


Abstract
This study concerns chimeric restriction enzymes that are hybrids between a zinc finger DNA-binding domain and the non-specific DNA-cleavage domain from the natural restriction enzyme FokI. Because of the flexibility of DNA recognition by zinc fingers, these enzymes are potential tools for cleaving DNA at arbitrarily selected sequences. Efficient double-strand cleavage by the chimeric nucleases requires two binding sites in close proximity. When cuts were mapped on the DNA strands, it was found that they occur in pairs separated by ∼4 bp with a 5′ overhang, as for native FokI. Furthermore, amino acid changes in the dimer interface of the cleavage domain abolished activity. These results reflect a requirement for dimerization of the cleavage domain. The dependence of cleavage efficiency on the distance between two inverted binding sites was determined and both upper and lower limits were defined. Two different zinc finger combinations binding to non-identical sites also supported specific cleavage. Molecular modeling was employed to gain insight into the precise location of the cut sites. These results define requirements for effective targets of chimeric nucleases and will guide the design of novel specificities for directed DNA cleavage in vitro and in vivo.
INTRODUCTION
Site-specific endonucleases are powerful tools for the manipulation of DNA sequences. Naturally occurring restriction enzymes have played a central role in the cloning and mapping of genes since their original isolation roughly three decades ago. Type II enzymes able to specifically cleave more than 140 different sites are now available commercially (1). Despite their diversity, these endonucleases have limited utility because their recognition sites are rather short (8 bp or less) and their specificity is not easily altered. The class of homing nucleases or meganucleases (2) recognizes longer sequences (∼20 bp), but shares the limitation of having rigid sequence requirements. For some applications it would be desirable to have enzymes that recognize specific sequences with good discrimination, but also have the ability to be manipulated to bind new, arbitrarily selected sequences.
We have developed a class of chimeric nucleases based on the linkage of a zinc finger DNA-binding domain to the DNA-cleavage domain (FN) from the Type IIs restriction enzyme FokI (3–6). Similar hybrids combine DNA-binding domains from natural and synthetic transcription factors to this or other non-specific cleavage domains (7–10). In these constructs, DNA cleavage is directed to sites recognized by the binding domains, thus proving the feasibility of manipulating the target specificity.
The Cys2His2 zinc fingers are of particular interest in this regard. Each individual finger contacts primarily three consecutive base pairs of DNA in a modular fashion (11,12; Fig. 1). By manipulating the number of fingers and the nature of critical amino acid residues that contact DNA directly, binding domains with novel specificities can be evolved and selected (13–21). In principle, a very broad range of DNA sequences can serve as specific recognition targets for zinc finger proteins. Chimeric nucleases with several different specificities based on zinc finger recognition have already been constructed and characterized (3,6,8,9).
Figure 1.

Schematic diagram of the chimeric nuclease. The three zinc fingers are shown in ribbon representation (12). The residues that provide the primary specificity-determining interactions with the DNA bases, at positions –1, 3 and 6 relative to the start of the α-helix of each finger, are indicated next to the bases they contact. The FokI endonuclease domain is C-terminal to the zinc fingers and separated from them by a 15 amino acid linker [(G4S)3; G, glycine, S, serine]. The specific sequences illustrated are for QQR (22,23).
In the present work, we examine in more detail the requirements for efficient DNA cleavage by two of these zinc finger–FN chimeras. Both Zif-QQR-FN (QQR) (22) and Zif-ΔQNK-FN (QNK) (6) have the general structure diagrammed in Figure 1, with the three finger DNA-binding domain at the N-terminus connected by a peptide linker to the nuclease domain at the C-terminus. Because of differences in several key residues in the middle finger, they recognize related, but distinct, sites: 5′-GGG GAA GAA for QQR (22,23) and 5′-GGG GCG GAA for QNK (6,24). Studies of these two chimeric nucleases were pursued in parallel, using similar, but not identical, procedures and substrates.
Both enzymes require two copies of the recognition site in close proximity to effect efficient double-strand cleavage, reflecting a requirement for dimerization of the cleavage domain. While natural FokI (25) must also dimerize, the need for neighboring paired binding sites is unique to the chimeric nucleases. A consequence of this requirement is that the chimeric enzymes have very high target specificity, since two designated 9 bp sequences must be bound. The results presented here will guide the future design of chimeric nucleases directed to specific targets. One potential application of these enzymes is site-specific cleavage of DNA in vivo with the goal of evaluating double-strand break repair or stimulating targeted recombination. The latter prospect is addressed in a separate study (Bibikova et al., manuscript in preparation).
MATERIALS AND METHODS
DNA substrates
For most QNK substrates, double-stranded oligodeoxyribonucleotides having the recognition site 5′-GGG GCG GAA were cloned into the BamHI site of pUC18. The parent plasmid for QQR substrates was pRW4 (26) and oligodeoxyribonucleotides containing the recognition site 5′-GGG GAA GAA were inserted into the unique XhoI site. QNK plasmids were transformed into Escherichia coli DH5α, QQR plasmids into E.coli XL-1 Blue and DNAs from individual colonies were characterized by DNA sequence analysis. The exact sequences of the inserts are given in Figure 5. Plasmid DNAs were purified using Qiagen columns (Qiagen, Valencia, CA).
Figure 5.


Maps of the cuts made on all substrates. (a) QNK substrates. (b) QQR substrates. The sequences of both strands are given in the region around the 9 bp recognition sites, which are indicated with a line between the strands. Cut sites corresponding to strong bands are indicated with an arrowhead, cuts of moderate intensity with a vertical line and weaker cuts with a dot.
The names of the plasmids reflect the structure of the inserts. pKS has a single QNK site, while pKT8 has two sites 8 bp apart in tail-to-tail inverted orientation, i.e. the G ends of the recognition site face each other. Similarly, pKT14, pKT28 and pKT48 carry inverted sites with the indicated separations. pQS carries a single copy of the QQR site; the pQTn series has tail-to-tail inverted sites with n bp between them; pQH10 has head-to-head inverted repeats 10 bp apart; pQD10 carries direct repeats separated by 10 bp.
Enzymes
Zif-QQR-FN and Zif-ΔQNK-FN were prepared as previously described (6). Briefly, the coding sequence for the chimeric nuclease was cloned into pET15b, so that it carries a His6 tag at its N-terminus, and was propagated in BL21 (DE3) cells that overproduce E.coli DNA ligase from a pACYC184 derivative. Expression of the nuclease was initiated by addition of IPTG to 0.7 mM to cells growing at 22°C in LB medium plus ampicillin, tetracycline and 100 µM ZnCl2. Harvested cells were disrupted by sonication or by passage twice through a French press and the clarified extract was passed over a His-bind column. The enzyme was eluted with 0.4 M imidazole and purified further on a heparin–Sepharose, then a gel filtration column (S-100 HR or Superdex-75). It was stored at –80°C in 40% glycerol (10% glycerol in some cases), 20 mM Tris, pH 7.9, 10 mM β-mercaptoethanol, 100 µM ZnCl2. In vitro reactions were typically performed in 20 µl containing 10 mM Tris, pH 8.5, 50 mM NaCl, 1 mM DTT, 100 µM ZnCl2, 50 µg/ml BSA, 100 µg/ml tRNA. QQR reactions used 50 ng of substrate DNA that had been linearized by PvuII digestion; QNK substrates were linearized with ScaI (Fig. 6) or with SspI (Figs 3a and 7) and used at 100 ng/reaction. Enzyme was added, followed by preincubation for 30 min at room temperature. MgCl2 was added to a final concentration of 10 mM and incubation was continued for 1 h at room temperature. Cleavage was monitored by electrophoresis in 1% agarose gels.
Figure 6.
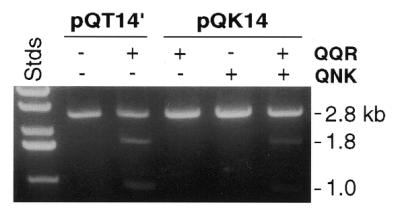
Cleavage of a hybrid site. pQT14′ has paired sites for QQR 14 bp apart, while pQK14 has one site each for QQR and QNK separated by 14 bp. DNA concentration, 2.5 nM; enzyme concentration, 10 nM each. Labels as in Figure 3a.
Figure 3.

Dependence of cleavage on separation between inverted sites. (a) QNK substrates with a single copy of the recognition site (pKS) or with 8, 14, 28 and 48 bp separations between tail-to-tail inverted sites, as indicated. Reactions contained 2.5 nM DNA and 10 nM enzyme. (b) QQR substrates with the separations indicated between inverted sites. Reactions and designations as in Figure 2a, with 0.7 nM DNA and either no enzyme (–) or QQR at 1.0 (a), 1.6 (b) or 5.0 nM (d). The band between 5.6 and 3.6 kb in the samples labeled 4 is an artifact of this particular plasmid preparation.
Figure 7.
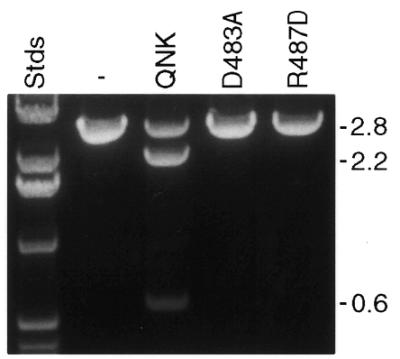
Dimerization mutants of QNK. Linear pKT8 was incubated with no enzyme (–), with QNK or with the D483A and R487D mutants, as indicated. All reactions were performed with 20 nM DNA and 40 nM protein. Labels as in Figure 3a.
Dimer interface mutants of QNK were constructed by PCR with primers that incorporate the desired mutations. For D483A, the forward primer was d(CAATTGGCCAAGCAGCTGAAATGCAACGATATGTCGAAGAAAATCAAACACG); the corresponding primer for R485D was d(CAATTGGCCAAGCAGATGAAATGCAAGATTATGTCGAAGAAAATCAAACACG). Each was used with the reverse primer d(TAGGATCCTCATTAAAAGTTTATCTCGCCGTTATT) from the C-terminus of the FN coding sequence. The resulting PCR products were cleaved with MscI, gel purified and used as reverse primers in a second round of PCR that included d(GAAGATCTTCGATCCCGCGAAATTAA), from the vector N-terminal to the QNK coding sequence, as the forward primer. The final PCR products were digested with NdeI and BamHI, gel purified and cloned into pET15b. Identities of individual clones were confirmed by DNA sequencing and the proteins were expressed and purified as described above.
Mapping cut sites
To label one strand of the QNK substrates, plasmids were cut at one end of the pUC18 polylinker with EcoRI. The DNA was treated with calf intestinal alkaline phosphatase (New England BioLabs, Beverly, MA) and then with T4 polynucleotide kinase (Boehringer Mannheim, Indianapolis, IN) and [γ-32P]ATP (Amersham Life Sciences, Arlington Heights, IL). After heat inactivation of the kinase, the DNA was digested with HindIII, which cuts at the other end of the polylinker. The resulting small fragment was purified from a 3% low melting point agarose gel. To label the other strand, the order of HindIII and EcoRI digests was reversed. One quarter of the labeled sample was subjected to each of the Maxam–Gilbert G and G+A reactions (27) and the remaining two quarters were used in reactions with or without QNK. After phenol/chloroform extraction and ethanol precipitation, the products were separated by electrophoresis in a 10% polyacrylamide sequencing gel.
For QQR reactions, a DNA fragment of ∼400 bp was amplified by PCR from each of the plasmid substrates. The primers, d(CAGGTAGATGACGACCATCAGG) and d(GGAATGGACGATATCCCGCAAG), correspond to sequences in pRW4 flanking the insertion site. This fragment was gel purified with a Qiaex II gel extraction kit (Qiagen) and used as a template for a second PCR, using the internal primers d(GGTTGGCATGGATTGTAGGCG) and d(TGTTAGATTTCATACACGGTGCC), to generate a fragment of ∼200 bp. To label each strand separately, one of the latter primers was treated with T4 polynucleotide kinase and [γ-32P]ATP. The PCR products were purified with a QIAquick PCR purification kit (Qiagen), then treated with QQR. Denatured reaction products were separated by electrophoresis in 6% polyacrylamide sequencing gels. Dideoxy sequencing reactions were performed for each substrate using a dsDNA cycle sequencing kit (Gibco BRL, Gaithersburg, MD) and the same labeled primers as for PCR. These were run on the same gels in lanes immediately adjacent to the nuclease cleavage products.
Molecular modeling
Coordinates for the zinc fingers were taken from the co-crystal structure of the DNA-binding domain of QNK bound to DNA (Protein Database accession no. 1MEY) (24). Coordinates for the FokI cleavage domain dimer include residues 387–579 from the structure of the protein alone (Protein Database accession no. 2FOK) (28). The cleavage domain dimer was docked to B-form DNA by eye, using the published model (28) as a guide. The zinc finger domains were placed at various positions along the DNA by aligning backbone phosphates from consecutive residues in the co-crystal structure (24) with corresponding phosphates on B-form DNA. All alignments were performed with the graphics program O and figures were prepared from these data using MolScript.
RESULTS
Binding site requirements for double-strand cleavage
Previous work with several zinc finger chimeric nucleases, including QQR, showed that they make cuts primarily to the left side of their recognition sequences, as depicted in Figure 1 (22). This was the expected location, given the orientation of the zinc fingers on the DNA and the structure of the chimeric protein. Some cleavage occurred on both strands, but the mapping of the sites was performed on denatured DNA and the efficiency of double-strand cleavage was not determined (22). Therefore, we focused our attention on the production of double-strand breaks.
We constructed and analyzed a collection of specifically designed plasmid substrates with variable numbers and orientations of the canonical recognition site for QQR. These were linearized and treated with QQR. At enzyme:substrate ratios close to 1, in order to achieve double-strand cleavage it was necessary to have at least two copies of the target oligonucleotide (Fig. 2a). A single copy of the recognition sequence (pQS) did not support cleavage. With 10 bp between paired sites, both tail-to-tail inverted repeats (pQT10) and direct repeats (pQD10) were effectively cut, while head-to-head inverted repeats (pQH10) were cleaved much less efficiently. Observed double-strand breaks mapped to the expected sites (Fig. 2a and data not shown).
Figure 2.
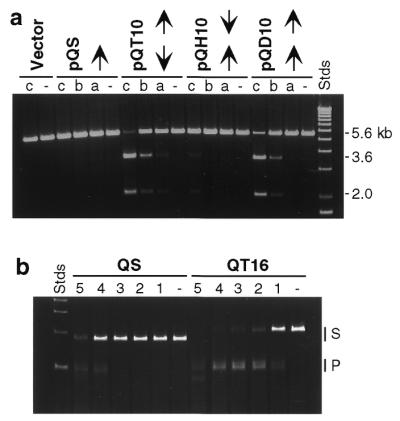
Substrate specificity of QQR. (a) Substrates with various binding site dispositions. pQS has a single copy of the canonical recognition site, indicated by the arrow. The remaining DNAs have two sites in tail-to-tail inverted (pQT10), head-to-head inverted (pQH10) and direct repeat (pQD10) orientations. The vector is pRW4 without an insert. Samples of DNA (0.7 nM, corresponding to 1.4 nM recognition sites in the cases with paired sites) were incubated without enzyme (–) or with QQR at 1.0 (a), 1.5 (b) or 3.0 nM (c). The locations of the 5.6 kb linear substrate DNAs and the 3.6 and 2.0 kb fragments expected from cleavage at the target site are indicated to the right of the figure. (b) Cleavage at higher enzyme concentrations. The substrates were PCR fragments from a single site plasmid (QS) and one with two inverted sites (QT16); DNA concentration ∼20 nM. QQR concentrations were 0 (–), 3.5 (1), 7 (2), 17.5 (3), 35 (4) and 50 nM (5). The locations of the substrate (S) and expected product (P) bands are indicated to the right. Faster migrating fragments are from cleavage at secondary sites. The Stds lane in each panel contains linear size standards.
At substantially higher enzyme:substrate ratios, both QQR and QNK made targeted cuts in DNAs that carried a single copy of the recognition site. In the comparison shown in Figure 2b, QS carries a single site, while QT16 has two in tail-to-tail orientation 16 bp apart. The DNAs were PCR fragments of ∼200 bp, identical to those used for mapping reactions (see below). QT16 was cleaved at all QQR concentrations tested and cleavage was essentially complete at an approximately 1:1 ratio of enzyme to sites (lane 4). In contrast, QS required ∼10-fold more enzyme to achieve comparable levels of cleavage (QS in lane 4 versus QT16 in lane 1), and this corresponds to a 20-fold higher ratio of enzyme to recognition sites. At the highest enzyme concentration used (lane 5), other sites began to be cleaved, perhaps reflecting binding of QQR to more distantly related sequences.
Influence of target site separation on cleavage efficiency
Paired inverted sites in the tail-to-tail orientation showed efficient double-strand cleavage when the sites were 10 or 16 bp apart (Fig. 2). To determine the upper and lower limits on distances that would allow cleavage, we examined a series of substrates for each chimeric nuclease in which variable amounts of essentially random DNA sequence were inserted between the recognition sites. For QNK, separations of 8, 14, 28 and 48 bp were tested (Fig. 3a). Under conditions that did not support cleavage at a single site (pKS), the 8, 14 and 28 bp separations allowed double-strand cleavage, while the 48 bp separation did not.
For QQR, we tested a larger collection of different separations, as shown in Figure 3b. When the paired sites were 4 bp apart, very little double-strand cleavage was observed and that only at the highest enzyme input. A separation of 6 bp led to good cleavage with QQR and this remained true for all distances tested up to 35 bp. The substrate with a separation of 40 bp, however, was essentially not cleaved. Thus, the upper limit for effective site separations is between 35 and 40 bp, in agreement with the observations for QNK.
Mapping cut sites on DNA strands
In principle, the requirement for two binding sites to achieve double-strand cleavage could reflect either of two underlying phenomena. (i) Each individual bound chimeric molecule might make an independent single-strand cut close to its binding site and two such cuts in proximity would be necessary to produce a double-strand break. In this view the upper limit on the distance between effective paired sites would be determined by the stability of the DNA duplex between nicks on the two strands. (ii) The cleavage domain of the chimeric nuclease might have to dimerize in order to act as an effective nuclease and when it does concerted breaks would be made in the two strands. Natural FokI dimerizes to cleave DNA (25) and it is reasonable to suspect that the cleavage domains in the context of the chimeric nuclease would do the same. In this case, the upper limit on effective site separation would reflect the maximum extension achievable by the peptide linker between the binding and cleavage domains.
We distinguished these possibilities by mapping the cut sites for QNK and QQR on a wide range of substrates at single nucleotide resolution. Model (i) predicts that single-strand cuts will be produced in fixed positions relative to each recognition site and that their locations will move apart as the distance between the sites is increased. Model (ii) predicts that cuts in the two strands will always be paired and, like FokI, they should produce a 5′ overhang of 4 bp.
To map the cuts made by QNK, a fragment carrying the paired sites, the intervening sequence and ∼50 bp of pUC18 was labeled on either end with 32P as described in Materials and Methods. After digestion with the enzyme, products were compared to G and G+A sequencing reactions of the same fragment (Fig. 4a). Maxam–Gilbert chemistry removes the designated base and leaves the preceding 3′-phosphate, while the chimeric nuclease leaves a 3′-hydroxyl. Both these properties increase the mobility of the Maxam–Gilbert fragments, so the alignment with the QNK products was adjusted by about 1.5 bands to identify the exact site of cleavage.
Figure 4.

Mapping cut sites on DNA strands. (a) QNK substrates. Lanes G and G+A contain Maxam–Gilbert sequencing reaction products of the end-labeled DNAs. Adjacent lanes have the same fragments (∼40 nM) treated without enzyme (–) or with QNK at 10 (+) or 100 nM (++). (b) QQR substrates. In each set, samples of a DNA fragment, labeled on one strand and treated with the nuclease, were run beside dideoxy sequencing reactions (GATC) initiated from a primer labeled at exactly the same position. DNAs (4 nM) were incubated without enzyme (–) or with QQR at 1.0 (a) or 3.0 nM (c). In both panels arrows indicate the positions and orientations of the 9 bp recognition sites.
With 8 bp between QNK sites (KT8), strong cuts were seen on both strands between the sites: a single cut on one strand, a strong and a secondary cut on the other strand (Fig. 4a). When mapped on the DNA sequence, the major cuts are 4 bp apart and result in a 5′ overhang (Fig. 5a). With KT14, five or six relatively strong cuts were made on each strand (Figs 4a and 5a). When mapped they overlap considerably, but may be interpreted as three clusters of paired cuts staggered by ∼4 bp, one near the middle of the intervening sequence and one near each end. With KT28, again a single strong cleavage site was seen on both strands near the middle of the space between binding sites with a 4 bp 5′ stagger. Minor bands were visible in all cases, indicating that the cut locations were not rigidly determined.
Cuts were also mapped on a DNA carrying a single recognition site for QNK (KS), using a high concentration of enzyme (Fig. 4a). Two groups of cuts were seen on each strand, similar to results obtained previously with other zinc finger chimeras (9). These cuts assemble on the DNA sequence into two clusters centered ∼4 and 13 bp from the 5′-end of the binding site (Fig. 5a). There is a general 5′ stagger in each cluster, although the distances between the cuts are not restricted to 4 bp. Similar locations were seen with KT48 at high QNK concentrations (Fig. 5a).
Also shown in Figure 5a are mapped cuts in two QNK substrates that were determined independently by the procedure described for QQR below. The major cuts in KT8′ are farther apart than seen in KT8, perhaps due to sequence preference of the FokI cleavage domain (see Discussion). KT12 showed paired, centered, strong cuts separated by a 4 bp 5′ stagger, plus one minor cut reflecting a 3 bp stagger.
To map cuts on QQR substrates, a PCR fragment of ∼200 bp from each plasmid was labeled on either end and reaction products were analyzed in parallel with dideoxy sequencing reactions on the same DNAs, using the same primers. At moderate QQR concentration essentially no nicks were produced in the vicinity of a single copy of the recognition site (not shown). At the same concentration single strong cuts were made in both strands between sites separated by 12 bp (QT12). When mapped onto the DNA sequence, these strong cuts were precisely 4 bp apart with a 5′ stagger (Fig. 5b). With QT16, cuts were made near one end or the other of the intervening sequence and the most prominent cuts occurred in pairs with a 4 bp 5′ stagger. QT30 provided the only case in which the strongest cuts were clearly separated by <4 bp. These paired cuts staggered by 3 bp were located at the center of the spacer.
Examining the full range of QQR substrates in Figure 5b, we see that paired nicks were always located between the binding sites and related by a 5′ stagger of ∼4 bp. With QT6, prominent cuts were made at the center of the interval with a 4 bp stagger. For QT8, the strongest cuts were centered and showed 5 or 6 bp staggers. QT10 showed alternative 4 bp staggers offset slightly from the center of the symmetrical intervening sequence and QT12 showed the centered 4 bp stagger described earlier. There were minor bands in each of these cases corresponding to slightly shorter or slightly longer 5′ staggers. With QT14, QT18 and QT20 the paired cuts on the two strands occurred not in the center of the intervening sequence, but close to either end, as described for QT16 above. While the major cuts were staggered by 4 bp in most cases, minor cut sites were also seen. In QT26, QT30 and QT35, the cut locations returned to the center of the interval, although weak cuts near the ends were seen with QT26. When the site separation reached 40 bp, no prominent nicks were seen, just as for the single site substrate.
These results support Model (ii), i.e. dimerization of the cleavage domain in the space between the binding sites.
Cleavage of paired non-identical sites
To achieve cleavage of an arbitrarily selected target, paired zinc finger binding sites would be chosen that would usually not be identical. To demonstrate that such a configuration also leads to effective dimerization and cleavage, we constructed a substrate having one site each for QNK and QQR 14 bp apart and in tail-to-tail inverted orientation (pQK14). This DNA was not cleaved by either enzyme alone at moderate enzyme concentration, but was cleaved by a mixture of the two (Fig. 6). The level of cleavage was comparable to that of pQT14 using the corresponding QQR enzyme alone. Thus, paired non-identical sites are effective cleavage targets when enzymes with both specificities are provided.
Dimerization mutants
The crystal structure of FokI reveals a dimer interface, where Asp483 of each cleavage domain monomer makes bidentate hydrogen bonds with Arg487 of the other (28). Simultaneous conversion of these two residues to Ala dramatically reduced the cleavage efficiency of the restriction endonuclease (25). In order to confirm the requirement for dimerization of the chimeric nucleases, we mutated each of these residues individually in QNK: Asp483 to Ala (D483A) and Arg487 to Asp (R487D). The corresponding proteins were purified and used to treat pKT8, which was readily cleaved by QNK. Both mutant enzymes were incapable of producing double-strand breaks (Fig. 7). Thus, dimerization is critical for the activity of the chimeric nucleases, just as for native FokI.
DISCUSSION
Double-strand cleavage requirements
The data presented here on in vitro cleavage by two chimeric nucleases demonstrate that the cleavage domain of the enzyme must dimerize to effect efficient double-strand incision of DNA. These results complement findings for natural FokI, which recognizes its binding site as a monomer (29,30), but requires dimerization to cleave DNA (25). In that case, the crystal structure of the enzyme showed a credible dimer interface (28); cleavage efficiency showed an exponential dependence on enzyme concentration; a separate cleavage domain could complement sub-optimal concentrations of the intact molecule; mutations in the dimer interface abolished cleavage (25). In addition to the need for an intact dimer interface, we showed that cleavage by the chimeric nucleases requires two copies of the binding site in close proximity and that paired cuts with a conserved stagger are made in the two strands, regardless of the disposition of the binding sites.
The structure of the FokI cleavage domain dimer resembles the active form of other restriction enzymes (28). It is highly plausible that nuclease activity depends on the structure achieved by dimerization and that single molecules are inactive. Both in the case of FokI (25) and our chimeric enzymes (J.Smith and S.Chandrasegaran, data not shown), results with dimerization mutants indicate that even targeted single-strand cleavage depends on dimerization.
The requirement for paired binding sites in close proximity distinguishes the chimeric nucleases from FokI. In the latter case, it appears that dimerization occurs between monomers bound to quite distant sites on the DNA (25). The fact that the chimeric nucleases and FokI exist as monomers in solution further differentiates them from several other Type II restriction endonucleases that must bind to two recognition sites to achieve full activity (31–34). In these cases the enzyme is naturally dimeric or tetrameric and association with the second site activates cleavage at one or both sites.
When a single recognition site supports cleavage at high concentrations of chimeric nuclease, we cannot distinguish among several possible explanations. Dimerization could occur between one DNA-bound and one unbound enzyme molecule, between two molecules bound to canonical sites on separate DNAs or between one enzyme bound to the canonical site and another that is weakly associated with nearby duplex through non-sequence-specific interactions. In the latter case, marginal energy contributions from zinc finger–DNA binding and the dimerization interface could combine to produce the necessary affinity and permit cleavage.
The dimer interface in the chimeric nucleases may be somewhat flexible, since cuts staggered by distances other than 4 bp were sometimes produced. Another manifestation of this flexibility may be a slight preference for cutting 3′ of a T (see Fig. 5). Native FokI cuts 9 and 13 bp from 5′-GGATG without apparent sequence preference (35,36).
Molecular modeling
In order to gain further insight into the requirements for recognition and cleavage by the chimeric nucleases, we constructed hypothetical molecular models using published coordinates for the separate domains (24,28). As envisioned by Wah et al. (28), the cleavage domain dimer sits like a saddle across the DNA duplex making close contacts in the major groove (Fig. 8). The N-termini of the monomers (A and B), which must be connected to the zinc finger domains through the flexible linker, are located on either side of the DNA at the base of the saddle, essentially in the positions of the stirrups. The two connecting points lie almost in a plane perpendicular to the DNA axis in our model, although the dimer may be angled into or across the major groove in reality.
Figure 8.

Molecular modeling. The DNA is represented as a space filling model and colored by atom type, from light gray to black. The cleavage domain dimer (aqua) is shown as a ribbon diagram; the N-termini of the two monomers are labeled A and B. The two zinc finger domains are also shown as ribbon diagrams, colored magenta (I) and purple (II), and the positions of their N- and C-termini are indicated. (a) Model of QT6, with binding domains 3 bp on either side of the center of the cleavage domain dimer. The cleavage domain dimer sits in the major groove largely behind the double helix as pictured, while the zinc finger domains wind through the major groove on either side. The distances between the closest pairs of connectable termini (I→B:II→A) are ∼20 Å. (b) Model of QT16 with the cleavage domain dimer centered between the binding sites, i.e. 8 bp on either side. The connecting points for both zinc finger domains are on the opposite side of the helix from the ends of the cleavage domains. (c) Model of QT16 with the cleavage dimer off-center, 3 bp from one binding site and 13 bp from the other. Now all the connecting points are on the same side of the duplex.
The zinc finger domains (I and II in Fig. 8) were placed in the major groove on either side of the cleavage dimer at separations and orientations corresponding to the experimental substrates. The distances that must be traversed by the peptide linker in order to join the binding and cleavage domains were estimated. Sometimes the connections could be made directly through space and in other cases a diversion was necessary to avoid clashing with the DNA. Both possible sets of connections were assessed: I→A:II→B and I→B:II→A.
There are 24 amino acids in the chimeric nuclease sequence between positions defined in the component crystal structures: three disordered at the end of the zinc finger domain, 15 in the intentional (Gly4Ser)3 linker, three as a result of cloning the linker and three disordered at the N-terminus of the cleavage domain. At full extension this segment could theoretically reach as far as 80–90 Å. The modeling has its limitations, since the positioning of the cleavage domain dimer on the DNA is hypothetical and no attempt was made to incorporate distortions of the DNA upon zinc finger binding or association with the cleavage domain dimer. Nonetheless, a number of conclusions could be drawn.
The modeling showed that with only 4 bp between inverted recognition sites, steric clash would occur between the binding and cleavage domains. This accounts for the observation that double-strand cleavage was very inefficient with QT4 (Fig. 3b). With a 6 bp separation, the C-termini of the zinc fingers project into the major groove in the area under the cleavage domain saddle and the binding and cleavage domains all have unobstructed access to the DNA (Fig. 8a). In practice, the QT6 substrate was readily cleaved by the nuclease.
With a separation of 16 bp, the observed cuts lay not in the center of the interval, but closer to either side, and this is rationalized by the modeling (Fig. 8b and c). If the cleavage domain dimer were centered between the binding sites, the termini of the domains that must be connected would lie on opposite sides of the DNA helix (Fig. 8b). When the domains are moved to the observed location of the cuts, centered 3 bp from one binding site and 13 bp from the other, the termini are all on the same side of the DNA (Fig. 8c) and the distances to be traversed by the linker are considerably shortened. Cuts ∼3 or 13 bp from the recognition sites were seen with a number of substrates (Fig. 5). In addition to placing the linker entirely on one side of the DNA, it is possible that the linker has two preferred conformations, one particularly compatible with extending each of the observed distances.
The modeling does not explain some of the limits to cleavage. For example, it is not clear why QT35 was cleaved, but QT40 was not, since the estimated distances are not much different for the two cases. The extension required for the linker in the case of direct repeats (QD10) is well within the range of those traversed for inverted repeats. From similar considerations, it is not clear why the head-to-head inverted repeat is cleaved inefficiently, since one pair of connections would, in principle, require the two linkers to stretch only 40 Å. There may be some features of the actual structure that our simplified model does not reflect.
In summary, the modeling has shown that, while the chimeric nucleases cleave DNAs with a wide range of separations between recognition sites, they prefer to cleave at specific positions that allow the linker between the DNA-binding and DNA-cleavage domains to remain entirely on one side of the DNA duplex. Although the linker is, in principle, sufficiently long to make the traverse around the duplex, this configuration is apparently disfavored. The situation is reminiscent of that for native FokI, which makes cuts approximately one helical turn away from its binding site, thus placing the binding and cleavage domains on the same side of the duplex. In this case the linker is thought to be an α-helix that is limited in its extensibility (30).
Applications of chimeric restriction enzymes
It was demonstrated previously that zinc finger chimeras can cut DNA in the vicinity of their specific recognition sites and their potential utility as highly specific cleavage reagents was noted (3–5,8). The current finding that paired binding sites are required for efficient dimerization and cleavage shows precisely what type of target will be susceptible to these enzymes. In order to cleave at an arbitrarily determined location, two 9 bp DNA sequences in inverted orientation and separated by 6–35 bp should be selected. Zinc finger combinations that bind these sequences specifically would then be derived by design or selection and linked to the FokI cleavage domain. Expression and purification of the two resulting chimeric nucleases would provide the desired site-specific cleavage reagent.
The feasibility of this approach is demonstrated by our studies with hybrid sites for QQR and QNK (Fig. 6). These particular enzymes are not ideal reagents in this regard, since both zinc finger domains, especially QNK (6,24), show affinity for DNA sequences other than their canonical recognition sites. Better discrimination has been achieved in several instances by selecting against binding to related sites during the evolution of novel specificities (19,21). Another possibility for improving selectivity is to include additional zinc fingers in the constructs that would bind additional DNA triplets (37,38).
Efficient double-strand cleavage with two chimeric nucleases with three zinc fingers involves the recognition of 18 bp of DNA. This provides a remarkable degree of specificity, since any particular 18 bp sequence is predicted to occur approximately once in 6.9 × 1010 (= 418) bp. This is ample specificity for locating a unique sequence in a mammalian genome (3 × 109 bp), assuming that each zinc finger domain discriminates well against related sites. A very promising application of these enzymes is the in vivo production of chromosomal breaks that could stimulate targeted homologous recombination. In a separate study (Bibikova et al., manuscript in preparation), we demonstrate that QQR and QNK are both capable of binding and cleaving their recognition sites in vivo, even when those sites are incorporated in chromatin. In appropriately constructed substrates, this cleavage leads to efficient homologous recombination.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Dr David Segal for his participation in the early phases of this project and to Jonathan Trautman for technical assistance. We thank Dr Jeremy Berg for advice on zinc finger recognition and Dr H.O. Smith for continuing interest in this project, which is an equal collaboration between the Chandrasegaran and Carroll laboratories. This work was supported in part by grants from the National Institutes of Health (GM53923) and the National Science Foundation (MCB 9415861) to S.C. and by grants from the National Institutes of Health (GM50739 and GM58504) and the University of Utah to D.C. Assistance was also provided by the Markey Center for Protein Biophysics and the Huntsman Cancer Institute at the University of Utah and by the Environmental Health Sciences Core Facility at Johns Hopkins University (supported by NIH grant ES03819). S.C. is a member of the Scientific Advisory Board of Sangamo Biosciences Inc.
REFERENCES
- 1.Roberts R.J. and Macelis,D. (1999) Nucleic Acids Res., 27, 312–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belfort M. and Roberts,R.J. (1997) Nucleic Acids Res., 25, 3379–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim Y.-G., Cha,J. and Chandrasegaran,S. (1996) Proc. Natl Acad. Sci. USA, 93, 1156–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim Y.-G., Kim,P.S., Herbert,A. and Rich,A. (1997) Proc. Natl Acad. Sci. USA, 94, 12875–12879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandrasegaran S. and Smith,J. (1999) Biol. Chem., 380, 841–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith J., Berg,J.M. and Chandrasegaran,S. (1999) Nucleic Acids Res., 27, 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim Y.-G. and Chandrasegaran,S. (1994) Proc. Natl Acad. Sci. USA, 91, 883–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang B., Schaeffer,C.J., Li,Q. and Tsai,M.-D. (1996) J. Protein Chem., 15, 481–489. [DOI] [PubMed] [Google Scholar]
- 9.Kim Y.-G., Smith,J., Durgesha,M. and Chandrasegaran,S. (1998) Biol. Chem., 379, 489–495. [DOI] [PubMed] [Google Scholar]
- 10.Nahon E. and Raveh,D. (1998) Nucleic Acids Res., 26, 1233–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pavletich N.P. and Pabo,C.O. (1991) Science, 252, 809–817. [DOI] [PubMed] [Google Scholar]
- 12.Berg J.M. and Shi,Y. (1996) Science, 271, 1081–1085. [DOI] [PubMed] [Google Scholar]
- 13.Desjarlais J.R. and Berg,J.M. (1992) Proc. Natl Acad. Sci. USA, 89, 7345–7349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desjarlais J.R. and Berg,J.M. (1993) Proc. Natl Acad. Sci. USA, 90, 2256–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choo Y. and Klug,A. (1994) Proc. Natl Acad. Sci. USA, 91, 11163–11167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jamieson A.C., Kim,S.-H. and Wells,J.A. (1994) Biochemistry, 33, 5689–5695. [DOI] [PubMed] [Google Scholar]
- 17.Rebar E.J. and Pabo,C.O. (1994) Science, 263, 671–673. [DOI] [PubMed] [Google Scholar]
- 18.Wu H., Yang,W.-P. and Barbas,C.F. III (1995) Proc. Natl Acad. Sci. USA, 92, 344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greisman H.A. and Pabo,C.O. (1997) Science, 275, 657–661. [DOI] [PubMed] [Google Scholar]
- 20.Isalan M., Klug,A. and Choo,Y. (1998) Biochemistry, 37, 12026–12033. [DOI] [PubMed] [Google Scholar]
- 21.Segal D.J., Dreier,B., Beerli,R.R. and Barbas,C.F. III (1999) Proc. Natl Acad. Sci. USA, 96, 2758–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim Y.-G., Shi,Y., Berg,J.M. and Chandrasegaran,S. (1997) Gene, 203, 43–49. [DOI] [PubMed] [Google Scholar]
- 23.Shi Y. and Berg,J.M. (1995) Science, 268, 282–284. [DOI] [PubMed] [Google Scholar]
- 24.Kim C.A. and Berg,J.M. (1996) Nature Struct. Biol., 3, 940–945. [DOI] [PubMed] [Google Scholar]
- 25.Bitinaite J., Wah,D.A., Aggarwal,A.K. and Schildkraut,I. (1998) Proc. Natl Acad. Sci. USA, 95, 10570–10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maryon E. and Carroll,D. (1989) Mol. Cell. Biol., 9, 4862–4871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maxam A.M. and Gilbert,W. (1977) Proc. Natl Acad. Sci. USA, 74, 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wah D.A., Bitinaite,J., Schildkraut,I. and Aggarwal,A.K. (1998) Proc. Natl Acad. Sci. USA, 95, 10564–10569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skowron P., Kaczorowski,T., Tucholski,J. and Podhajska,A. (1993) Gene, 125, 1–10. [DOI] [PubMed] [Google Scholar]
- 30.Wah D.A., Hirsch,J.A., Dorner,L.F., Schildkraut,I. and Aggarwal,A.K. (1997) Nature, 388, 97–100. [DOI] [PubMed] [Google Scholar]
- 31.Kruger D.H., Barcak,G.J., Reuter,M. and Smith,H.O. (1988) Nucleic Acids Res., 16, 3997–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conrad M. and Topal,M.D. (1989) Proc. Natl Acad. Sci. USA, 86, 9707–9711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oller A.R., Vanden Broek,W., Conrad,M. and Topal,M.D. (1991) Biochemistry, 30, 2543–2549. [DOI] [PubMed] [Google Scholar]
- 34.Wentzell L.M., Nobbs,T.J. and Halford,S.E. (1995) J. Mol. Biol., 248, 581–595. [DOI] [PubMed] [Google Scholar]
- 35.Sugisaki H. and Kanazawa,S. (1981) Gene, 16, 73–78. [DOI] [PubMed] [Google Scholar]
- 36.Kim S.C., Skowron,P.M. and Szybalski,W. (1996) J. Mol. Biol., 258, 638–649. [DOI] [PubMed] [Google Scholar]
- 37.Liu Q., Segal,D.J., Ghiara,J.B. and Barbas,C.F. III (1997) Proc. Natl Acad. Sci. USA, 94, 5525–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim J.-S. and Pabo,C.O. (1998) Proc. Natl Acad. Sci. USA, 95, 2812–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]