooth muscle cells (PASMCs) highly express the transcription factor hypoxia-inducible factor-1α (HIF-1α), yet the role of PASMC HIF-1α in the development of PAH remains controversial. To study the role of SMC HIF-1α in the pulmonary vascular response to acute and chronic hypoxia, we used a gain-of-function strategy to stabilize HIF-1α in PASMC by generating mice lacking prolyl hydroxylase domain (PHD) 1 and 2 in SM22α-expressing cells. This strategy increased HIF-1α expression and transcriptional activity under conditions of normoxia and hypoxia. Acute hypoxia increased right ventricular systolic pressure (RVSP) in control, but not in SM22α-PHD1/2−/− mice. Chronic hypoxia increased RVSP and vascular remodeling more in control SM22α-PHD1/2+/+ than in SM22α-PHD1/2−/− mice. In vitro studies demonstrated increased contractility and myosin light chain phosphorylation in isolated PHD1/2+/+ compared with PHD1/2−/− PASMC under both normoxic and hypoxic conditions. After chronic hypoxia, there was more p27 and less vascular remodeling in SM22α-PHD1/2−/− compared with SM22α-PHD1/2+/+ mice. Hypoxia increased p27 in PASMC isolated from control patients, but not in cells from patients with idiopathic pulmonary arterial hypertension (IPAH). These findings highlight an SM22α-expressing cell-specific role for HIF-1α in the inhibition of pulmonary vasoconstriction and vascular remodeling. Modulating HIF-1α expression in PASMC may represent a promising preventative and therapeutic strategy for patients with PAH.
NEW & NOTEWORTHY In a mouse model wherein hypoxia-inducible factor 1 alpha (HIF-1α) is stabilized in vascular smooth muscle cells, we found that HIF-1α regulates vasoconstriction by limiting phosphorylation of myosin light chain and regulates vascular remodeling through p27 induction. These findings highlight a cell-specific role for HIF-1α in the inhibition of pulmonary vasoconstriction and vascular remodeling.
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a disease characterized by an increase in vascular tone due to excesses in vasoconstriction and vascular remodeling (1, 2). Evidence supports a central role for the transcription factor hypoxia-inducible factor-1α (HIF-1α) in modulating pulmonary vascular tone (3). However, how pulmonary artery (PA) SMC-specific HIF-1α contributes to the development of PAH remains unclear.
HIF-1α is highly expressed in pulmonary artery smooth muscle cells (PASMCs), under both normoxic and hypoxic conditions (4). Previously we have shown that mice with a constitutive SM22α-specific deletion of HIF-1α exhibit significantly higher right ventricular systolic pressure (RVSP) compared with control (Cre−) littermates under normoxia and with exposure to either acute or chronic hypoxia (5). The increase in pulmonary arterial pressure in the SM22α-HIF-1α−/− mice was due to an increase in PASMC myosin light chain phosphorylation (pMLC) and vascular tone in the absence of vascular remodeling. These data, derived from murine models, are supported by results from cells and lung tissues from patients with idiopathic pulmonary arterial hypertension (IPAH) wherein PASMC HIF-1α expression was decreased, whereas myosin light chain kinase (MLCK) activity, pMLC expression, and contractility were all increased compared with controls (6).
However, other studies have reported seemingly contradictory results. In a mouse model of hypoxia-induced pulmonary hypertension (PH) wherein HIF-1α was deleted in SMC using an inducible Cre driver, pulmonary vascular remodeling and pulmonary arterial pressures were attenuated compared with controls (7). Further, in a mouse model of hypoxia-induced PH, constitutive deletion of CD146 in PDGFRβ-expressing cells decreased HIF-1α expression and attenuated pulmonary vascular remodeling and PH (8).
Each of these findings, however divergent, underscore the importance of an SM22α-expressing cell-specific role for HIF-1α in regulating pulmonary vascular tone and remodeling. Therefore, to more definitively elucidate the role of HIF-1α in PASMC, we created mice with a constitutive SM22α-mediated deletion of the genes encoding prolyl hydroxylase domain 1 and 2 (PHD1/2) proteins to stabilize HIF-1α expression in pulmonary vascular SMC. With the deletion of PHD1/2 in SM22α-expressing cells, we hypothesized that: 1) HIF-1α expression would be increased in PASMC, and 2) the pulmonary vascular response to both acute and chronic hypoxia would be markedly attenuated.
MATERIALS AND METHODS
SM22α-PHD1/2+/+ and SM22α-PHD1/2−/− Mice
Transgenic mice with selective deletion of PHD1 and PHD2 in SMC (SM22α-PHD1/2−/−) were created by crossbreeding SM22α promoter-driven Cre recombinase mice with PHD1flox/flox and PHD2flox/flox mice (PHD1/2fl/fl mice). The PHD1fl/fl and PHD2fl/fl mice were kindly provided by Dr. Amato Giaccia, Stanford University (9). The PHD1fl/fl and PHD2fl/fl mice were on a mixed C57BL/6-FVB genetic background. The SM22α-Cre mice were obtained from The Jackson Laboratory. The SM22α-Cre mice were originally derived on a 129S5 background, and then backcrossed to C57BL/6J mice for > 15 generations. All studies used only littermate controls. The Institutional Animal Care and Use Committee (IACUC) at Stanford University approved all the procedures and protocols governing the care and use of laboratory animals. Offspring were genotyped by PCR for the detection of Cre recombinase, floxed PHD1, and floxed PHD2 genes. Littermates that did not express the Cre recombinase gene, yet expressed floxed PHD1 and floxed PHD2 genes (PHD1fl/fl;PHD2fl/fl) were studied (SM22α-PHD1/2+/+) in comparison to littermates that did express the Cre recombinase, floxed PHD1, and floxed PHD2 genes (SM22α-PHD1/2−/−).
Genotyping
To identify the Cre recombinase gene, primers (forward: 5′- CCATCTGCCACCAGCCAG-3′; reverse: 5′- TCGCCATCTTCCAGCAGG-3′) were used to generate a 281 bp PCR product. To identify the floxed PHD1 gene (exon 3 is floxed), primers (forward 5′- TGGGCGCTGCATCACCTGTATCT-3′; reverse 5′- ACTGGTGAAGCCTGTAGCCTGTC-3′) were used to amplify two DNA fragments at ∼840 bp and 393 bp, representing PHD1flox/flox and PHD1WT/WT mice, respectively. To identify the floxed PHD2 gene (exon 2 is floxed), primers (forward 5′- CAAATGGAGATGGAAGATGC-3′; reverse 5′- TCAACTCGAGCTGGAAACC-3′) were used to amplify two DNA fragments at ∼840 bp and 389 bp, representing PHD2flox/flox and PHD2WT/WT mice, respectively.
Primary Cell Culture
Pulmonary artery smooth muscle cells (PASMC) were isolated from SM22α-PHD1/2−/− mice and littermate control (SM22α-PHD1/2+/+) mice using a modified elastase/collagenase digestion protocol (5). Pulmonary artery tissues, pooled from 5 mice of mixed gender per genotype, were digested in a dispersion medium containing 40 µmol/L CaCl2, 0.5 mg/mL elastase (Worthington Biochemical), 0.5 mg/mL collagenase (Worthington Biochemical), 0.2 mg/mL soybean trypsin inhibitor (Worthington Biochemical), and 2 mg/mL albumin for 30 min at 37°C. After filtration with 100 µm cell strainers, cells were incubated with Dynabeads (Invitrogen) coated with CD31 antibody (BD Biosciences) for 30 min, to deplete EC expressing CD31. The remaining SMCs were collected through centrifugation at 225 g for 5 min at 4°C and cultured in DMEM containing 10% FBS and 1% antibiotic-antimycotic solution. Isolation of cells was performed multiple times (n = 5 replicates per genotype). To confirm isolation of PASMC, cells were stained for α-smooth muscle actin (α-SMA, 1:200, A2547 Sigma), SM22α (1:200, ab14106 Abcam), Lumican (1:100, NBP2-76847 Novus Biologicals), and nuclei (Vectashield Mounting Medium with DAPI, Vector Laboratories) expression by immunofluorescence. The number of PASMC expressing α-SMA and SM22α was assessed as a percentage of total cells per high-powered field (HPF) at 200x magnification with 10 fields counted per sample; both PHD1/2+/+ and PHD1/2−/− PASMC were 100% positive for α-SMA and SM22α expression. Cells at passages 2–8 were used for experiments.
Human PASMC was obtained through the pulmonary hypertensive breakthrough initiative (PHBI). Cells from control (n = 4) and IPAH (n = 4) patients at passages 3–8 were used for experiments. PASMC were cultured in SmBM (Lonza) containing 5% fetal bovine serum (FBS), 0.1% insulin, 0.2% human fibroblastic growth factor-B (hFGF-B), 0.1% human epidermal growth factor (hEGF), and 0.1% gentamicin/amphotericin (GA-1000). All cells were examined for the expression of SMC markers: α-SMA, MHC11, SM22α, and Calponin (6).
Western Immunoblotting
For protein analysis, cells were lysed in 0.5% NP-40 lysis buffer [50 mM Tris-HCl pH 7.5, 150 mM NaCl, 2.5 mM EDTA, 0.5% NP-40, 1 mM Na3VO4, 1 mM PMSF, 10 µg/mL aprotinin, 10 µg/mL leupeptin, and 5 µM HIF Prolyl Hydroxylase Inhibitor (Calbiochem)]. Protein content was quantified using the Pierce BCA Protein Assay Kit (Thermo Scientific). Ten micrograms of protein/sample were subjected to SDS-PAGE analysis using 4–12% gradient or 8% gels (Invitrogen). Separated proteins were transferred onto immobilon-P membranes (Millipore), incubated with primary antibodies as indicated [HIF-1α 14179 Cell Signaling (5), HIF-2α ab8365 Abcam (6), HIF-3α sc-28707 Santa Cruz (6), PHD1 ab108980 Abcam (9), PHD2 NB100-2219 Novus (9), PHD3 NB100-139 Novus (9), Phosphorylated MLC ab76092 Abcam (5), MLC M4401 Sigma (5), PGK1 sc-130335 Santa Cruz (10), Rho kinase 9029 Cell Signaling (11), Myosin phosphatase 2634 Cell Signaling (12), MLCK ab76092 Abcam (6), p27 3688 Cell Signaling (13), VEGF-A ab46154 Abcam, and β-actin A5441 Sigma (5)], and then incubated with the respective horseradish peroxidase (HRP)-conjugated secondary antibodies (Cell Signaling), followed by detection with ECL reagents (GE Healthcare). Graphs represent quantification of protein expression by densitometry with results represented relative to β-actin expression (relative expression). All antibodies used in this study have been previously validated.
Immunocytochemistry
To visualize nuclear HIF-1α expression, subconfluent cultures of isolated PASMC were subjected to normoxic (21% O2) or hypoxic (1% O2, 1 h) conditions, fixed in 3% paraformaldehyde/PBS for 30 min, blocked and permeabilized in VSVG blocking solution (0.1% Triton X-100, 15 mg/mL glycine, and 2.5% FBS in PBS) for 1 h, incubated with HIF-1α (1:50, GTX127309 GeneTex) antibody for 1 h, incubated with Alexa Fluor 568 α-Rb IgG antibody (1:200, A10042 Invitrogen) and Alexa Fluor 488 Phalloidin (1:40, A12379 Invitrogen) for 1 h, and then mounted with Vectashield Mounting Medium with DAPI (Vector Laboratories). Results shown represent the ratio of nuclear HIF-1α over cytoplasmic HIF-1α as measured by ImageJ software with 5 fields at ×200 magnification assessed per sample from four replicate experiments.
To visualize p27 expression in human PASMC, subconfluent cultures of PASMC isolated from control and idiopathic pulmonary arterial hypertensive (IPAH) patients were subjected to normoxic (21% O2) or hypoxic (5% O2, 2 days) conditions, fixed in 3% paraformaldehyde/PBS for 30 min, blocked and permeabilized in VSVG blocking solution for 1 h, incubated with p27 antibody (1:100, 3688S Cell Signaling) for 1 h, followed by incubation with Alexa Fluor 488 α-Rb IgG antibody (1:200, A11008 Invitrogen) for 1 h, and then mounted with Vectashield Mounting Medium with DAPI (Vector Laboratories). To assess protein expression, 10 random fields at 200x magnification per sample were examined. p27 fluorescence intensities were quantified using the following equation: Corrected Total Cell Fluorescence (CTCF) = Integrated Density – (Area of Selected Cell × Mean Fluorescence of Background) as measured by ImageJ software. All images for control and experimental groups were collected at the same time and under the same conditions.
Dual Luciferase Assay
PASMC in 60 mm dishes were grown to 80% confluence, co-transfected with 5 µg of HRE-luciferase (26731 Addgene) (14) and 0.5 µg of renilla luciferase (PIS1, 12179 Addgene) constructs using Lipofectamine LTX with Plus Reagent (Thermo Fisher). 72 h post-transfection, cells were lysed in a passive lysis buffer from the Luciferase Assay System (Promega). Prior to harvest, samples were incubated in normoxia (21% O2) or hypoxia (1% O2, 1 h) conditions. Lysates were incubated with Luciferase Assay Reagent (Promega) and luciferase activity was measured using a GloMax Luminometer (Promega).
Hemodynamic Assessments
To measure vasoreactivity, adult SM22α-PHD1/2+/+ and SM22α-PHD1/2−/− littermate mice matched for age and gender were used in each group. For the acute RVSP studies, mice were anesthetized with 1.5–2.0% isofluorane and right ventricular systolic pressure (RVSP) measurements were obtained using a 1.4 F Millar catheter (Millar Instruments) at baseline (21% O2, 10 min), during acute hypoxia (10% O2, 10 min), and with recovery to baseline (21% O2, 10 min) (n = 6–10 mice per group). For the chronic RVSP studies, mice were anesthetized with 1.5–2.0% isofluorane and RVSP measurements were obtained as described above using mice subjected to normoxic (21% O2) or hypoxic (10% O2) conditions for 24-28d (n = 7 mice per group). There was no difference in the duration of hypoxia exposure between genotypes.
Fulton’s Index
After 24–28 days of chronic hypoxia, hearts were removed and perfused with 1 mL PBS and then dried for 5 min between gauze pads. Each dried heart was weighed, then the right ventricle (RV) and left ventricle plus ventricular septum (LV + Septum) were disjointed and weighed separately (n = 9–22 mice per group). Right ventricular hypertrophy (RVH) was expressed as RV/(LV+Septum).
Collagen Gel Contraction Assay
Collagen gel contraction assays were performed as previously described (6). In brief, 150 µL of collagen (PureCol EZ Gel Bovine Collagen Solution Type I in DMEM-F12, 5 mg/mL, Advanced BioMatrix) was aliquoted per well of a 48-well plate. After 1 h of polymerization, 1 × 105 PASMC were seeded in triplicate per well in 300 µL of smooth muscle basic medium plus supplements [5% FBS with 0.1% insulin, 0.2% basic human fibroblast growth factor, 0.1% human epidermal growth factor, and 0.1% gentamicin/amphotericin (GA-1000)] (Smooth Muscle Growth Medium-2, Lonza). After 1 h of incubation, the sides of the collagen wells were gently detached from the walls of the wells by use of a sterile 200 µL pipette tip to initiate cell-mediated collagen contraction. Cells were then incubated at 21% O2 (normoxia) or 5% O2 (hypoxia) for 18 h. After 18 h, collagen gels were fixed in 3% paraformaldehyde in PBS for 30 min. Collagen gels were stained with picrosirius red (ScyTek Laboratories) for imaging. In brief, fixed collagen gels were incubated with picrosirius red stain for 1 h, rinsed with acidified H2O, and stored in PBS.
To increase collagen gel contraction sensitivity, the collagen gel contraction assay protocol was modified to increase the ratio of cells seeded to collagen (1:1). In brief, 50 µL of collagen (PureCol EZ Gel Bovine Collagen Solution Type I in DMEM-F12, 5 mg/mL, Advanced BioMatrix) was aliquoted per well of a 96-well round bottom plate. After 1 h of polymerization, 5 × 104 PASMC were seeded in triplicate per well in 100 µL of smooth muscle basic medium plus supplements [5% FBS with 0.1% insulin, 0.2% basic human fibroblast growth factor, 0.1% human epidermal growth factor, and 0.1% gentamicin/amphotericin (GA-1000)] (Smooth Muscle Growth Medium-2, Lonza). After 1 h of incubation, the sides of the collagen wells were gently detached from the walls of the wells by use of a sterile 200 µL pipette tip to initiate cell-mediated collagen contraction. Cells were then incubated at 21% O2 (normoxia) or 5% O2 (hypoxia) for 18 h. For the U46619 studies, cells were incubated with 5 nM, 50 nM, 100 nM, or 400 nM of U46619 for 18 h. After 18 h, collagen gels were fixed in 3% paraformaldehyde in PBS for 15 min.
The degree of contraction was assessed by measuring the area of the gels with ImageJ software. The cross-sectional diameter of each gel was measured (two diameter measurements per sample). The following formula was used to determine the elliptical area: Area = (3.14) x (a/2) × (b/2), where a and b represent separate diameter measurements. Results shown represent the collagen gel surface area relative to the area of no cell control (set at 1.0). All images for control and experimental groups were collected at the same time and under the same conditions.
Lung Fixation
Mice were euthanized by CO2 inhalation. A cannula was inserted into the trachea and lungs were inflation fixed with 10% formalin at a constant pressure of 25 cm for 5 min. Afterwards, the lungs were placed in 10% formalin for 24 h, followed by 70% ethanol. Each lung was embedded in paraffin and cut into 4 µm sections for further examination.
Modified Movat Pentachrome Staining
To visualize vascular remodeling of pulmonary arteries, pulmonary tissues were deparaffinized and rehydrated, incubated in Verhoeff’s elastic stain for 30 min, 2% ferric chloride for 50 s, 5% sodium thiosulfate for 1 min, 3% acetic acid for 3 min, 1% Alcian Blue for 30 min, van Gieson’s stain for 1 min, dehydrated and cleared, then mounted with Permount (Fisher Scientific). All images for control and experimental groups were collected at the same time and under the same conditions via blind analysis.
Immunohistochemistry
To visualize von Willebrand Factor (VWF) and α-SMA expression, pulmonary tissues were deparaffinized and rehydrated, subjected to Universal Antigen Retrieval Solution (RD Systems) for 30 min at 95°C to unmask antigens, 0.25% Triton X-100/PBS solution for 30 min to permeabilize cells, 100 mM glycine solution for 20 min to quench autofluorescence, Sea Block Blocking Solution (Thermo Scientific) for 40 min, Fc Receptor Blocker Solution (Innovex Biosciences) for 30 min to block macrophages, Mouse Detective (Biocare Medical) for 30 min to block mouse IgG, primary antibodies (VWF 1:50 ab7356 Millipore, α-SMA 1:400 A2547 Sigma) for overnight at 4°C, secondary antibodies (Alexa Fluor 488 α-Rb IgG and Alexa Fluor 568 α-M IgG 1:200, A11008 and A11031, respectively, Invitrogen) for 1 h at RT, 0.1% Tween-20/PBS solution for 3x10min to wash, 1 µg/mL Hoechst solution for 5 min, and then mounted in 70% glycerol/PBS solution.
To visualize p27 and α-SMA expression, pulmonary tissues were deparaffinized and rehydrated, subjected to Citrate Buffer pH 6 (Vector Laboratories) for 45 min at 95°C to unmask antigens, 0.5% Triton X-100/PBS solution for 40 min to permeabilize cells, Sea Block Blocking Solution (Thermo Scientific) for 40 min, Mouse Detective (Biocare Medical) for 30 min to block mouse IgG, primary antibodies (p27 1:50 3688 Cell Signaling, α-SMA 1:200 A2547 Sigma) for overnight at 4°C, secondary antibodies (Alexa Fluor 488 α-M IgG and Alexa Fluor 568 α-Rb IgG 1:200, A11029 and A10042, respectively, Invitrogen) for 2 h at RT, 0.1% Tween-20/PBS solution for 3 × 10 min to wash, 1 µg/mL Hoechst solution for 5 min, and then mounted in 70% glycerol/PBS solution. Results shown represent the number of p27-positive vessels as a percentage of total vessels per HPF at ×400 magnification with 10 fields counted per sample. All images for control and experimental groups were collected at the same time and under the same conditions via blind analysis.
Pulmonary Artery Muscularization
To assess vascular remodeling, a modified Movat pentachrome staining procedure was used to evaluate pulmonary artery (PA) muscularization in vessels less than 150 µm in diameter as analyzed by Metamorph software (Molecular Devices). Representative images are at 100x magnification, calibration bar 100 µm. Quantification of PA muscularization was assessed in 10 random fields per mouse (n = 5 mice per group) using the following equation: Medial Thickness Index (%) = [(areaexternal − areainternal)/areaexternal] × 100. Areaexternal and areainternal represent the areas within the external and internal boundaries of the SMC medial layer (elastic fibers, blue-black; nuclei, black; collagen, magenta; red blood cells, yellow; glycosylated proteins, blue).
To assess differences in vessel muscularization, pulmonary tissues were stained with von Willebrand Factor (VWF, red), α-SMA (green), and nuclei (blue) to identify PA. PA was categorized into non-muscularized (NM), partially muscularized (PM), or fully muscularized (FM) arteries based on α-SMA staining and medial layer thickness using Metamorph software (Molecular Devices). In brief, vessels less than 150 µm in diameter were analyzed in 10 random fields per mouse at ×200 magnification, calibration bar of 50 µm (n = 4 mice per group). Duplicate measurements were taken per artery and averaged with over 100 vessels assessed per mouse. Vessels were categorized as NM (VWF-positive vessels), PM (α-SMA-positive vessels with a medial layer less than 20 µm in diameter), or FM (α-SMA-positive vessels with a medial layer equal to or greater than 20 µm in diameter). Results shown represent the number of NM, PM, or FM arteries as a percentage of the total number of arteries measured. All images for control and experimental groups were collected at the same time and under the same conditions via blind analysis.
Cell Cycle Analysis
Subconfluent cultures of PASMC [normoxic, hypoxic (1 h), and hypoxic (3d)] were trypsinized, fixed, and stained with propidium iodide (PI), per manufacturer’s instructions [Propidium Iodide Flow Cytometry Kit (ab139418 Abcam)], to detect cell cycle phase distribution by flow cytometry analysis.
Statistical Analysis
All experiments were performed a minimum of three times unless otherwise indicated. Statistical analysis was performed with the GraphPad Prism 9.0 software. Results are presented as the means ± SD. Statistical significance was assessed with Student’s t test, one-way ANOVA, or two-way ANOVA followed by Tukey’s multiple comparisons test. A P value of < 0.05 was taken as the threshold level for statistical significance.
RESULTS
HIF-1α is Increased in PASMC Isolated from SM22α-PHD1/2−/− Mice
The smooth muscle-specific protein SM22α is expressed in the pulmonary vasculature beginning at E13.5-E14.5 and persists throughout adulthood (15–17). SM22α promoter-driven Cre recombinase transgenic mice have been used to address the role of SMC-specific proteins during the later stages of development and in the adult animal (18–20). For this study, SM22α-Cre recombinase mice were crossed with PHD1 and PHD2 floxed mice to generate mice deficient in SMC PHD1/2 (SM22α-PHD1/2−/−). PHD proteins initiate the degradation of HIF-1α by hydroxylating two proline residues (P402 and P564) within the oxygen-dependent degradation domain (ODDD) (21). Three PHD isoforms have been identified: PHD1, PHD2, and PHD3. Evidence strongly suggests that PHD2 has more influence on HIF-1α degradation than other isoforms (22, 23), some functional redundancy may exist as PHD1 activity has also been shown to promote HIF-1α degradation independent of and in concert with PHD2 (22).
To confirm the loss of PHD1 and PHD2 and the stabilization of HIF-1α in the pulmonary vascular SMC of the SM22α-PHD1/2−/− mice, PASMCs were isolated from the pulmonary arteries of the PHD1/2 mice. Isolated cells from each genotype were 100% positive for α-SMA and SM22α expression and uniformly negative for lumican, a fibroblast-specific marker, confirming SMC identity (Fig. 1A). Cells were then incubated under normoxia (21% O2) or hypoxia (1% O2) and protein expression was evaluated (Fig. 1B). PHD1 and PHD2 protein expression was significantly decreased in PASMC derived from SM22α-PHD1/2−/− mice compared with SM22α-PHD1/2+/+ mice (Fig. 1C). PHD3 protein was expressed in both genotypes independent of oxygen concentration. HIF-1α protein expression was increased in PHD1/2−/− PASMC compared with PHD1/2+/+ PASMC under normoxic conditions, and was further enhanced under hypoxic conditions (Fig. 1D). Both HIF-2α and HIF-3α were minimally expressed in the isolated PASMC; and, expression did not vary with hypoxia indicating that the loss of PHD1 and PHD2 expression did not cause an appreciable increase in either HIF-2α or HIF-3α in PASMC. These data are consistent with our previously reported results showing that HIF-2α expression was minimal and HIF-3α expression was undetectable in human PASMC (6). Stabilized HIF-1α localizes to the nucleus where it initiates the transcription of target genes (24, 25). Nuclear expression of HIF-1α was increased in PHD1/2−/− compared with PHD1/2+/+ PASMC (Fig. 1E). With hypoxia, HIF-1α nuclear localization increased in both cell populations, but was substantially greater in PHD1/2−/− cells compared with PHD1/2+/+ cells (Fig. 1E).
Figure 1.
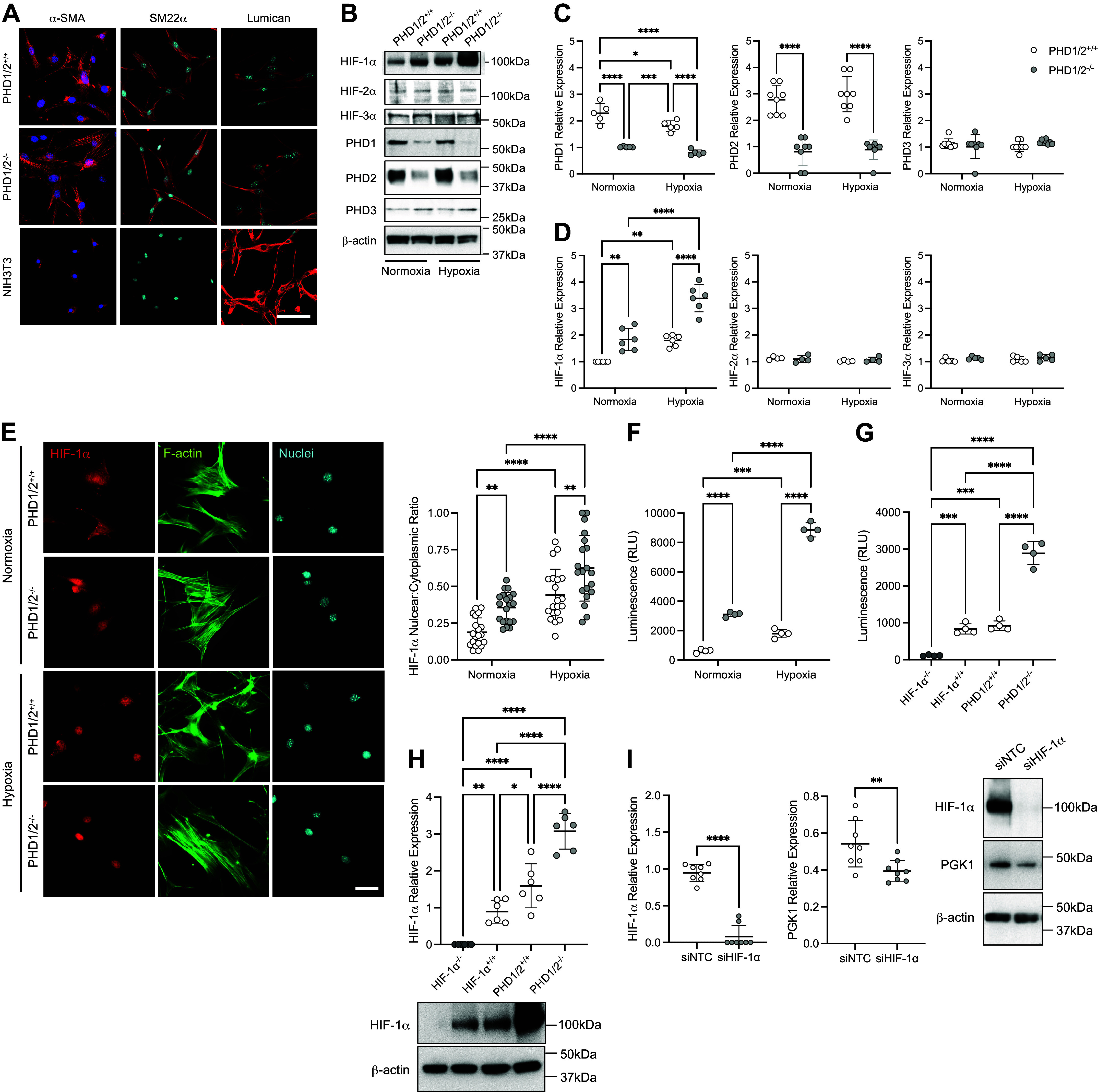
Characterization of HIF-1α in PASMC isolated from SM22α-PHD1/2+/+ and SM22α-PHD1/2−/− mice. Mice expressing Cre recombinase (Cre+) under the control of the SM22α promoter or without (Cre−) were crossed with PHD1flox/flox; PHD2flox/flox mice to generate SM22α-PHD1/2−/− and SM22α-PHD1/2+/+ mice, respectively. A: expression of α-SMA, SM22α, and Lumican in isolated PASMC (n = 3 replicate experiments). α-SMA (red), SM22α (red), Lumican (red), nuclei (blue); ×100 magnification, calibration bar 100 µm. B: protein expression profile of isolated PASMC exposed to normoxic (21% O2) or hypoxic (1% O2, 1 h) conditions. C: quantification of PHD1, PHD2, and PHD3 expression in isolated PASMC (n = 5–8 replicate experiments). D: quantification of HIF-1α, HIF-2α, and HIF-3α expression in PASMC (n = 4–6 replicate experiments). E: nuclear HIF-1α expression in PHD1/2 PASMC (n = 4 replicate experiments with five fields analyzed for each). HIF-1α (red), F-actin (green), nuclei (blue); ×200 magnification, calibration bar 50 µm. F: HIF-1α transcriptional activity in PASMC under normoxic and hypoxic conditions (n = 4 replicate experiments performed in triplicate). G: HIF-1α transcriptional activity in PASMC isolated from SM22α-HIF-1α−/−, SM22α-HIF-1α+/+, SM22α-PHD1/2−/−, and SM22α-PHD1/2+/+ mice (n = 4 replicate experiments performed in triplicate). H: HIF-1α protein expression in HIF-1α and PHD1/2 PASMC (n = 6 replicate experiments). I: expression of Phosphoglycerate kinase 1 (PGK1), a target of HIF-1α, in siHIF-1α-transfected PHD1/2−/− PASMC (n = 8 replicate experiments). Graphs represent the means ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, as indicated. P values were measured by two-way ANOVA (C, D, E, F), one-way ANOVA (G, H), and unpaired t test (I). HIF-1α, hypoxia-inducible factor-1α; PHD, prolyl hydroxylase domain; PASMC, pulmonary artery smooth muscle cell; SMA, smooth muscle actin.
HIF-1α Activity is Increased in PASMC Isolated from SM22α-PHD1/2−/− Mice
To determine if the increase in HIF-1α nuclear localization in the PHD1/2−/− cells correlated with an increase in HIF-1α transcriptional activity, luciferase assays were performed. PASMC were co-transfected with a reporter for HIF-1α transcriptional activity, HRE-luciferase, which contains three copies of the hypoxia response element (HRE) of the PGK-1 gene upstream of the luciferase gene (14) and a control for transfection efficiency (PIS1). PHD1/2−/− cells expressed significantly greater luciferase activity compared with PHD1/2+/+ cells (Fig. 1F). Hypoxia increased the luciferase activities in both cell types, but was ∼ three-fold higher in the PHD1/2−/− cells compared with the PHD1/2+/+ PASMC.
To further assess the contribution of HIF-1α transcriptional activity in PASMC, luciferase assays were performed with PASMC isolated from SM22α-HIF-1α mice (5). PASMC null for HIF-1α (HIF-1α−/−), expressed minimal luciferase activity (Fig. 1, G and 1H). In contrast, PASMC with constitutively active HIF-1α (PHD1/2−/−) displayed ∼ three-fold greater luciferase activity than cells expressing HIF-1α+/+ and PHD1/2+/+. These results demonstrate that stabilized HIF-1α in the PHD1/2−/− cells is considerably more active than cells expressing wild-type HIF-1α. Moreover, PHD1/2−/− cells were transfected with siHIF-1α to knock-down HIF-1α expression, and then examined for the expression of Phosphoglycerate kinase 1 (PGK1), a classical target of HIF-1α (Fig. 1I) (10). With the loss of HIF-1α, PGK1 is decreased. These results provide further support for an SM22α cell-specific role for HIF-1α.
Hypoxic Pulmonary Vasoconstriction and Hypoxia-Induced Pulmonary Hypertension Are Attenuated in SM22α-PHD1/2−/− Mice
The pulmonary vascular response to acute hypoxia was determined in SM22α-PHD1/2+/+ and SM22α-PHD1/2−/− mice. Right ventricular systolic pressures (RVSP) were measured during 10 min of acute hypoxia (10% O2) and then upon return to 21% O2 (Fig. 2A and Table 1). Acute hypoxia increased RVSP in the SM22α-PHD1/2+/+ mice, but not in the SM22α-PHD1/2−/− mice, consistent with an absence of hypoxia-induced vasoconstriction. Upon return to normoxia, RVSP decreased to baseline values in the SM22α-PHD1/2+/+ mice.
Figure 2.
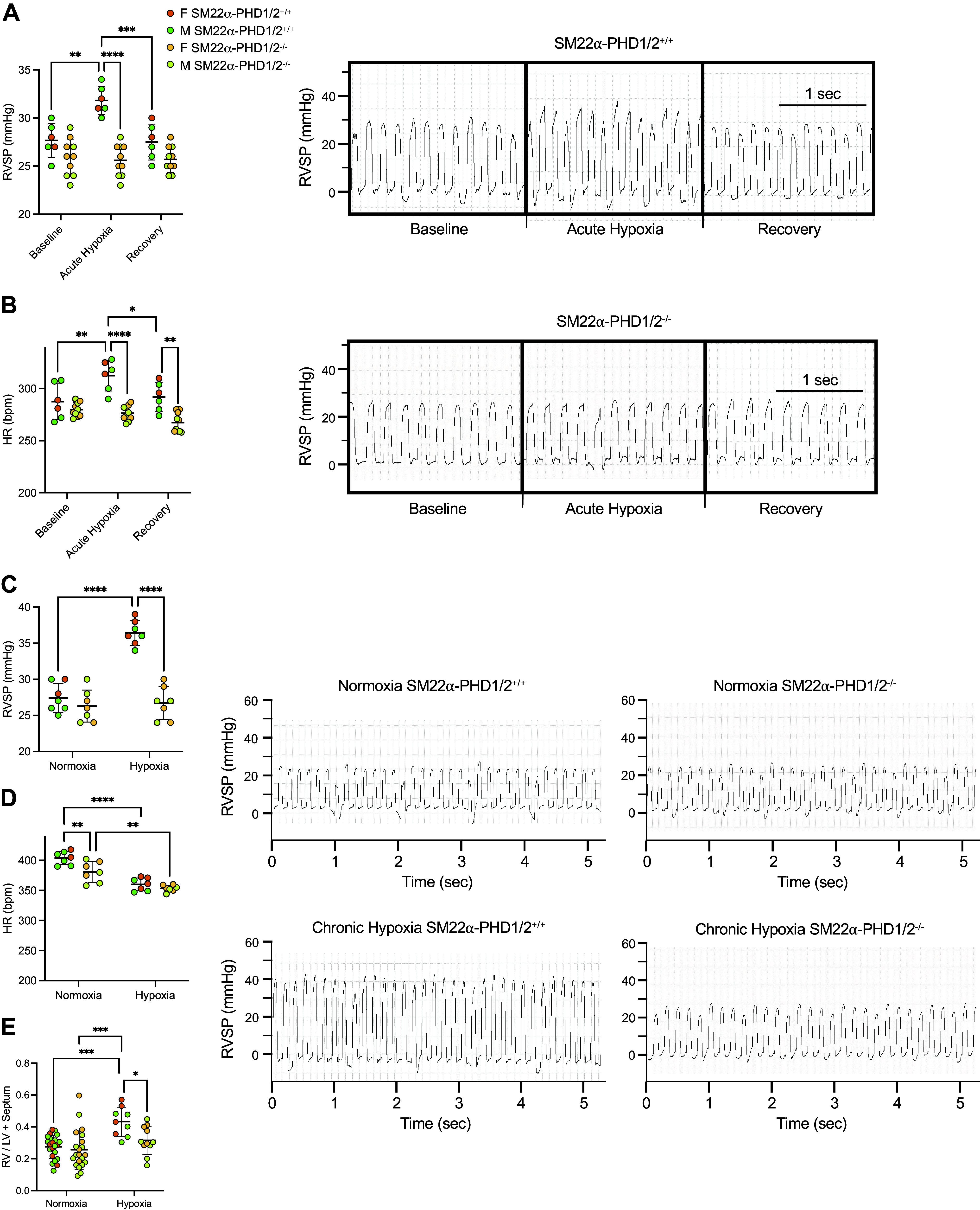
SMC HIF-1α activity is protective for mice exposed to hypoxia. A: pulmonary arterial pressures (PAPs) of SM22α-PHD1/2 mice induced by acute hypoxia. SM22α-PHD1/2 mice were subjected to acute hypoxia (10% O2) for 10 min [n = 6–10 mice per group, with n = 4 males (M) and n = 2 females (F) for SM22α-PHD1/2+/+ and n = 5 males and n = 5 females for SM22α-PHD1/2−/− mice]. Representative tracings from SM22α-PHD1/2+/+ and SM22α-PHD1/2−/− mice are shown. B: heart rates (HRs) of SM22α-PHD1/2 mice induced by acute hypoxia. C: PAP of SM22α-PHD1/2 mice in response to chronic hypoxia. PAPs were measured in SM22α-PHD1/2 mice subjected to normoxic conditions (21% O2) or hypoxic conditions (10% O2) for 24–28 days [n = 7 mice per group, with n = 5 males and n = 2 females for SM22α-PHD1/2+/+, n = 4 males and n = 3 females for SM22α-PHD1/2−/− mice (normoxia), and n = 3 males and n = 4 females for SM22α-PHD1/2 mice (chronic hypoxia)]. Representative tracings from SM22α-PHD1/2+/+ and SM22α-PHD1/2−/− mice exposed to chronic hypoxia are shown. D: HR of SM22α-PHD1/2 mice in response to chronic hypoxia. E: hypoxia-induced right ventricular hypertrophy (RVH) of SM22α-PHD1/2 mice (n = 9–22 mice per group). Graphs represent the means ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, as indicated. P values were measured by two-way ANOVA. HIF-1α, hypoxia-inducible factor-1α; PHD, prolyl hydroxylase domain; SMC, smooth muscle cell; SMA, smooth muscle actin.
Table 1.
Acute hypoxia studies of SM22α-PHD1/2 mice
| Baseline |
Acute Hypoxia |
Recovery |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Age, wk | Male, % | RVSP, mmHg | HR, beats/min | RVSP, mmHg | HR, beats/min | RVSP, mmHg | HR, beats/min | |
| SM22α-PHD1/2+/+ | 6 | 12.1 ± 0.7 | 66.6 | 27.0 ± 1.0 | 287.4 ± 25.5 | 31.6 ± 1.7§ | 313.4 ± 25.5 | 27.5 ± 0.5⌘ | 292.1 ± 22.5 |
| SM22α-PHD1/2−/− | 10 | 12.6 ± 0.1 | 50.0 | 26.0 ± 0.6 | 280.2 ± 7.9 | 25.6 ± 1.0** | 276.5 ± 10.8 | 25.7 ± 0.6 | 269.0 ± 10.7 |
Values are means ± SE. wk, week; RVSP, right ventricular systolic pressure; mmHg, millimeters of mercury; HR, heart rate; beats/min, beats per minute. **P < 0.01, PHD1/2−/− vs. PHD1/2+/+; §P < 0.05, acute hypoxia vs. baseline; ⌘P < 0.05, recovery vs. acute hypoxia; n = 6 mice, SM22α-PHD1/2+/+; n = 10 mice, SM22α-PHD1/2−/−. P values were measured by two-way ANOVA.
To address the effects of chronic hypoxia on pulmonary arterial pressures of SM22α-PHD1/2+/+ and SM22α-PHD1/2−/− mice, RVSP was measured (Fig. 2B and Table 2) after prolonged hypoxia exposure. RVSP measurements did not differ between SM22α-PHD1/2+/+ and SM22α-PHD1/2−/− mice under normoxic conditions. However, after chronic hypoxia (10% O2, 24–28 days) RVSP was significantly increased in SM22α-PHD1/2+/+, but not in SM22α-PHD1/2−/− mice. Consistent with the RVSP results, Fulton’s index [right ventricle (RV) mass/left ventricle (LV) plus ventricular septum (S) mass] increased in SM22α-PHD1/2+/+, but not in SM22α-PHD1/2−/− mice after chronic hypoxia (Fig. 2C).
Table 2.
Chronic hypoxia studies of SM22α-PHD1/2 mice
| n | Age, mo | Male, % | RVSP, mmHg | HR, beats/min | Weight Gain, g | |
|---|---|---|---|---|---|---|
| Normoxia | ||||||
| SM22α-PHD1/2+/+ | 7 | 4.7 ± 0.9 | 71.4 | 29.0 ± 1.0 | 404.3 ± 24.1 | +2.55 ± 0.41 |
| SM22α-PHD1/2−/− | 7 | 5.2 ± 1.1 | 71.4 | 26.9 ± 1.0 | 374.6 ± 29.8 | +2.03 ± 0.37 |
| Chronic hypoxia | ||||||
| SM22α-PHD1/2+/+ | 7 | 4.6 ± 1.0 | 57.1 | 36.9 ± 1.0§ | 360.1 ± 20.1 | −1.13 ± 0.57§§§ |
| SM22α-PHD1/2−/− | 7 | 5.3 ± 0.9 | 42.9 | 26.9 ± 1.9*** | 354.7 ± 19.2 | −0.90 ± 0.49§§§ |
Values are means ± SE. mo, month; RVSP, right ventricular systolic pressure; mmHg, millimeters of mercury; HR, heart rate; beats/min, beats per minute; g, grams. ***P < 0.001, PHD1/2−/− vs. PHD1/2+/+; §P < 0.05, §§§P < 0.001, chronic hypoxia vs. normoxia; n = 7 mice, normoxia SM22α-PHD1/2+/+; n = 7 mice, normoxia SM22α-PHD1/2−/−; n = 7 mice, hypoxia SM22α- PHD1/2+/+; n = 7 mice, hypoxia SM22α-PHD1/2−/−; n = 18–23, weight gain studies. P values were measured by two-way ANOVA.
The heart rates for the SM22α-PHD1/2−/− mice remained relatively lower than their SM22α-PHD1/2+/+ counterparts regardless of condition (Tables 1 and 2). The lack of an observed change in blood pressure for the SM22α-PHD1/2−/− mice under acute hypoxic conditions may reflect an increase in blood flow. As such, it is difficult to draw conclusions regarding the hypoxic pulmonary vasoconstriction (HPV) status of these mice based on heart rate alone.
MLC Phosphorylation and Contractility Are Decreased in PHD1/2−/− PASMC
Previous work from our laboratory demonstrated that the loss of HIF-1α increased MLC phosphorylation in murine PASMC (5). Given that phosphorylation of MLC facilitates myosin and actin interaction to augment vascular SMC contraction (26), we evaluated the effect of HIF-1α expression on PASMC contractility. Consistent with our previous studies (5,6), loss of HIF-1α increased contractility and pMLC expression in PASMC (Fig. 3A, 3B, and 3C). In contrast, PASMC isolated from PHD1/2−/− mice expressing stabilized HIF-1α demonstrated a decrease in pMLC expression and contraction (Fig. 3, A–C). Collectively, these results highlight the relationship between HIF-1α, pMLC, and SMC contraction wherein HIF-1α expression inhibits the phosphorylation of MLC and PASMC contractility.
Figure 3.
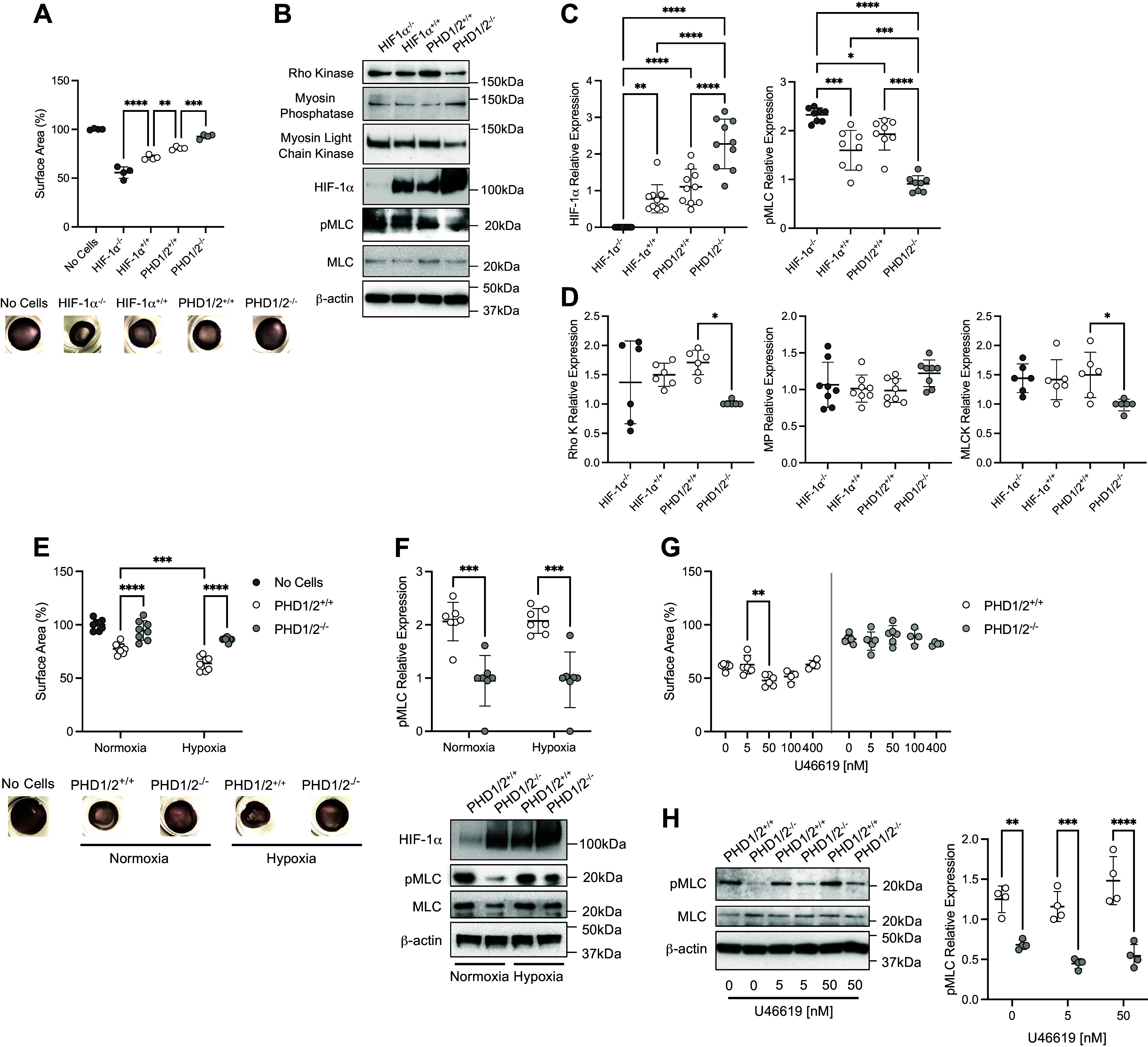
Vascular tone is decreased in PHD1/2−/− PASMC. A: collagen gel contraction assays of isolated PASMC from SM22α-HIF-1α and SM22α-PHD1/2 mice (n = 4 replicate experiments performed in triplicate). Representative images of collagen gels are shown. B: protein expression profile of isolated HIF-1α and PHD1/2 PASMC. C: quantification of HIF-1α and pMLC expression in isolated PASMC (n = 8–10 replicate experiments). D: quantification of Rho kinase (Rho K), myosin phosphatase (MP), and myosin light chain kinase (MLCK) expression in PASMC (n = 6–8 replicate experiments). E: collagen gel contraction assays of PHD1/2 PASMC under normoxic and hypoxic (5% O2, 18 h) conditions (n = 8 replicate experiments performed in triplicate). Representative images of collagen gels are shown. F: quantification of pMLC expression in PHD1/2 PASMC under normoxic and hypoxic conditions (n = 7 replicate experiments). G: collagen gel contraction assays of PHD1/2 PASMC exposed to U46619, a Thromboxane A2 agonist (n = 4–6 replicate experiments performed in triplicate). H: quantification of pMLC expression in PHD1/2 PASMC exposed to U46619 (n = 4 replicate experiments). Graphs represent the means ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, as indicated. For the collagen gel contraction assays, all samples are statistically significant in comparison to “No Cells” control. P values were measured by one-way ANOVA (A, C, D) and two-way ANOVA (E, F, G, H). HIF-1α, hypoxia-inducible factor-1α; PHD, prolyl hydroxylase domain; PASMC, pulmonary artery smooth muscle cell.
MLCK and Rho Kinase Expression Are Decreased in PASMC from SM22α-PHD1/2−/− Mice
To further examine the mechanistic link between HIF-1α and pMLC, the expression of key regulators upstream of pMLC was analyzed in the isolated cells. Myosin light chain kinase (MLCK) directly phosphorylates Ser19/20 of MLC promoting myosin association with actin filaments and subsequent contraction (26). Myosin phosphatase (MP) terminates SMC contraction by dephosphorylating MLC (12). Rho kinase promotes MLC activity indirectly by phosphorylating MP (11).
There was no significant difference in Rho kinase, MLCK, and MP protein levels between the HIF-1α+/+ and PHD1/2+/+ cells (Fig. 3, B and D). However, Rho kinase and MLCK were decreased in PHD1/2−/− compared with PHD1/2+/+ PASMC. The loss of Rho kinase and MLCK expression may partially explain the decrease in pMLC or SMC contraction found in the PHD1/2−/− cells.
HIF-1α stabilization in the PHD1/2−/− cells decreased contractility and pMLC expression under both normoxic and hypoxic conditions (Fig. 3, E and F). These decreases were quantitatively similar independent of oxygen concentration. These results suggest that the link between stabilized HIF-1α activity and the reduced expression of pMLC is not hypoxia-sensitive.
To determine whether HIF-1α stabilization in PASMC affects contractile response generally as opposed to solely hypoxia, collagen gel contraction assays were performed with the thromboxane A2 mimetic U46619 (27). PASMC from PHD1/2+/+, but not PHD1/2−/− mice contract in response to increasing U46619 concentration (range 5–100 nM) (Fig. 3G). pMLC was increased in PHD1/2+/+ cells treated with U46619 (Fig. 3H). Altogether these results suggest that the contractile capacity of PASMC from PHD1/2−/− mice is severely constrained.
Hypoxia-Induced Pulmonary Vascular Remodeling is Decreased in SM22α-PHD1/2−/− Mice
To assess the effect of HIF-1α stabilization on chronic hypoxia-induced pulmonary vascular remodeling, wall thickness was measured in small arteries (Fig. 4A). Medial wall thickness did not differ between the two genotypes under normoxic conditions. Chronic hypoxia significantly increased pulmonary arteriole wall thickness in SM22α-PHD1/2+/+ compared with SM22α-PHD1/2−/− mice.
Figure 4.
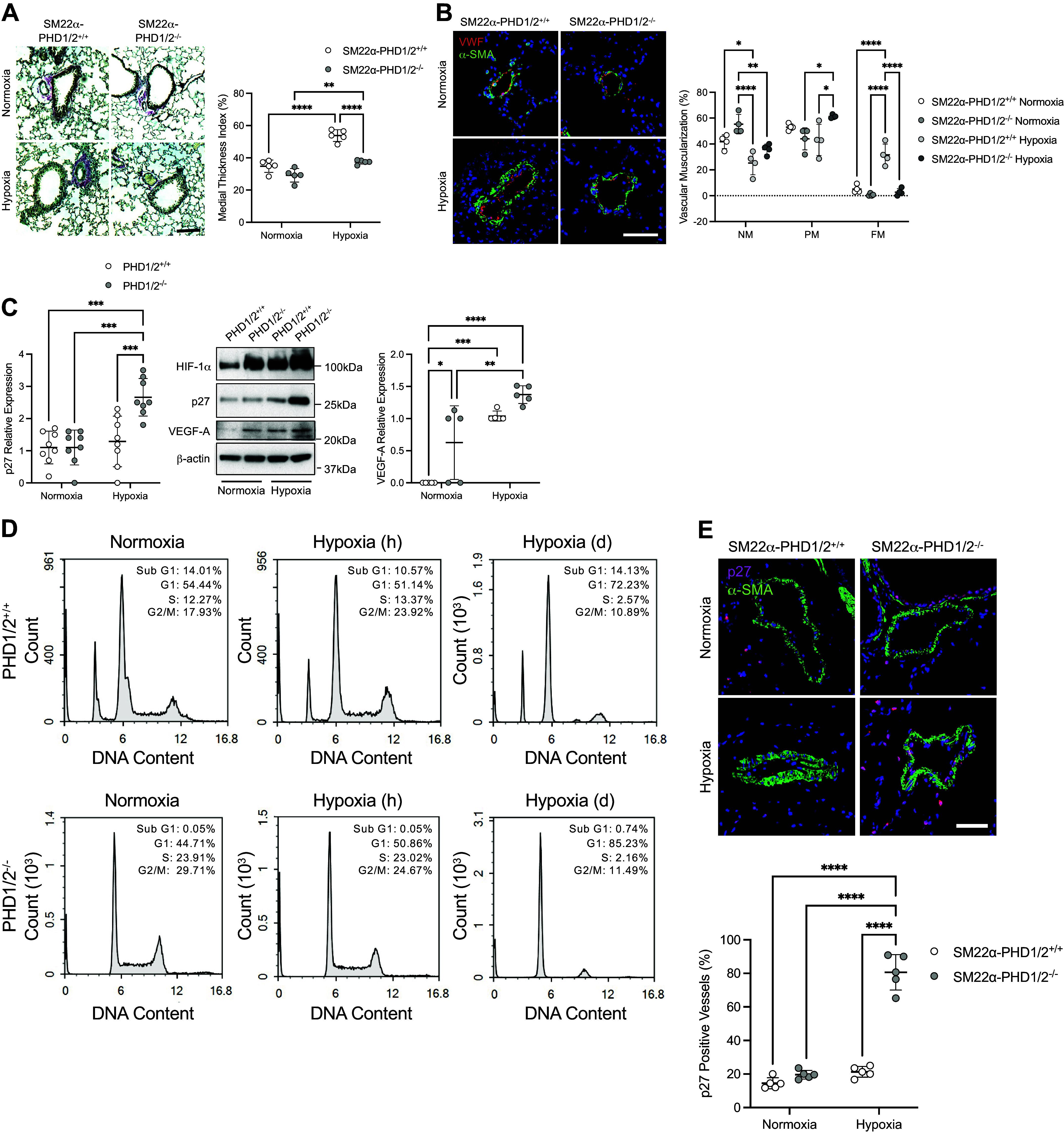
Vascular remodeling is attenuated in SM22α-PHD1/2−/− mice exposed to hypoxia. A: quantification of vascular remodeling as assessed by medial thickness index (MTI). Pulmonary tissues of SM22α-PHD1/2 mice were stained with a modified Movat pentachrome stain to visualize artery thickness (n = 5 mice per group). Representative images are shown; elastic fibers (blue-black), nuclei (black), collagen (magenta), glycosylated proteins (blue), and red blood cells (yellow); ×100 magnification, calibration bar 100 µm. B: assessment of arteriole muscularization in SM22α-PHD1/2 mice under normoxic and chronic hypoxic conditions. Non-muscularized (NM), partially muscularized (PM), and fully muscularized (FM) arteries less than 150 µm in diameter were measured (n = 4 mice per group). NM, VWF-positive (red) arterioles; PM, α-SMA-positive (green) arterioles with a medial layer less than 20 µm; FM, α-SMA-positive (green) arterioles with a medial layer of 20 µm or greater; ×200 magnification, calibration bar 50 µm. C: p27 and VEGF-A expression in isolated PASMC from SM22α-PHD1/2 mice under normoxic (21% O2) and hypoxic (1% O2, 24 h) conditions (n = 5–8 replicate experiments). D: cell cycle analysis of isolated PASMC from SM22α-PHD1/2 mice under normoxic (21% O2), hypoxic (5% O2, 1 h), and hypoxic (5% O2, 3 days) conditions (n = 3 replicate experiments). Number of cells (Count) is plotted as a function of DNA Content. E: expression of p27 in pulmonary tissues of SM22α-PHD1/2 mice (n = 5 mice per group). Representative images are shown; ×400 magnification, calibration bar 50 µm. Graphs represent the means ± SD, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 as indicated. P values were measured by two-way ANOVA. HIF-1α, hypoxia-inducible factor-1α; PHD, prolyl hydroxylase domain; PASMC, pulmonary artery smooth muscle cell; SMA, smooth muscle actin; VEGF-A, vascular endothelial growth factor-A.
To further evaluate differences in vascular remodeling, the muscularization of peripheral arteries was assessed (Fig. 4B). Under normoxic conditions, both SM22α-PHD1/2+/+ and SM22α-PHD1/2−/− mice had similar percentages of non-muscularized (NM), partially muscularized (PM), and fully muscularized (FM) vessels. Although chronic hypoxia altered the arteriole muscularization profile for both genotypes, increases in the proportion of vessels with FM were greater in SM22α-PHD1/2+/+ compared with SM22α-PHD1/2−/− mice. Altogether these results demonstrate that hypoxia causes less pulmonary vascular remodeling in SM22α-PHD1/2−/− compared with SM22α-PHD1/2+/+ mice.
p27 Expression Is Increased in Hypoxic PASMC from SM22α-PHD1/2−/− Mice
To examine the molecular mechanism by which HIF-1α stabilization constrains vascular remodeling in the SM22α-PHD1/2−/− mice, isolated PASMC were assessed for the expression of p27 (Fig. 4C). Under hypoxic conditions PHD1/2−/− SMC express p27, a key inhibitor of cell proliferation and a hypoxia-associated target of HIF-1α (13, 28–31). As a control for the inducement of HIF-1α activity, PHD1/2 SMC was examined for vascular endothelial growth factor-A (VEGF-A) expression, a known downstream target of HIF-1α. VEGF-A was increased in PHD1/2−/− compared with PHD1/2+/+ cells. With hypoxia, both cell types increased expression of VEGF-A, respectively. Flow cytometry demonstrated PHD1/2−/− PASMC G1 arrest with prolonged exposure to hypoxia (Fig. 4D). Consistent with the in vitro data, p27 expression was greater in lung tissue from SM22α-PHD1/2−/− compared with the SM22α-PHD1/2+/+ hypoxia-exposed mice (Fig. 4E). To assess if the cell cycle arrest induced by p27 leads to apoptosis, isolated PASMC were examined for activated caspase 3 expression. Caspase 3 is a transcriptional target of HIF-1α (32). Active caspase 3 expression was undetectable in both genotypes (data not shown), indicating that the p27 activity in the PHD1/2−/− PASMC does not lead to SMC apoptosis. Altogether, these results suggest the limited vascular remodeling in the SM22α-PHD1/2−/− mice is associated with p27-induced cell cycle arrest rather than apoptosis.
Hypoxia-Induced Expression of p27 Is Absent in PASMC from Patients with IPAH
To demonstrate clinical relevance of p27 activity in PAH, PASMC isolated from control and idiopathic pulmonary arterial hypertensive (IPAH) patients were examined for p27 expression (Fig. 5, A and B). Cells were incubated under normoxic (21% O2) or hypoxic (5% O2) conditions for 2 days. Hypoxia-induced p27 expression in PASMC isolated from control patients, but not patients with IPAH. Short-term hypoxic exposure (1–3 h) did not induce p27 expression in control or IPAH PASMC. Altogether these results suggest that the loss of p27 expression in IPAH PASMC may contribute to the vascular remodeling associated with PAH.
Figure 5.
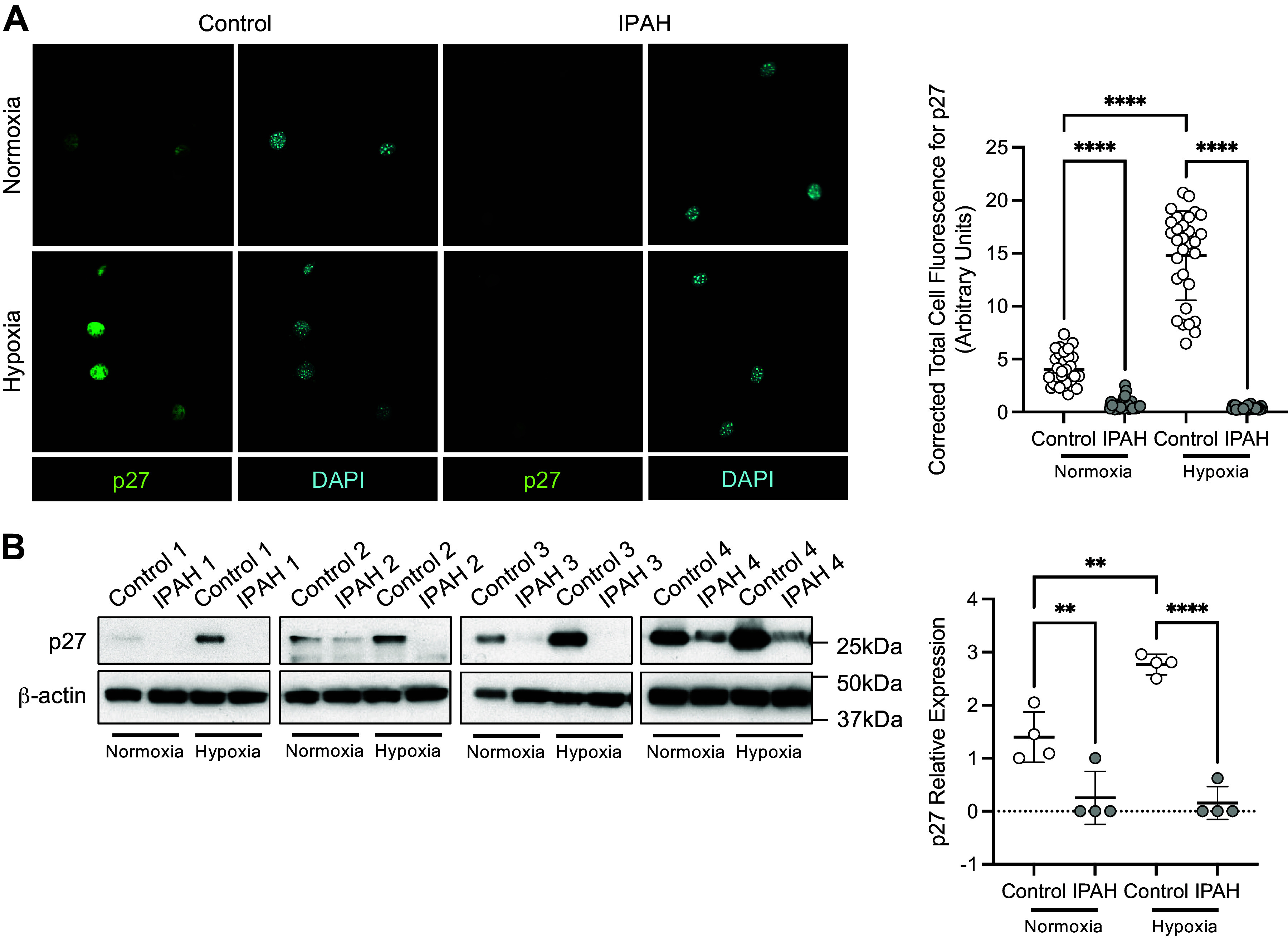
PASMC isolated from patients with IPAH demonstrate a loss of hypoxia-induced p27 expression. A: p27 expression in PASMC from control and IPAH patients (n = 3 replicate experiments with 10 fields analyzed for each). p27 (green), nuclei (blue); ×200 magnification, calibration bar 50 µm. B: p27 protein expression and quantification in human PASMC exposed to normoxic (21% O2) or hypoxic (5% O2, 2 days) conditions (n = 4 replicate experiments). Graphs represent the means ± SD, **P < 0.01, ****P < 0.0001, as indicated. P values were measured by two-way ANOVA. IPAH, idiopathic pulmonary arterial hypertension; PASMC, pulmonary artery smooth muscle cell.
DISCUSSION
The present study sought to further elucidate the role of PASMC HIF-1α in the development of hypoxia-induced pulmonary hypertension. We show that SM22α-specific deletion of PHD1 and PHD2 stabilizes PASMC HIF-1α protein expression in mice and promotes the nuclear accumulation and activity of HIF-1α. There was no change in PASMC HIF-2α protein expression, likely owing to its limited expression in SMC. Constitutive stabilization of HIF-1α in PASMC resulted in decreased pMLC and contractility. These results support the concept that in PASMC the transcription factor HIF-1α functions to constrain contractility by inhibiting MLC activity. Indeed, the SM22α-PHD1/2−/− mice were relatively hypoxia-insensitive and did not demonstrate hypoxic pulmonary vasoconstriction. Moreover, in chronic hypoxia studies, vascular remodeling was markedly attenuated in the SM22α-PHD1/2−/− mice in association with an increase in p27 expression, suggesting that HIF-1α may mitigate pulmonary vascular remodeling under hypoxic conditions by inducing expression of p27. The hypoxic induction of p27 may underlie the attenuated pulmonary vascular response in the SM22α-PHD1/2−/− mice.
These results point to a SM22α cell-specific role for HIF-1α in limiting the pulmonary vascular contractile response to acute and chronic hypoxia. Evidence suggests that cell-dependent alterations of HIF-1α and HIF-2α contribute to PAH. In pulmonary artery endothelial cells (PAECs), there is a consensus that increased HIF-2α causes PAH and vascular remodeling (33–36). These studies describe an endothelial cell-specific role for HIF-2α in promoting the development of PAH. Hypoxia-induced PH mouse models have demonstrated that HIF-1α haploinsufficiency mitigates the increase in RVSP and vascular remodeling initially with exposure to chronic hypoxia (7, 8, 37). Taken together, these studies highlight SM22α cell, context, and isoform-specific roles for HIF-1α in PAH.
Experiments demonstrated that in PASMC isolated from mice with deletion of PHD1 and PHD2, loss of HIF-1α via silencing RNA, decreased PGK1 expression, a classical HIF target. These experiments provide further support for an SM22α cell-specific role for HIF-1α in the regulation of vascular tone. The present findings are consistent with prior reports from our laboratory demonstrating that loss of HIF-1α in PASMC potentiated hypoxia-induced pulmonary hypertension and the pulmonary vascular response to acute hypoxia. Taken together, the gain- and loss-of-function approaches argue compellingly for an SM22α cell specific role for HIF-1α in mediating the pulmonary vascular response to both acute and chronic hypoxia.
Since HIF-1α in the lung differs by cell type with the highest level of expression in PASMC (4), cell-specific loss has been undertaken to gain a more nuanced understanding of the role of HIF in the pulmonary circulation. In PAECs, there is a consensus that increasing HIF-2α causes profound PH and vascular remodeling. In PASMC, consensus has been more elusive. A prior report from our laboratory, using the same promoter as in the present report, demonstrated that HIF-1α loss accentuated the response to either acute or chronic hypoxia, and increased pulmonary vascular tone even in normoxia (5). Though the present report addresses hypoxia-induced pulmonary hypertension with the use of a constitutive, as opposed to an inducible, deletion of PHD1 and PHD2 in SM22α-expressing cells there is potential that the current findings result, in part, from structural or functional alterations associated with lifelong, SM22α-expressing cell-specific changes in prolyl hydroxylase and HIF expression.
In contrast, in two subsequent reports wherein cell-specific loss of HIF-1α was accomplished using an inducible strategy, the response to hypoxia was limited (7, 8), consistent with the prior study in HIF-1α haploinsufficient mice (37). However, in both studies, residual SMC HIF-1α gene expression persisted. In Ball et al., SMC HIF-1α protein expression or activity following hypoxia was not interrogated (7). In Luo et al.(8), CD146 deletion in PDGFRβ-expressing cells mitigated pulmonary vascular remodeling and the development of PH in chronic hypoxic mice. Interestingly, with the ablation of CD146, there was a corresponding decrease in SMC HIF-1α expression, albeit incomplete. Given the residual HIF-1α expression in each of the reports (7, 8, 37), as opposed to the complete loss of HIF-1α in SM22α expressing cells (5), we postulate that hypoxia may have increased HIF-1α expression and activity. Thus, the seemingly dichotomous results may derive from the effect of partial versus complete loss of HIF-1α in PASMC.
SMC proliferation is a key determinant of the vascular remodeling that characterizes PAH. In our model, chronic hypoxia did not increase the muscularization of the pulmonary arteries in the SM22α-PHD1/2−/− mice, suggesting an absence of the SMC proliferation characteristic of PAH. We focused on a potential role for p27 given its function in limiting cell cycle progression, and prior reports establishing p27 as a major determinant of PASMC proliferation (38). Further, hypoxia-inducible p27 expression can prevent the development of PH in mice. Moreover, in the absence of p27 hypoxia-induced vascular remodeling is increased (13, 29–31, 39–42). The promoter region of p27 includes three core HRE consensus sequences (CGTG), suggesting that p27 may be a direct transcriptional target of HIF-1α. Yet, further experiments are needed to definitively demonstrate that p27 is a direct HIF-1α target. Taken together, these results point to a potential role for SMC HIF-1α in restraining hypoxia-induced pulmonary vascular remodeling by inducing the cell cycle inhibitor p27. SM22α-expressing cell-specific loss of HIF-1α in PASMC then, might be expected to potentiate the response to chronic hypoxia (5). Indeed, IPAH PASMC did not induce p27 expression in response to hypoxia.
The results from these studies also point to a role for HIF-1α in SM22α-expressing cells in the response to acute hypoxia. Hypoxic pulmonary vasoconstriction facilitates ventilation and perfusion matching and minimizes intrapulmonary shunting and subsequent hypoxemia (43, 44). Hypoxia induces pulmonary vasoconstriction via K+ channel inactivation on PASMC and subsequent entry of extracellular calcium through voltage-operated calcium channels (45, 46). Several of the oxygen-sensing molecules in PASMC, including voltage-gated K+ channels (Kv) and a subunit of the calcium-sensitive K+ channel, are downstream targets of HIF-1α (47, 48). Constitutive stabilization of HIF-1α may increase β1 subunit expression of the calcium-sensitive K+ channel and thereby decrease sensitivity to hypoxia (49). The observation that the loss of HIF-1α in SM22α-expressing cells potentiates hypoxia-induced increases in both PASMC cytosolic calcium and contractility supports this overall notion (6). Thus, distinct HIF-1α targets in SM22α-expressing cells may mitigate the response to acute hypoxia. Notwithstanding the dramatically attenuated hypoxia-induced increase in RVSP in mice with PHD1 and 2 deletions in SM22α-expressing cells relative to those with preserved PHD1 and 2 expression, the differences in heart rates between the two groups limit the conclusions regarding hypoxic pulmonary vasoconstriction. Although the differences in pressure were significant, the experiments did not include measurements of cardiac output. Hence, if pulmonary blood flow was lower in mice lacking PHD1/2 in SM22α-expressing cells, then it is possible that despite the marked differences in RVSP, pulmonary vascular resistance might have increased similarly in both groups. That said, the data from contraction gel assays with PASMC studied in vitro offers strong support for the proposition that in the absence of PHD1 and 2, the contractile response to hypoxia is attenuated.
Though the present approach provides SM22α-expressing cell-specific insight into the effect of constitutive HIF-1α stabilization in PASMC, there are substantial limitations to the findings. First, SM22α is expressed in multiple mesenchymal cell types during late embryonic and early fetal life, which raises the potential for off-target losses of PHD1 and PHD2 in cells such as pericytes and myofibroblasts. Moreover, the constitutive deletion of PHD1 and PHD2 does not preclude potential off-target effects. Although several HIF targets were interrogated, given the ubiquity of HIF targets, many were unaddressed. There is a potential that loss of PHD1 and PHD2 might more directly affect the expression of molecules linked to vascular remodeling and reactivity, though such effects remain unknown. Although the present findings were based on prolonged hypoxic exposure, a well-accepted murine model for pulmonary hypertension, there may be specific implications for pulmonary hypertension associated with chronic hypoxia, or Group 3 pulmonary hypertension such as might occur at high altitude or in the context of pulmonary parenchymal disease (1, 2, 44, 47).
In addition, in the SM22α-PHD1/2 mouse model, HIF-1α stabilization is not complete, as exposure to hypoxia further stabilizes HIF-1α expression in the SM22α-PHD1/2−/− mice. We found that deletion of all three isoforms in SM22α-expressing cells resulted in fetal demise. Thus, we opted for a strategy wherein only two isoforms, PHD1 and PHD2 were deleted, with PHD3 expression preserved. With only two isoforms deleted, pups were viable and thrived. Regardless, the observation that hypoxia increased HIF-1α protein despite deletion of PHD1 and PHD2 implies an important role for PHD3 in stabilization of HIF, at least in the absence of PHD1 and PHD2. It may be the case that with constitutive deletion of PHD1 and PHD2, the functional significance of PHD3 is amplified. The increase in HIF protein with hypoxia, considered in concert with the fetal lethality for mice with all PHD isoforms deleted in SM22α-expressing cells argues for some substantial degree of functional overlap between the three isoforms.
Interestingly, though normoxic PASMC isolated from SM22α-PHD1/2−/− mice have relatively similar levels of HIF-1α protein as hypoxic PASMC from SM22α-PHD1/2+/+ mice, the mice differ phenotypically. This suggests that the absolute levels of HIF-1α expression in PASMC do not, independently, account for the physiologic and signaling differences noted with hypoxia. These findings suggest that the effects of incompletely described downstream prolyl hydroxylase targets in hypoxia may underlie the phenotypic differences between genotypes. This construct is consistent with prior observations from our laboratory detailing a role for HIF-1α in PASMC in maintaining low pulmonary vascular resistance in normoxia (5, 6), though the finding has not been present in alternative models (7). Hypoxia may heighten already present differences and manifest with an attenuated pulmonary vascular response.
In conclusion, we show that augmenting PASMC HIF-1α activity reduces pulmonary arterial pressure by constraining hypoxic vasoconstriction and vascular remodeling. This work provides mechanistic data surrounding SM22α-expressing cell-specific, HIF-1α determined, regulation of normal and elevated pulmonary vascular tone and muscularization (Fig. 6). Our previous study demonstrated that the loss of PASMC HIF-1α caused an increase in pulmonary vascular tone, not only with hypoxia but under normoxic conditions (5). As shown in this study, an increase in the stabilization of SMC HIF-1α does not further decrease normal pulmonary vascular tone, as the vascular tone may already be as low as possible. The data presented herein addresses the role of increased HIF-1α expression and activity in SM22α-expressing cells in the context of hypoxia, both acute and chronic, a stimulus known to increase pulmonary vascular tone. These findings highlight the importance of establishing a causal link between HIF-1α and p27 expression. Successfully modulating HIF-1α activity in PASMC or manipulating the expression of p27 may represent a feasible strategy to prevent or treat PAH.
Figure 6.

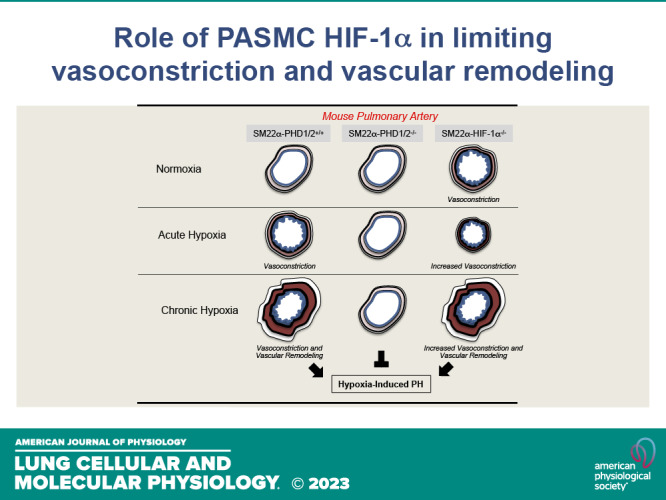
Role for PASMC HIF-1α in limiting vasoconstriction and vascular remodeling of pulmonary arteries through induction of p27. A: in SM22α-genetically altered mouse pulmonary arteries: (i) under normoxia and without PASMC HIF-1α pMLC levels are increased, with wild-type and stabilized PASMC HIF-1α pMLC relative levels are decreased; (ii) under acute hypoxia and without PASMC HIF-1α pMLC levels are further increased, with wild-type PASMC HIF-1α pMLC relative levels are increased, and with stabilized PASMC HIF-1α pMLC levels remain low; (iii) under chronic hypoxia and without PASMC HIF-1α pMLC levels remain high and p27 is not induced, with wild-type PASMC HIF-1a pMLC levels increase and p27 is induced, and with stabilized PASMC HIF-1α pMLC levels remain low, and p27 is highly expressed. B: in IPAH pulmonary arteries pMLC levels are increased, and p27 is not induced in response to hypoxia, and relative to control pulmonary arteries. HIF-1α, hypoxia-inducible factor-1α; IPAH, idiopathic pulmonary arterial hypertension; PASMC, pulmonary artery smooth muscle cell; pMLC, myosin light chain phosphorylation.
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
This work has been supported by the National Institutes of Health grant HL-060784 (to D.N.C.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.A.B., C.M.A., and D.N.C. conceived and designed research; E.A.B., R.I., and X.C. performed experiments; E.A.B. and R.I. analyzed data; E.A.B., R.I., and D.N.C. interpreted results of experiments; E.A.B. and R.I. prepared figures; E.A.B. drafted manuscript; E.A.B. and D.N.C. edited and revised manuscript; D.N.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Amato Giaccia (Stanford University) for kindly providing the PHD1/2fl/fl mice. Flow cytometry analysis for this project was done on instruments in the Stanford Shared FACS Facility.
REFERENCES
- 1. Grover RF, Reeves JT. Experimental induction of pulmonary hypertension in normal steers at high altitude. Med Thorac 19: 543–550, 1962. doi: 10.1159/000192263. [DOI] [PubMed] [Google Scholar]
- 2. Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99: 675–691, 2006. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- 3. Semenza GL. Oxygen sensing, homeostasis, and disease. N Engl J Med 365: 537–547, 2011. [Erratum in N Engl J Med 365: 968, 2011]. doi: 10.1056/NEJMra1011165. [DOI] [PubMed] [Google Scholar]
- 4. Yu AY, Frid MG, Shimoda LA, Wiener CM, Stenmark K, Semenza GL. Temporal, spatial, and oxygen-regulated expression of hypoxia-inducible factor-1 in the lung. Am J Physiol Lung Cell Mol Physiol 275: L818–L826, 1998. doi: 10.1152/ajplung.1998.275.4.L818. [DOI] [PubMed] [Google Scholar]
- 5. Kim YM, Barnes EA, Alvira CM, Ying L, Reddy S, Cornfield DN. Hypoxia inducible factor-1α in pulmonary artery smooth muscle cells lowers vascular tone by decreasing myosin light chain phosphorylation. Circ Res 112: 1230–1233, 2013. doi: 10.1161/CIRCRESAHA.112.300646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barnes EA, Chen CH, Sedan O, Cornfield DN. Loss of smooth muscle cell hypoxia inducible factor-1α underlies increased vascular contractility in pulmonary hypertension. FASEB J 31: 650–662, 2017. doi: 10.1096/fj.201600557R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, Beussink L, Dettman RW, Berkelhamer SK, Steinhorn RH, Shah SJ, Schumacker PT. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1α. Am J Respir Crit Care Med 189: 314–324, 2014. doi: 10.1164/rccm.201302-0302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luo Y, Teng X, Zhang L, Chen J, Liu Z, Chen X, Zhao S, Yang S, Feng J, Yan X. CD146-HIF-1α hypoxic reprogramming drives vascular remodeling and pulmonary arterial hypertension. Nat Commun 10: 3551, 2019. [Erratum in Nat Commun 10: 4098, 2019]. doi: 10.1038/s41467-019-11500-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Taniguchi CM, Miao YR, Diep AN, Wu C, Rankin EB, Atwood TF, Xing L, Giaccia AJ. PHD inhibition mitigates and protects against radiation-induced gastrointestinal toxicity via HIF2. Sci Transl Med 6: 236ra64, 2014. doi: 10.1126/scitranslmed.3008523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Firth JD, Ebert BL, Pugh CW, Ratcliffe PJ. Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: similarities with the erythropoietin 3’ enhancer. Proc Natl Acad Sci USA 91: 6496–6500, 1994. doi: 10.1073/pnas.91.14.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J Biol Chem 271: 20246–20249, 1996. doi: 10.1074/jbc.271.34.20246. [DOI] [PubMed] [Google Scholar]
- 12. Yoshida M, Yagi K. Purification and characterization of a phosphoprotein phosphatase that dephosphorylates myosin and the isolated light chain from chicken gizzard smooth muscle. J Biochem 99: 1027–1036, 1986. doi: 10.1093/oxfordjournals.jbchem.a135566. [DOI] [PubMed] [Google Scholar]
- 13. Gardner LB, Li Q, Park MS, Flanagan WM, Semenza GL, Dang CV. Hypoxia inhibits G1/S transition through regulation of p27 expression. J Biol Chem 276: 7919–7926, 2001. doi: 10.1074/jbc.M010189200. [DOI] [PubMed] [Google Scholar]
- 14. Emerling BM, Weinberg F, Liu JL, Mak TW, Chandel NS. PTEN regulates p300-dependent hypoxia-inducible factor 1 transcriptional activity through Forkhead transcription factor 3a (FOXO3a). Proc Natl Acad Sci USA 105: 2622–2627, 2008. doi: 10.1073/pnas.0706790105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moessler H, Mericskay M, Li Z, Nagl S, Paulin D, Small JV. The SM22 promoter directs tissue-specific expression in arterial but not in venous or visceral smooth muscle cells in transgenic mice. Development 122: 2415–2425, 1996. doi: 10.1242/dev.122.8.2415. [DOI] [PubMed] [Google Scholar]
- 16. Li L, Miano JM, Mercer B, Olson EN. Expression of the SM22alpha promoter in transgenic mice provides evidence for distinct transcriptional regulatory programs in vascular and visceral smooth muscle cells. J Cell Biol 132: 849–859, 1996. doi: 10.1083/jcb.132.5.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hoggatt AM, Simon GM, Herring BP. Cell-specific regulatory modules control expression of genes in vascular and visceral smooth muscle tissues. Circ Res 91: 1151–1159, 2002. doi: 10.1161/01.res.0000047508.30800.4f. [DOI] [PubMed] [Google Scholar]
- 18. Wang KC, Chen PS, Chao TH, Luo CY, Chung HC, Tseng SY, Huang TY, Lin YL, Shi GY, Wu HL, Li YH. The role of vascular smooth muscle cell membrane-bound thrombomodulin in neointima formation. Atherosclerosis 287: 54–63, 2019. doi: 10.1016/j.atherosclerosis.2019.05.019. [DOI] [PubMed] [Google Scholar]
- 19. Wan F, Letavernier E, Abid S, Houssaini A, Czibik G, Marcos E, Rideau D, Parpaleix A, Lipskaia L, Amsellem V, Gellen B, Sawaki D, Derumeaux G, Dubois-Randé JL, Delcroix M, Quarck R, Baud L, Adnot S. Extracellular calpain/calpastatin balance is involved in the progression of pulmonary hypertension. Am J Respir Cell Mol Biol 55: 337–351, 2016. doi: 10.1165/rcmb.2015-0257OC. [DOI] [PubMed] [Google Scholar]
- 20. Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, Hammer RM, Herz J, Kuhn M. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc Natl Acad Sci USA 99: 7142–7147, 2002. doi: 10.1073/pnas.102650499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107: 43–54, 2001. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 22. Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem 279: 38458–38465, 2004. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 23. Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J 22: 4082–4090, 2003. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 5: 343–354, 2004. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 25. Kaelin WG Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell 30: 393–402, 2008. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 26. Ikebe M, Hartshorne DJ. Phosphorylation of smooth muscle myosin at two distinct sites by myosin light chain kinase. J Biol Chem 260: 10027–10031, 1985. [PubMed] [Google Scholar]
- 27. Scornik FS, Toro L. U46619, a thromboxane A2 agonist, inhibits KCa channel activity from pig coronary artery. Am J Physiol Cell Physiol 262: C708–C713, 1992. doi: 10.1152/ajpcell.1992.262.3.C708. [DOI] [PubMed] [Google Scholar]
- 28. Sharma SS, Pledger WJ. The non-canonical functions of p27(Kip1) in normal and tumor biology. Cell Cycle 15: 1189–1201, 2016. doi: 10.1080/15384101.2016.1157238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang G, Reisdorph R, Clark RE Jr, Miskimins R, Lindahl R, Miskimins WK. Cyclin dependent kinase inhibitor p27(Kip1) is upregulated by hypoxia via an ARNT dependent pathway. J Cell Biochem 90: 548–560, 2003. doi: 10.1002/jcb.10621. [DOI] [PubMed] [Google Scholar]
- 30. Horrée N, Gort EH, van der Groep P, Heintz APM, Booijs M, van Diest PJ. Hypoxia-inducible factor 1α is essential for hypoxic p27 induction in endometrioid endometrial carcinoma. J Pathol 214: 38–45, 2008. doi: 10.1002/path.2244. [DOI] [PubMed] [Google Scholar]
- 31. Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS. Hypoxia-inducible factor 1α is essential for cell cycle arrest during hypoxia. Mol Cell Biol 23 359–369, 2003. doi: 10.1128/MCB.23.1.359-369.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Van Hoecke M, Prigent-Tessier AS, Garnier PE, Bertrand NM, Filomenko R, Bettaieb RA, Marie C, Beley AG. Evidence of HIF-1 functional binding activity to caspase-3 promoter after photothrombotic cerebral ischemia. Mol Cell Neurosci 34: 40–47, 2007. doi: 10.1016/j.mcn.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 33. Brusselmans K, Compernolle V, Tjwa M, Wiesener MS, Maxwell PH, Collen D, Carmeliet P. Heterozygous deficiency of hypoxia-inducible factor-2α protects mice against pulmonary hypertension and right ventricular dysfunction during prolonged hypoxia. J Clin Invest 111: 1519–1527, 2003. doi: 10.1172/JCI15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cowburn AS, Crosby A, Macias D, Branco C, Colaco RD, Southwood M, Toshner M, Crotty Alexander LE, Morrell NW, Chilvers ER, Johnson RS. HIF2α-arginase axis is essential for the development of pulmonary hypertension. Proc Natl Acad Sci USA 113: 8801–8806, 2016. doi: 10.1073/pnas.1602978113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dai Z, Li M, Wharton J, Zhu MM, Zhao YY. Prolyl-4 hydroxylase 2 (PHD2) deficiency in endothelial cells and hematopoietic cells induces obliterative vascular remodeling and severe pulmonary arterial hypertension in mice and humans through hypoxia-inducible factor-2α. Circulation 133: 2447–2458, 2016. doi: 10.1161/CIRCULATIONAHA.116.021494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kapitsinou PP, Rajendran G, Astleford L, Michael M, Schonfeld MP, Fields T, Shay S, French JL, West J, Haase VH. The endothelial prolyl-4-hydroxylase domain 2/hypoxia-inducible factor 2 axis regulates pulmonary artery pressure in mice. Mol Cell Biol 36: 1584–1594, 2016. doi: 10.1128/MCB.01055-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JSK, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. J Clin Invest 103: 691–696, 1999. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fouty BW, Grimison B, Fagan KA, Le Cras TD, Harral JW, Hoedt-Miller M, Sclafani RA, Rodman DM. p27(Kip1) is important in modulating pulmonary artery smooth muscle cell proliferation. Am J Respir Cell Mol Biol 25: 652–658, 2001. doi: 10.1165/ajrcmb.25.5.4592. [DOI] [PubMed] [Google Scholar]
- 39. Gao YF, Zhu XD, Shi DM, Jing ZC, Li L, Ma D, Fan ZX, Li J, Wang YW, Wu BX. The effects of atorvastatin on pulmonary arterial hypertension and expression of p38, p27, and Jab1 in rats. Int J Mol Med 26: 541–547, 2010. doi: 10.3892/ijmm_00000497. [DOI] [PubMed] [Google Scholar]
- 40. Yu L, Quinn DA, Garg HG, Hales CA. Gene expression of cyclin-dependent kinase inhibitors and effect of heparin on their expression in mice with hypoxia-induced pulmonary hypertension. Biochem Biophys Res Commun 345: 1565–1572, 2006. doi: 10.1016/j.bbrc.2006.05.060. [DOI] [PubMed] [Google Scholar]
- 41. Yu L, Quinn DA, Garg HG, Hales CA. Cyclin-dependent kinase inhibitor p27Kip1, but not p21WAF1/Cip1, is required for inhibition of hypoxia-induced pulmonary hypertension and remodeling by heparin in mice. Circ Res 97: 937–945, 2005. doi: 10.1161/01.RES.0000188211.83193.1a. [DOI] [PubMed] [Google Scholar]
- 42. Luo Y, Zhang B, Dong HY, Liu Y, Li ZC, Dong MQ, Gao YQ. Prevention of hypoxic pulmonary hypertension by hypoxia-inducible expression of p27 in pulmonary artery smooth muscle cells. Gene Ther 21: 751–758, 2014. doi: 10.1038/gt.2014.49. [DOI] [PubMed] [Google Scholar]
- 43. Euler USv, Liljestrand G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand 12: 301–320, 1946. doi: 10.1111/j.1748-1716.1946.tb00389.x. [DOI] [Google Scholar]
- 44. Sylvester JT, Shimoda LA, Aaronson PI, Ward JPT. Hypoxic pulmonary vasoconstriction. Physiol Rev 92: 367–520, 2012. [Erratum in Physiol Rev 94: 989, 2014]. doi: 10.1152/physrev.00041.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Post JM, Hume JR, Archer SL, Weir EK. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. Am J Physiol Cell Physiol 262: C882–C890, 1992. doi: 10.1152/ajpcell.1992.262.4.C882. [DOI] [PubMed] [Google Scholar]
- 46. Yuan XJ, Goldman WF, Tod ML, Rubin LJ, Blaustein MP. Hypoxia reduces potassium currents in cultured rat pulmonary but not mesenteric arterial myocytes. Am J Physiol Lung Cell Mol Physiol 264: L116–L123, 1993. doi: 10.1152/ajplung.1993.264.2.L116. [DOI] [PubMed] [Google Scholar]
- 47. Weir EK, López-Barneo J, Buckler KJ, Archer SL. Mechanisms of disease acute oxygen-sensing mechanisms. N Engl J Med 353: 2042–2055, 2005. doi: 10.1056/NEJMra050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ahn YT, Kim YM, Adams E, Lyu SC, Alvira CM, Cornfield DN. Hypoxia-inducible factor-1α regulates KCNMB1 expression in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 302: L352–L359, 2012. doi: 10.1152/ajplung.00302.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guignabert C, Tu L, Izikki M, Dewachter L, Zadigue P, Humbert M, Adnot S, Fadel E, Eddahibi S. Dichloroacetate treatment partially regresses established pulmonary hypertension in mice with SM22α-targeted overexpression of the serotonin transporter. FASEB J 23: 4135–4147, 2009. doi: 10.1096/fj.09-131664. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.