

Summary
Pancreatic cancer (PC) is a lethal disease and associated with metabolism dysregulation. Nogo-B is related to multiple metabolic related diseases and types of cancers. However, the role of Nogo-B in PC remains unknown. In vitro, we showed that cell viability and migration was largely reduced in Nogo-B knockout or knockdown cells, while enhanced by Nogo-B overexpression. Consistently, orthotopic tumor and metastasis was reduced in global Nogo knockout mice. Furthermore, we indicated that glucose enhanced cell proliferation was associated to the elevation expression of Nogo-B and nuclear factor κB (NF-κB). While, NF-κB, glucose transporter type 1 (GLUT1) and sterol regulatory element-binding protein 1 (SREBP1) expression was reduced in Nogo-B deficiency cells. In addition, we showed that GLUT1 and SREBP1 was downstream target of NF-κB. Therefore, we demonstrated that Nogo deficiency inhibited PC progression is regulated by the NF-κB/GLUT1 and SREBP1 pathways, and suggested that Nogo-B may be a target for PC therapy.
Subject areas: Molecular biology, Cell biology, Cancer
Graphical abstract
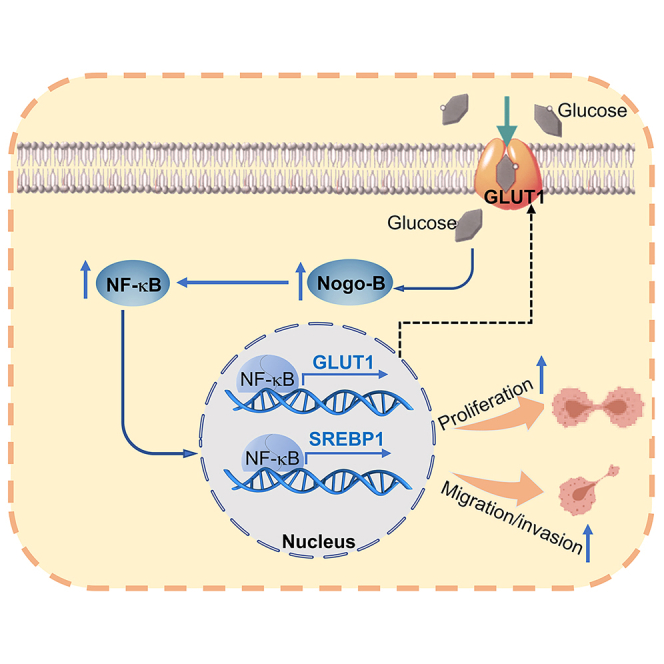
Highlights
-
•
Inhibition of Nogo-B reduces PC progress
-
•
NF-κB expression is involved in Nogo-B-mediated PC development
-
•
GLUT1 and SREBP1 are downstream targets of NF-κB
-
•
Nogo-B deficiency reduced PC via inhibiting NF-κB/GLUT1 and SREBP1 pathways
Molecular biology; Cell biology; Cancer
Introduction
Pancreatic cancer (PC) is a global malignancy, and one of the most lethal diseases. More worryingly, PC is expected to overtake colorectal cancer and trail only lung cancer as the leading cause of cancer-related deaths by 2040.1 Due to the vague symptoms and lack of early detection screening for PC, this disease is usually only discovered in its advanced stages, often after distant metastasis has occurred. As a result, the mortality rate of PC is extremely high. Chemotherapy and surgery are the main strategies for PC treatment. Unfortunately, only 15–20% patients are eligible to undergo the appropriate surgery. Despite complete resection, local or distant recurrence of PC is usually relapse within two years after surgery.2 In addition, the most recommended chemotherapy regimen for PC treatment is gemcitabine plus nab-paclitaxel and FOLFIRINOX, but acquired resistance is common.3,4 Therefore, the search for crucial regulatory molecules or mechanisms involved in PC development holds significant clinical value and offers far-reaching societal benefits.
The progression of PC is complex, and metabolic rewiring plays a central role in this process. High glucose levels can contribute to the development and progression of PC.5 Increased glucose metabolism not only provides energy for tumor growth but also supports other biological processes necessary for cancer cell survival and proliferation. Additionally, elevated glucose levels can stimulate inflammation and induce insulin resistance, both of which are linked to an elevated risk of PC. Previous study indicated that high level of glucose transporter type 1 (GLUT1) in pancreatic cancer is associated with poor prognosis.6,7,8 Mutations in kirsten rat sarcoma viral oncogene homolog, an oncogenic gene, upregulate GLUT1 expression, which can further promote glucose uptake and utilization, then regulates tumor cells metabolism, and provides energy for cells proliferation.7,9 In addition, GLUT1 was shown to be upregulated and ultimately facilitates pancreatic cancer cells proliferation, invasion, and metastasis.10 Moreover, the GLUT1 downstream proteins, such as hexokinase 2 (HK2), pyruvate kinase M2 (PKM2) and lactate dehydrogenase (LDH) also play key roles in multiple cancers. It has been shown that HK2 overexpression is related to poor prognosis in multiple cancers, including colorectal cancer, PC, prostate cancer, hepatocellular carcinoma and other types of tumor.11 PKM2 can promote cell survival, invasion and tumor angiogenesis in pancreatic ductal adenocarcinoma.12,13 LDH is overexpressed in PC and serum LDH levels can predict PC. Moreover, inhibition of LDH can be a stagey for PC treatment.14,15 In addition to glycolysis, the energy from the lipids, can also provide cell proliferation and promote cancer progression. It has been shown that the risk of PC is positively correlated with lipid uptake.16 In PC cells, sterol regulatory element-binding protein 1 (SREBP1) and fatty acid synthase (FASN) levels were highly expressed. Knockdown of SREBP1 in a PC mouse model can significantly reduce the viability of PC cells.17,18 Therefore, maintaining healthy blood glucose and lipids levels may play a role in reducing the risk of PC or slowing its progression.
Reticulon 4, also named as Nogo, known as a neurite outgrowth inhibitor (Nogo), is an endoplasmic reticulum-associated protein that consists of three main protein isoforms, Nogo-A, Nogo-B and Nogo-C. Nogo-A and Nogo-C are predominantly expressed in the central nervous system.19 Nogo-B is expressed in most tissues and plays a key role in vascular remodeling, cell migration, proliferation and epithelial-mesenchymal transition (EMT).20 In recent years, the involvement of Nogo-B has been demonstrated in various types of cancer. Nogo-B can promote the proliferation of hepatocellular carcinoma cells by regulating Interleukin-6/signal transducer and activator of transcription 3 signaling pathway and development of nonalcoholic fatty liver cancer by regulating the reprogramming of lipid metabolism.21 In colon cancer tissues, the expression of Nogo-B was significantly higher than that in precancerous tissues, and inhibition of Nogo-B not only promoted apoptosis in colon cancer cells, but also regulated EMT capacity in tumor tissues and suppressed tumor development. In addition, overexpression of Nogo-B promotes cervical cancer development.22 Our previous studies have shown that Nogo-B can be upregulated in both high-fat and high-glucose diet, which was associated with enhanced glucose levels. Inhibition of Nogo-B has therapeutic effects on disorders of glycolipid metabolism.23,24 Therefore, we wanted to figure out whether Nogo-B can regulate PC by the both glucose and lipid pathways.
In the present study, we found that Nogo is upregulated in PC and closely related to unfavorable clinical outcomes. Further studies revealed that nuclear factor κB (NF-κB) can be inhibited by Nogo inhibition, which contributed to reducing GLUT1 and SREBP1 expression, ultimately reducing the proliferation, invasion, and metastasis of PC cells. Based on these results, we demonstrate the role and mechanism of Nogo-B in promoting PC progression, and propose that Nogo-B may be promising therapeutic target for PC.
Results
Nogo-B expression is elevated in PC tissues and correlated with worse prognosis of PC patients
It has been reported that Nogo plays essential roles in multiple cancers. To explore the role of Nogo on PC, we firstly analyzed Nogo levels using the The Cancer Genome Atlas (TCGA) database. We found that Nogo expression was higher in PC than adjacent tissue (Figure 1A). In addition, the prognostic value of Nogo was analyzed using the Gene Expression Profiling Interactive Analysis (GEPIA) dataset. As shown in Figure 1B, high expression of Nogo predicted a poorer prognosis of PC. Furthermore, the results of Immunohistochemistry (IHC) staining results indicated that expression of Nogo-B in PC tissues was significantly higher than that in adjacent tissues (Figure 1C). The above results indicate that Nogo-B expression is positively related to PC progression.
Figure 1.

Nogo-B is upregulated in PC
(A) Expression of Nogo in PC and adjacent tissues was collected from TCGA and GTEx (n = 179 for PC tissues, n = 171 for adjacent tissues).
(B) Association between Nogo expression and PC prognosis was analyzed by Kaplan-Meier analysis based on GEPIA data. (n = 178).
(C) Expression of Nogo-B in cancer tissues and adjacent non-cancerous tissues (n = 30) were detected by IHC staining, and IHC scores were quantified by ImageJ software, scale bar: 100 μm. The data represent the mean ± SD, ∗p < 0.05, ∗∗p < 0.01.
Knockdown of Nogo-B inhibits the proliferation, migration but promotes apoptosis of PC cells in vitro
To investigate the role of Nogo-B in proliferation and migration of PC cells, we firstly transfected Pan02 or Bxpc-3 cells with control or Nogo-B siRNA. The results of scratch assay indicated that knockdown of Nogo-B inhibited the ability of migration (Figure 2A). In addition, crystalline violet staining and MTT assay showed that knockdown of Nogo-B reduced cell proliferation (Figures 2B–2D). Furthermore, we constructed Nogo knockout (KO) cell line using CRISPR-cas9 technology, and named as KO#1, KO#2. Similar to the results of knockdown experiments, we found that the abilities of proliferation and migration were inhibited in KO#1 and KO#2 cells (Figures 2E–2G). To explore whether overexpression of Nogo-B showed the opposite effects on cell proliferation and migration. We transfected PC cells with Nogo-B overexpression (Nogo-B-OE) plasmid, and found that cell migration and proliferation was increased in Nogo-B overexpressed cells (Figures 2H and 2I). At molecular levels, we examined apoptosis and EMT-related proteins. As shown in Figure 2J, knockdown of Nogo-B resulted in enhanced expression of E-Cadherin (CDH1), but reduced N-cadherin (CDH2), indicating reduced EMT pathway. Furthermore, proliferation-related protein proliferating cell nuclear antigen (PCNA) was reduced in Nogo-B knockdown cells, apoptosis-related protein B cell leukemia/lymphoma 2 (Bcl-2) was reduced, while BCL2-associated X protein (BAX) was induced in Nogo-B knockout cells. However, Nogo-B overexpressed cells showed the opposite effects on the proliferation- and apoptosis-related protein expression (Figure 2J–2L). Moreover, we showed the phosphorylated-AKT (p-AKT) levels were reduced in Nogo-B knockdown cells (Figure 2J), indicating that Nogo-B regulated cell proliferation was related to AKT pathway.
Figure 2.

Nogo-B is corrected with PC cells proliferation and migration
(A–D) Pan02 and Bxpc-3 cells were transfected with Nogo-B siRNA for 24 h, then treated in serum free (SF) medium for 12 h (A), 7 days (B and C) or indicated time (D). Cell migration, proliferation was assessed using scratch assay (A), crystalline violet staining (B and C) and MTT method (D).
(E–G) Nogo-B knockout cell line using CRISPR-cas9 technology was named as KO#1, KO#2. Cells were cultured in SF medium for 12 h (E and F) or 7 days (G) then used to detect cell migration or proliferation by scratch assay (E) or crystalline violet staining (G).
(J–L) Knockdown/knockout or overexpressed cells were treated in SF medium for 24 h, protein expression of proliferation and apoptosis-related genes was determined by Western blot with quantitative analysis of band intensity. Scale bar: 100 μm (A, E and H), 2 mm (B and I), 1 cm (G). The data represent the mean ± SD, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n = 3.
Nogo-B deficiency reduced PC progression in vivo
Based on the in vitro results, we speculated that inhibition of Nogo-B can reduce the development of PC in vivo. We constructed an orthotopic transplantation model of PC using Pan02 cells (Ctrl) or Nogo-B knockout Pan02 cells (KO) injected to pancreas of wild type (WT) or global Nogo knockout (Nogo−/−) mice. During the experiment, body weight was recorded daily. Mice were euthanized 13 days after cells injection, tumor in pancreas, spleen and liver samples were collected. As shown in Figure 3A, we found that pancreas tumor volume and weight in the WT/Ctrl group was larger and higher than the other three groups. Furthermore, we found that Nogo−/− mice injected with Nogo-B knockout Pan02 cells showed the smallest tumor volume and lightest tumor weight, indicating the fundamental role of Nogo-B in PC progression. However, mice body weight among these groups showed little difference (Figure S1A). Previous studies have shown that orthotopic transplantation PC model can induce metastasis. Similar to the results of tumor in pancreas tissues, we found that both Nogo-B knockout in mice and cells alleviated liver metastasis (Figure 3B). In addition, pancreas tumor tissue sections were performed TUNEL staining to detect cell apoptosis. Compared with WT/Ctrl group, apoptotic cells were enhanced in the other groups (Figure 3C). Furthermore, we conducted IHC staining to detect the expression of EMT markers. Consistent with the in vitro results, we found that Nogo-B knockout increased CDH1 levels, while reduced CDH2 levels (Figure 3D). These data suggest that inhibition of Nogo-B largely ameliorated the proliferation and metastasis of PC in vivo.
Figure 3.

Knockout of Nogo-B inhibits PC proliferation and metastasis in vivo
(A) Orthotopic tumors were excised 13 days after inoculation with the indicated Pan02 cells or Nogo KO Pan02 cells (left panel) and the tumors were weighted (right panel).
(B) Ectopic tumors in liver were photographed (left panel) and the number of tumors were counted (right panel).
(C) Orthotopic tumor sections were conducted with TUNEL staining with quantitative analysis of apoptosis cells, TUNEL positive cells were indicated by the white arrows.
(D) CDH1 and CDH2 expression in orthotopic tumor sections was detected by IHC staining with quantitative analysis of MD. Scale bar: 1 cm (B), 100 μm (C), 50 μm (D). The data represent the mean ± SD, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n = 6.
High levels of glucose promotes proliferation and migration of PC cells
High glucose levels can contribute to the development and progression of PC. In order to clarify whether Nogo-B is involved in glucose-related PC progression, we incubated Pan02 cells in medium contain low (5 mM) or high (25 mM) concentration of glucose, which was wildly used for determination the role of glucose in biology.17 Consistent with previous study, we found that 25 mM glucose promoted cell proliferation and migration (Figures 4A and 4B). Moreover, pro-proliferation- and anti-apoptotic-related genes were positively regulated by glucose. We indicated that Vimentin, PCNA, Bcl-2 protein levels were enhanced by glucose, while BAX was reduced (Figure 4C). More importantly, we indicated that Nogo-B protein expression was enhanced by the treatment of high levels of glucose (Figure 4C). Then, we used normal and Nogo-B KO cells for further investigation. As shown in Figure 4D, the high glucose-enhanced ability of proliferation was largely reduced in Nogo-B KO cells. Consistently, proliferation- and apoptosis-related protein expression was largely attenuated in Nogo-B KO cells (Figure 4E). Therefore, we indicated that high glucose promoted PC progression is positively related to Nogo-B expression.
Figure 4.

High glucose promotes tumor growth in PC cells through Nogo-B
Pan02 or Nogo-B KO Pan02 (KO#1) cells were treated with indicated concentration of glucose for 24 h. Cell proliferative and migratory abilities were determined by MTT (A), scratch assay (B) and crystalline violet staining (D). EMT, proliferation and apoptosis-related genes expression was detected by Western blot with quantitative analysis of band intensity (C and E). Scale bar: 100 μm (B), 2 mm (D). The data represent the mean ± SD, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n = 3.
Nogo-B promotes PC progression through inducing NF-κB pathway
Studies have shown that NF-κB plays important role in PC development.25 We and other studies have demonstrated that NF-κB can be induced by Nogo-B.23,26 In this study, our results showed that high glucose promoted NF-κB expression. Whereas, NF-κB levels were reduced in Nogo-B KO cells. Whereas, glucose had little effects on NF-κB expression in Nogo-B KO cells (Figure 5A), indicating inhibition of Nogo-B expression reduced cell proliferation may be related to NF-κB inhibition. Next, normal or Nogo-B KO cells were treated with BAY (an inhibitor of NF-κB) or transfected with NF-κB overexpression plasmid. Our results revealed that the abilities of migration and proliferation were reduced by BAY, while enhanced by NF-κB overexpression plasmid in Pan02 cells. While the regulation of BAY on cell migration and proliferation was largely reduced in KO#1 cells. Compared to KO#1 cells, the abilities of cell proliferation and migration were enhanced in NF-κB overexpressed KO#1 cells (Figures 5B and 5C). Similarly, PCNA protein expression was enhanced by NF-κB overexpression, while reduced by the BAY treatment (Figures 5D, 5E, and S2). Taken together, we indicated that Nogo-B regulated PC progression is dependent on NF-κB.
Figure 5.

Nogo-B promotes PC progression through activation NF-κB pathway
Pan02 or Nogo-B KO Pan02 (KO#1) cells were treated with indicated concentration of glucose for 24 h (A) or transfected with NF-κB overexpression plasmid or Bay for 24 h, then treated with indicated concentration of glucose for 24 h (B, D and E) or 7 days (C). Protein expression was detected by Western blot with quantitative analysis of band intensity (A, D and E), colony forming migratory abilities were assessed by scratch assay (B) and crystal violet staining (C). Scale bar: 100 μm (B), 2 mm (C). The data represent the mean ± SD, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n = 3.
NF-κB promotes glucose uptake by upregulating GLUT1 and SREBP1
Metabolic deregulation is a hallmark of multiple types of cancers. The glucose and lipid metabolism pathways were shown to be involved in PC. GLUT1, a master transporter of glucose, and SREBP1, a main regulator of lipid metabolism, can promote PC development.17,27 Our results showed that high glucose was able to promote the expression of GLUT1, and the glycolysis related proteins HK2, PKM2, LDHA, as well as SREBP1 in Pan02 cells (Figure 6A). While the enhanced expression of GLUT1, HK2, PKM2, LDHA, and SREBP1 was largely blocked in Nogo-B knockout cells. Moreover, we found that GLUT1 and SREBP1 expression was reduced in Nogo mice or WT mice injected with Nogo-B KO cells (Figure S3). To investigate whether NF-κB is involved in regulation of GLUT1 and SREBP1, we treated cells with BAY or transfection of NF-κB overexpression plasmid. We found that both GLUT1 and SREBP1 expression was reduced by NF-κB inhibitor, while increased in NF-κB overexpressed cells. More importantly, we indicated that FASN, a target of SREBP1, as well as HK2, PKM2, LDHA, downstream targets of GLUT1 was also regulated by NF-κB (Figure 6B, 6C, S4, and S5). Furthermore, we transfected Pan02 cells with GLUT1 or SREBP1 siRNA, and found that knockdown of GLUT1 or SREBP1 reduced the ability of cell migration and proliferation (Figures 6D and 6E). In addition, the glucose enhanced role of cell migration and proliferation was largely blocked in cells with GLUT1 or SREBP1 knockdown. Therefore, we indicated that expression of GLUT1 and SREBP1 are involved in Nogo-B regulated cell proliferation.
Figure 6.

NF-κB directly regulates GLUT1 and SREBP1
(A–C) Pan02 or Nogo-B KO Pan02 (KO#1) cells were treated with indicated concentration of glucose for 24 h (A). Cells were pre-treated with Bay (B) or transfected with NF-κB overexpression plasmid (C) for 24 h, then received indicated concentration of glucose treatment for 24 h. After treatment, protein expression was detected by Western blot with quantitative analysis of band intensity. Pan02 or Nogo-B KO Pan02 (KO#1) cells were transfected with GLUT1 siRNA or SREBP1 siRNA for 24 h, then treated with indicated concentration of glucose for 24 h (D) or 7 days (E). Scale bar: 2 mm (E). The data represent the mean ± SD, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; n = 3.
Consistent with the protein expression, we found that mRNA levels of GLUT1 and SREBP1 were enhanced by high glucose treatment. Compared to normal cells, GLUT1 and SREBP1 mRNA expression was largely reduced in Nogo-B KO cells. In addition, high glucose had little effects on GLUT1 and SREBP1 mRNA expression in Nogo-B KO cells (Figures 7A and 7B). Furthermore, we constructed promoters for GLUT1 and SREBP1, and found that high glucose induced the activities of GLUT1 and SREBP1 promoters in Pan02 cells, while had little effects in KO#1 cells (Figures 7C and 7D). By completing a sequence alignment analysis, we found two putative NF-κB response element (NRE) in the proximal region of the GLUT1 promoter, and three NRE in the proximal region of the SREBP1 promoter. Therefore, we constructed normal GLUT1 and SREBP1 promoters and the promoters with NRE mutation (pGLUT1-Mut1/2 or pSREBP1-Mut1/2/3), respectively (Figures 7E and 7G). Compared with normal GLUT1 promoter, high glucose had little effects on GLUT1-Mut1 promoter, indicating the critical role of NRE in region of −365∼−356 of GLUT1 promoter (Figure 7F). We also found that the activities of SREBP1-Mut3 promoter did not be regulated by high glucose (Figure 7H). Indeed, the results of chromatin immunoprecipitation (ChIP) assay suggest that the binding of NF-κB in promoters of GLUT1 and SREBP1 was mediated by NRE1 and NRE3, respectively. Also, high glucose induced the binding ability of NF-κB to NRE in Pan02 cells, while had little effects in KO#1 cells (Figures 7I, 7J, and S6). Therefore, we indicated that high glucose increased GLUT1 and SREBP1 expression by activating the promoter.
Figure 7.

NF-κB through promoter effects to GLUT1 and SREBP1
(A and B) Pan02 or Nogo-B KO Pan02 (KO#1) cells were treated with indicated concentration of glucose for 24 h. mRNA expression of GLUT1, SREBP1 was determined by qRT-PCR.
(C–H) Cells were transfected with indicated plasmids for 12 h. After stabilization for 4 h, cells were treated with the indicated concentrations of glucose for 24 h. Cell lysates were collected for determination the activities of firefly and Renilla luciferases (Luc) using the Dual-Luciferase Reporter Kit.
(I and J) Cells were treated with the indicated concentration of glucose for 24 h. Cellular chromatin DNA was extracted, IgG (negative control) or NF-κB antibody was used for immunoprecipitation. PCR was conducted with the corresponding primers for the NF-κB response element in GLUT1 or SREBP1 promoter. The data represent the mean ± SD, ∗p < 0.05 compared with control (n = 3). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Discussion
The pathogenesis of PC is complex and remains unclear. Metabolic alterations and the complex microenvironment are largely related to the progress of PC. The primary treatment options for PC are chemotherapy and surgery, while the suitable patients for surgery are as low as 15%–20% at diagnosis. Therefore, it is still an urgent for exploration biomarkers or strategies for PC treatment. From the database of PC, high Nogo-B expression is associated with poor prognosis in patients. We also observed that Nogo-B expression is much higher in PC tissues than that in the adjacent normal tissues by IHC staining and the database of PC. In vitro and in vivo results showed that inhibition of Nogo-B reduces the development of PC. At molecular levels, we showed that Nogo-B promotes NF-κB expression, then increases the levels of SREBP1 and GLUT1. Therefore, our study suggests that Nogo-B may be a potential prognostic marker, and therapeutic target for PC, which provides strategy for PC diagnose and treatment.
Growing evidence confirms the pivotal role of Nogo-B in types of cancers, including hepatocellular carcinoma, nasopharyngeal, cervical and colorectal cancers.22,28,29 Our group has demonstrated that Nogo-B is a master regulator for metabolic diseases, such as high-fat induced obesity and high glucose-caused fatty liver.23,24 Previous study indicated that Nogo-B can promote Non-alcoholic fatty liver disease-associated hepatic carcinoma, which was related to high-fat, high-carbohydrate diet caused metabolic dysfunction.30 It is known that obesity, nutrition, and type 2 diabetes are risk factors for PC.31,32,33 In this study, we indicated that Nogo-B is elevated in PC tissues and correlated with worse prognosis of PC patients (Figure 1). Moreover, we indicated that inhibition the expression of Nogo-B reduces cell viability and proliferation in vitro, and orthotopic tumor progression and metastasis in vivo (Figures 2 and 3). More importantly, we indicated that high glucose-induced cell proliferation is also related to Nogo-B levels (Figure 4). Therefore, we showed that Nogo-B plays a fundamental role in PC progression. Our group has found some strategies for Nogo-B inhibition, such as siRNA, natural products and PPARγ agonists,23,24,34,35 which might be a promising therapeutic strategy for PC due the inhibition of Nogo-B. Moreover, we also indicated that Nogo-B might be an effective target for development chemical compound to treat PC.
Glucose is a main fuel for tumorigenesis. GLUT1 is widely expressed in multiple tissues and a major glucose transmembrane transport protein that mediates the uptake of glucose and plays a critical role in multiple cancers.36 GLUT1 levels were elevated in many malignant tumors and correlated with poor clinical outcomes in patients.37,38 Post-transcriptional regulation, such as palmitoylation, can promote GLUT1 plasma membrane localization, which increases glycolysis, thereby promoting glioblastoma tumorigenesis.39 In recent study, the researchers have shown that GLUT1 not only plays an important role in Forkhead box D1-enhanced PC progression, but also consider as a potential target for chemotherapy-resistant PC.10,40 In addition to glucose, lipids generation can also promote the progression of tumor. SREBP1, a main regulator of lipogenesis, is associated with a variety of cancers, such as hepatocellular carcinoma, gastric cancer, breast cancer, colorectal cancer and renal cell cancer.41,42,43,44,45 However, another study indicated that found that fatty acids synthesis-related genes elevation in hepatocellular carcinoma samples was not related to enhancement of mature SREBP1, but due to the excessive m6A modification.46 It has been reported high glucose promoted PC development both in vitro and in vivo by enhancing SREBP1 expression, which was mediated by the autophagy pathway.17 In this study, we found that inhibition of Nogo-B can reduce both GLUT1 and SREBP1 levels, which contributed to the inhibition of PC progression (Figure 6).
Previous studies have shown that GLUT1 can be regulated by a series of molecules. In brain tumor, NF-κB signaling can be activated by α-Ketoglutarate, then promotes GLUT1 expression.8 GLUT1 expression can be induced by hypoxia-inducible factor 1α and c-Myc, and reduced by peroxisome proliferator-activated receptor α.47 It can also be regulated by PI3K/AKT pathway. In cancer cells, SREBP1 can be upregulated by long chain acyl CoA synthetase 4, SET domain-containing protein 8 and other molecules.42,44 In colorectal cancer, SREBP1 promotes the invasion of cancer cells is accompanied with NF-κB activation.45 Studies have shown that NF-κB expression was upregulated in PC cells and tissues, and is associated with poor prognosis in PC patients.48,49 In our previous study, we have shown that Nogo-B deficiency promotes NF-κB degradation. Other study indicated that Nogo-B downregulation significantly reduced toll-like receptor 4 protein expression and inhibited the activation of NF-κB pathway.23,50 In this study, we demonstrated that NF-κB expression was enhanced in high glucose environment, which was largely reduced in Nogo-B deficiency cells. Moreover, inhibition NF-κB expression can largely reduce cell proliferation. Furthermore, we also found that both GLUT1 and SREBP1 can be activated by NF-κB, and identified a response element in GLUT1 and SREBP1 promoter (Figure 7).
In conclusion, we indicated that Nogo-B can promote PC progression both in vitro and in vivo. We also found that high glucose-enhanced cell proliferation is positively related to Nogo-B expression. In this study, we indicated that glycolipid metabolism is regulated by Nogo-B, which promotes PC development, contribued by the regulating GLUT1 and SREBP1 expression. More importantly, we demonstrated that NF-κB is involved in Nogo-B regulated PC, which is also a major regulator of GLUT1 and SREBP1 by directly binding to their promoters. Therefore, we suggest that Nogo-B may be a potential prognostic marker for PC diagnose, and a promising candidate for drug developing.
Limitations of the study
Nogo-B has a variety of functions, we mainly focused on the role of regulating the NF-κB/GLUT1 and SREBP1 pathways, other pathways in promote PC development is still needed to be further analyzed. In addition, GLUT1 and SREBP1 can both promote PC development, whether any interaction between them is still unclear. Thirdly, we only showed the role of the Nogo-B for PC progression in high glucose environment in cells, the PC mouse model needed further conducted. Fourthly, it is still unclear whether Nogo-B might be a biomarker of PC as we didn’t detect Nogo-B expression in any liquid biopsies.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-AKT antibody | Proteintech Group | Cat# 10176-2-AP; RRID:AB_2224574 |
| anti-p-AKT antibody | Proteintech Group | Cat# 66444-1-Ig; RRID:AB_2782958 |
| anti-Bcl2 antibody | Proteintech Group | Cat# 26593-1-AP; RRID:AB_2818996 |
| anti-SREBP1 antibody | Proteintech Group | Cat# 14088-1-AP; RRID:AB_2255217 |
| anti-BAX antibody | Proteintech Group | Cat# 50599-2-Ig; RRID:AB_2061561 |
| anti-Vimentin antibody | Proteintech Group | Cat# 10366-1-AP; RRID:AB_2273020 |
| anti-PCNA antibody | Proteintech Group | Cat# 10205-2-AP; RRID:AB_2160330 |
| anti-NF-κB p65 antibody | Proteintech Group | Cat# 10745-1-AP; RRID:AB_2178878 |
| Fluorescein (FITC)–Conjugated Affinipure Goat anti-Rabbit IgG | Proteintech Group | Cat# SA00003-2; RRID:AB_2890897 |
| Horseradish Peroxidase (HRP)-Conjugated α-Tubulin | Proteintech Group | Cat# HRP-66031; RRID:AB_2687491 |
| anti-CDH1 antibody | Abclonal | Cat# A3044; RRID:AB_2764849 |
| anti-CDH2 antibody | Abclonal | Cat# A19083; RRID:AB_2862575 |
| anti-GLUT1 antibody | Abclonal | Cat# A6982; RRID:AB_2767538 |
| anti-FASN antibody | Abclonal | Cat# A0461; RRID:AB_2757202 |
| anti-HK2 antibody | Affinity | Cat# BF0283; RRID:AB_2833836 |
| anti-PKM2 antibody | Affinity | Cat# AF5234; RRID:AB_2837720 |
| anti-LDHA antibody | Affinity | Cat# DF6280; RRID:AB_2838246 |
| anti-Nogo-B antibody | Santa | Cat# sc-271878; RRID:AB_10709573 |
| Bacterial and virus strains | ||
| Subcloning Efficiency DH5α | ThermoFisher | Cat# 18265017 |
| Biological samples | ||
| Pancreatic cancer patients tissue sections | Anhui Provincial Cancer Hospital | N/A |
| pancreatic orthotopic tumors | This paper | N/A |
| mice pancreatic tissue sections | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Crystalline Violet | Solaibao Technology | Cat# IC0600 |
| MTT | Sigma-Aldrich | Cat# 298-93-1 |
| Mouse Nogo-B siRNA | GENEWIZ | N/A |
| Critical commercial assays | ||
| TUNEL staining kit | KeyGEN BioTECH | Cat# KGA702 |
| DAB | KeyGEN BioTECH | Cat# KGP1045-100 |
| Duo-Lite Luciferase Assay System | Vazyme | Cat# DD1205-01 |
| HiScript II Q Select RT SuperMix for qPCR | Vazyme | Cat# R233-01 |
| AceQ qPCR SYBR Green Master Mix | Vazyme | Cat# Q131-02 |
| Liposomal nucleic acid transfection reagent | Yisheng Biotechnology | Cat# 40802ES03 |
| Experimental models: Cell lines | ||
| Pan02 | OriCell | Cat# M2-0501 |
| Bxpc-3 | ATCC | Cat# CRL-1687 |
| Experimental models: Organisms/strains | ||
| Wild type C57 BL/6J mice | GemPharmatech | Cat# N000295 |
| RTN4-KO mice | GemPharmatech | Cat# T004776 |
| Oligonucleotides | ||
| Nogo-B Forward (5’ to 3’): GGTGTTTGGTGCCAGCTTAT | This paper | N/A |
| Nogo-B Reverse (5’ to 3’): TGAATGGGTGGCCTTCATCT | This paper | N/A |
| NF-κB Forward (5’ to 3’): CACCGGATTGAAGAGAAGCG | This paper | N/A |
| NF-κB Reverse (5’ to 3’): AAGTTGATGGTGCTGAGGGA | This paper | N/A |
| SREBP1 Forward (5’ to 3’): GCTGCTGACCGACATCGAA | This paper | N/A |
| SREBP1 Reverse (5’ to 3’): GGGTGGGTCAAATAGGCCAG | This paper | N/A |
| GLUT1 Forward (5’ to 3’): CAGCTGTCGGGTATCAATGC | This paper | N/A |
| GLUT1 Reverse (5’ to 3’): TCCAGCTCGCTCTACAACAA | This paper | N/A |
| SREBP1-NRE1 Forward (5’ to 3’): CTAACCTCTCAGCTGGCTCA | This paper | N/A |
| SREBP1-NRE1 Reverse (5’ to 3’): TGGGAGCAGAGAACAGAAAAC | This paper | N/A |
| SREBP1-NRE2 Forward (5’ to 3’): CCACTGCAGGCCTGTATCT | This paper | N/A |
| SREBP1-NRE2 Reverse (5’ to 3’): CTAGACCCAGCCCAGACATC | This paper | N/A |
| SREBP1-NRE3 Forward (5’ to 3’): CTAGGCGAGCTGTCAGGAT | This paper | N/A |
| SREBP1-NRE3 Reverse (5’ to 3’): ATCTCGGCCAGTGTCTGTTC | This paper | N/A |
| GLUT1-NRE1 Forward (5’ to 3’): ACCAAGCGCTACCATTTTGC | This paper | N/A |
| GLUT1-NRE1 Reverse (5’ to 3’): CCACAGTCTGGCTCTGGATG | This paper | N/A |
| GLUT1-NRE2 Forward (5’ to 3’): TGACGATCTGAGCTACGGGG | This paper | N/A |
| GLUT1-NRE2 Reverse (5’ to 3’): CTCGGCAGCATCCCTACC | This paper | N/A |
| si-m-Srebp1_001 | RiboBio | Cat# siG101028133544-1-5 |
| si-m-Slc2a1_001 | RiboBio | Cat# siG141127092029-1-5 |
| si-r-Nogo-B_001 | RiboBio | Cat# siG171107091412-1-5 |
| siR NC #1 | RiboBio | Cat# siN0000001-1-5 |
| Software and algorithms | ||
| GraphPad Prism 8 | GraphPad | N/A |
| Photoshop CS6 | Adobe | N/A |
| ImageJ | National Institutes of Health | N/A |
Resource availability
Lead contact
The relevant experimental reagents, experimental methods and related data of this study can be obtained by contacting Xiaoxiao Yang (yangxiaoxiao@hfut.edu.cn).
Materials availability
The plasmids used in this study are available from the lead contact. This study did not generate new unique reagents.
Data and code availability
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Clinical samples and public datasets
Clinical tissue samples from 30 patients were obtained from The First Affiliated Hospital of University of Science and Technology of China (2022-N(H)-050) (detailed information was shown in Table S1). All participating patients provided informed consent for the collection of tissue samples. For each patient, all specimens were fixed and embedded in paraffin. All experiments involving human samples and clinical data were approved by the Ethics Committee of The First Affiliated Hospital of University of Science and Technology of China. Tissue sections were conducted immunohistochemical staining for determination Nogo-B expression. The mean density was calculated by Image J and graded into four ranges (intensity score): no staining (0), light brown staining (1), brown staining (2) and dark brown staining (3), and defined brown staining. The number of positively stained cells was divided into four ranges (percentage score): < 5% (0), 5–30% (1), 31–70% (2), > 70% (3). The final staining score was calculated using the formula: overall score = intensity score × percentage score. A final score ≤ 3 was defined as low Nogo-B expression, and > 3 as high Nogo-B staining.
RNA-sequencing expression profiles and corresponding clinical information for pancreatic cancer were downloaded from the TCGA dataset (https://portal.gdc.com) and current-release (V8) GTEx datasets (https://www.gtexportal.org/home/datasets). Statistical analyses were performed using R software v4.0.3 (n = 179 for PC tissues, n = 171 for adjacent tissues). Association between Nogo expression and PC prognosis was analyzed by Kaplan-Meier analysis based on GEPIA data (http://gepia.cancer-pku.cn/index.html) (n = 178).
Animals and in vivo studies
C57BL/6J mice and Nogo global knockout mice (C57BL/6J background) were obtained from GemPharmatech (Nanjing, China). All animal experiments were approved by the Ethics Committee of Hefei University of Technology (No. HFUT20190323001) and conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (publication No. 8023, revised 1978). Mice were free access to normal food and water, and allowed to acclimatize to environment for at least 7 days before experiment. A total of 1 × 106 stably transfected cells with Nogo-B knockout expression were injected orthotopic into the mouse pancreas (6 mice per group, male, ∼8 weeks old). At the end of the experiment, all mice were euthanized. Tumors were collected for further study and tumor weights were measured.
Cell culture
Pan02 (mouse pancreatic cancer cell line) or Bxpc-3 (human pancreatic cancer cell line) cells were purchased from ATCC and cultured in 1640 medium or DMEM medium containing 10% fetal bovine serum and 1% penicillin/streptomycin, respectively. Cells were cultured at low passage numbers and passaged consistently using Trypsin-EDTA 0.25% and tested regularly for mycoplasma contamination (all cells used for experiments were mycoplasma negative at the time of testing).
Method details
Immunohistochemical, immunofluorescence and TUNEL staining
After collection, fresh tissues were fixed overnight in 4% paraformaldehyde at 4°C, dehydrated using graded ethanol or 30% sucrose solutions, and embedded in paraffin or optimal cutting temperature compounds, respectively. The embedded tissues were then cut into 5 μm sections for further staining. Prior to staining, 5 μm paraffin sections were dewaxed in xylene and hydrated using graded ethanol solutions. Tissue sections were conducted immunohistochemical or immunofluorescence staining for determination protein expression. Alternatively, process tissue sections using the TUNEL Apoptosis Detection Kit. All images were captured with a Leica microscope. The mean density of the images was quantified by segmentation color threshold analysis using Photoshop CS6 and normalized by the values of control mice.
MTT assays
MTT assay was used to determine cell viability. Cells were cultured in 96-well plate received indicated treatment. After treatment, cells were added with MTT solution (20 μL/well, 3 mg/mL) for 4 h at 37°C, then washed with PBS twice. 150 μL dimethyl sulfoxide (DMSO, Sigma-Aldrich) was added to dissolve formazan. The absorbance was then measured at 550 nm using a Multi-Mode Microplate Reader (BioTek Synergy H1; Vermont, USA).
Wound healing assay
Cells were inoculated in 24-well plate and incubated at 37°C until cell density reached 80-90%. Cells were scraped using a 200 μL pipette wall scraper and washed with PBS at least three times. Photographs were taken at 0 h, 12 h and 24 h, and to calculate the wound healing rate was calculated with the formula: (wound width at 0 h - wound width at 12 or 24 h)/wound width at 0 h.
Colony formation assay
About 2000 cells were inoculated in 6-well plate. After 1 week treatment, cells were stained with Crystal Violet Staining Solution. Crystal Violet Staining Solution is prepared from 20 mL of saturated ethanol solution of crystalline violet (2 g of crystalline violet dissolved in 20 mL of 95% ethanol solution) and 80 mL of 1% aqueous ammonium oxalate. Images were captured with a camera. Then stained cells were incubated with 1 mL of 33% aqueous acetic acid solution for 10 min, the supernate was collected and measured at 570 nm using a Multi-Mode Microplate Reader (BioTek Synergy H1; Vermont, USA).
Knockdown of gene expression by siRNA transfection
Pan02 or Bxpc-3 cells were transfected with siRNA (40 nM/well) or the corresponding negative control siRNA (siCtrl) using LipofectamineTM RNAiMAX in SF medium according to the manufacturer’s instruction. After incubation in transfection medium for 24 h, cells were treated in complete medium for another 12 h, followed by received the indicated treatment.
Generation of a CRISPR-Cas9-mediacted Nogo-B knockout Pan02 cell line
A Nogo-B genome knockout Pan02 cell line was generated using the CRISPR-Cas9 technology.51 The guide RNA was designed using the online CRISPR Design Tool (http://tools.genome-engineering.org/). The target sequence of Nogo-B is 5′-CTCCGCTAGGGGGCTCGCGC-3′, which locates in the first exon 63 bp downstream of the start codon. Pan02 cells were transfected with pSpCas9 (BB)-2A-Puro empty vector or Cas9-Nogo-B plasmid for 24 h in SF medium, then changed to complete medium for another 2∼3 days, followed by treated with puromycin (3 μg/mL) for 3-5 days until no cell died. The rest cells were used for selection of mono clones and confirmed by Western blot. Pan02 cells lacking Nogo-B expression were named as Nogo-KO cells, short as KO#1 or KO#2.
Western blot
Total protein was extracted from cells or mouse tissue using a lysis buffer. Protein concentration was determined using the BCA method. Expression of Nogo-B, AKT, BCL2, BAX, SREBP1, Vimentin, PCNA, NF-κB, p-AKT, α-Tubulin, GAPDH, CDH1, CDH2, FASN, SREBP1, GLUT1, HK2, PKM2 and LDHA proteins as determined by Western blot. Images were scanned by Clinx ChemiCapture (CLINX; Shanghai, China).
Quantitative real-time PCR
Total RNA was isolated from cell or tissue samples using Trizol reagent (Biosharp, BS259A). cDNA was synthesized by reverse transcription reagent (Vazyme). mRNA levels were determined by quantitative real-time PCR using SYBR Green PCR reagent (Vazyme) with indicated primers (key resources table), and normalized with GAPDH mRNA in the corresponding samples.
Construction of normal or mutated GLUT1 and SREBP1 promoter(s), and determination the promoter activities
Promoters of murine SREBP1 (-471∼+115) and GLUT1 (-467 ∼+66), and indicated mutations (as shown in Figures 6E and 6G) were constructed with pGL4.10 luciferase reporter vector by GENEWIZ. Mutant promoters were named as SREBP1-Mut1/2/3 or GLUT1-Mut1/2.
Cells in 48-well plate at ∼70% confluence were transfected with promoter and pGL4.70 plasmids for 12 h in SF medium, then treated in complete medium for 12 h. After stabilization, cells were received indicated treatment, then the activities Firefly and Renilla luciferase in cell lysates were measured using a dual luciferase reporter assay system (Vazyme; Nanjing, China).
ChIP assay for determination DNA binding activity
To determine the binding activity of NF-κB protein with SREBP1 and GLUT1 promoter, we treated Pan02 and KO#1 cells with different glucose concentrations for 24 h. The protein and DNA complex was crosslinked with 1% formaldehyde for 10 min, and then glycine (0.125 M) was added to stop the crosslinking. Nuclear protein was extracted by a Nuclear Protein Extraction Kit (KeyGen Biotech; Nanjing, China). Chromatin was then broken into 200-1000 bp fragments by ultrasound (Qsonica Q700; Newtown, USA). After crosslink reversal, input PCR was performed with DNA extracted from sonicated chromatin. Same amount of chromatin as input from each sample was used for immunoprecipitation using NF-κB rabbit monoclonal antibody or IgG (as a negative control antibody). PCR was conducted using the indicated primers shown in key resources table.
Quantification and statistical analysis
Data are expressed as mean ± SD, and all experiments were repeated independently at least three times. First, all data were analyzed using Graphpad Prism 8 software, and the data obeyed a normal distribution. Analysis was then performed by one-way or two-way ANOVA. Correlation coefficients were calculated using the Pearson correlation test, and P values were calculated. A value was considered significant if P < 0.05 (n≥3).
Acknowledgments
This work was supported by the Anhui Provincial Natural Science Foundation grant 2308085QA31, the Fundamental Research Funds for the Central Universities (JZ2022HGTA0314, JZ2022HGQA0139) to L. L. Wei, 2019 Liaoning Social Science Planning Fund Project L19ASH003 to Y. X. Zhu, Anhui Provincial Key Research and Development Project (2022i01020004) to C.Z. Liao and Technology and Innovation Commission of Shenzhen Municipality grant JCYJ20220530113609020 to B. T. Zhang.
Author contributions
Conception and design: X.X.Y.; Experiments: T.X.W., M.Z., X.Y.G. and Y.P.; Statistical analysis: W.J.C., G.D.S., H.R.R., T.X.W. and M.Z.; Analysis and interpretation of data: C.Z.L., H.M.X., Q.S.L., T.X.W. and M.Z.; Review and/or revision of the manuscript: X.X.Y., T.X.W., Y.X.Z., Y.L.C., B.T.Z. and L.L.W. All the authors have read the authorship agreement of the journal and that the manuscript has been reviewed by and approved by all named authors.
Declaration of interests
The authors declare no competing interests.
Published: April 12, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109741.
Contributor Information
Lingling Wei, Email: linglingwei@hfut.edu.cn.
Yuanli Chen, Email: chenyuanli@hfut.edu.cn.
Xiaoxiao Yang, Email: yangxiaoxiao@hfut.edu.cn.
Supplemental information
References
- 1.Halbrook C.J., Lyssiotis C.A., Pasca di Magliano M., Maitra A. Pancreatic cancer: advances and challenges. Cell. 2023;186:1729–1754. doi: 10.1016/j.cell.2023.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang X., Zhang G., Tang T.Y., Gao X., Liang T.B. Personalized pancreatic cancer therapy: from the perspective of mRNA vaccine. Mil. Med. Res. 2022;9:53. doi: 10.1186/s40779-022-00416-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conroy T., Hammel P., Hebbar M., Ben Abdelghani M., Wei A.C., Raoul J.L., Choné L., Francois E., Artru P., Biagi J.J., et al. FOLFIRINOX or Gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 2018;379:2395–2406. doi: 10.1056/NEJMoa1809775. [DOI] [PubMed] [Google Scholar]
- 4.Von Hoff D.D., Ervin T., Arena F.P., Chiorean E.G., Infante J., Moore M., Seay T., Tjulandin S.A., Ma W.W., Saleh M.N., et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013;369:1691–1703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng C., Ru P., Geng F., Liu J., Yoo J.Y., Wu X., Cheng X., Euthine V., Hu P., Guo J.Y., et al. Glucose-mediated N-glycosylation of SCAP is essential for SREBP-1 activation and tumor growth. Cancer Cell. 2015;28:569–581. doi: 10.1016/j.ccell.2015.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kitasato Y., Yasunaga M., Okuda K., Kinoshita H., Tanaka H., Okabe Y., Kawahara A., Kage M., Kaida H., Ishibashi M. Maximum standardized uptake value on 18F-fluoro-2-deoxy-glucose positron emission tomography/computed tomography and glucose transporter-1 expression correlates with survival in invasive ductal carcinoma of the pancreas. Pancreas. 2014;43:1060–1065. doi: 10.1097/mpa.0000000000000185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parks S.K., Chiche J., Pouysségur J. Disrupting proton dynamics and energy metabolism for cancer therapy. Nat. Rev. Cancer. 2013;13:611–623. doi: 10.1038/nrc3579. [DOI] [PubMed] [Google Scholar]
- 8.Wang X., Liu R., Qu X., Yu H., Chu H., Zhang Y., Zhu W., Wu X., Gao H., Tao B., et al. α-Ketoglutarate-activated NF-κB signaling promotes compensatory glucose uptake and brain tumor development. Mol Cell. 2019;76:148–162.e147. doi: 10.1016/j.molcel.2019.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Yun J., Rago C., Cheong I., Pagliarini R., Angenendt P., Rajagopalan H., Schmidt K., Willson J.K.V., Markowitz S., Zhou S., et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai K., Chen S., Zhu C., Li L., Yu C., He Z., Sun C. FOXD1 facilitates pancreatic cancer cell proliferation, invasion, and metastasis by regulating GLUT1-mediated aerobic glycolysis. Cell Death Dis. 2022;13:765. doi: 10.1038/s41419-022-05213-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ciscato F., Ferrone L., Masgras I., Laquatra C., Rasola A. Hexokinase 2 in Cancer: A prima donna playing multiple characters. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22094716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li C., Zhao Z., Zhou Z., Liu R. PKM2 promotes cell survival and invasion under metabolic stress by enhancing warburg effect in pancreatic ductal adenocarcinoma. Dig. Dis. Sci. 2016;61:767–773. doi: 10.1007/s10620-015-3931-2. [DOI] [PubMed] [Google Scholar]
- 13.Azoitei N., Becher A., Steinestel K., Rouhi A., Diepold K., Genze F., Simmet T., Seufferlein T. PKM2 promotes tumor angiogenesis by regulating HIF-1α through NF-κB activation. Mol. Cancer. 2016;15:3. doi: 10.1186/s12943-015-0490-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng C.S., Tan H.Y., Wang N., Chen L., Meng Z., Chen Z., Feng Y. Functional inhibition of lactate dehydrogenase suppresses pancreatic adenocarcinoma progression. Clin. Transl. Med. 2021;11 doi: 10.1002/ctm2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tas F., Aykan F., Alici S., Kaytan E., Aydiner A., Topuz E. Prognostic factors in pancreatic carcinoma: serum LDH levels predict survival in metastatic disease. Am. J. Clin. Oncol. 2001;24:547–550. doi: 10.1097/00000421-200112000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Philip B., Roland C.L., Daniluk J., Liu Y., Chatterjee D., Gomez S.B., Ji B., Huang H., Wang H., Fleming J.B., et al. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology. 2013;145:1449–1458. doi: 10.1053/j.gastro.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou C., Qian W., Li J., Ma J., Chen X., Jiang Z., Cheng L., Duan W., Wang Z., Wu Z., et al. High glucose microenvironment accelerates tumor growth via SREBP1-autophagy axis in pancreatic cancer. J. Exp. Clin. Cancer Res. 2019;38:302. doi: 10.1186/s13046-019-1288-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun Y., He W., Luo M., Zhou Y., Chang G., Ren W., Wu K., Li X., Shen J., Zhao X., Hu Y. SREBP1 regulates tumorigenesis and prognosis of pancreatic cancer through targeting lipid metabolism. Tumour Biol. 2015;36:4133–4141. doi: 10.1007/s13277-015-3047-5. [DOI] [PubMed] [Google Scholar]
- 19.Long S.L., Li Y.K., Xie Y.J., Long Z.F., Shi J.F., Mo Z.C. Neurite outgrowth inhibitor B receptor: A versatile receptor with multiple functions and actions. DNA Cell Biol. 2017;36:1142–1150. doi: 10.1089/dna.2017.3813. [DOI] [PubMed] [Google Scholar]
- 20.Acevedo L., Yu J., Erdjument-Bromage H., Miao R.Q., Kim J.E., Fulton D., Tempst P., Strittmatter S.M., Sessa W.C. A new role for Nogo as a regulator of vascular remodeling. Nat. Med. 2004;10:382–388. doi: 10.1038/nm1020. [DOI] [PubMed] [Google Scholar]
- 21.Zhu B., Chen S., Hu X., Jin X., Le Y., Cao L., Yuan Z., Lin Z., Jiang S., Sun L., Yu L. Knockout of the Nogo-B gene attenuates tumor growth and metastasis in hepatocellular carcinoma. Neoplasia. 2017;19:583–593. doi: 10.1016/j.neo.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao W., Zhou S., Xu H., Li H., He G., Liu Y., Qi Y. Nogo-B promotes the epithelial-mesenchymal transition in HeLa cervical cancer cells via Fibulin-5. Oncol. Rep. 2013;29:109–116. doi: 10.3892/or.2012.2069. [DOI] [PubMed] [Google Scholar]
- 23.Wang X., Yang Y., Zhao D., Zhang S., Chen Y., Chen Y., Feng K., Li X., Han J., Iwakiri Y., et al. Inhibition of high-fat diet-induced obesity via reduction of ER-resident protein Nogo occurs through multiple mechanisms. J. Biol. Chem. 2022;298 doi: 10.1016/j.jbc.2022.101561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang S., Guo F., Yu M., Yang X., Yao Z., Li Q., Wei Z., Feng K., Zeng P., Zhao D., et al. Reduced Nogo expression inhibits diet-induced metabolic disorders by regulating ChREBP and insulin activity. J. Hepatol. 2020;73:1482–1495. doi: 10.1016/j.jhep.2020.07.034. [DOI] [PubMed] [Google Scholar]
- 25.Kemp S.B., Carpenter E.S., Steele N.G., Donahue K.L., Nwosu Z.C., Pacheco A., Velez-Delgado A., Menjivar R.E., Lima F., The S., et al. Apolipoprotein E promotes immune suppression in pancreatic cancer through NF-κB-mediated production of CXCL1. Cancer Res. 2021;81:4305–4318. doi: 10.1158/0008-5472.Can-20-3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gong P., Jia H.Y., Li R., Ma Z., Si M., Qian C., Zhu F.Q., Sheng-Yong L. Downregulation of Nogo-B ameliorates cerebral ischemia/reperfusion injury in mice through regulating microglia polarization via TLR4/NF-kappaB pathway. Neurochem. Int. 2023;167 doi: 10.1016/j.neuint.2023.105553. [DOI] [PubMed] [Google Scholar]
- 27.Nagarajan A., Dogra S.K., Sun L., Gandotra N., Ho T., Cai G., Cline G., Kumar P., Cowles R.A., Wajapeyee N. Paraoxonase 2 facilitates pancreatic cancer growth and metastasis by stimulating GLUT1-mediated glucose transport. Mol. Cell. 2017;67:685–701.e6. doi: 10.1016/j.molcel.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J., Zhong Q., Zhang H., Liu S., Li S., Xia T., Xiao Z., Chen R., Ye Y., Liang F., et al. Nogo-B promotes invasion and metastasis of nasopharyngeal carcinoma via RhoA-SRF-MRTFA pathway. Cell Death Dis. 2022;13:76. doi: 10.1038/s41419-022-04518-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawaguchi N., Tashiro K., Taniguchi K., Kawai M., Tanaka K., Okuda J., Hayashi M., Uchiyama K. Nogo-B (Reticulon-4B) functions as a negative regulator of the apoptotic pathway through the interaction with c-FLIP in colorectal cancer cells. Biochim. Biophys. Acta, Mol. Basis Dis. 2018;1864:2600–2609. doi: 10.1016/j.bbadis.2018.04.018. [DOI] [PubMed] [Google Scholar]
- 30.Tian Y., Yang B., Qiu W., Hao Y., Zhang Z., Yang B., Li N., Cheng S., Lin Z., Rui Y.C., et al. ER-residential Nogo-B accelerates NAFLD-associated HCC mediated by metabolic reprogramming of oxLDL lipophagy. Nat. Commun. 2019;10:3391. doi: 10.1038/s41467-019-11274-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitcomb D.C., Shelton C.A., Brand R.E. Genetics and genetic testing in pancreatic cancer. Gastroenterology. 2015;149:1252–1264.e4. doi: 10.1053/j.gastro.2015.07.057. [DOI] [PubMed] [Google Scholar]
- 32.Bardeesy N., DePinho R.A. Pancreatic cancer biology and genetics. Nat. Rev. Cancer. 2002;2:897–909. doi: 10.1038/nrc949. [DOI] [PubMed] [Google Scholar]
- 33.Dooley J., Lagou V., Goveia J., Ulrich A., Rohlenova K., Heirman N., Karakach T., Lampi Y., Khan S., Wang J., et al. Heterogeneous effects of calorie content and nutritional components underlie dietary influence on pancreatic cancer susceptibility. Cell Rep. 2020;32 doi: 10.1016/j.celrep.2020.107880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gong K., Zhang Z., Chen S.S., Zhu X.R., Wang M.Y., Yang X.Y., Ding C., Han J.H., Li Q.S., Duan Y.J. 6-Methyl flavone inhibits Nogo-B expression and improves high fructose diet-induced liver injury in mice. Acta Pharmacol. Sin. 2023;44:2216–2229. doi: 10.1038/s41401-023-01121-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang S., Yu M., Guo F., Yang X., Chen Y., Ma C., Li Q., Wei Z., Li X., Wang H., et al. Rosiglitazone alleviates intrahepatic cholestasis induced by α-naphthylisothiocyanate in mice: The role of circulating 15-deoxy-Δ(12,14) -PGJ(2) and Nogo. Br. J. Pharmacol. 2020;177:1041–1060. doi: 10.1111/bph.14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J., Ye C., Chen C., Xiong H., Xie B., Zhou J., Chen Y., Zheng S., Wang L. Glucose transporter GLUT1 expression and clinical outcome in solid tumors: a systematic review and meta-analysis. Oncotarget. 2017;8:16875–16886. doi: 10.18632/oncotarget.15171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ancey P.B., Contat C., Boivin G., Sabatino S., Pascual J., Zangger N., Perentes J.Y., Peters S., Abel E.D., Kirsch D.G., et al. GLUT1 expression in tumor-associated neutrophils promotes lung cancer growth and resistance to radiotherapy. Cancer Res. 2021;81:2345–2357. doi: 10.1158/0008-5472.Can-20-2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao H., Wang J., Yan W., Cui Y., Chen Z., Gao X., Wen X., Chen J. GLUT1 regulates cell glycolysis and proliferation in prostate cancer. Prostate. 2018;78:86–94. doi: 10.1002/pros.23448. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Z., Li X., Yang F., Chen C., Liu P., Ren Y., Sun P., Wang Z., You Y., Zeng Y.X., Li X. DHHC9-mediated GLUT1 S-palmitoylation promotes glioblastoma glycolysis and tumorigenesis. Nat. Commun. 2021;12:5872. doi: 10.1038/s41467-021-26180-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y., Tang S., Shi X., Lv J., Wu X., Zhang Y., Wang H., He J., Zhu Y., Ju Y., et al. Metabolic classification suggests the GLUT1/ALDOB/G6PD axis as a therapeutic target in chemotherapy-resistant pancreatic cancer. Cell Rep. Med. 2023;4 doi: 10.1016/j.xcrm.2023.101162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X., Liu Y., Han A., Tang C., Xu R., Feng L., Yang Y., Chen L., Lin Z. The NQO1/p53/SREBP1 axis promotes hepatocellular carcinoma progression and metastasis by regulating Snail stability. Oncogene. 2022;41:5107–5120. doi: 10.1038/s41388-022-02477-6. [DOI] [PubMed] [Google Scholar]
- 42.Chen J., Ding C., Chen Y., Hu W., Yu C., Peng C., Feng X., Cheng Q., Wu W., Lu Y., et al. ACSL4 reprograms fatty acid metabolism in hepatocellular carcinoma via c-Myc/SREBP1 pathway. Cancer Lett. 2021;502:154–165. doi: 10.1016/j.canlet.2020.12.019. [DOI] [PubMed] [Google Scholar]
- 43.Zhang N., Zhang H., Liu Y., Su P., Zhang J., Wang X., Sun M., Chen B., Zhao W., Wang L., et al. SREBP1, targeted by miR-18a-5p, modulates epithelial-mesenchymal transition in breast cancer via forming a co-repressor complex with Snail and HDAC1/2. Cell Death Differ. 2019;26:843–859. doi: 10.1038/s41418-018-0158-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li X., Liu Z., Xia C., Yan K., Fang Z., Fan Y. SETD8 stabilized by USP17 epigenetically activates SREBP1 pathway to drive lipogenesis and oncogenesis of ccRCC. Cancer Lett. 2022;527:150–163. doi: 10.1016/j.canlet.2021.12.018. [DOI] [PubMed] [Google Scholar]
- 45.Gao Y., Nan X., Shi X., Mu X., Liu B., Zhu H., Yao B., Liu X., Yang T., Hu Y., Liu S. SREBP1 promotes the invasion of colorectal cancer accompanied upregulation of MMP7 expression and NF-κB pathway activation. BMC Cancer. 2019;19:685. doi: 10.1186/s12885-019-5904-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Y., Cai J., Yang X., Wang K., Sun K., Yang Z., Zhang L., Yang L., Gu C., Huang X., et al. Dysregulated m6A modification promotes lipogenesis and development of non-alcoholic fatty liver disease and hepatocellular carcinoma. Mol. Ther. 2022;30:2342–2353. doi: 10.1016/j.ymthe.2022.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.You M., Jin J., Liu Q., Xu Q., Shi J., Hou Y. PPARα promotes cancer cell Glut1 transcription repression. J. Cell. Biochem. 2017;118:1556–1562. doi: 10.1002/jcb.25817. [DOI] [PubMed] [Google Scholar]
- 48.Khurana N., Dodhiawala P.B., Bulle A., Lim K.H. Deciphering the role of innate immune NF-κB pathway in pancreatic cancer. Cancers. 2020;12 doi: 10.3390/cancers12092675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nomura A., Gupta V.K., Dauer P., Sharma N.S., Dudeja V., Merchant N., Saluja A.K., Banerjee S. NFκB-mediated invasiveness in CD133(+) pancreatic TICs is regulated by autocrine and paracrine activation of IL1 signaling. Mol. Cancer Res. 2018;16:162–172. doi: 10.1158/1541-7786.Mcr-17-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao D., Xue C., Yang Y., Li J., Wang X., Chen Y., Zhang S., Chen Y., Duan Y., Yang X., Han J. Lack of Nogo-B expression ameliorates PPARγ deficiency-aggravated liver fibrosis by regulating TLR4-NF-κB-TNF-α axis and macrophage polarization. Biomed. Pharmacother. 2022;153 doi: 10.1016/j.biopha.2022.113444. [DOI] [PubMed] [Google Scholar]
- 51.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.