

Abstract
Osteogenesis imperfecta (OI) commonly involving defects in COL1A1 and COL1A2 is a rare hereditary disease of bone fragility affecting 6–7 per 100,000 population. On the other hand, hypopituitarism is a separate entity that manifests with reduced levels of pituitary hormones. The cooccurrence of these two is seldom reported previously in literature as a deviation from Occam's razor. Here, we reported a case of pathological fracture in a 31-year-old male who had blue sclera and secondary adrenal insufficiency, hypogonadotropic hypogonadism, and growth hormone deficiency along with primary autoimmune hypothyroidism. Diagnosis of OI was suggested by heterozygous missense variant in exon 40 of the COL1A2 gene (chr7: g.94423092G > A; Depth: 99×) that resulted in the amino acid substitution of Serine for Glycine at codon 847. Replacement of glucocorticoid, levothyroxine, and testosterone was started along with antiresorptive therapy for better bone health outcomes.
Keywords: Pathological fracture, Hypopituitarism, Osteogenesis imperfecta
Graphical abstract

Highlights
-
•
Hypopituitarism and Osteogenesis imperfecta (OI) can rarely coexist.
-
•
OI may lead to a difficult birth process resulting in anatomical defects in the Sella.
-
•
The majority of documented cases are of Asian origin, particularly in the males.
1. Introduction
Osteogenesis imperfecta (OI) is a rare hereditary disease of connective tissue mostly due to mutation of COL1A1 and COL1A2 genes presenting with pathological fracture and skeletal deformities like flat midface and triangular facies, scoliosis or kyphosis, and chest wall deformities including pectus excavatum, carinatum, and barrel chest and also may lead to secondary damages, such as deafness, dentinogenesis imperfecta, macrocephaly, hydrocephalus, syringomyelia, and basilar invagination, and may have short stature sometimes with a normal growth velocity (Forlino et al., 2011; Marini et al., 1993).
Combined pituitary hormone deficiency is a condition either acquired or due to transcription factor defects, in which the pituitary gland produces insufficient hormones, including adrenocorticotropic hormone (ACTH), follicle-stimulating hormone (FSH), luteinizing hormone (LH), growth hormone (GH), prolactin production (PRL), and/or thyroid stimulating hormone (TSH) (Fang et al., 2016).
This report documented a rare coexistence of two different entities in an adult male presenting with pathological fracture.
2. Case presentation
A 31-year-old male patient presented with a history of fracture of the right femur shaft 1.5 years ago after a fall while running [Fig. 1] which had been managed by open reduction and internal fixation. The implanted nail broke off following a fall while cycling at the usual speed 4 months ago and had to be fixed operatively. On further query, there were histories of poor height gain and poor development of sexual characteristics noticed from 15 years of age. He denies any symptoms of polydipsia or polyuria. The patient did not seek any medical attention for those symptoms earlier. He was born by assisted breech delivery without any perinatal complications. He achieved developmental milestones as per his peer group. There was no other relevant history that could be contributory and no familial history of pathological fracture.
Fig. 1.

Skiagram of the right femur showing shaft fracture.
On examination, mild pallor was present. His weight was 50 kg at the 10th–25th percentile and the height was 148.5 cm, less than the 3rd percentile as per the Indian height chart at 18 years of age. The arm span was 160 cm and the upper and lower segment ratio was 0.94. Axillary and pubic hair was absent [Fig. 2A]. His voice was high-pitched. The testes were palpable at the scrotum with a volume of 3 mL for each, the stretched penile length was 8 cm, below −2.5 standard deviation (SD) for his age [Fig. 2B]. His sexual maturity rating was prepubertal. He had barrel-shaped chest and also blue sclera in both eyes [Fig. 2C]. His dental and auditory examination findings were normal. Apart from these, no other remarkable clinical findings were there.
Fig. 2.

A – Image of the patient showing no secondary hair growth, B – Genitalia of the patient at prepubertal stage, C – Suggestive of blue sclera.
The routine baseline investigations showed hemoglobin of 8.3 g% with mean corpuscular volume (MCV) of 76, white blood count of 6700 per millimeter3 with 49 % neutrophils, 42 % lymphocytes, 3 % eosinophils, 5 % monocytes, and no basophils, platelet count was 3.9 lacs. Serum sodium was 133 mEq/L, and potassium was 5.6 mEq/L. Serum alkaline phosphatase (ALP) level was 279 IU/L, serum calcium was 9.2 mg/dL, serum phosphate was 3.4 mg/dL, and serum 25-hydroxy Vitamin D levels was 34.7 ng/mL. His hormonal profile has been summarized in Table 1. His bone age was between 12 and 14 years [Fig. 3]. In the Dual X-ray Absorptiometry (DXA) scan, the Z-score was (−2.06) and bone mineral density (BMD) was 0.687 g/cm2 for the lumber, also the Z-score was (−4.07) and BMD was 0.506 g/cm2 for the left femoral neck.
Table 1.
Hormonal evaluation of the patient. ACTH – Adrenocorticotropic hormone, FSH – Follicular stimulating hormone, IGF1 – Insulin-like growth factor 1, LH – Luteinizing hormone, TPO – Thyroid peroxidase, TSH – Thyroid stimulating hormone, T4 – Thyroxine,
| Investigations (units) | Patient's value | Reference range |
|---|---|---|
| Morning serum cortisol (μg/dL) | 2.2 | 5–25 |
| Plasma ACTH (pg/mL) | 16.7 | <120 |
| LH (mIU/mL) | 0.16 | 0.8–7.6 |
| FSH (mIU/mL) | 1.08 | 0.7–11.1 |
| Testosterone (ng/dL) | 25 | 227–1030 |
| Prolactin (ng/mL) | 19.87 | 1.5–14.7 |
| Free T4 (ng/dL) | 0.82 | 0.89–1.8 |
| TSH (μIU/mL) | 24.21 | 0.35–4.6 |
| IGF1 (ng/mL) | 36.9 | 37–275 (age specific) |
| Anti-TPO antibody (IU/mL) | 271.0 | <34 |
Fig. 3.

Skiagram of the left hand indicating delayed bone age in comparison with the patient's chronological age.
Pituitary MRI [Fig. 4] was suggestive of a partial empty sella with a very thin and flattened pituitary gland in the floor of the sella. The rest was filled with CSF signal intensity, the posterior pituitary bright spot was not well visualized, and the pituitary stalk was normal.
Fig. 4.
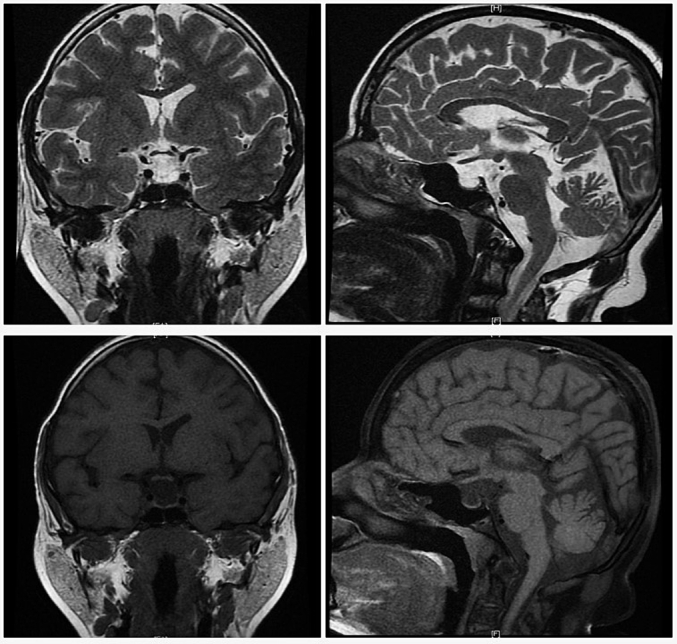
T2 (upper panel) and T1 (lower panel) weighted magnetic resonance imaging reflecting partial empty sella and absence of pituitary bright spot.
The whole exome sequencing was suggestive of a heterozygous missense variant in exon 40 of the COL1A2 gene (chr7: g.94423092G > A; Depth: 99×) that resulted in the amino acid substitution of Serine for Glycine at codon 847.
3. Discussion
The individual who was referred to the Endocrinology department for evaluation of pathological fracture was also found to have central hypogonadism as suggested by eunuchoid body habitus, and prepubertal sexual maturation according to Tanner staging which was confirmed by LH and testosterone reports at the age of 31 years. This patient did not encounter any fracture earlier than the described one as per clinical details found, such late-onset fracture has also been described in a milder variety of OI and with other types of mutations (Stathopoulos et al., 2014; Muñoz-Miro et al., 2022; Selina et al., 2023).
Along with that, he had secondary adrenal insufficiency and growth hormone deficiency evidenced by low IGF1 level. The bone age was significantly delayed as per his chronological age. Besides this, he had primary hypothyroidism of autoimmune etiology. Thus, with available hormonal profiles and pituitary imaging, poor bone health due to hypopituitarism was considered as the possible etiology of pathological fracture. Hence, he was started on glucocorticoid, levothyroxine replacement, and testosterone intramuscular injections (50 mg 4 weekly) sequentially.
Our patient had blue sclera which is rarely found in PROP-1 mutation-related hypopituitarism and commonly in osteogenesis imperfecta; a whole exome sequencing was therefore sent which pointed to the latter (Rosenbloom et al., 1999).
Osteogenesis imperfecta is a heterogeneous group of inherited connective tissue disorders mainly (90 %) caused by structural or quantitative mutations in the collagen genes COL1A1 and COL1A2 (Marini and Blissett, 2013). Apart from these two, alterations in >13 genes have been detected in association with OI have been summarized in Table 2 (Wang et al., 2019; Unger et al., 2023).
Table 2.
Genetic associations of osteogenesis imperfecta other than collagen type I chain. BMP1 – Bone morphogenetic protein 1, CCD134 – Coiled-Coil Domain Containing 134, CREB3L1 – cAMP responsive element binding protein 3 like 1, CRTAP – Cartilage-associated protein, FKBP10 – FKBP prolyl isomerase 10, IFITM5 – Interferon-induced transmembrane protein 5, KDELP2 – KDEL endoplasmic reticulum protein retention receptor 2, LEPRE1/P3H1 – Leprecan/Prolyl 3-hydroxylase 1, MBTPS2 – Membrane-bound transcription factor peptidase, site 2, MESD – Mesoderm development LRP chaperone, PLOD2 – Procollagen-lysine,2-oxoglutarate 5-dioxygenase 2, PPIB – Peptidylprolyl isomerase B, SERPINF1 – Serpin family F member 1, SERPINH1 – Serpin family H member 1, SP7 – Sp7 transcription factor, SPARC – Secreted protein acidic and cysteine-rich, TENT5 A – Terminal nucleotidyl transferase 5 A, TMEM38B – Transmembrane protein 38B, WNT1 – Wnt Family Member 1.
| Processes | Genes |
|---|---|
| Collagen processing | BMP1, CCD134, KDELP2 |
| Collagen modification | CRTAP, LEPRE1/P3H1, PPIB, TMEM38B, KDELP2 |
| Collagen folding and cross-linking | SERPINH1, FKBP10, PLOD2 |
| Bone mineralization | IFITM5, SERPINF1, TENT5A |
| Osteoblast development with collagen insufficiency | SP7, WNT1, CREB3L1, SPARC, MESD |
| Regulated Intramembrane Proteolysis | MBTPS2 |
On the other hand, hypopituitarism may be acquired or due to transcription factor defects like POU1F1, PROP1, HESX1, OTX2, LHX3, LHX4, and GLI2 (Fang et al., 2016). In our case, no transcription defect was found in the genetic study.
Hypopituitarism can occur in association with breech presentation also which is highly prevalent in patients with OI (24–37 %) probably due to large head size and decreased intrauterine movement of the fetus (Cubert et al., 2001). As found in the literature, breech presentation at birth has been associated with pituitary stalk interruption syndrome (Wang et al., 2019), hypoplastic pituitary and its stalk (though there was an associated pituitary transcription factor defect) (Hemwong et al., 2020) [Table 3], where Kitamura T et al. described pituitary stalk interruption syndrome in their case after vacuum extraction during delivery (Kitamura et al., 2023). Similarly, our case had history of assisted breech delivery, and partial empty sella was found in MRI. Thus, it can be hypothesized that assisted and difficult labor processes during the birth of fetuses carrying a genetic mutation of OI may result in hypopituitarism in the future. Familial inheritance is an important feature of OI, though there was no family history of pathological fracture or other characteristic features of OI in the family concerning the present case. Previously reported cases were associated with the history of fractures in parents and siblings without any evidence of pituitary hormone deficit.
Table 3.
Summary of all reported cases of hypopituitarism associated with osteogenesis imperfecta. ACTH – Adrenocorticotropic hormone, FSH – Follicular stimulating hormone, GH – Growth hormone, LH – Luteinizing hormone, MRI – Magnetic resonance imaging, OI – Osteogenesis imperfecta, TSH – Thyroid stimulating hormone.
| Cases and countries | Age of presentation and gender | Pituitary hormone deficit | OI features | Sellar MRI findings | Genetics of hypopituitarism | Genetics of OI |
|---|---|---|---|---|---|---|
|
Plachel et al., 2015 Austria |
33 years, Male | FSH, LH, GH | Low-velocity fractures of spine and long bones since childhood, hearing impairment, periodontal disease | Pituitary hypoplasia | Not mentioned | Not mentioned |
|
Steele et al., 2015 United Kingdom |
15 years, Male | TSH, FSH, LH, GH | Blue sclera, dentinogenesis imperfecta, spinal fracture | Not mentioned | Not mentioned | Not mentioned |
|
Arliena and Zaini, 2018 Malaysia |
2.2 years, Male | ACTH, TSH, GH | Blue sclera, diagnosed as having OI at 3 months of age | Hypoplastic pituitary and stalk | Not mentioned | Not mentioned |
|
de Carlos et al., 2019 Brazil |
7 years, Male | ACTH, TSH, FSH, LH, GH | Blue sclera, left femur fracture at 4 days of age | Empty sella | Not mentioned | Not mentioned |
|
Wang et al., 2019 China |
19 years, Male | ACTH, TSH, FSH, LH, GH | Blue sclera, left ulna fracture at 2 years of age | Pituitary stalk interruption syndrome | Not found | COL1A1, COL1A2 |
| Hemwong et al., 2020Thailand | 6 years, Male | TSH, GH | Blue sclera, dentinogenesis imperfecta, deformed limbs at birth, pectus carinatum, and flat feet at 3 years of age | Hypoplastic pituitary and stalk | LHX4 | COL1A2 |
|
Kitamura et al., 2023 Japan |
46 years, Male | ACTH, FSH, LH, GH | Blue sclera, peripheral and vertebral fracture during childhood and adolescent | Pituitary stalk interruption syndrome | Not found | COL1A1 |
| Present case India |
31 years, Male | ACTH, FSH, LH, GH | Blue sclera, right femur fracture at 30 years of age | Partially empty sella, absent bright spot, and normal stalk | Not found | COL1A2 |
Androgen deficiency hampers the process of bone mineralization, while GH deficiency also deteriorates bone density (Finkelstein et al., 1987; Holmes et al., 1994). Thus, appropriate replacement of deficient hormones is necessary for bone health in hypopituitarism. Though the patient was aware of his delayed growth and pubertal development, his reluctance to seek medical attention earlier due to lack of knowledge and shamefulness of disclosing symptoms of hypogonadism became a double trouble for his bone health on the background of OI and resulted in a serious fracture. Along with glucocorticoid (at physiological dose), levothyroxine, and testosterone replacement, the patient was started on annual zoledronic acid infusion to reduce his risk of fracture (Land et al., 2007).
Another thing to note is that the patient had significantly lower BMD in the femoral neck (0.506 g/cm2) than the spine (0.687 g/cm2). In the case of hypogonadism, the spine is more severely affected due to more trabecular bone mass (Brandi, 2009), that altered bone loss could be due to the presence of OI (associated with COL1A2 mutation) along with hypogonadism where long bones are affected more (Kocijan et al., 2013).
The major limitations of our report were that correlation with primary autoimmune hypothyroidism was not found, genetic screening of OI in the family could not be done, and GH replacement in adult age was not possible due to our limited resources in the hospital setting and the patient's low economic background. Interestingly, the reason behind the male preponderance of all the documented cases (including the present one) is still unrevealed.
This is the tenth reported case globally and the first one from India with such rare coexistence. Very little data are available for the first two cases of OI associated with hypopituitarism, one had vertebral fractures and deformity and another had right femur fractures (Scott et al., 1971). Though there are hypotheses regarding the causation of hypopituitarism in OI, no definite explanation has been developed. Moreover, apart from one case with a pituitary transcription factor defect and the collagen mutation, others did not have any. Except for the three cases found in the online database, all were of Asian origin and surprisingly all reported cases were male [Table 3], thus whether demography and gender play a role in the association should be sought. More research is necessary to determine the exact mechanism of such association with variable hormone deficiency and to address the gender predisposition. Missed-out diagnosis, and thus delay in proper management may invite hazards.
Funding
Nil.
CRediT authorship contribution statement
Rajdeep Basu: Writing – review & editing, Writing – original draft. Soumik Goswami: Writing – review & editing, Conceptualization. Nilanjan Sengupta: Supervision, Project administration. Arjun Baidya: Supervision. Sunetra Mondal: Writing – review & editing, Conceptualization. Kumar Swapnil: Methodology, Investigation. Rajat Deb: Methodology, Investigation. Vibhu Ranjan Khare: Methodology, Investigation. Joydip Datta: Methodology, Investigation.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
The written informed consent had been obtained from the patient for the publication of the report.
Data availability
Data will be made available on request.
References
- Arliena A., Zaini A.A. Infant with osteogenesis imperfecta and panhypopituitarism: a case report [internet] Bioscientifica. 2018;89 https://abstracts.eurospe.org/hrp/0089/hrp0089p3-p284 (abstracts.eurospe.org. [cited 2024 Jan 12]. Available from) [Google Scholar]
- Brandi M.L. Microarchitecture, the key to bone quality. Rheumatology (Oxford) 2009;48(supplement 4):iv3–iv8. doi: 10.1093/rheumatology/kep273. [DOI] [PubMed] [Google Scholar]
- Cubert R., Cheng E.Y., Mack S., Pepin M.G., Byers P.H. Osteogenesis imperfecta: mode of delivery and neonatal outcome. Obstet. Gynecol. 2001;97:66–69. doi: 10.1097/00006250-200101000-00014. [DOI] [PubMed] [Google Scholar]
- de Carlos G., Marques J., Naves L. SUN-440 osteogenesis imperfecta and hypopituitarism: a life-long consequences case report. J Endocr Soc. 2019;3(Suppl. 1) doi: 10.1210/js.2019-SUN-440. (SUN-440. Apr 30, PMCID: PMC6553194) [DOI] [Google Scholar]
- Fang Q., George A.S., Brinkmeier M.L., Mortensen A.H., Gergics P., Cheung L.Y., et al. Genetics of combined pituitary hormone deficiency: roadmap into the genome era. Endocr. Rev. 2016;37(6):636–675. doi: 10.1210/er.2016-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein J.S., Klibanski A., Neer R.M., Greenspan S.L., Rosenthal D.I., et al. Osteoporosis in men with idiopathic hypogonadotropic hypogonadism. Ann. Intern. Med. 1987;106:354–361. doi: 10.7326/0003-4819-106-3-. [DOI] [PubMed] [Google Scholar]
- Forlino A., Cabral W.A., Barnes A.M., Marini J.C. New perspectives on osteogenesis imperfecta. Nat. Rev. Endocrinol. 2011;7:540–557. doi: 10.1038/nrendo.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemwong N., Phokaew C., Srichomthong C., Tongkobpetch S., Srilanchakon K., Supornsilchai V., et al. A patient with combined pituitary hormone deficiency and osteogenesis imperfecta associated with mutation in LHX4 and COL1A2. J. Adv. Res. 2020;21:121–127. doi: 10.1016/j.jare.2019.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes S.J., Economou G., Whitehouse R.W., Adams J.E., Shalet S.M. Reduced bone mineral density in patients with adult onset growth hormone deficiency. J. Clin. Endocrinol. Metab. 1994;78:669–674. doi: 10.1210/jcem.78.3.8126140. [DOI] [PubMed] [Google Scholar]
- Kitamura T., Ishihara Y., Kusakabe Toru, Tsuiki M., Nanba Kazutaka, Hiroshima-Hamanaka Kaho, et al. A case of osteogenesis imperfecta caused by a COL1A1 variant, coexisting with pituitary stalk interruption syndrome. Endocr. J. 2023;70(8):839–846. doi: 10.1507/endocrj.EJ22-0564. (Jan 1) [DOI] [PubMed] [Google Scholar]
- Kocijan R., Muschitz C., Fratzl-Zelman N., Haschka J., Dimai H.P., Trubrich A., et al. Femoral geometric parameters and BMD measurements by DXA in adult patients with different types of osteogenesis imperfecta. Skeletal Radiol. 2013;42(2):187–194. doi: 10.1007/s00256-012-1512-4. (Feb) [DOI] [PubMed] [Google Scholar]
- Land C., Rauch F., Travers R., Glorieux F.H. Osteogenesis imperfecta type VI in childhood and adolescence: effects of cyclical intravenous pamidronate treatment. Bone. 2007;40:638–644. doi: 10.1016/j.bone.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Marini J.C., Blissett A.R. New genes in bone development: what’s new in osteogenesis imperfecta. J. Clin. Endocrinol. Metab. 2013;98:3095–3103. doi: 10.1210/jc.2013-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini J.C., Bordenick S., Heavner G., Rose S., Chrousos G.P. Evaluation of growth hormone axis and responsiveness to growth stimulation of short children with osteogenesis imperfecta. Am. J. Med. Genet. 1993;45:261–264. doi: 10.1002/ajmg.1320450223. [DOI] [PubMed] [Google Scholar]
- Muñoz-Miro H., Lugo E., Carlo S., Ramírez N. COL1A2 (p.Gly322Ser) mutation causes late-onset osteogenesis imperfecta: a case report. Cureus [Internet] 2022;14(10) doi: 10.7759/cureus.30172. (Oct 1 [cited 2023 Sep 21]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plachel F., Renner U., Kocijan R., Muschitz C., Lomoschitz F., Resch H. Osteogenesis imperfecta type III and hypogonadotropic hypogonadism result in severe bone loss: a case report. Wiener medizinische Wochenschrift. 2015;165 doi: 10.1007/s10354-015-0367-4. [DOI] [PubMed] [Google Scholar]
- Rosenbloom A.L., Almonte A.S., Brown M.R., Fisher D.A., Baumbach L., Parks J.S. Clinical and biochemical phenotype of familial anterior hypopituitarism from mutation of the PROP1 gene. J. Clin. Endocrinol. Metab. 1999;84:50–57. doi: 10.1210/jcem.84.1.5366. [DOI] [PubMed] [Google Scholar]
- Scott C.I., Mengel M.C., Lawrence G.D., Schultz K.T., Edgar P.J. Osteogenesis imperfecta and hypopituitarism in two unrelated males. Birth Defects Orig. Artic. Ser. 1971;7:259–262. [PubMed] [Google Scholar]
- Selina A., Kandagaddala M., Kumar V., Cleave S., Danda S., Madhuri V. SERPINF1 gene variants causing late-onset progressive deforming osteogenesis imperfecta – a study of 18 patients from India. Bone Rep. 2023;(18) doi: 10.1016/j.bonr.2023.101690. (101690–0, Jun 1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopoulos K.D., Koromila T., Paschalis E.P., Soultanis K., Atsali E., Bournazos I., et al. Late onset presentation of osteogenesis imperfecta with additional mutation on GNAS gene: case report. Bone Abstr. 2014;3:388. [Google Scholar]
- Steele C., Sultan W., Bishop N., Ehtisham S. Short stature in osteogenesis imperfecta: consider alternate diagnosis. Bone. 2015;4:181. (doi:10.153/boneabs.4.P181) [Google Scholar]
- Unger S., Ferreira C.R., Mortier G.R., Ali H., Bertola D.R., Calder A., Cohn D.H., Cormier-Daire V., Girisha K.M., Hall C., Krakow D., Makitie O., Mundlos S., Nishimura G., Robertson S.P., Savarirayan R., Sillence D., Simon M., Sutton V.R., Warman M.L., Superti-Furga A. Nosology of genetic skeletal disorders: 2023 revision. Am. J. Med. Genet. A. 2023;191(5):1164–1209. doi: 10.1002/ajmg.a.63132. (May) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Zhang M., Guan H., Wang X. Osteogenesis imperfecta due to combined heterozygous mutations in both COL1A1 and COL1A2, coexisting with pituitary stalk interruption syndrome. Front. Endocrinol. 2019;10:193. doi: 10.3389/fendo.2019.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.