

Abstract
The RE1-silencing transcription factor (REST)/neuron-restrictive silencer factor (NRSF) can repress transcription of a battery of neuronal differentiation genes in non-neuronal cells by binding to a specific consensus DNA sequence present in their regulatory regions. However, REST/NRSF–/– mice suggest that the absence of REST/NRSF-dependent repression alone is not sufficient for the expression of these neuronal differentiation genes and that the presence of other promoter/enhancer-specific activators is required. Here we describe the construction of a recombinant transcription factor, REST-VP16, by replacing repressor domains of REST/NRSF with the activation domain of a viral activator VP16. In transient transfection experiments, REST-VP16 was found to operate through RE1 binding site/neuron-restrictive enhancer element (RE1/NRSE), activate plasmid-encoded neuronal promoters in various mammalian cell types and activate cellular REST/NRSF target genes, even in the absence of factors that are otherwise required to activate such genes. Efficient expression of REST-VP16 through adenoviral vectors in NT2 cells, which resemble human committed neuronal progenitor cells, was found to cause activation of multiple neuronal genes that are characteristic markers for neuronal differentiation. Thus, REST-VP16 could be used as a unique tool to study neuronal differentiation pathways and neuronal diseases that arise due to the deregulation of this process.
INTRODUCTION
Mammalian neuronal stem cells have been isolated that can be converted into neurons and other cell types under various growth conditions (1–5). The neuronal differentiation pathways were previously thought to be regulated primarily through positive regulators. Several genes encoding such regulators and their cellular interactions were identified through analysis of mammalian and non-mammalian embryogenesis, regeneration, repair and disease (6–11). However, the mechanism responsible for initiating these processes as well as the exact sequence of such pathways are not known. The transcription factor RE1-silencing transcription factor (REST)/neuron-restrictive silencer factor (NRSF) was identified to be the first global neuronal repressor and potentially one of the critical regulators of neurogenesis (12,13).
REST/NRSF is a DNA-binding protein and has been found to be responsible for silencing the transcription of most neuronal differentiation genes by binding to a 23 bp consensus sequence (RE1 binding site/neuron-restrictive silencer element or RE1/NRSE), which is present at the upstream promoter–enhancer region of these genes (12–17). The estimated 116 kDa molecular weight protein contains a DNA-binding domain with eight zinc-finger regions and two inhibitory domains (16). REST/NRSF has been found to be expressed in most, if not all, non-neuronal cells including neuroblasts in vivo (12,13). These studies revealed that REST/NRSF is not expressed at high levels in differentiated neurons during embryogenesis. In fact, using a mouse REST probe, the presence of REST in most non-neuronal cells but not in neurons has been found in mouse embryos between the ages of 11.5 and 13.5 days. However, later studies found it to be expressed in mature neurons in adults (18,19), suggesting a complex role for REST/NRSF depending on the cellular and physiological environment. REST/NRSF-dependent promoter repression requires interaction with several cofactors, such as CoREST, mSin3A and histone deacetylase complex (HDAC), and requires histone deacetylase activity (20–23). CoREST was found to bind to the C-terminal repressor domain, while sin3A and HDAC bind to the N-terminal repressor domain. Based on the expression pattern of msin3A and CoREST, it has been suggested that while mSin3A is required constitutively for REST/NRSF-dependent repression, CoREST is required for more specialized repressor functions (24). Gene deletion studies with REST/NRSF–/– mice indicate that the absence of REST/NRSF in vivo causes expression of only one of the REST/NRSF target genes, the neuron-specific tubulin gene, in a subset of non-neuronal tissue followed by embryonic lethality (25). This lack of REST/NRSF does not cause activation of other REST/NRSF target genes. This indicated that the absence of REST/NRSF-dependent repression alone is not sufficient to activate multiple REST/NRSF target genes in these cell types and suggested that such a process requires relief from other repression mechanisms and/or the presence of other promoter/enhancer-specific positive activators.
To examine this question, we constructed a regulator that not only counters REST/NRSF repression but also activates REST/NRSF-dependent promoters, even in the absence of either its cofactors (CoREST, mSin3 or HDAC) or other promoter-specific activators. We constructed two recombinant transcription factors (REST1-VP16 and REST-VP16) by replacing different repressor domains of REST with the strong activation domain of the viral activator VP16. In transient transfection assays, we found that REST-VP16 binds to the same DNA-binding site as REST but functions as an activator instead of a repressor of neuronal genes. To increase the transfection efficiency, we constructed adenoviral vectors encoding REST-VP16. In this study, we used NT2 cells, which are derived from teratocarcinoma and resemble human committed neuronal progenitor cells (26). Here we find that adenovirus-mediated expression of REST-VP16 alone can cause expression of multiple neural differentiation genes in NT2 cells, indicating that REST-VP16 can be used as a tool for this purpose.
MATERIALS AND METHODS
Recombinant DNA constructs
The first expression vector encoding the mutant protein pREST1-VP16 was constructed by inserting the HindIII–XmnI fragment of pREST-Express (encoding the DNA-binding domain of REST; 12) into the EcoRI–BglII site of pCRF3 (Novagen, Madison, WI). This caused the REST DNA-binding domain including the N-terminal repressive domain to be in-frame with the VP16 activation domain flanked by the 5′-HindIII and the 3′-BamHI sites. The latter fragment was then inserted into the HindIII–ApaI site of the expression vector pcDNA 3.1 (Invitrogen, Carlsbad, CA). The resulting construct encodes the recombinant gene under the control of a powerful CMV promoter and an SV40-polyadenylation signal. The second expression vector, pREST-VP16, was constructed by replacing ClaI–HindIII fragment of pREST1-VP16 by ClaI–HindIII fragment of p73 (12). The resulting construct deletes both the N- and C-terminal repressor domains (Fig. 1A). The test plasmids pNaCh (pSDK7) and pNaChΔRE (pMB4) are described by Kraner et al. (15). Construction of pT.luc was described by Majumder et al. (27). pRE.T.luc and pREm.T.luc were constructed by inserting a BglII–EcoRI fragment containing dimers of either RE1 and mutated RE1 sequences (12), respectively, into the BglII–EcoRI site of p2025 (27,28), which placed them in front of adenovirus major late promoter TATA box. The BglII–HindIII fragments of the resulting plasmid containing the RE or REm sequence plus the TATA box were inserted into the same sites of pGL2-Basic (Promega, Madison, WI). All constructs were sequenced to confirm their identity.
Figure 1.
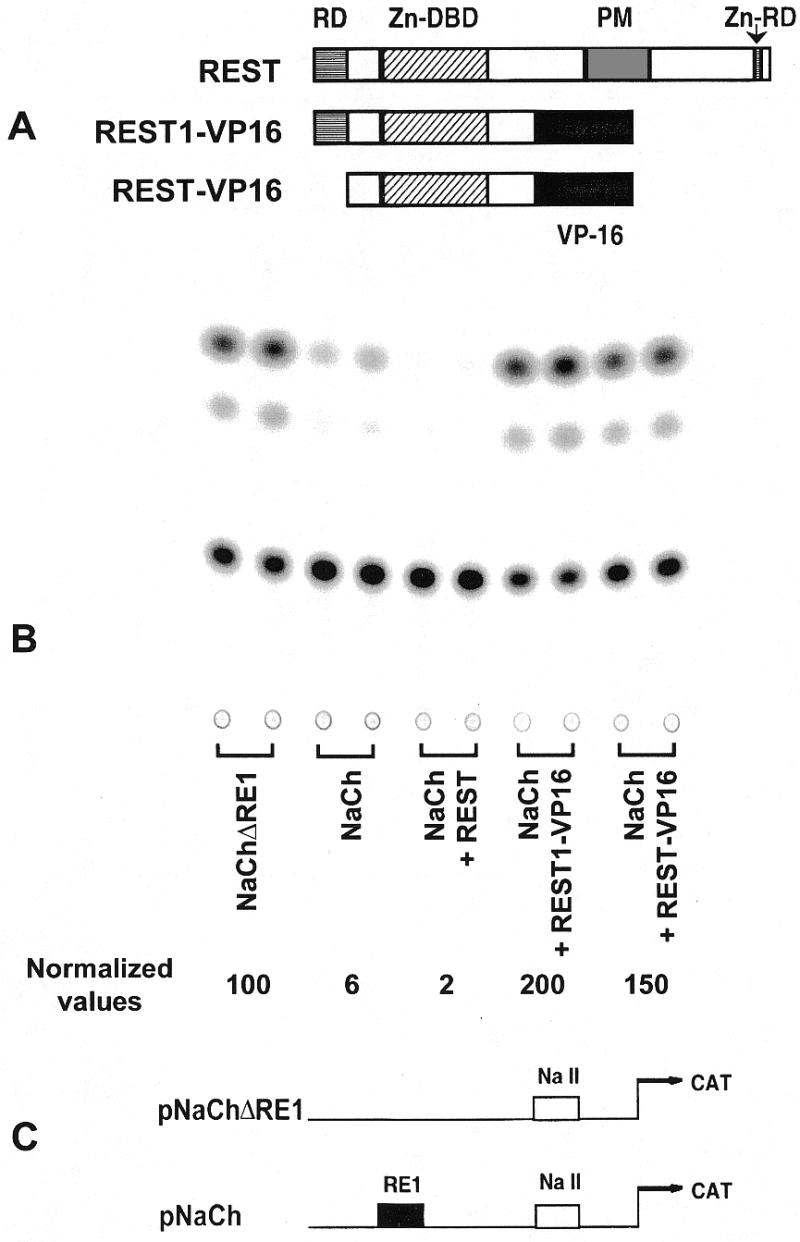
REST1-VP16 and REST-VP16 counter REST/NRSF-mediated repression of neural promoters. (A) The transcriptional repressor REST/NRSF contains a DNA-binding domain (Zn-DBD), consisting of eight zinc finger motifs, that bind to the consensus RE1 (or NRSE) sequence. It contains two distinct repressor domains (RD): one within the first 73 amino acids at the N-terminus, and the other within another zinc finger motif (Zn) at the C-terminus (16). It also contains a proline-rich motif of unknown function (PM). The recombinant construct, REST1-VP16, was constructed by replacing the C-terminal sequence of REST beyond the Zn-DBD by the strong activation domain of the viral activator VP16. It still contains the N-terminal repressor domain. REST-VP16 was constructed by deleting the N-terminal domain of REST1-VP16, and it does not contain either of the two repressor domains. REST1-VP16 and REST-VP16 bind to the same DNA-binding site as REST/NRSF but function as activators instead of a repressor as determined by transfection analysis in HeLa cells (B). Here, the promoter activity is expressed as CAT reporter gene activity. The two reporter plasmids to assay REST-mediated repression of promoters are shown (C): pNaCH, containing the neuronal sodium channel type II promoter/enhancer (NaCh) elements that include REST binding site (RE1/NRSE sequence). These elements are placed in front of the CAT reporter gene. pNaCh is acted upon by functional REST/NRSF (12). pNaChΔRE1, containing the CAT reporter gene under the control of the NaCh without the RE1 sequence. Functional REST/NRSF cannot bind to this promoter/enhancer element and, thus, cannot repress its activity (12). Therefore, in the presence of functional REST/NRSF, pNaChΔRE1 would produce higher activity than pNaCh (B).
Cell culture, transfection and reporter gene assay
HeLa and Daoy medulloblastoma cells were purchased from ATCC. NT2 cells were purchased from Stratagene (La Jolla, CA) and cultured as described by Lawinger et al. (29). Transfection of plasmid DNA in cells was carried out in one of several ways: electroporation, transfection with Fugene 6 kit (Boehringer Mannheim, Indianapolis, IN) or transfection with MBS kit (Stratagene). Electroporation conditions are described elsewhere (29,30). Fugene 6 and MBS transfections were done essentially as per the manufacturer’s instructions. Following transfection, cells were usually incubated for 48 h, harvested and assayed for luciferase, β-galactosidase or bacterial chloramphenicol acetyl transferase (CAT) reporter gene activity as described (27,29).
RT–PCR, western blot and immunocytochemistry
RT–PCR assay was carried out from total cellular mRNA using primers and conditions as described previously (29). Western blotting analyses were carried out as described (29) using the ECL kit from Amersham (Piscataway, NJ). The antibodies used were anti-synapsin (Chemicon International, Temecula, CA), anti-class III β-tubulin (Babco, Richmond, CA), anti-neurofilament 200 (Sigma, St Louis, MO) and anti-actin (Amersham). Immunocytochemistry was carried out using the avidin–biotin immunoperoxidase method employing 3,3′-diaminobenzidine as chromogen (ABC kit, Vector Laboratories, Burlingame, CA) (31,32).
Adenoviral vectors and adenoviral infection
Ad vectors used in this study are replication-deficient, E1 and E3-deleted recombinant human serotype 5 adenovirus. We constructed four adenoviral vectors: Ad.REST-VP16, Ad.REST, Ad.Gal4-VP16, and the E1 and E3-deleted control virus Ad.ΔE1.3 (Ad). Another vector encoding GFP (Ad.GFP) was purchased from Microbix (Ontario, Canada). All of the inserts were under the control of CMV promoter/enhancer elements, except the Gal4-VP16, which used RSV promoter/enhancer elements. To construct the adenoviral shuttle vectors pAd.REST-VP16 and pAd.REST, the DNA fragments containing the REST-VP16 or REST coding sequences were released with restriction enzymes HindIII and NotI and then inserted into the respective sites in the polylinker region of pAd.CMV as described by Guo et al. (33). To construct pAd.RSV-Gal4-VP16, a HindIII DNA fragment containing the coding sequence for GAL4-VP16 fusion protein was released from pSGVP (34) and inserted into the HindIII site of pAd.RSV vector (33). The inserted transgenes in these shuttle vectors were confirmed with PCR or DNA sequencing. Finally, recombinant adenoviruses were made by cotransfection of the each shuttle vector with pBHG10 (35) into human 293 cells, and recombinant plaques were isolated. All viruses were amplified in human 293 cells, then purified and stored as described previously (33). Viral titers were determined as plaque forming units (p.f.u./ml) using a plaque assay in 293 cells. Cells were seeded at a known density to ensure reproducible infections. The multiplicity of infection (m.o.i.) listed in all experiments is based on p.f.u.. Medium was aspirated off the cells and they were re-fed with a reduced volume of medium (5 ml for 15 cm plates and 3 ml for 10 cm plates). The appropriate amount of virus was added to the plates, which were then gently swirled to spread the virus evenly. Proper pipetting of concentrated virus was insured through the creation of dilutions in serum-free media. Cells were incubated at 37°C for 30 min before additional media was added to the plates to bring the volume up (15 ml for 15 cm plates and 5 ml for 10 cm plates) and plates were returned to the 37°C incubator overnight.
RESULTS
REST1-VP16 and REST-VP16 counter REST/NRSF-mediated repression and activate neuronal promoters from transfected plasmids
Mutational analysis revealed that the REST protein contains a DNA-binding domain with zinc-finger motifs flanked by two distinct repressor domains: one within the first 73 amino acids at the N-terminus and the other within another zinc finger motif at the C-terminus (16; Fig. 1A). To counter REST/NRSF activity, we constructed two recombinant molecules, REST1-VP16 and REST-VP16, by replacing different repressor domains of REST/NRSF with the strong activation domain of the viral activator VP16 (Fig. 1A). Thus, REST1-VP16 and REST-VP16 would be expected to bind to the same DNA-binding site as REST/NRSF but function as activators instead of repressors. REST1-VP16 retained the N-terminal repressor domain of REST, but REST-VP16 did not retain the N-terminal or the C-terminal repressor domains of REST. The recombinant constructs were placed under the control of CMV promoter/enhancer elements and an SV40 polyadenylation signal.
To assay REST/NRSF-dependent promoter repression, we used two reporter plasmid DNAs (Fig. 1C): (i) pNaCH (originally called pSDK7; 15), containing a bacterial CAT reporter gene under the control of sodium channel type II promoter/enhancer (NaCh) elements. NaCh is a mammalian native neuronal promoter/enhancer element that contains the RE1/NRSE sequence and is acted upon by functional REST/NRSF (12). (ii) pNaChΔRE1 (originally called pMB4; 15), containing the CAT reporter gene under the control of the ‘minimal’ NaCh without the RE1/NRSE sequence. Functional REST/NRSF has been found to have no repressive effect on the promoter activity of this plasmid (12). Thus, the presence of functional REST/NRSF in a system would produce a lower CAT activity from pNaCh as compared to pNaChΔRE1.
To examine REST/NRSF-mediated repression of neuronal promoters, human HeLa cells were used. HeLa cells have been shown to contain endogenous REST/NRSF protein that binds to RE1/NRSE sequence (12,13). Cells were transfected by electroporation with the reporter plasmids pNaCh or pNaChΔRE1. Cells were co-transfected with an internal standard plasmid, pST.luc, which contains the luciferase (luc) reporter gene under the control of an Sp1-dependent promoter (36,37). pST.luc expresses high level of the luciferase reporter gene in mammalian cells. The total amount of plasmid DNA transfected into cells was maintained at the same level in each experiment. Transfected cells, in triplicate, were incubated for 48 h before harvesting, and the cell extract was then analyzed for CAT and luciferase activities. The mean values for CAT activities were normalized to the corresponding mean values for luciferase activities as described previously (30). Figure 1B shows a typical CAT assay; the average normalized values are also shown. The CAT activity from pNaCh was found to be ∼15-fold lower than that from pNaChΔRE1, indicating that the endogenous REST present in HeLa cells can repress the neuronal NaCh promoter through its RE1/NRSE sequence under our experimental conditions. Furthermore, REST-dependent repression of the NaCh promoter could still be increased when these cells were cotransfected with the expression vector pREST encoding the REST protein. Thus, in our hands, the plasmids pNaCh and pNaChΔRE1 can be used to detect REST/NRSF-dependent neuronal promoter activity as has been previously shown (12).
Cotransfection pNaCh with either REST1-VP16 or REST-VP16 in HeLa cells was found to block REST-dependent repression and activate the promoter 33- and 25-fold, respectively (Fig. 1B). In contrast, neither REST1-VP16 nor REST-VP16 stimulates the promoter activity from pNaChΔRE1 (data not shown), suggesting that these recombinant molecules activate the promoter through the RE1/NRSE sequence. Since REST-VP16 did not contain either of the repressor domains, it was expected to stimulate the sodium channel promoter more strongly than REST1-VP16, which still contained the N-terminal repressor domain. However, in this reporter gene assay, REST1-VP16 stimulated the sodium channel promoter slightly more than REST-VP16. Several explanations could be proposed for this discrepancy. It is possible that the VP16 activation domain was strong enough to counter the repressive function of the N-terminal domain, that the conformation of the recombinant protein REST-VP16 is less favorable than that of REST1-VP16, or that REST1-VP16 protein has a higher affinity for the sodium channel promoter elements present in the context of the plasmid encoding the reporter gene. The last assumption appears to be the likely explanation, because REST-VP16, but not REST1-VP16, was found to be more potent in activating cellular neuronal genes (see below). In summary, both REST1-VP16 and REST-VP16 were found to counter REST-dependent repression and to stimulate neuronal promoters from transfected plasmids.
REST1-VP16 and REST-VP16 can stimulate a TATA box basal promoter through the RE1/NRSE sequence
To further assess the effect of REST1-VP16 and REST-VP16 directly through the RE1/NRSE sequence, we constructed synthetic promoters (Fig. 2) containing a luciferase reporter gene under the control of a TATA box alone (pT.luc) (36,37) or a TATA box plus two copies of either the wild-type RE1/NRSE sequence (pRE.T.luc) or the mutated RE1/NRSE sequence (pREm.T.luc) (12,15). HeLa cells were cotransfected with pT.luc, pRE.T.luc or pREm.T.luc in the presence and absence of various expression vectors, encoding no foreign insert (vector), Gal4-VP16 (containing the VP16 activation domain attached to Gal4 DNA-binding domain), REST1-VP16 or REST-VP16. These cells were processed and analyzed as described above. The plasmid pβ-gal was used as an internal control in these experiments. As shown in Figure 2, the basal promoter containing the TATA box alone could not be stimulated by any of the expression vectors. In contrast, the TATA box in the presence of the RE1/NRSE sequence could be stimulated by expression vectors encoding REST1-VP16 and REST-VP16 at 59- and 25-fold, respectively. Such stimulation through the RE1/NRSE sequence was not observed in the presence of the vector alone, or the vector encoding Gal4-VP16, indicating that the stimulation was not caused by the VP16 domain alone. Furthermore, REST1-VP16 and REST-VP16 could not stimulate the TATA box promoter when the RE1/NRSE sequence was substituted by a mutant sequence (pREm.T.luc) that does not bind REST/NRSF (12). Taken together, these data show that both REST1-VP16 and REST-VP16 can specifically stimulate a basal promoter through the RE1/NRSE sequence using the VP16 activation domain. Interestingly, in this reporter gene assay, REST1-VP16 stimulated the sodium channel promoter more efficiently than REST-VP16 as was seen before with pNaCh transfection experiments (Fig. 1).
Figure 2.

REST1-VP16 and REST-VP16 stimulate basal promoters through RE1/NRSE sequence. HeLa cells were transfected with the reporter plasmid pT.luc, pRE.T.luc or pREm.T.luc in the presence and absence of the expression vector alone, or the expression vector encoding REST, REST1-VP16, REST-VP16 or Gal4-VP16 and analyzed as described in Materials and Methods. Promoter activity is expressed as relative luciferase units (RLU).
REST-VP16 activates neuronal differentiation marker genes even in the absence of their normal activating factors
Construction of REST-VP16 was based on the hypothesis that it would accomplish two objectives: compete with the endogenous REST/NRSF for binding to the RE1/NRSE sequence through the DNA-binding domain and activate attached promoters through the VP16 activation domain, even in the absence of activators that are otherwise required to activate these promoters/enhancers. To examine this hypothesis, we transfected various expression vectors in Daoy cells (Fig. 3). These are medulloblastoma cells and are believed to be arrested in their neuronal differentiation pathways (38). Using immunocytochemical analysis, they have been found not to express the neuronal differentiation marker gene synapsin, which is another REST/NRSF target gene (39) and, therefore, served as a useful system for this purpose. After transfection of Daoy cells, immunocytochemical analysis was carried out to detect the expression of synapsin. Expression of synapsin was not observed in untransfected control cells or in cells transfected with pcDNA3.1 (vector), pREST, pGal4-VP16 or with the dominant negative mutant pREST-DBD (originally called p73; 12). Since REST-DBD contains only the DNA-binding domain of REST/NRSF, it can block the repressive function of endogenous REST/NRSF by competing for binding to its consensus sequence (12) but it cannot activate promoters unless the cells also contain the corresponding activators. Thus, Daoy cells appear not to contain the activators necessary to express the synapsin gene. However, synapsin could be expressed in these cells when they were transfected with pREST-VP16 and to some extent by REST1-VP16 (still carrying one of the two repressor domains; see above). Similarly, expression of synapsin could be stimulated in NT2 cells by REST-VP16 but not REST.DBD (data not shown). These results support the hypothesis that REST-VP16 can activate neuronal differentiation genes even in the absence of activators which are otherwise required to express these genes.
Figure 3.
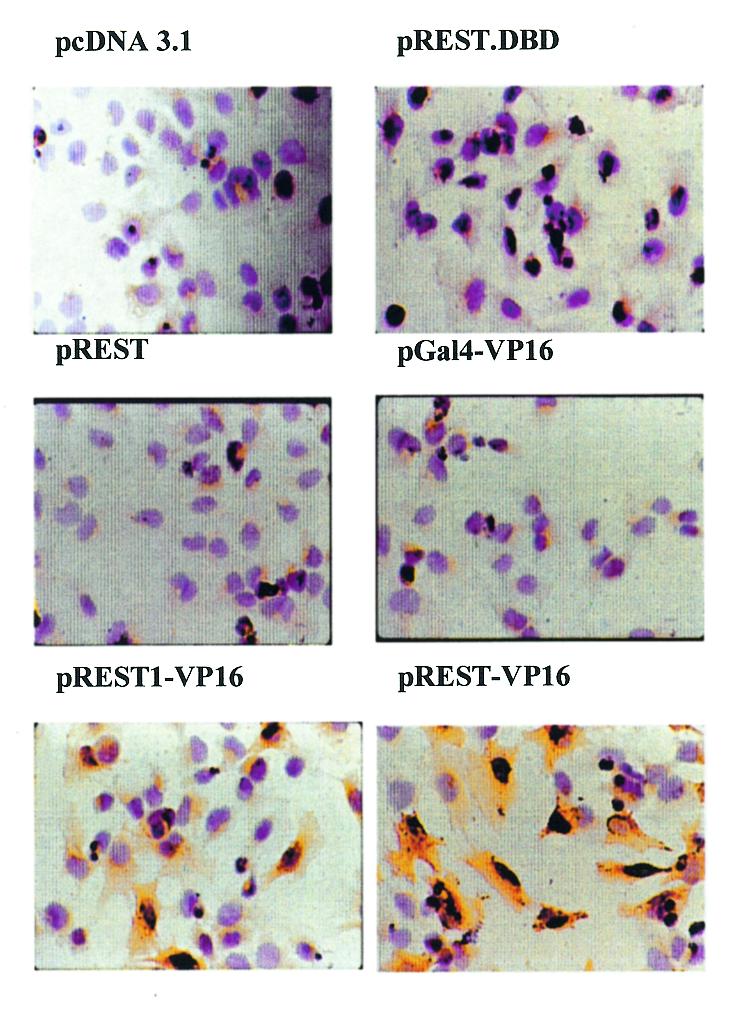
REST-VP16 activates cellular neuronal differentiation genes even in the absence of their normal activating factors. Daoy cells were transfected with pcDNA 3.1, pREST-DBD, pREST, pGal4-VP16, pREST1-VP16 or pREST-VP16, and analyzed by immunocytochemistry using anti-synapsin antibodies. Expression of synapsin was observed only in cells infected with pREST-VP16 and to a lower extent with pREST1-VP16.
NT2 cells express low level of endogenous REST/NRSF activity
As discussed above, NT2 cells resemble committed central nervous system neuronal progenitor cells. In the presence of retinoic acid under culture conditions, NT2 cells can be differentiated into mature neurons and to cause expression of multiple REST/NRSF target genes, indicating that they contain the proper cellular machinery to express such genes, and provide a well-characterized system to study such processes. To determine if these cells express endogenous REST/NRSF-dependent repression, they were transfected with pNaCh or pNaChΔRE and the promoter activity was determined as described above. HeLa cells, which express very high endogenous REST activity, and PC12 cells, which express no or very low level (12,14,15), were used as positive and negative control cells, respectively. Results shown in Figure 4A and B indicate that under these conditions, the REST/NRSF-dependent repression of the NaCh promoter was 1 (no repression) for PC12 cells and 17 for HeLa cells. In contrast, the relative repression was 2.2 for NT2 cells. Thus, NT2 cells express endogenous REST/NRSF-dependent repression at a much lower level than HeLa cells. To determine if this repression could be countered and the NaCh promoter stimulated by REST-VP16, pNaCh was transfected in NT2 cells by itself or in the presence of pcDNA3.1 (Vector), pGFP, pREST, pREST.DBD, pGal4-VP16 or pREST-VP16 and analyzed as before. As shown in Figure 4C, co-expression of the vector DNA or pGal4-VP16 had little effect on the level of NaCh promoter activity in NT2 cells whereas expression of REST lowered the activity to 0.2 indicating that the endogenoous level of REST-dependent repression was not at the saturating amount. Expression of REST.DBD could stimulate the promoter ∼3-fold, bringing the promoter activity to a level that is similar to that of pNaChΔRE. This indicated that REST.DBD could compete with the endogenous REST-dependent repression, but could not stimulate the promoter further. In contrast, REST-VP16 stimulated the NaCh promoter 27-fold indicating that it can not only successfully compete with the endogenous REST activity but can also stimulate the promoter additionally through its VP16 activation domain.
Figure 4.

NT2 cells show low level of endogenous REST/NRSF-dependent promoter repression (A and B). NT2, PC12 and HeLa cells were transfected with pNaCh or pNaChΔRE and analyzed as described in Materials and Methods. The data are represented as percent promoter activity (A; pNaChΔRE = 100%) and REST-dependent repression (B; pNaChΔRE/pNaCh; no repression is denoted as a relative value of 1). REST-VP16 can counter REST/NRSF-dependent repression and stimulate neuronal promoters (C). NT2 cells were transfected with pNaCh in the presence of pcDNA3.1, pGFP, pREST, pREST.DBD, pGal4-VP16 and pREST-VP16 and the NaCh promoter activity was determined as described in Materials and Methods.
REST-VP16 activates multiple neuronal differentiation marker genes in NT2 cells
Expression of REST-VP16 in NT2 cells was found to be ∼20% (data not shown). To increase the efficiency of REST-VP16 expression in these cells, we constructed adenoviral vectors encoding the CMV promoter alone (Ad), or the CMV promoter expressing green fluorescent protein (Ad.GFP), REST (Ad.REST), Gal4-VP16 (Ad.Gal4-VP16) and REST-VP16 (Ad.REST-VP16). These constructs were verified by PCR analysis using primers specific for junction sequences. Expression of functional REST and REST-VP16 proteins from these viruses was also verified by the reporter gene assay using plasmids pNaCh and pNaChΔRE1 as described in Figure 1 (data not shown).
Using an RT–PCR assay, previously we found that NT2 cells express the neuronal stem cell marker nestin but not the differentiation marker glutamate receptor (GluR) (29). When these cells were infected with Ad, Ad.GFP and Ad.REST-VP16 at an m.o.i. of 25, incubated for 48 h and assayed for the expression of REST/NRSF target genes at the RNA level by RT–PCR assay (Fig. 5A), they were found to express GluR in response to Ad.REST-VP16. GluR was not expressed in uninfected cells or cells infected with Ad or Ad.GFP. Expression of GAPDH was used as internal control gene in these experiments. Similar expression of GluR in NT2 cells was also observed when these cells were differentiated in the presence of retinoic acid (26,29). Furthermore, expression of other neuronal differentiation genes such as acetylcholine receptor β2 (AchRB2), SCG10, sodium channel type II also appeared to be upregulated specifically in response to REST-VP16. Interestingly, NeuroD3, the expression of which was observed in immature neurons but not in mature neurons (40), was also downregulated in response to REST-VP16. The role of REST-VP16 in this process is not clear at present.
Figure 5.
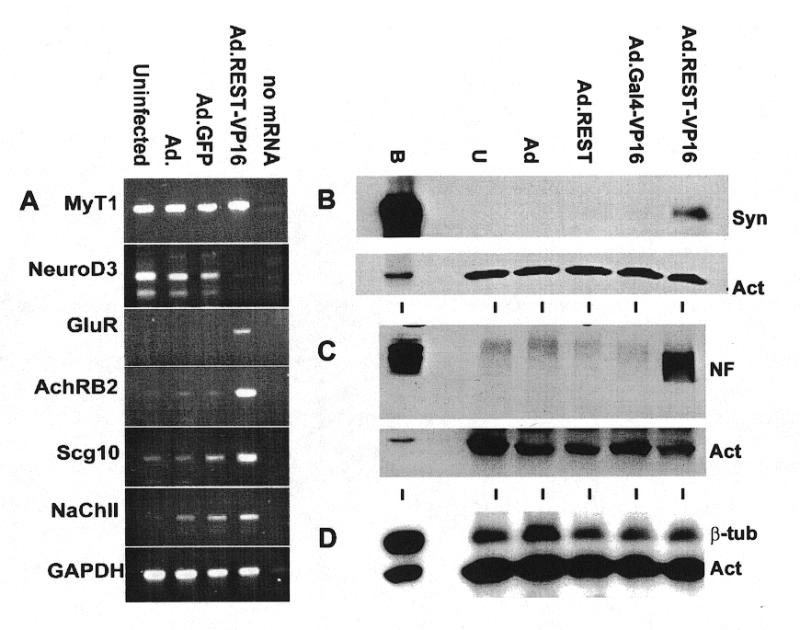
Ad.REST-VP16 activates neuronal differentiation marker genes in NT2 cells. Uninfected NT2 cells and NT2 cells infected with Ad, Ad.GFP or Ad.REST-VP16 were assayed for the expression of neuronal markers MyT1, NeuroD3, GluR, AchRB2, Scg10 and NaCh genes by RT–PCR (A). GAPDH was used as internal control gene. A lane with no mRNA was also used as a negative control. Uninfected NT2 cells and NT2 cells infected with Ad, Ad.GFP, Ad.REST or Ad.REST-VP16 were also analyzed by immunoblotting assays using anti-synapsin (B), anti-neurofilament 200 (C) and anti-class III β-tubulin antibodies (D). Anti-actin antibody was used as an internal control in these assays.
To determine if the efficient expression of REST-VP16 via adenoviral vectors in NT2 cells caused activation of REST/NRSF-regulated neuronal differentiation marker genes at the protein level, cells were infected with Ad, Ad.REST, Ad.Gal4-VP16 or Ad.REST-VP16 and analyzed by western blotting using anti-synapsin, anti-neurofilament 200 and anti-neuronal β-tubulin antibodies. Uninfected cells were used as a negative control and human brain extract was used as a positive control. Expression of the housekeeping gene actin as determined by anti-actin was used as an internal control in these experiments. Results shown in Figure 5B indicated that uninfected NT2 cells as well as cells infected with various control viruses (devoid of REST-VP16) showed no expression of synapsin and this level of expression was strongly stimulated by the adenoviral expression of Ad.REST-VP16. Likewise, uninfected cells and cells infected with control viruses did not express neurofilament 200, and the expression of this neuronal marker gene was again activated in cells infected with Ad.REST-VP16 (Fig. 5C). Uninfected NT2 cells, which are committed neuronal progenitor cells, already expressed neuronal β-tubulin and this level of expression remained unaltered by any of these viruses (Fig. 5D). One of the possibilities why REST-VP16 could not stimulate the level of β-tubulin expression any further in these cells is that this gene is probably expressed to its full capacity in untransfected cells and REST-VP16 can not cause additional stimulation. This view is supported by the observation that Ad.REST-VP16 could stimulate expression of both synapsin and β-tubulin in Daoy cells (data not shown). Taken together, these results indicate that the efficient expression of REST-VP16 through adenoviral vectors stimulate the expression of REST/NRSF-regulated neuronal differentiation genes that are otherwise not expressed in uninfected NT2 cells.
DISCUSSION
Differentiation of stem cells into mature neurons in the mammalian system is believed to take place by complex interactions among a large number of cell-intrinsic and cell-extrinsic factors (1–5,9–11,41). In a simplistic view, inhibition of factors such as the bone morphogenic protein (BMP) by BMP antagonists would initiate neuronal induction, the beginning of the nervous system. The resulting cells would then undergo cell-type specification to eventually form neurons or glial cells because of the selection of one fate over the other. The neuronal pathway can be thought to take place in four steps corresponding to the expression and activity of specific gene products. Thus, these gene products can also be utilized as stage-specific markers. In this scheme, the stem cells, which can multiply and make their own kind under one set of conditions and can differentiate into a different form under other conditions, express p75 and nestin. The neuronal determination step is characterized by the action of the basic helix–loop–helix (bHLH) proteins: MASH, MATH, neuroD3/neurogenin, etc. This is followed by the commitment step where genes such as NeuroD1/2, Myt1 or neurofilament 150 are expressed. Experiments with the frog system have shown that whereas the role of bHLH proteins of the determination step can be inhibited by neighboring cells (lateral inhibition) through the interaction of X-Notch and X-Delta gene products, those of the commitment step such as NeuroD are not under such inhibitory regulation. This suggests that there is a progressive restriction of cell fate as they move along the differentiation pathway. The final differentiation step is characterized by the expression of genes such as SCG10, sodium channel type II, synapsin, GluR and acetylcholine receptor. It is mostly this group of terminal differentiation genes that are direct targets of REST/NRSF-mediated transcriptional repression. REST-VP16 can directly stimulate transcription of these neuronal differentiation genes, bypassing the requirement of upstream signals even in cells that do not contain their normal promoter/enhancer-specific activators. Thus, REST-VP16 is a powerful molecule and may be utilized to examine the effect of activating expression of neuronal differentiation genes on various aspects of neurogenesis, for example, the interrelationship among various regulators, cell cycle exit and generic versus subtype-specific differentiation. In addition, REST/NRSF may also be helpful as a unique tool in studying tumors that arise as a result of the deregulation of normal neurogenesis pathways, such as medulloblastoma.
Although REST/NRSF is a critical repressor of multiple neuronal differentiation genes, as described above, its absence in REST/NRSF–/– mice causes expression only of the neuronal βIII tubulin gene in non-neuronal cells and embryonic lethality. Interestingly, ectopic expression of a dominant negative form, which can compete with REST/NRSF for binding to the RE1/NRSE (similar to REST.DBD) and, thus, block its repression in chicken embryos, can cause expression or upregulation of genes encoding βIII tubulin, Ng-CAM and SCG10 in both non-neuronal cells and neuronal progenitor cells (25). Several possibilities exist for this apparent difference in the pattern of gene expression including requirement of cell type-specific factors and chromatin structure (25).
Experiments described here with NT2 cells, which resemble human committed neuronal progenitor cells and which also express very low level of endogenous REST/NRSF activity, reveal that they also express βIII tubulin gene but not the other differentiation genes like synapsin, neurofilament 200 or GluR (Fig. 5). Expression of the latter group of genes in NT2 cells required the presence of REST-VP16-mediated activation. Similarly, countering REST/NRSF-dependent repression alone by expressing REST.DBD in Daoy human medulloblastoma cells, which do express endogenous REST/NRSF activity (data not shown), did not cause expression of synapsin. Expression of synapsin in these cells required the additional presence of VP16-mediated promoter/enhancer activation (Fig. 3). Thus, in mammals, there appears to be two classes of neuronal differentiation genes. One is represented by βIII tubulin, whose expression depends mostly on the release of REST/NRSF-dependent repression. Apparently, various non-neuronal as well as neuronal cell types contain all the other promoter/enhancer-specific activators that are required for their expression. The other is represented by synapsin, GluR, neurofilament 200 etc., whose expression not only requires the absence of REST/NRSF-dependent repression, but also the presence of promoter/enhancer-specific activators. Experiments shown here in NT2 and Daoy cells indicate that undifferentiated mammalian neuronal cells contain neither such activators nor any other major repression mechanisms than the REST/NRSF activity. Thus, in mammalian neuronal progenitor cells, in the absence of REST/NRSF-dependent repression, it is the regulated and temporal expression of the activators that appears to control expression of different neuronal differentiation genes and their eventual differentiation into mature neurons.
Although expression of REST-VP16 caused expression of multiple neuronal differentiation genes in NT2 cells, it did not cause neuronal differentiation under these conditions. There are several possible explanations. For example, neuronal differentiation may require expression of genes other than REST/NRSF-target genes. Another possibility is that the period of expression of REST-VP16 was not sufficient for these cells to acquire neurite outgrowth, which could be a rate-limiting factor. A third possibility is that REST-VP16 activates multiple neuronal genes at once, and thereby interferes with the ordered expression of these genes. Stable cell lines expressing inducible levels of REST-VP16 may circumvent the latter two problems. A fourth possibility is that although REST-VP16 activates multiple neuronal differentiation genes, it cannot block the expression of stem cell marker genes. We are currently making transgenic mice expressing REST-VP16 through various promoter/enhancer constructs to answer such questions.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Gail Mandel for her generous gift of pREST-Express, p73, pBS.REST and pSDK7. This work was supported in part by grants to S.M. from the Pediatric Brain Tumor Foundation of the US, Association for Research of Childhood Cancer and the National Institutes of Health (GM53454 and CA81255). L.R. was supported by a Translational Research Award from the American Brain Tumor Association.
REFERENCES
- 1.McKay R. (1997) Science, 276, 66–71. [DOI] [PubMed] [Google Scholar]
- 2.Johansson C.B., Momma,S., Clarke,D.L., Risling,M., Lendahl,U. and Frisen,J. (1999) Cell, 96, 25–34. [DOI] [PubMed] [Google Scholar]
- 3.Bjornson C.R., Rietze,R.L., Reynolds,B.A., Magli,M.C. and Vescovi,A.L. (1999) Science, 283, 534–537. [DOI] [PubMed] [Google Scholar]
- 4.Edlund T. and Jessell,T.M. (1999) Cell, 96, 211–224. [DOI] [PubMed] [Google Scholar]
- 5.Morrison S.J., White,P.M., Zock,C. and Anderson,D.J. (1999) Cell, 96, 737–749. [DOI] [PubMed] [Google Scholar]
- 6.Mandel G. and McKinnon,D. (1993) Annu. Rev. Neurosci., 16, 323–345. [DOI] [PubMed] [Google Scholar]
- 7.St-Jacques B. and McMahon,A.P. (1996) Curr. Opin. Genet. Dev., 6, 439–444. [DOI] [PubMed] [Google Scholar]
- 8.Rowitch D.H., Danielian,P.S., Lee,S.M., Echelard,Y. and McMahon,A.P. (1997) Cold Spring Harb. Symp. Quant. Biol., 62, 535–544. [PubMed] [Google Scholar]
- 9.Brunet L.J., McMahon,J.A., McMahon,A.P. and Harland,R.M. (1998) Science, 280, 1455–1457. [DOI] [PubMed] [Google Scholar]
- 10.Mariani F.V. and Harland,R.M. (1998) Development, 125, 5019–5031. [DOI] [PubMed] [Google Scholar]
- 11.McMahon J.A., Takada,S., Zimmerman,L.B., Fan,C.M., Harland,R.M. and McMahon,A.P. (1998) Genes Dev., 12, 1438–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chong J.A., Tapia-Ramirez,J., Kim,S., Toledo-Aral,J.J., Zheng,Y., Boutros,M.C., Altshuller,Y.M., Frohman,M.A., Kraner,S.D. and Mandel,G. (1995) Cell, 80, 949–957. [DOI] [PubMed] [Google Scholar]
- 13.Schoenherr C.J. and Anderson,D.J. (1995) Science, 267, 1360–1363. [DOI] [PubMed] [Google Scholar]
- 14.Maue R.A., Kraner,S.D., Goodman,R.H. and Mandel,G. (1990) Neuron, 4, 223–231. [DOI] [PubMed] [Google Scholar]
- 15.Kraner S.D., Chong,J.A., Tsay,H.-J. and Mandel,G. (1992) Neuron, 9, 37–44. [DOI] [PubMed] [Google Scholar]
- 16.Tapia-Ramirez J., Eggen,B.J., Peral-Rubio,M.J., Toledo-Aral,J.J. and Mandel,G. (1997) Proc. Natl Acad. Sci. USA, 94, 1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schoenherr C.J., Paquette,A.J. and Anderson,D.J. (1996) Proc. Natl Acad. Sci. USA, 93, 9881–9886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koenigsberger C., Chicca,J.J., Amourex,M.C., Edelman,G.M. and Jones,F.S. (2000) Proc. Natl Acad. Sci. USA, 97, 2291–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Palm K., Metsis,M. and Timmusk,T. (1999) Brain Res. Mol. Brain Res., 72, 30–39. [DOI] [PubMed] [Google Scholar]
- 20.Andres M.E., Burger,C., Peral-Rubio,M.J., Battaglioli,E., Anderson,M.E., Grimes,J., Dallman,J., Ballas,N. and Mandel,G. (1999) Proc. Natl Acad. Sci. USA, 96, 9873–9878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roopra A., Sharling,L., Wood,I.C., Briggs,T., Bachfischer,U., Paquette,A.J. and Buckley,N.J. (2000) Mol. Cell. Biol., 20, 2147–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naruse Y., Aoki,T., Kojima,T. and Mori,N. (1999) Proc. Natl Acad. Sci. USA, 96, 13691–13696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang Y., Myers,S.J. and Dingledine,R. (1999) Nature Neurosci., 2, 867–872. [DOI] [PubMed] [Google Scholar]
- 24.Grimes J.A., Nielsen,S.J., Battaglioli,E., Miska,E.A., Speh,J.C., Berry,D.L., Atouf,F., Holdener,B.C., Mandel,G. and Kouzarides,T. (2000) J. Biol. Chem., 31, 9461–9467. [DOI] [PubMed] [Google Scholar]
- 25.Chen Z.F., Paquette,A.J. and Anderson,D.J. (1998) Nature Genet., 20, 136–142. [DOI] [PubMed] [Google Scholar]
- 26.Younkin D.P., Tang,C.M., Hardy,M., Reddy,U.R., Shi,Q.Y., Pleasure,S.J., Lee,V.M. and Pleasure,D. (1993). Proc. Natl Acad. Sci. USA, 90, 2174–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Majumder S., Miranda,M. and DePamphilis,M.L. (1993) EMBO J., 12, 1131–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smale S.T. and Baltimore,D. (1989) Cell, 57, 103–113. [DOI] [PubMed] [Google Scholar]
- 29.Lawinger P., Rastelli,L., Zhao,Z. and Majumder,S. (1999) J. Biol. Chem., 274, 8002–8011. [DOI] [PubMed] [Google Scholar]
- 30.Majumder S. and DePamphilis,M.L. (1994) Mol. Cell. Biol., 14, 4258–4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tohiyama T., Lee,V.M., Rorke,L.B., Marvin,M., McKay,R.D. and Trojanowski,J.Q. (1992) Lab. Invest., 66, 303–313. [PubMed] [Google Scholar]
- 32.Fuller G.N. (1996) In Parham,D.M. (ed.), Pediatric Neoplasia: Morphology and Biology. Lippincott-Raven Publishers, Philadelphia, pp. 153–205.
- 33.Guo Z.S., Wang,L.H., Eisensmith,R.C. and Woo,S.L. (1996) Gene Ther., 3, 802–810. [PubMed] [Google Scholar]
- 34.Guo Z.S. and DePamphilis,M.L. (1992) Mol. Cell. Biol., 12, 2514–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bett A.J., Haddara,W., Prevec,L. and Graham,F.L. (1994) Proc. Natl Acad. Sci. USA, 91, 8802–8806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Majumder S., Zhao,Z., Kaneko,K. and DePamphilis,M.L. (1997) EMBO J., 16, 1721–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Majumder S. (1997) In Cid-Arregui,A. and Garcia-Carranca,A (eds), Microinjection and Transgenesis: Strategies and Protocols. Springer-Verlag, Heidelberg, pp. 323–349.
- 38.He X.M., Skapek,S.X., Wikstrand,C.J., Friedman,H.S., Trojanowski,J.Q. and Bigner,D.D. (1989) J. Neuropathol. Exp. Neurol., 48, 48–68. [DOI] [PubMed] [Google Scholar]
- 39.Trojanowski J.Q., Tohyama,T. and Lee,V.M. (1992) Mol. Chem. Neuropathol., 17, 121–135. [DOI] [PubMed] [Google Scholar]
- 40.Katayama M., Mizuta,I., Sakoyama,Y., Kohyama-Koganeya,A., Akagawa,K., Uyemura,K. and Ishii,K. (1997) Exp. Cell Res., 236, 412–417. [DOI] [PubMed] [Google Scholar]
- 41.Sasaki Y. (1998) Neuron, 21, 455–458. [DOI] [PubMed] [Google Scholar]