

Abstract
Observational studies in Parkinson’s disease (PD) deeply characterize relatively small numbers of participants. The Molecular Integration in Neurological Diagnosis Initiative seeks to characterize molecular and clinical features of every PD patient at the University of Pennsylvania (UPenn). The objectives of this study are to determine the feasibility of genetic characterization in PD and assess clinical features by sex and GBA1/LRRK2 status on a clinic-wide scale. All PD patients with clinical visits at the UPenn PD Center between 9/2018 and 12/2022 were eligible. Blood or saliva were collected, and a clinical questionnaire administered. Genotyping at 14 GBA1 and 8 LRRK2 variants was performed. PD symptoms were compared by sex and gene groups. 2063 patients were approached and 1,689 (82%) were enrolled, with 374 (18%) declining to participate. 608 (36%) females were enrolled, 159 (9%) carried a GBA1 variant, and 44 (3%) carried a LRRK2 variant. Compared with males, females across gene groups more frequently reported dystonia (53% vs 46%, p = 0.01) and anxiety (64% vs 55%, p < 0.01), but less frequently reported cognitive impairment (10% vs 49%, p < 0.01) and vivid dreaming (53% vs 60%, p = 0.01). GBA1 variant carriers more frequently reported anxiety (67% vs 57%, p = 0.04) and depression (62% vs 46%, p < 0.01) than non-carriers; LRRK2 variant carriers did not differ from non-carriers. We report feasibility for near-clinic-wide enrollment and characterization of individuals with PD during clinical visits at a high-volume academic center. Clinical symptoms differ by sex and GBA1, but not LRRK2, status.
Subject terms: Parkinson's disease, Genetic testing, Translational research, Neurological manifestations
Introduction
Parkinson’s disease (PD) affects >6 million people worldwide, making it the second most common neurodegenerative disease, affecting 1–3% of people over age 651–3. The disorder is characterized by bradykinesia plus tremor or rigidity4; PD can also affect mood, cognition, sleep, and autonomic function, collectively referred to as non-motor symptoms5.
The occurrence of motor complications such as fluctuations in medication responsiveness, dyskinesia, and dystonia, as well as non-motor symptoms, varies from person to person and over the disease course. The frequency and impact of motor and non-motor symptoms of PD have been demonstrated in research cohorts, where they have been shown to vary with sex, age, and with variants in GBA1 or LRRK2, two common PD risk genes6–13.
The real-world experience of patients with PD may differ from what has been described in research studies, which may suffer from selection bias, recruiting those individuals with the fewest barriers to participation14–16. Indeed, minorities and women are under-represented in PD research cohorts, consistent with this bias17. A clinic-wide approach to assessments of motor and non-motor features, capturing all patients at the time of their clinical visit, may address this selection bias to give a more accurate real-world accounting of PD symptom frequency. We hypothesized that interest in research enrollment would be high among PD patients when typical barriers to enrollment such as transportation and time were reduced, and that sex- and gene-defined groups would report different frequencies in motor and non-motor symptoms of PD and medication use at the clinic-wide level.
Here we report the results of a clinic-wide research recruitment at a single academic center. The Molecular Integration in Neurological Diagnosis (MIND) initiative aimed to approach every patient with PD seen at the University of Pennsylvania (UPenn) Parkinson’s Disease and Movement Disorders Center (PDMDC). Patients were approached for enrollment at their clinical visit, and enrolled participants provided information about their PD symptoms, a blood or saliva sample, as well as optional consent to allow for access to their medical record, recontact in the future for additional studies for which they may be eligible, and future use of their blood or saliva samples for additional research. We present our findings from 1689 participants enrolled, representing 82% of the 2063 patients approached for enrollment. Variants in GBA1 and LRRK2 were identified, and clinical symptoms were analyzed by sex and gene status. Our study demonstrates feasibility of near-clinic-wide research enrollment and clinical and genetic characterization.
Results
Participant enrollment
Participants were enrolled between September 7, 2018, and December 23, 2022. During this period, 19 Movement Disorders neurologists or nurse practitioners completed in-person or telemedicine patient visits for 3010 unique PD patients at the PDMDC. Of 3010 established PD patients, 1 (0.03%) did not meet enrollment criteria (<21 years old), and 946 (31%) were not approached due to logistical reasons (staffing, room availability, unsuccessful patient contact within window followed by patient death or no longer following at UPenn, visits too late in the day for same-day sample processing, among others). 2063 patients were successfully captured at the time of their visit and offered enrollment in the MIND cohort, representing 69% of the UPenn PDMDC PD population. Three hundred and seventy-four patients (18% of patients offered enrollment) declined enrollment, while 1689 patients (82% of PD patients offered enrollment or 56% of all established PD patients) consented to enrollment. A summary of enrollment as well as clinical data, blood and saliva collection, and optional consent can be seen in Fig. 1.
Fig. 1. Summary of enrollment in the molecular integration in neurological diagnosis (MIND) Parkinson’s Disease cohort.

PDMDC = Parkinson’s Disease and Movement Disorders Center at the University of Pennsylvania. EMR = Electronic Medical Record. *Participants were not offered enrollment due to logistical reasons (staffing, room availability, unsuccessful patient contact within window followed by patient death or no longer following at UPenn, visits too late in the day for same-day sample processing, among others).
Demographics and cohort characteristics
Table 1 provides a summary of cohort demographics stratified by gene status. Among 1689 PD participants, 159 GBA1 and 44 LRRK2-variant carriers were identified. Three individuals (<1% of cohort) carried variants in both GBA1 and LRRK2 (2 GBA1 N409S/LRRK2 G2019S carriers, and one GBA1 N409S/LRRK2 homozygous G2019S variant carrier). Three (<1%) LRRK2 G2019S homozygotes were identified (including the GBA1 N409S co-carrier/LRRK2 G2019S), and 3 (<1%) GBA1 homozygotes were identified (1 E365K/E365K, 1 N409S/N409S, 1 T408M/T408M). One participant carried the GBA1 H294Q and D448H variants, and 1 Rec1 carrier (carrying the L483P, A495P, and Val499= variants) was identified. See Supplementary Fig. 1 and Supplementary Table 2 for summaries of GBA1 and LRRK2 testing results.
Table 1.
Cohort demographics
| Whole Cohort | GBA1/LRRK2 Negativeb | GBA1 Carriers | p | LRRK2 Carriers | p | |
|---|---|---|---|---|---|---|
| N (%) | 1689 | 1483 | 159c | 44c | ||
| Variant, n (%) | ||||||
| GBA1 Risk | 94 (59) | |||||
| GBA1 Mild | 42 (26) | |||||
| GBA1 Severe | 23 (14) | |||||
| LRRK2 G2019S | 41 (93)d | |||||
| Sex, n (%)a | ||||||
| Female | 608 (36) | 517 (35) | 67 (42) | 0.081 | 23 (51) | 0.048 |
| Male | 1080 (64) | 965 (65) | 92 (58) | 21 (49) | ||
| Ethnicity, n (%) | ||||||
| Hispanic or Latino | 34 (2) | 31 (2) | 2 (1) | nt | 1 (2) | nt |
| Not Hispanic or Latino | 1654 (98) | 1451 (98) | 157 (99) | 43 (98) | ||
| Unknown or not reported | 1 (<1) | 1 (<1) | 0 (0) | 0 (0) | ||
| Race, n (%) | ||||||
| White | 1532 (91) | 1334 (90) | 155 (98) | nt | 40 (91) | nt |
| Black or African American | 78 (5) | 76 (5) | 2 (1) | 0 (0) | ||
| Am Indian/Alaska Native | 1 (<1) | 1 (<1) | 0 (0) | 0 (0) | ||
| Asian | 56 (3) | 54 (4) | 0 (0) | 2 (4) | ||
| More than one race | 9 (<1) | 6 (<1) | 2 (1) | 1 (2) | ||
| Other | 5 (<1) | 4 (<1) | 0 (0) | 1 (2) | ||
| Unknown or not reported | 8 (<1) | 8 (1) | 0 (0) | 0 (0) | ||
| Age, mean (SD) | ||||||
| At diagnosis | 62 (11) | 62 (11) | 61 (10) | 0.215 | 61 (11) | 0.347 |
| At enrollment | 69 (9) | 69 (9) | 67 (9) | 0.049 | 70 (9) | 0.683 |
| Clinical Presentation at Onset | ||||||
| Tremor | 865 (51) | 764 (52) | 77 (49) | 0.482 | 22 (50) | 0.482 |
| Gait Disorder | 191 (11) | 170 (11) | 18 (11) | 2 (5) | ||
| Mixed | 120 (7) | 101 (7) | 16 (10) | 3 (7) | ||
| Neither | 513 (31) | 448 (30) | 48 (30) | 17 (38) | ||
| 1st Degree Family History | ||||||
| None | 1401 (83) | 1241 (84) | 128 (80) | 0.225 | 30 (68) | 0.002 |
| PD | 155 (9) | 122 (8) | 20 (13) | 12 (27) | ||
| Other NDD | 108 (6) | 99 (7) | 8 (5) | 1 (2) | ||
| Both | 25 (2) | 21 (1) | 3 (2) | 1 (2) | ||
GBA1 (N = 3) and LRRK2 (N = 2) homozygotes and 1 individual carrying two different variants in GBA1 were included.
a1 missing/unreported. Nt not tested.
bIncludes 28 cases without DNA available. Box indicates corrected p < 0.05 relative to GBA1/LRRK2 Negative group. P values after Benjamini-Hochberg multiple testing correction are reported.
cThree individuals carried variants in both GBA1 and LRRK2 (all were GBA1 N409S/LRRK2 G2019S) and are excluded. NDD neurodegenerative disease.
d3 LRRK2 G2385R carriers were identified (6% of LRRK2 carriers).
Significant differences among groups are indicated in bold type.
GBA1 and LRRK2 Variant Carriers: Sex and Family History
While the entire MIND cohort was enriched with males (64% of whole cohort), LRRK2 G2019S carriers with PD differed in the proportion of males to females compared to non-carriers (exact p = 0.024, corrected p = 0.048); 49%, 95% CI 26–93, of LRRK2 G2019S carriers were males. LRRK2 G2019S carriers (N = 44) were more likely to report having a 1st degree family member with PD (exact p = 0.001, corrected p = 0.002) than GBA1 non-carriers or LRRK2 non-carriers (N = 1483). GBA1 carriers had a lower age at study enrollment compared to GBA1 non-carriers or LRRK2 non-carriers (z = −2.42, exact p = 0.025, corrected p = 0.049). No differences were detected between GBA1 carriers and GBA1 non-carriers or LRRK2 non-carriers in frequency of sex, age at diagnosis, presentation at onset, and family history of neurodegenerative diseases. No differences were detected between LRRK2 carriers and GBA1 non-carriers or LRRK2 non-carriers in age at diagnosis or enrollment or clinical presentation at onset (Table 1).
Parkinson’s disease motor and non-motor features on a clinic-wide scale
Participants were asked to report on their current or prior history of 12 different motor and non-motor symptoms of PD. Among enrolled participants, 1680 (99% of cohort) responses were collected. The most frequently reported symptoms were fatigue (69% currently experiencing), anxiety (50%), and vivid dreaming (47%), while the least common symptoms were compulsive behaviors (9%), psychosis (15%) and depression (37%, Fig. 2).
Fig. 2. Motor and non-motor Parkinson’s disease symptoms.
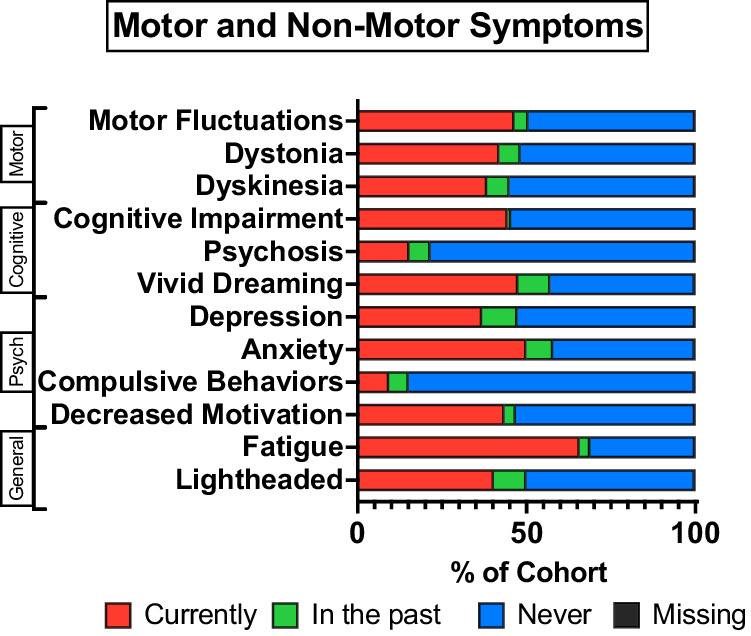
Self-reported motor and non-motor symptoms from N = 1689 Parkinson’s Disease participants. Two missing due to incomplete clinical questionnaire. Psych, Psychological.
Females and males differ in the frequency of motor and non-motor symptoms
Females (N = 608), accounting for 36% of the MIND cohort, reported a higher frequency of current or prior dystonia (53%, 95% CI = 49–57, exact p = 0.003, corrected p = 0.012) and anxiety (64%, 95% CI = 60–68, exact p = 0.0002, corrected p = 0.002) compared to males (N = 1080, 46%, 95% CI = 43–49 and 55%, 95%CI = 52–58, respectively). Additionally, females reported a lower frequency of current or past cognitive impairment (40%, 95% CI = 36–44, exact p = 0.0003, corrected p = 0.002) and vivid dreaming (53%, 95% CI = 49–57, exact p = 0.004, corrected p = 0.012) compared to males (49%, 95% CI = 46–52, and 60%, 95% CI = 57–63, respectively). The frequencies of depression and lightheadedness did not differ between females and males after correction for multiple testing (exact p = 0.03 and 0.03, corrected p = 0.06 and 0.07, respectively). All other motor and non-motor symptom frequencies did not differ between females and males (Fig. 3A).
Fig. 3. Motor and non-motor Parkinson’s disease symptoms by sex, GBA1, and LRRK2 variant status.

Responses grouped as ever (currently or in the past) or never experiencing a symptom. Heterozygote and homozygote carriers are combined. GBA/LRRK2 co-carriers omitted (N = 3). *p < 0.05, **p < 0.01. Error bars represent 95% CI. Panel A compares sexes, Panel B compares groups by GBA1 status, and Panel C compares groups by LRRK2 status.
GBA1 variant carriers report a higher frequency of depression and anxiety than non-carriers
Carriers of variants in GBA1 (N = 159), accounting for 9% of the MIND cohort, reported a higher frequency of current or past depression (63%, 95% CI = 55–70, exact p = 0.00003, corrected p = 0.0004) or anxiety (69%, 95% CI = 61–75, exact p = 0.006, corrected p = 0.036), compared to GBA1/LRRK2 non-carriers (N = 1483, 46%, 95% CI = 43–48, and 57%, 95% CI = 55–60, respectively). The frequency of psychosis did not differ between GBA1 carriers and those not carrying a variant in GBA1 after correcting for multiple testing (exact p = 0.02, corrected p = 0.054). Other motor and non-motor symptoms did not differ between GBA1 carriers and those not carrying a variant in GBA1 (Fig. 3B). Among GBA1 variant carriers, no differences in motor and non-motor symptoms were detected comparing individuals carrying risk, mild, or severe GBA1 variants (categories detailed in Supplementary Table 1 and Supplementary Fig. 2).
LRRK2 variant carriers did not differ from non-carriers in motor and non-motor symptoms
No differences in motor and non-motor symptoms were detected between individuals carrying the G2019S LRRK2 variant (N = 44, 3% of cohort) and those not carrying a variant in GBA1 or LRRK2 (N = 1483, Fig. 3C). The frequency of vivid dreaming comparing LRRK2 variant carriers with GBA1/LRRK2 non-carriers did not differ after correcting for multiple testing (exact p = 0.02, corrected p = 0.20).
Use of common PD medications on a clinic-wide scale
All active prescriptions for PD-related medications at the enrollment visit were extracted from the medical record (N = 1674, 15 participants did not consent to future access to their medical record). Medications were grouped by mechanism of action, comprising all formulations of levodopa, dopamine agonists, monoamine oxidase-B inhibitors, catechol-o-methyltransferase inhibitors, anticholinergics, amantadine, adenosine A2A antagonists, acetylcholinesterase inhibitors, and antipsychotics (including pimavanserin, quetiapine, and clozapine). Females were less likely to be prescribed a PD-related medication (8%, 95% CI = 6–11 of females not taking a PD medication, compared to 5%, 95% CI = 4–6 of males, exact p = 0.006, corrected p = 0.030), while males were prescribed acetylcholinesterase inhibitors more frequently than females (12%, 95% CI = 10–14 versus 5%, 95% CI = 4–7, respectively, exact p = 0.000006, corrected p = 0.00006). The prescription frequencies of all other medications did not differ between males and females, or between GBA1 or LRRK2 variant carriers and non-carriers (Table 2).
Table 2.
Medication summary
| Whole Cohort | Male | Female | GBA1/LRRK2 Negative | GBA1 Carrier | LRRK2 Carrier | |
|---|---|---|---|---|---|---|
| N (%) | 1674a | 1070 | 604 | 1469 | 158b | 44b |
| None | 104 (6) | 53 (5) | 51 (8) | 88 (5) | 15 (1) | 1 (<1) |
| Levodopa | 1455 (87) | 944 (88) | 511 (85) | 1281 (87) | 134 (85) | 38 (86) |
| DA agonist | 356 (21) | 219 (21) | 137 (23) | 310 (21) | 28 (18) | 17 (39) |
| MAOB-I | 462 (28) | 311 (29) | 151 (25) | 409 (28) | 36 (23) | 16 (36) |
| COMT Inh | 86 (5) | 58 (5) | 28 (5) | 73 (5) | 9 (6) | 4 (9) |
| Anticholinergic | 31 (2) | 23 (2) | 8 (1) | 26 (2) | 5 (3) | 0 (0) |
| Amantadine | 327 (20) | 201 (19) | 126 (21) | 288 (20) | 27 (17) | 11 (25) |
| A2A Ant | 10 (1) | 8 (1) | 2 (<1) | 9 (1) | 0 (0) | 1 (2) |
| AChEI | 159 (10) | 127 (12) | 32 (5) | 136 (9) | 22 (14) | 1 (2) |
| Antipsychotic | 86 (5) | 51 (5) | 35 (6) | 73 (5) | 11 (7) | 2 (5) |
aN = 15 did not permit access to the electronic medical record.
bThree individuals carried variants in both GBA1 and LRRK2 (all were GBA1 N409S/LRRK2 G2019S) and are excluded. DA Dopamine, MAOB-I Monoamine oxidase B inhibitor, COMT catechol-o-methyltransferase, A2A Ant Adenosine A2A receptor antagonist, AChEI Acetylcholinesterase inhibitor. Box indicates corrected p < 0.05.
Significant differences among groups are indicated in bold type.
Discussion
Here we report the summary results of the MIND PD Cohort Study, which aimed to characterize molecular and clinical features of every PD patient in a large academic movement disorders center. A total of 2063 of 3010 unique PD patients who received care at the PDMDC between September 7, 2018, and December 23, 2022, were successfully approached at their clinical visit, and 1689 (82% of those approached) enrolled in the MIND cohort. The overwhelming majority of enrollees consented to access to the EMR (99% of 1689 enrolled), contact for future studies and possible genetic disclosure (99% of 1689 enrolled), and use of biosamples for future studies (96% of 1689 enrolled). Among enrollees, 159 (9%) harbored a GBA1 variant, and 44 (3%) harbored a LRRK2 variant.
Despite the unexpected transition of an active movement disorders practice to telemedicine-based visits during the COVID-19 pandemic, our all-comers approach continued to be highly efficient for enrolling participants, minimizing selection bias based on sex or self-described race. Continuing the trends reported in our interim analysis18, the percentage of participants who were female (36%) and those who identified as non-White (9%) were significantly higher than for prior genetic research studies from our same clinical center pre-dating this all-comers-enrollment strategy (32% female and <4% non-White in prior studies)19. Our findings suggest that a clinic-wide approach to participant capture at the time of the office visit minimizes barriers to research participation, mitigating selection bias. With 82% of all PD individuals approached consenting to enrollment in a single-visit, our study also provides evidence for widespread interest in research participation when barriers are reduced.
The existing landscape of PD cohort studies comprises “deep and narrow” cohorts vs. “shallow and wide” cohorts. In the former situation, exemplified by the international Parkinson’s Progression Marker Initiative (PPMI) De Novo PD cohort20, the Penn-based U19 cohort21, and others22–24, a small fraction of a total clinic population is extensively characterized. In the latter situation, exemplified by Fox Insight25 or the 23andMe PD cohort26, thousands of individuals are enrolled, but PD status is not clinically confirmed and information such as medication use is self-reported only. The MIND cohort is unique in capturing clinically confirmed PD patients, with EMR-confirmed medication use, while maintaining broad capture of a total clinical population. Indeed, of 3010 possible PD patients who received clinical care from 19 providers over a span of 52 months, the MIND study enrolled 1689 (56% of total clinic).
As a consequence, the MIND study provides a global picture of PD at the clinic-wide level, highlighting intriguing sex- and gene-based differences. For example, our finding that women with PD are more likely to report anxiety, but less likely to report cognitive impairment or vivid dreaming than men extends prior work suggesting sex-based differences in PD non-motor symptoms7–9, reviewed in27,28. Similarly, we find significant sex differences comparing PD individuals who do versus do not carry the LRRK2 G2019S variant. Our finding supports prior work in targeted populations enriched for LRRK2 variants29–31, suggesting sex-based differences in LRRK2-associated PD (or, more precisely, lack of sex-based differences generally observed in PD in this genetically-defined group). We note, however, that an important consideration in these cases is the breadth of our current study. Specifically, the sex-based differences in LRRK2 G2019S PD are found in a clinic-wide study vs. one designed to evaluate a targeted genetic population. Similarly, prior reports of sex-based differences in symptoms among PD individuals have generally studied many fewer patients (ranging from 85 to 569 individuals in a recent review of this topic)28. Likewise, genetic screening in large cohort studies is often limited to few variants, and those variants are often overrepresented in specific ancestral populations that can confound clinical-genetic associations11. Clinical testing, historically a large source of genetic information, is often limited to certain populations (young onset or ancestral populations) limiting the implications beyond these groups. Despite this limitation, prior research and clinical genetic screening efforts have reported differences in age at onset, rate of disease progression, and motor and non-motor complications in GBA1 and LRRK2 carriers compared to non-carriers32,33. An unbiased approach to genetic and molecular characterization limits ascertainment bias and improves external validity for these prior studies. Thus, the clinic-wide capture utilizing multiple gene variant screening afforded by the MIND study greatly amplifies the importance of these sex- and gene-based differences. Moreover, differences are small, such that it may be difficult to predict based on phenotypic characteristics which individual PD patients carry LRRK2 or GBA1 variants. Thus, global screening may be preferred, and our study demonstrates feasibility.
We acknowledge our study’s limitations. First, while we have captured greater than 50% of our entire clinic population, it is still possible that the MIND study suffers from selection bias. We note, however, that frequency for sex (36% women) among MIND enrollees does not differ from frequencies for the total clinic population (38% women for 3010 possible PD patients seen at the PDMDC during MIND enrollment period). Regarding race, while we have more than doubled the representation of participants self-identifying as non-White, compared to historical Penn-based PD cohorts (9% compared to 4%), 21% of PDMDC patients identify as non-White, representing a potential for bias in these results as well as an opportunity for ongoing enrollment to address this disparity. Second, clinical features described in this study are by self-report only, which could impact some of our findings. For example, GBA1 carriers were not more likely to report cognitive impairment or psychosis in this study, deviating from prior reports34. In contrast, our significant findings relating to anxiety, for example, pertain to subjective, but important, symptoms, thus increasing the likelihood that our survey-based approach captures accurate information. Third, as a single-site study, it is unclear whether the high efficiency in enrollment seen here will be widely reproducible. We note, however, that the PDMDC clinical site encompasses the practices of 19 movement disorders specialists, and more than one-third of the MIND cohort enrollment period was impacted by the COVID-19 pandemic, offering confidence that rates of uptake for the global capture approach reported here are robust to practice style and can withstand even an unforeseen, once-in-a-century event.
In summary, we demonstrate that a clinic-wide approach to capturing genetic and clinical data at the time of the clinical visit is feasible in a busy clinical setting. As such, the MIND study offers a model for how genetic information might enter the clinic at scale.
Methods
Participant recruitment and enrollment
This study was approved by the University of Pennsylvania Institutional Review Board, and informed consent was obtained at study enrollment. Clinical research coordinators screened the electronic medical record (EMR) and called eligible patients in advance of the PD&MDC visit. Patients were recruited from the movement disorders clinic at the Parkinson’s Disease and Movement Disorders Center (PDMDC) and enrolled on the day of their clinical visit. Patients were approached for enrollment at their office visit if they met the following inclusion criteria: (1) greater than or equal to 21 years of age, (2) a clinical diagnosis of PD by a movement disorder specialist, and (3) able to provide informed consent. Patients were excluded if they were unable to consent to research or were a part of a vulnerable population (e.g., pregnant and lactating women, prisoners). All patients with PD seen at the PDMDC were eligible if they met the inclusion and exclusion criteria above. Participants completed one 20–30-min study visit, where patients reviewed and signed an informed consent form, completed a brief clinical questionnaire, and provided a blood or saliva sample. The informed consent form included three optional consent statements that allowed the study staff to: (1) access the EMR for future research, (2) contact participants to offer additional research studies, and (3) store plasma and DNA as part of the UPenn Integrated Neurodegenerative Disease Database biobank. Due to the SARS-CoV-2 pandemic or by preference, some participants were enrolled remotely beginning September 2020: coordinators called eligible patients around the time of their telehealth visit, and if the patient was interested, they scheduled a 20–30-minute virtual study appointment. A saliva collection kit with pre-paid return shipment materials was sent to the patient’s home address. Patients met with the study staff through a secure audio video platform (Bluejeans or Zoom), reviewed and signed an electronic version of the informed consent form in REDCap, answered the clinical questionnaire, and completed an observed saliva collection. The saliva sample was securely packaged in a return envelope and returned to the study team for DNA extraction and genetic testing.
Study visit and electronic medical record extraction
Trained clinical research coordinators conducted the study visits as previously described18. Participants completed a questionnaire to ascertain demographic information, family history, and clinical symptoms, as previously described18. Data were directly entered into a REDCap electronic database hosted at UPenn18 or by paper at participant request. For all participants that allowed EMR access, medication records were extracted by matching the medical record number to determine current use of PD-related medications. Blood was drawn by peripheral venipuncture into sterile EDTA vacutainer tubes and processed on the day of collection according to previously published protocols35. Briefly, 4 ml blood were used for DNA extraction, and remaining blood was centrifuged, and plasma was aliquoted and frozen at –80 °C for future studies. Due to the SARS-CoV-2 pandemic or by preference, some participants were enrolled remotely by providing a saliva sample beginning in September 2020. Saliva samples were obtained in an Oragene-DNA OG 500 collection kit (DNA Genotek, Ontario Canada) and incubated at 55 °C for at least 2 h up to overnight to inactivate nucleases. DNA was extracted in a semi-automated QuickGene 610 L using the DNA whole blood kit following the manufacturer’s protocol (Autogen, Holliston, MA). For all participants who allowed EMR access, medication records were extracted by matching the medical record number to determine use of PD-related medications at the time of the enrollment visit.
Genetic testing
All participants were screened for 14 GBA1 and 8 LRRK2 variants on an expanded MIND assay (v2, Supplementary Table 1). The assay was designed as described previously with modifications to add 10 additional GBA1 variants18. Briefly, twenty-two SNPtype assays were designed by D3 Assay Design tool (Standard BioTools, San Francisco, CA). Allele-specific PCR was performed using either Flex Six or 48.48 genotyping Dynamic Array integrated fluidic circuits (IFCs) (Standard BioTools), and genotyping was carried out using the Biomark™ HD system (Standard BioTools) according to the manufacturer’s protocol. Specific target amplification (STA) was performed to enrich target sequences flanking the 22 SNPs using 40 ng genomic DNA and 50 nM of each STA and locus-specific primer pairs. Pre-amplified DNA was mixed with 2X Fast Probe Master mix (Biotium Inc., Fremont, CA), 20X SNP Type sample loading reagents, 60X SNP Type reagent, and ROX in 5 μl. Sample mix and 10X SNP Type assay mix was added into assay and sample inlets on the IFC, respectively, and loaded and mixed in the IFC controller. PCR was performed on the BioMark HD System under the following cycling conditions: thermal mix for 30 min at 70 °C followed by 10 min at 25 °C, hot start for 5 min at 95 °C, touchdown PCR for 15 s at 95 °C, 45 s from 64 °C–61 °C dropping 1 °C per cycle, and 15 s at 72 °C, and additional 34 cycles of 15 s at 95 °C, 45 s at 60 °C, and 15 s at 72 °C, and cooling for 10 s at 25 °C. The genotype call data were analyzed using BioMark Genotyping Analysis software which calculated the FAM and VIC relative fluorescence intensities and automatically called genotypes by the k-means clustering algorithm. Automatic genotype calls were reviewed in individual plots after cluster analysis. Positive calls were confirmed by either Taqman SNP genotyping assay or Sanger sequencing.
Statistical analysis
Statistical analysis was performed in Stata and in R-Studio36. Frequencies between groups were compared using χ2 or Fisher Exact Tests, while continuous variables were compared using the non-parametric Kruskal–Wallis Test. Benjamini-Hochberg correction for multiple testing was applied to all analyses. We chose the Benjamini-Hochberg procedure because it limits the false discovery rate, and the Bonferroni method is likely too conservative for outcomes that are not entirely independent37.
Supplementary information
Acknowledgements
The authors would like to acknowledge our patients for their generous participation in this study. T.F.T. and A.S.C.P. had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. T.F.T. conducted all data analysis. This study was supported by the NIH (R01 NS115139, P30 AG072979, U19 AG062418), and the Penn Center for Precision Medicine. T.F.T. is additionally supported by the NIH/NINDS (K23 NS114167). A.S.C.P. is additionally supported by the Parker Family Chair. Funding organizations did not participate in the design or conduct of this study.
Author contributions
T.F.T. Study design, data analysis design and execution, writing of the first manuscript draft and editing, data collection. W.H. Study design, data collection, editing of manuscript. N.A. Study design, data collection, editing of manuscript. D.B. Data collection, editing of manuscript. J.R. Study design, data collection, editing of manuscript. E.S. Genetic data collection and analysis, editing of manuscript. H.Z. Data collection, editing of manuscript. R.A.P. Study design, data collection, editing of manuscript. N.H. Study design, data collection, editing of manuscript. R.Z. Study design, data collection, editing of manuscript. E.M.B. Data collection, editing of manuscript. I.A. Data collection, editing of manuscript. J.J. Data collection, editing of manuscript. M.Sp. Clinical data collection, editing of manuscript. A.D. Clinical data collection, editing of manuscript. W.W.A. Clinical data collection, editing of manuscript. N.D. Clinical data collection, editing of manuscript. A.H. Clinical data collection, editing of manuscript. A.L. Clinical data collection, editing of manuscript. H.H. Clinical data collection, editing of manuscript. M.St. Clinical data collection, editing of manuscript. D.W. Clinical data collection, editing of manuscript. P.V. Clinical data collection, editing of manuscript. A.W.W. Clinical data collection, editing of manuscript. A.S. Clinical data collection, editing of manuscript. S.X.X. Data analysis review, editing of manuscript. V.V.D. Genetic data collection, study design, data analysis design, editing of manuscript. A.S.C.P. Data collection, study design, data analysis design, editing of manuscript, overall supervision.
Data availability
The datasets used and/or analyzed during the current study are found in the Supplementary and have been submitted to DbGaP (https://submit.ncbi.nlm.nih.gov/dbgap/regsys/54461/). The full protocol and participant informed consent form are available on Nature Protocol Exchange38. Additional supporting materials including standard operating procedures are available from the corresponding author on reasonable request.
Code availability
The R code used is found in the Supplementary.
Competing interests
T.F.T. No relevant disclosures or COIs. T.F.T. has received grants from the NIH (K23-NS114167), and research support from the Parkinson Foundation and The Michael J Fox Foundation. He has received consulting fees and honoraria from Sanofi Genzyme, Bial, and the Parkinson Foundation. W.H. No relevant disclosures or COIs. N.A. No relevant disclosures or COIs. D.B. No relevant disclosures or COIs. J.R. No relevant disclosures or COIs. E.S. No relevant disclosures or COIs. H.Z. No relevant disclosures or COIs. R,A.P. No relevant disclosures or COIs. N.H. No relevant disclosures or COIs. R.Z. No relevant disclosures or COIs. E.M.B. No relevant disclosures or COIs. I.A. No relevant disclosures or COIs. J.J. No relevant disclosures or COIs. M.Sp. No relevant disclosures or COIs. A.D. No relevant disclosures or COIs. A.D. has received clinical trial funding from Teva, Lundbeck, Cerevel, Eli Lilly, and Annovis Bio, royalties from UpToDate and Elsevier and serves on advisory boards for Laboratorios LAM, Supernus, Abbvie. W.A.A. No relevant disclosures or COIs. W.A.A. has received grant from the American Academy of Neurology (CRTS in Neurodisparities). N.D. No relevant disclosures or COIs. N.D. receives grant support from the NIH (R01NS125294, U19AG062418), Michael J Fox Foundation, Parkinson Foundation, Genentech (clinical trial). She has received honoraria from Mediflix and consulting fees from Genentech. She has reviewed medical records for Post & Schell (law firm). A.H. No relevant disclosures or COIs. A.H. has received grants from the National Eye Institute and the Michael J. Fox Foundation and clinical trial support from Biogen and Biohaven. A.L. No relevant disclosures or COIs. H.H. No relevant disclosures or COIs. M.St. No relevant disclosures or COIs. M.St. has received consulting fees from Neuroderm, Alexion, Biogen and Mediflix. D.W. No relevant disclosures or COIs. P.V. No relevant disclosures or COIs. A.W.W. No relevant disclosures or COIs. A.S. No relevant disclosures or COIs. S.X.X. No relevant disclosures or COIs. V.V.D. No relevant disclosures or COIs. A.S.C.P. No relevant disclosures or COIs. A.S.C.P. has received funding from the NIH, the Chan Zuckerberg Initiate Neurodegeneration Challenge, the AHA/Allen Institute Brain Health Initiative, and the Parker Family Chair. She also receives royalties from a patent relating to AAV-mediated approaches to treating frontotemporal dementia.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41531-024-00690-6.
References
- 1.Tanner CM, Goldman SM. Epidemiology of movement-disorders. Curr. Opin. Neurol. 1994;7:340–345. doi: 10.1097/00019052-199408000-00011. [DOI] [PubMed] [Google Scholar]
- 2.de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5:525–535. doi: 10.1016/S1474-4422(06)70471-9. [DOI] [PubMed] [Google Scholar]
- 3.Marras C, et al. Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis. 2018;4:21. doi: 10.1038/s41531-018-0058-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Postuma RB, et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015;30:1591–1601. doi: 10.1002/mds.26424. [DOI] [PubMed] [Google Scholar]
- 5.Martinez-Martin P, Rodriguez-Blazquez C, Kurtis MM, Chaudhuri KR. The impact of non-motor symptoms on health-related quality of life of patients with Parkinson’s disease. Mov. Disord. 2011;26:399–406. doi: 10.1002/mds.23462. [DOI] [PubMed] [Google Scholar]
- 6.Chaudhuri KR, Healy DG, Schapira AH. Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol. 2006;5:235–245. doi: 10.1016/S1474-4422(06)70373-8. [DOI] [PubMed] [Google Scholar]
- 7.Picillo M, et al. Sex-related longitudinal change of motor, non-motor, and biological features in early Parkinson’s disease. J. Parkinsons Dis. 2022;12:421–436. doi: 10.3233/JPD-212892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Solla P, et al. Gender differences in motor and non-motor symptoms among Sardinian patients with Parkinson’s disease. J. Neurol. Sci. 2012;323:33–39. doi: 10.1016/j.jns.2012.07.026. [DOI] [PubMed] [Google Scholar]
- 9.Martinez-Martin P, et al. Gender-related differences in the burden of non-motor symptoms in Parkinson’s disease. J. Neurol. 2012;259:1639–1647. doi: 10.1007/s00415-011-6392-3. [DOI] [PubMed] [Google Scholar]
- 10.Petrucci S, et al. GBA‐related Parkinson’s disease: dissection of genotype–phenotype correlates in a large italian cohort. Mov. Disord. 2020;35:2106–2111. doi: 10.1002/mds.28195. [DOI] [PubMed] [Google Scholar]
- 11.Sidransky E, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Eng. J. Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mata IF, et al. GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson’s disease. Mov. Disord. 2016;31:95–102. doi: 10.1002/mds.26359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alcalay RN, et al. Neuropsychological performance in LRRK2 G2019S carriers with Parkinson’s disease. Parkinsonism Relat. Disord. 2015;21:106–110. doi: 10.1016/j.parkreldis.2014.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Macleod AD, et al. Age-related selection bias in Parkinson’s disease research: are we recruiting the right participants? Parkinsonism Relat. Disord. 2018;55:128–133. doi: 10.1016/j.parkreldis.2018.05.027. [DOI] [PubMed] [Google Scholar]
- 15.Hamedani, A. G. et al. Adjusting for underrepresentation reveals widespread underestimation of Parkinson’s disease symptom burden. Mov. Disord.10.1002/mds.29507 (2023). [DOI] [PMC free article] [PubMed]
- 16.Aamodt WW, Willis AW, Dahodwala N. Racial and ethnic disparities in Parkinson disease. Neurol. Clin. Pract. 2023;13:e200138. doi: 10.1212/CPJ.0000000000200138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider MG, et al. Minority enrollment in Parkinson’s disease clinical trials. Parkinsonism Relat. Disord. 2009;15:258–262. doi: 10.1016/j.parkreldis.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tropea TF, et al. Whole clinic research enrollment in Parkinson’s disease: the Molecular Integration in Neurological Diagnosis (MIND) study. J. Parkinsons Dis. 2021;11:757–765. doi: 10.3233/JPD-202406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morley JF, et al. Genetic influences on cognitive decline in Parkinson’s disease. Mov. Disord. 2012;27:512–518. doi: 10.1002/mds.24946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marek K, et al. The Parkinson Progression Marker Initiative (PPMI) Prog. Neurobiol. 2011;95:629–635. doi: 10.1016/j.pneurobio.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tropea TF, et al. APOE, thought disorder, and SPARE-AD predict cognitive decline in established Parkinson’s disease. Mov. Disord. 2018;33:289–297. doi: 10.1002/mds.27204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yarnall AJ, et al. Characterizing mild cognitive impairment in incident Parkinson disease: the ICICLE-PD study. Neurology. 2014;82:308–316. doi: 10.1212/WNL.0000000000000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mollenhauer B, et al. Nonmotor and diagnostic findings in subjects with de novo Parkinson disease of the DeNoPa cohort. Neurology. 2013;81:1226–1234. doi: 10.1212/WNL.0b013e3182a6cbd5. [DOI] [PubMed] [Google Scholar]
- 24.Williams-Gray CH, et al. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J. Neurol. Neurosurg. Psychiatry. 2013;84:1258–1264. doi: 10.1136/jnnp-2013-305277. [DOI] [PubMed] [Google Scholar]
- 25.Smolensky L, et al. Fox insight collects online, longitudinal patient-reported outcomes and genetic data on Parkinson’s disease. Sci. Data. 2020;7:67. doi: 10.1038/s41597-020-0401-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nalls MA, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18:1091–1102. doi: 10.1016/S1474-4422(19)30320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cerri S, Mus L, Blandini F. Parkinson’s disease in women and men: what’s the difference? J. Parkinsons Dis. 2019;9:501–515. doi: 10.3233/JPD-191683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Russillo MC, et al. Sex differences in Parkinson’s disease: from bench to bedside. Brain Sci. 2022;12:917. doi: 10.3390/brainsci12070917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Orr-Urtreger A, et al. The LRRK2 G2019S mutation in Ashkenazi Jews with Parkinson disease: Is there a gender effect? Neurology. 2007;69:1595–1602. doi: 10.1212/01.wnl.0000277637.33328.d8. [DOI] [PubMed] [Google Scholar]
- 30.Marder K, et al. Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology. 2015;85:89–95. doi: 10.1212/WNL.0000000000001708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alcalay RN, et al. Parkinson disease phenotype in Ashkenazi jews with and without LRRK2 G2019S mutations. Mov. Disord. 2013;28:1966–1971. doi: 10.1002/mds.25647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malek N, et al. Features of GBA-associated Parkinson’s disease at presentation in the UK Tracking Parkinson’s study. J. Neurol. Neurosurg. Psychiatry. 2018;89:702–709. doi: 10.1136/jnnp-2017-317348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toffoli M, et al. Phenotypic effect of GBA1 variants in individuals with and without Parkinson’s disease: the RAPSODI study. Neurobiol. Dis. 2023;188:106343. doi: 10.1016/j.nbd.2023.106343. [DOI] [PubMed] [Google Scholar]
- 34.Neumann J, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain. 2009;132:1783–1794. doi: 10.1093/brain/awp044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toledo JB, et al. A platform for discovery: the University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimer’s Dementia. 2014;10:477–484.e1. doi: 10.1016/j.jalz.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Racine JS, RStudio: A. Platform-Independent I. D. E. for R and Sweave. J. Appl. Econom. 2012;27:167–172. doi: 10.1002/jae.1278. [DOI] [Google Scholar]
- 37.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 1995;57:289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 38.Tropea, T. F. et al. Molecular Integration in Neurological Diagnosis (MIND) Parkinson’s Disease Inception Cohort Study Protocol. PROTOCOL (Version 1) available at Protocol Exchange [10.21203/rs.3.pex-2505/v1] (2024).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are found in the Supplementary and have been submitted to DbGaP (https://submit.ncbi.nlm.nih.gov/dbgap/regsys/54461/). The full protocol and participant informed consent form are available on Nature Protocol Exchange38. Additional supporting materials including standard operating procedures are available from the corresponding author on reasonable request.
The R code used is found in the Supplementary.