


Abstract
Helicobacter pylori is a gram-negative bacterium, which colonizes the gastric mucosa of humans and is implicated in a wide range of gastroduodenal diseases. The genomic sequences of two H.pylori strains, 26695 and J99, have been published recently. About two dozen potential restriction–modification (R-M) systems have been annotated in both genomes, which is far above the average number of R-M systems in other sequenced genomes. Here we describe a functional analysis of the 16 putative Type II R-M systems in the H.pylori J99 genome. To express potentially toxic endonuclease genes, a unique vector was constructed, which features repression and antisense transcription as dual control elements. To determine the methylation activities of putative DNA methyltransferases, we developed polyclonal antibodies able to detect DNA containing N6-methyladenine or N4-methylcytosine. We found that <30% of the potential Type II R-M systems in H.pylori J99 strain were fully functional, displaying both endonuclease and methyltransferase activities. Helicobacter pylori may maintain a variety of functional R-M systems, which are believed to be a primitive bacterial ‘immune’ system, by alternatively turning on/off a subset of numerous R-M systems.
INTRODUCTION
Bacterial restriction–modification (R-M) systems are traditionally divided into three major types: I, II and III. The designations are based on enzyme subunit composition, co-factor requirements, DNA specificity characteristics and reaction products (1). The Type II R-M systems are the simplest. They usually have two independent polypeptides: a restriction endonuclease which cleaves DNA and a corresponding DNA methyltransferase (methylase), which protects endogenous DNA from endonuclease digestion by methylating the endonuclease recognition sequence (2). Type II restriction endonucleases recognize specific sequences and cleave precisely either within or very close to their recognition sequences. They are key enzymes in biotechnology and molecular biology, and new specificities remain desirable (3,4).
The traditional approach to screen for restriction endonucleases was to grow small cultures of individual strains, prepare cell extracts and then test the crude cell extracts for their ability to produce specific fragments on small DNA molecules (5). Using this approach, about 12 000 strains have been screened worldwide to yield the current harvest of more than 3000 restriction endonucleases (6). Roughly, one in four of all strains examined, using a biochemical approach, shows the presence of a Type II restriction enzyme. This is much lower than the average number of putative R-M systems predicted from a computer analysis of sequenced microbial genomes (Table 1).
Table 1. Potential DNA R-M systems in sequenced microbial genomes based on the computational identification of putative methylase genes and the presence of adjacent ORFs of unknown function.
| Organism | Genome size (Mb) | Type I | Type II | Type III | Orphan M | mR | Total |
|---|---|---|---|---|---|---|---|
| Aeropyrum pernix | 1.67 | 7 | 7 | ||||
| Aquifex aeolicus | 1.55 | 1 | 1 | ||||
| Archaeoglobus fulgidus | 2.18 | 1 | 2 | 1 | 4 | ||
| Bacillus subtilis | 4.21 | 2 | 1 | 1 | 4 | ||
| Borrelia burgdorferi | 1.44 | 2 | 2 | ||||
| Campylobacter jejuni | 1.64 | 1 | 4 | 1 | 6 | ||
| Chlamydia muridarum | 1.07 | no candidates | 0 | ||||
| Chlamydia trachomatis | 1.05 | no candidates | 0 | ||||
| Chlamydia pneumoniae AR39 | 1.23 | no candidates | 0 | ||||
| Deinococcus radiodurans | 2.65 | 4 | 3 | 7 | |||
| Escherichia coli | 4.60 | 1 | 2 | 3 | |||
| Haemophilus influenzae | 1.83 | 2 | 3 | 1 | 1 | 7 | |
| Helicobacter pylori 26695 | 1.66 | 3 | 14 | 2 | 3 | 22 | |
| Helicobacter pylori J99 | 1.64 | 3 | 16 | 2 | 3 | 23 | |
| M. thermoautotrophicum | 1.75 | 1 | 1 | 3 | 5 | ||
| Methanococcus jannaschii | 1.66 | 3 | 8 | 11 | |||
| Mycobacterium tuberculosis | 4.40 | 1 | 1 | 2 | |||
| Mycoplasma genitalium | 0.58 | 1 | 1 | 2 | |||
| Mycoplasma pneumoniae | 0.81 | 1 | 1 | 2 | |||
| Neisseria meningitidis serotype A | 2.18 | 3 | 7 | 2 | 1 | 13 | |
| Neisseria meningitidis serotype B | 2.27 | 1 | 4 | 1 | 1 | 7 | |
| Pyrococcus abyssi | 1.77 | 1 | 4 | 5 | |||
| Pyrococcus horikoshii | 1.74 | 3 | 1 | 4 | |||
| Rickettsia prowazekii | 1.10 | no candidates | 0 | ||||
| Synechocystis species | 3.57 | 1 | 3 | 1 | 5 | ||
| Thermatoga maritima | 1.80 | 1 | 1 | ||||
| Treponema pallidum | 1.16 | 1 | 1 | ||||
| Ureaplasma urealyticum | 0.71 | 1 | 1 | 2 | 4 |
The column labeled ‘Orphan M’ contains solitary genes that share similarity with one component of an R-M system, but are missing others. The dam methylase of E.coli would be an example. The column labeled ‘mR’ contains genes that share similarity with methylation-dependent restriction systems.
More than two dozen bacterial and archaeal genomes have been completely sequenced within the last 5 years (7; Table 1). The complete sequences of these genomes have revealed a remarkable fact: >80% of the genomes appear to have at least one DNA R-M system and 75% of these genomes appear to contain multiple R-M systems, most of which have never been assayed biochemically (Table 1). The extreme case is Helicobacter pylori J99 which contains almost two dozen R-M systems, of which 16 appear to be Type II (8,9).
The availability of these complete genome sequences has opened new avenues to screen for Type II restriction endonucleases. First, bioinformatics methods can be used to identify methyltransferase genes and nearby open reading frames (ORFs) are candidate endonuclease genes. Those candidate genes can then be cloned and their gene products tested biochemically. However, characterizing restriction endonuclease genes in vivo by cloning and expression can be very challenging, because the gene products are cytotoxic when expressed in vivo unless the endogenous genomic DNA is completely protected by methylation (10). This means that usually both R and M genes must be cloned together or, more reliably, the M gene must be cloned and expressed first to provide a safe recipient.
In principle an R gene could be safely expressed under the control of an inducible promoter such as the T7 promoter in the absence of protective methylation. However, while expression can be repressed >100-fold in the T7 expression system (11), the cloning and expression of extremely toxic genes, such as those for restriction endonucleases, often cannot be achieved presumably because of low level expression from read-through transcripts (H.Kong and R.J.Roberts, unpublished observations). While the idea of using opposing promoters to modulate gene expression has been described previously (12,13), it has not been successfully employed as a method to clone a toxic gene. One system, which relies upon conditional expression of a gene encoding spectinomycin resistance, has proved to be a useful genetic selection for genes encoding proteins capable of exhibiting transcriptional repressor-like activity (14–16). These studies showed that transcriptional inactivation of a gene can be achieved with an antisense promoter. Here we describe a new cloning vector, pLT7K, which combines repression and an antisense promoter as dual control elements to repress basal expression but nevertheless permits inducible expression of a cloned gene. We have used this vector to clone some of the extremely toxic genes in sequenced genomes and describe here the analysis of the putative Type II restriction endonuclease genes in H.pylori J99 and the functional characterization of their companion DNA methylase genes using novel antibodies. Finally we discuss the biological significance of the numerous R-M systems in H.pylori.
MATERIALS AND METHODS
Genomic DNA and plasmid vector
Helicobacter pylori J99 genomic DNA was kindly provided by Dr Martin Blaser’s laboratory at Vanderbilt University (Nashville, TN). Plasmid pLT7K (Fig. 1) was constructed as follows. The segment encoding the replicative functions (encoded by rop and ori) in plasmid pLT7K is derived from pBR322 (17). The gene encoding β-lactamase (bla) confers ampicillin resistance, and has been altered to remove a recognition site for the PstI restriction endonuclease. The gene encoding kanamycin resistance is flanked by restriction enzyme sites suitable for cloning. The cI857 gene encodes a mutant form of the repressor protein, cI857 (18). The λ cI857 allele and PR promoter were subcloned from the pGW7 (G. G. Wilson, New England Biolabs, Beverly, MA) derivative, pJIH1 (gift of R. E. Webster, Duke University, Durham, NC). The lacI gene was derived from pTYB1 (New England Biolabs).
Figure 1.
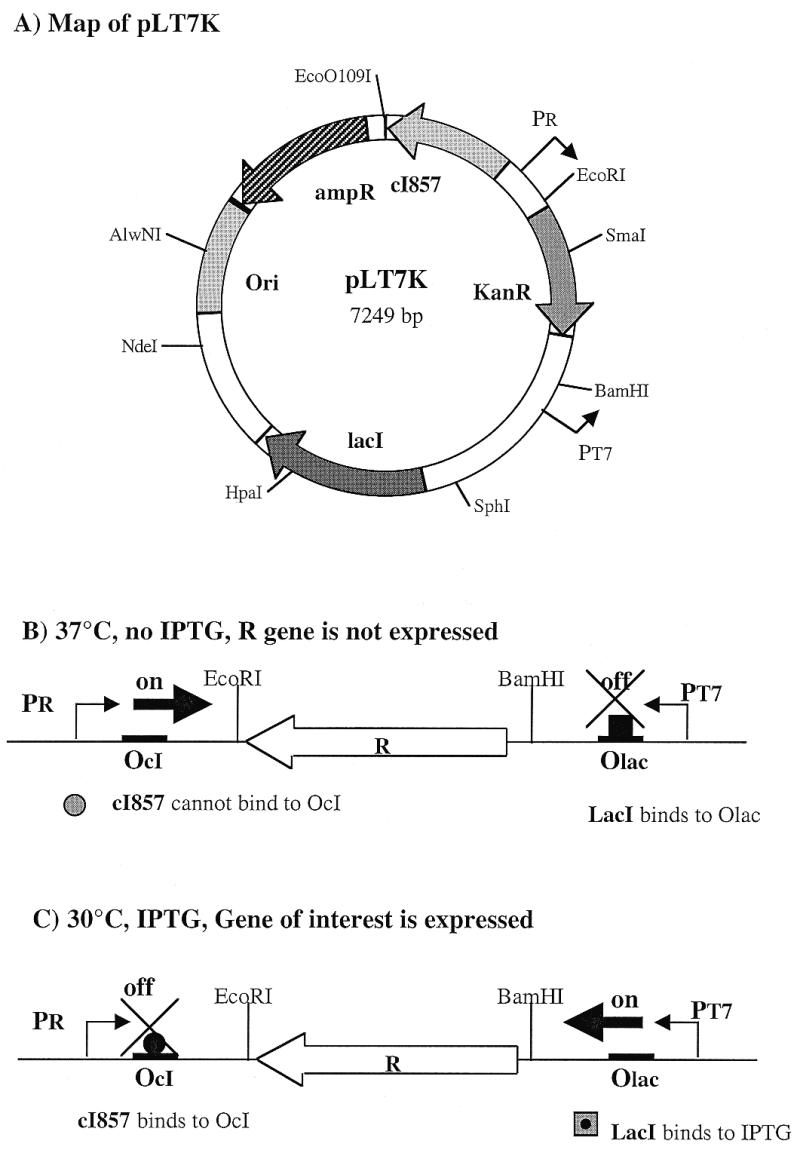
Map of plasmid pLT7K. Ori, origin of replication (colE1 derived from pBR322); ampR, β-lactamase gene (ampicillin resistance gene); KanR, kanamycin resistance gene which is flanked by multiple restriction sites suitable for cloning; cI857, the gene for a mutant form of the λ repressor; lacI, the gene for the lactose repressor protein, LacI; PR, bacteriophage λ major rightward promoter; PT7, bacteriophage T7 promoter; OcI, λ operator bound by λ CI repressor; Olac, lac operator bound by LacI.
Bacterial strains and growth
Escherichia coli DB24 is derived from E.coli GM4714 (19), with an additional mutation at the dcm locus via P1 transduction [dam-16::Kan trp-31 his-1 fhuA2 rpsL104 D(lacZ)r1 glnV44 xyl-7 mtl-2 metR1 mcr-62 argG6 D(mcrB-hsd-mrr)114 dcm-6 zed-501::Tn10]. Escherichia coli ER2502 [F– l– fhuA2 ara-14 leu Δ(gpt-proA)62 lacY1 glnV44 galK2 rpsL20 endA1 R(zgb210::Tn10 )Tet S xyl-5 mtl-1 Δ(mcrC-mrr)HB101] and ER2566 {F– l– fhuA2 [lon] ompT lacZ::T7 gene1 gal sulA11 Δ(mcrC-mrr)114::IS10 R(mcr-73::miniTn10–TetS)2 R(zgb-210::Tn10–TetS) endA1 [dcm]} were obtained from E. Raleigh and M. Sibley (New England Biolabs). Escherichia coli ER2566-pLysP is E.coli ER2566 which contains a mutant T7 lysozyme gene in the plasmid pACYC184. Escherichia coli strains were grown in LB broth (10 g Bacto-tryptone, 5 g bacto-yeast extract, 10 g NaCl per liter, adjusted to pH 7.5 with NaOH) or LB plates containing 1.5% agar at 37°C. Liquid cultures were shaken at 200 r.p.m. For expression of endonuclease genes cloned in pLT7K, and transformed into E.coli ER2566-pLysP, liquid cultures were grown at 37°C until OD600 = 0.8, and then transferred to a 30°C shaker for 30 min. Isopropylthiogalactoside (IPTG) (Sigma, St Louis, MO) was added to cultures to a final concentration of 0.4 mM. After 30 min at 30°C, rifampicin (Sigma) was added (final concentration of 200 µg/ml) to inhibit the host E.coli RNA polymerase. Induced cultures were grown at 30°C for a further 2 h, and then the E.coli cells were collected by centrifugation.
Identification of putative R-M genes
First we compared the H.pylori genome sequence to sequences in our REBASE collection (6), which includes sequences of identified and putative R-M components from public sequence databases [GenBank (20), DBEST (21), SwissProt (22) and PIR (23)]. In addition, some as yet unpublished sequences, generated at New England Biolabs, are also in this database. Different versions of the BLAST program (24) were used in the comparison, BLASTP for detecting matches of amino acid sequences of annotated H.pylori ORFs to REBASE sequences, and BLASTX to detect matches of translated DNA segments to the amino acid sequences of REBASE. The latter search enabled us to detect matches that extend beyond frameshifts and stop codons. Since the sequences of Type II methylases possess a moderate level of sequence conservation, this phase of the computer analysis identified all the putative methylases of H.pylori. In contrast, restriction endonucleases only share sequence similarity when they have similar recognition specificities (25). Therefore, few putative restriction enzymes could be identified at this step of sequence analysis.
The search for additional endonuclease candidates is based on the principle of ‘guilt by association’. Endonuclease and methylase genes in a given R-M system are usually located next to each other. Therefore an unknown gene, with no homolog on GenBank, next to a putative methylase gene is a candidate endonuclease gene. We extracted the neighboring ORFs of each putative H.pylori methylase gene, and queried the sequence databases with them, using NIH’s Email BLAST server (blast@ncbi.nlm.nih.gov ). Matches to known proteins eliminated a neighbor from consideration. Matches to unknown proteins also exclude a neighbor from the potential restriction enzyme set unless the unknown homolog is associated with a methylase. In short, methylase neighbors without similarity to other sequences in sequence databases are selected as potential restriction endonucleases.
Cloning and expression of putative endonuclease genes
The putative endonuclease genes were amplified by PCR using H.pylori J99 genomic DNA and a pair of primers corresponding to the 5′ and 3′ sequences of each gene. In addition to the coding region, the 5′-primer also contains a ribosome binding site plus a BamHI restriction site and the 3′-primer contains an XbaI cloning site. The amplified genes were cloned into plasmid pLT7K at the BamHI and XbaI sites such that expression was driven by the T7 promoter. The recombinant plasmid DNA was transformed into E.coli ER2502, which lacks T7 RNA polymerase, and plasmid DNA was purified. The nucleotide sequence of each putative endonuclease gene was checked to verify that no mutation had been introduced during PCR and cloning. Recombinant plasmids containing the wild-type putative endonuclease gene were then transformed into E.coli ER2566 pLysP strain, which contains the T7 RNA polymerase gene. The culture was first grown at 37°C without IPTG. When the culture reached mid-log phase, expression of the putative endonuclease gene was induced by equilibrating the culture at 30°C for 30 min, and then adding IPTG to a final concentration of 0.4 mM. This treatment turns off the PR promoter and turns on the T7 promoter (Fig. 1), enabling high level transcription of the putative endonuclease gene.
After 2–3 h induction, the E.coli culture was harvested. The cells were ruptured by sonication, and centrifuged at 31 000 g for 10 min at 4°C. Restriction endonuclease activity was determined by assaying the supernatant for DNA cleavage activity in vitro (5). Extracts were incubated with bacteriophage λ DNA, phage T7 DNA, and adenovirus-2 DNA in NEBuffers 2 and 4 (New England Biolabs) at 37°C for 15 min and the digestion products were analyzed by agarose gel electrophoresis.
Purification of restriction endonucleases
All operations were performed at 4°C unless otherwise noted. Escherichia coli cells were resuspended in buffer A (20 mM Tris–HCl pH 7.5, 0.1 mM Na2EDTA, 1 mM dithiothreitol) containing 50 mM NaCl. The suspension was sonicated. Following cell rupture, the supernatant was applied to a Heparin–Sepharose column equilibrated in buffer A. Endonuclease protein was eluted with a linear gradient of 0.05 to 1 M NaCl in buffer A. Endonuclease activity was assayed by digestion of λ DNA with aliquots of selected fractions. Fractions that showed cleavage activity were pooled, dialyzed against storage buffer (10 mM Tris–HCl pH 7.4, 100 mM NaCl, 1 mM dithiothreitol, 0.1 mM Na2EDTA, 50% glycerol), and kept at –20°C. Additional columns, DEAE–Sepharose and Resource-Q, were used to purify Hpy99I (encoded by JHP755).
Characterization of restriction endonuclease activity
The recognition sequence of all active endonucleases were determined by DNA mapping and computer analysis using the programs MAPSORT and GAP (Genetics Computer Group software package, Madison, WI). The locations of the cleavage sites by the endonuclease were mapped by double digestion of plasmid DNA (pBR322 or pUC19) with known restriction endonucleases and the endonuclease of interest. The mapping results were compared with data obtained using known restriction endonucleases.
Dot blot assay for methylation activity
Methylation activity was measured in a dot blot assay using two rabbit primary antibodies raised against DNA with N6-methyladenine (m6A) and N4-methylcytosine (m4C). Total DNAs were isolated from clones expressing individual H.pylori methylase genes. Sixteen DNA samples were spotted onto a nitrocellulose BA85 membrane and fixed by UV cross-linking (1.2 mJ/cm2 for 30 s). The membrane was then incubated in a 3% non-fat dry milk solution. After washing, the membrane was incubated with the primary antibodies at room temperature for 1 h. A secondary anti-rabbit antibody conjugated with horseradish peroxidase (HRP) (New England Biolabs) was then added to the membrane. Positive signals were detected using a phototope-HRP Western Detection Kit (New England Biolabs). DNA dots were visualized by developing the X-ray film which captured the light emitted by destabilized LumiGLO substrates.
RESULTS
A cloning vector for toxic genes
Plasmid pLT7K was designed for the cloning and expression of genes encoding products, in this case restriction endonucleases, that are toxic to E.coli. The gene of interest is cloned between two opposing promoters (T7 and λ-PR) that have distinct and independent control elements (Fig. 1). The bacteriophage T7 promoter is placed adjacent to a lac operator, which controls its activity, and is used to express the target gene. The expression from this promoter is blocked by the repressor, but can be induced by IPTG. The λ-PR promoter is used as an antisense promoter to suppress any basal level expression from the T7 promoter. The λ-PR promoter is regulated using a mutant form of the λ CI repressor that is temperature sensitive (17). When cells are grown in a suppressed condition (37°C, no IPTG; Fig. 1B), the expression of the target gene is prevented because the mutant λ repressor cI857 is inactive permitting antisense transcription from the λ-PR promoter to occur. The T7 promoter is inhibited by the lac repressor. When the cells are grown in an induced condition (30°C, +IPTG; Fig. 1C), expression of the target gene is elevated because the low temperature allows the cI857 repressor to suppress the antisense PR promoter while transcription from the T7 promoter is enabled by IPTG induction. We have tested the pLT7K vector in cloning and expression of various restriction endonucleases, such as EcoRI, HindIII, NlaIII, DraIII, PacI and SwaI, in the absence of their cognate methylases. All of these toxic endonuclease genes can be cloned into E.coli ER2566 pLysP and, following induction, endonuclease activities could be detected in crude cell extracts (data not shown). This shows that the pLT7K vector permits the controllable expression of extremely toxic genes in E.coli, at least for a few generations, and can be used to clone putative restriction endonuclease genes in the absence of methylation, thereby allowing an assay for their function.
Functional analysis of putative endonuclease genes
Sixteen putative Type II R-M systems have been identified in H.pylori J99 based on the original annotation plus our own sequence analysis detailed in the Materials and Methods (Table 2). All of the putative endonuclease genes were cloned into the pLT7K vector and expressed in E.coli. The DNA cleavage activities of these recombinant clones were assessed by digestion of DNA substrates with proteins encoded by the putative endonuclease genes. Four clones, derived from ORFs JHP46, JHP630, JHP755 and JHP1049, showed active endonuclease activities by this assay. The JHP46 gene was expressed well in E.coli and complete digestion of test DNA was achieved (Fig. 2). In this case, the endonuclease encoded by the JHP46 gene was identified as a Tsp45I (GTSAC) isoschizomer by analyzing the digestion pattern and this was further confirmed by double digestion of λ DNA with a crude extract of JHP46 and Tsp45I endonuclease. This enzyme is named Hpy99II (Table 2).
Table 2. Type II R-M systems in H.pylori J99 strain.
| JHP (ORF) | Annotation | Putative function | Function confirmed | Name |
|---|---|---|---|---|
| 42 | – | ENase | – | |
| 43 | 37% with M.DpnA, N6-A | MTase | +, m6A | M.Hpy99V |
| 45 | 45% with M.Tsp45I, N6-A | MTase | +, m6A | M.Hpy99II |
| 46 | – | ENase | +, (Tsp45I) | Hpy99II |
| 84 | 51% with R.MboI | ENase | Inactive ENase | |
| 85 | 60% with M.MboI, N6-A | MTase | +, m6A | M.Hpy99VI |
| 247 | – | ENase | – | |
| 248 | 28% with M.ScaI, N4-C | MTase | +, m4C | M.Hpy99VIII |
| 430 | 40% with M.VspI, N6-A | MTase | Inactive MTase | |
| 431 | – | ENase | – | |
| 433 | 29% with M.FokI, N6-A | MTase | Inactive MTase | |
| 434 | – | ENase | – | |
| 435 | 37% with M.HphI, 5-C | MTase | +, m5C | M.Hpy99XI |
| 436 | – | ENase | – | |
| 629 | 38% with M.CfrBI, N4-C | MTase | +, m4C | M.Hpy99IV |
| 630 | 25% with R.CfrBI | ENase | +, (BsaJI) | Hpy99IV |
| 755 | – | ENase | +, (new) | Hpy99I |
| 756 | 45% with BamHI, N4-C | MTase | +, m4C | M.Hpy99I |
| 845 | – | ENase | – | |
| 846 | 33% with M.HindII, N6-A | MTase | Inactive MTase | |
| 1012 | 31% with M.CviQI, N6-A | MTase | Inactive MTase | |
| 1013 | – | ENase | – | |
| 1049 | – | ENase | +, (HhaI) | Hpy99III |
| 1050 | 36% with M.Bsp6I, 5-C | MTase | + | M.Hpy99III |
| 1131 | 60% with M.NlaIII, N6-A | MTase | + | M.Hpy99X |
| 1270 | – | ENase | Inactive ENase | |
| 1271 | 62% with M.HifI, N6-A | MTase | +, m6A | M.Hpy99IX |
| 1364 | 31% with BcgI.B | S | – | |
| 1365 | 28% with BcgI.A | RM | Inactive RM | |
| 1442 | 37% with MboII | ENase | Inactive ENase |
MTase, DNA methylase; ENase, restriction endonuclease; S, specificity subunit. The active R-M systems are highlighted in bold type. Inactive ENase or MTase indicates an ORF with similarity to a known endonuclease or methylase but no function was detected. +, function detected; –, no function detected.
Figure 2.
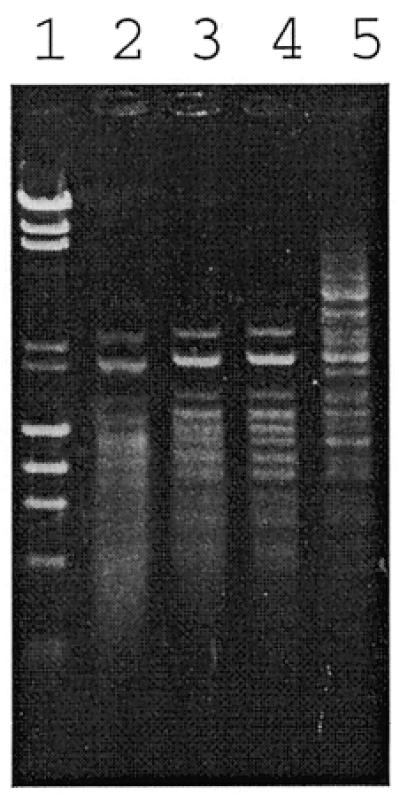
Agarose gel (1%) showing the electrophoresis patterns of bacteriophage λ DNA digested by different amounts of restriction endonuclease, Hpy99II, encoded by JHP46. Lane 1, bacteriophage λ DNA + HindIII, φX174 + HaeIII, size standard; lane 2, λ DNA digested with 9 µl Hpy99I; lane 3, λ DNA digested with 3 µl Hpy99I; lane 4, λ DNA digested with 1 µl Hpy99I; lane 5, λ DNA digested with 0.3 µl Hpy99I.
In the case of JHP1049, only weak activity was detected in crude extracts, and so the endonuclease encoded by JHP1049 was purified using heparin column chromatography. Good endonuclease activity was detected in the active fractions and this endonuclease, Hpy99III, was identified as an HhaI (GCGC) isoschizomer (Fig. 3).
Figure 3.
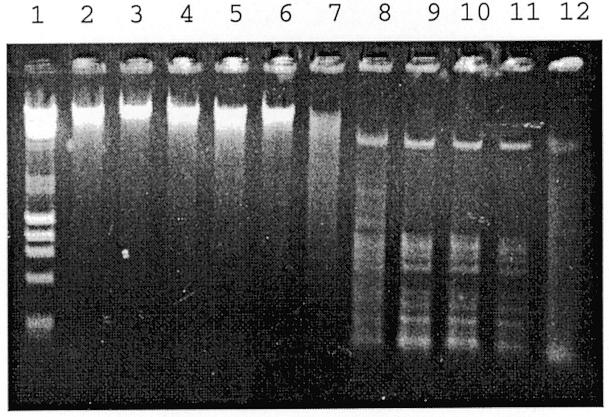
Agarose gel (1%) showing the electrophoresis patterns of λ DNA digested by restriction endonuclease Hpy99III, encoded by JHP1049, from consecutive fractions of a heparin column. Lane 1, λ DNA + HindIII, φX174 + HaeIII, size standard; lanes 2–12, λ DNA digested with 5 µl of Hpy99III eluted from a heparin column.
Similarly, the endonuclease activity of Hpy99IV (JHP630) was purified by heparin column chromatography and identified as a BsaJI (CCNNGG) isoschizomer. The endonuclease activity of JHP755 was purified by multiple column chromatographies (DEAE, Heparin, Resource-Q). Its cleavage activity was identified as a new restriction endonuclease Hpy99I, which recognizes the sequence CGWCG (the Hpy99I recognition sequence was identified by Q.Xu, R.Morgan, and M.Blaser, unpublished observation). Among 16 putative endonuclease genes, only the four genes mentioned above showed DNA cleavage activities in our assays. The rest displayed no detectable cleavage activities even when tested on additional DNA substrates such as phage T7 DNA and the G-C rich adenovirus-2 DNA (Table 2).
Characterization of DNA methylase activities
In Type II R-M systems, a methylase, which usually is encoded next to the restriction endonuclease, is responsible for methylating the host to prevent the endonucleolytic digestion of the endogenous DNA. Thus, after a restriction endonuclease is characterized, the methylation activity of its corresponding methylase can easily be determined by digesting the DNA, isolated from cells containing the relevant methylase gene, with the identified endonuclease. Since we have identified four active endonucleases in J99, we were able to test the functional activities of their corresponding DNA methylase genes by a simple digestion experiment. As expected, DNA isolated from cells expressing the JHP45 gene product is resistant to Hpy99II. The same is true for JHP629 (resistant to BsaJI), JHP756 (resistant to Hpy99I encoded by JHP755) and JHP1050 (resistant to HhaI).
The protein encoded by JHP1131 (Table 2) shares 93% sequence identity with the H.pylori methylase, M.HpyI (26) and 60% identity with M.NlaIII methylase (27). Genomic DNA isolated from H.pylori J99 cells containing the JHP1131 gene is resistant to the NlaIII endonuclease, showing that JHP1131 encodes an active methylase.
Since the majority of the H.pylori R-M systems examined showed no detectable endonuclease activity, it was difficult to assay the DNA methylase activity using the endonuclease digestion method as described above because we did not know the recognition specificity of these putative methylase genes. To circumvent this problem, we have developed two polyclonal antibodies raised against m6A or m4C.
We have cloned all the putative DNA methylase genes into pUC19 under the constitutively expressed lactose promoter. The recombinant plasmids were transformed into E.coli DB24, a strain that is deficient in the EcoKI restriction system, all of the known E.coli methylases, and the known methylated DNA restriction systems. To examine the enzymatic activity of these potential m6A and m4C methylases, we isolated total DNAs from cells carrying each of the H.pylori methylase genes and the methylation status of each sample was determined using the two methylated DNA specific antibodies in a dot blot assay as described in the Materials and Methods. A total of eight positive signals were detected from 13 DNA samples. The m6A antibody interacted with five DNA samples (JHP85, 1271, 43, 45 and 244) plus the m6A-methylase M.Tsp45I as a positive control (J.Wayne and S.-Y.Xu, unpublished data; S.Stickel and R.J.Roberts, unpublished data; Fig. 4A). The ORF of JHP244 was originally annotated as a Type II methylase (9). However it shares significantly higher sequence similarity with a Type III methylase than with a Type II. It is omitted from this analysis. The m4C antibody reacted with three of the samples (JHP629, 248 and 756), as well as a control DNA sample from a known m4C-methylase, M.BglII (Fig. 4B; 28; B.P.Anton, D.F.Heiter and J.E.Brooks, unpublished results). The two antibodies are very specific. The m6A antibody specifically reacted with the DNA modified by the N6-adenine methylase M.Tsp45I, and the m4C antibody only reacted with DNA modified by the N4-cytosine methylase M.BglII.
Figure 4.

Dot blot assay to measure methylase activities using rabbit primary antibodies that react specifically with DNA containing m6A or m4C modifications. The dot blot assay was performed as described in Materials and Methods. The ORF from which each DNA sample was derived is marked above each set of dots. (A) Methylase dot blot assay using m6A antibody with dilution of 1:750 000. (B) Methylase dot blot assay using m4C antibody with dilutions of 1:150 000. Three dilutions of DNA samples were spotted: 0.45, 0.15 and 0.05 µg of DNA (top to bottom on each panel).
Comparable antibodies against 5-methylcytosine (m5C) are not available and this is probably due to the fact that mammalian DNA also contains m5C modifications. To examine the methylation activity of the m5C methylase encoded by JHP435 ORF, we digested plasmid DNA isolated from cells expressing the JHP435 gene with 49 restriction endonucleases which recognize 4 or 5 bp sequences. Plasmid DNA containing JHP435 was resistant to only one restriction endonuclease, HpyCH4IV (R.Morgan and Q.Xu, unpublished results), suggesting that JHP435 encoded an active methylase which forms m5C within the sequence ACGT.
DISCUSSION
Helicobacter pylori R-M systems
We tested the restriction and modification activities of all putative Type II R-M systems from H.pylori J99 and the results are summarized graphically in Table 2. Among the 16 completely tested Type II R-M systems, only four are fully functional in that they contain both active endonucleases and methylases (Table 2, in bold type). The endonuclease activities from the native H.pylori J99 have been purified, and three endonucleases [isoschizomers of Tsp45I and HhaI, and the new specificity of Hpy99I (GCWGC)] have been found and characterized by purifying the endonuclease activities from native H.pylori J99 cells (Q.Xu and R.Morgan, personal communication). The genes encoding these three endonucleases have all been identified in this study. Thus, the pLT7K cloning/expression method is sensitive enough to detect all endonuclease activities that were identified in native H.pylori cells by conventional screening methods. The genome-based screening method described in this study has two advantages: (i) it is more sensitive (the BsaJI activity encoded by JHP630 was not detected by conventional methods); and (ii) it can save the fermentation of large amounts of microbes which could be difficult to grow such as Methanococcus jannaschii or are potentially dangerous pathogens such as H.pylori.
In addition to the four fully active Type II R-M systems, six putative R-M system genes encode active DNA methylases but failed to show detectable endonuclease activity (Table 2). We found frameshift and/or missense mutations in some of these inactive, putative endonuclease genes. For example, JHP84 shares 51% sequence identity with the Type II ENase MboI but contains three frameshift mutations in its coding region which explains why it is inactive. A second example concerns JHP1270, which showed no detectable endonuclease activity when it was expressed in pLT7K in E.coli. The corresponding gene, HP1351, in another sequenced H.pylori strain showed significant endonuclease activity when expressed in E.coli (L.Lin and H.Kong, unpublished result). The proteins, encoded by JHP1270 and HP1351, are both 290 amino acids long and share 93% sequence identity. Missense mutations are presumably responsible for the loss of endonuclease activity in JHP1270.
The remaining putative R-M systems lack any detectable restriction and modification activities. They may be ones that are evolving into oblivion or they may be in a temporarily inactive state.
Why does H.pylori contain so many R-M systems?
Helicobacter pylori is naturally competent and prone to take in DNA from the environment (29). In addition, bacteriophages are known to infect H.pylori (30). Thus, multiple R-M systems might be needed to protect its genome integrity. Our results showed that the majority of the R-M systems in H.pylori lack complete functional activity. Thus, numerous R-M systems may be needed to compensate for the propensity of these systems to accumulate inactivating mutations in H.pylori.
Why are so many of the H.pylori R-M systems completely or partially inactive? Clearly, in the absence of a continuous positive selection, missense and frameshift mutations can accumulate and inactivate the R-M systems. In addition, the phenomenon of phase variation can be responsible for the inactivation of some of the Type II R-M genes. Short tandem repeat sequences as well as homopolymeric tracts in DNA are subject to loss or gain of a repeat unit, presumably through slipped-strand mispairing during DNA replication. This results in frameshifting, which can alternately activate or inactivate genes (31). In the case of the H.pylori genome, 27 putative genes which contain simple sequence repeats have been identified and may be subject to phase variation. Three groups have been identified: lipopolysaccharide (LPS) biosynthesis, cell-surface-associated proteins and DNA R-M systems (32). For example, the ORF JHP1364, one of the inactive R-M systems in H.pylori J99 which encodes the BcgI-like S subunit (Table 2), contains a string of 14 G residues that may be subject to phase variation (33). In order for phase variation to work most effectively, both endonuclease and methylase genes have to be turned off. This is not a problem for BcgI-like R-M systems, because the endonuclease and methylase activities are located in the same polypeptide (34). Indeed, repeat sequences are found in genes that encode both Type I (JHP416) and Type III (JHP1411) R-M systems, which contain both endonuclease and methylase activities in one enzyme complex (33). A typical Type II R-M system contains two separate activities on two independent proteins. Sequential inactivation steps could be used to turn off both activities: the endonuclease gene might be inactivated followed by inactivation of the methylase gene. Such a mechanism might account for the inactive MboII endonuclease isoschizomer encoded by the ORF JHP1442 (Table 2). In this case, a homopolymeric tract of eight A residues is present in the middle of the ORF, resulting in a premature termination of translation (33). Thus, ORF JHP1442 is an inactive, orphan endonuclease gene and no corresponding methylase genes are found nearby (33). This could be explained as follows: JHP1442 gained an additional A in an original repeat of seven A residues and became a truncated, inactive endonuclease (33); without the selection pressure the corresponding methylase genes were deleted in a subsequent event, leaving an orphan, truncated endonuclease gene.
Biological benefits can result from phase variation and mutation. For instance, H.pylori cells can escape the human immune response by changing surface antigens such as LPS and cell-surface-associated proteins. Additionally, the population of H.pylori cells could maintain a variety of R-M systems, which are believed to be a primitive bacterial ‘immune’ system, through phase variation and mutation. At any one time just a small percentage of the population might have one of the hibernating systems in a fully active configuration. In this way the population can maintain a defensive system such that bacteriophages or free DNA, such as plasmids, that successfully take over one cell type will be restricted when entering other cells.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr Martin Blaser and Qing Xu for providing H.pylori J99 genomic DNA. We thank Qing Xu and Richard Morgan for sharing their research results on H.pylori R-M systems. We also acknowledge Drs Ira Schildkraut, Elisabeth Raleigh, Francine Perler and Donald Comb for constructive discussions. This work was partially supported by NIH grant GM56535 (to R.J.R.).
REFERENCES
- 1.Wilson G.G. and Murray,N.E. (1991) Restriction and modification systems. Annu. Rev. Genet., 25, 585–627. [DOI] [PubMed] [Google Scholar]
- 2.Roberts R.J. (1990) Restriction enzymes and their isoschizomers. Nucleic Acids Res., 18, 2331–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith H.O. and Wilcox,K.W. (1970) A restriction enzyme from Haemophilus influenzae. I. Purification and general properties. J. Mol. Biol., 51, 379–391. [DOI] [PubMed] [Google Scholar]
- 4.Roberts R.J., Breitmeyer,J.B., Tabachnik,N.F. and Myers,P.A. (1975) A second specific endonuclease from Haemophilus aegyptius. J. Mol. Biol., 91, 121–123. [DOI] [PubMed] [Google Scholar]
- 5.Schildkraut I.S. (1984) Screening for and characterizing restriction endonucleases. In Setlow,J.K. and Hollaender,A. (eds), Genetic Engineering, Principles and Methods, Vol. 6. Plenum Press, New York, NY, pp. 117–140.
- 6.Roberts R.J. and Macelis,D. (2000) REBASE-restriction enzymes and methylases. Nucleic Acids Res., 28, 306–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pallen M.J. (1999) Microbial genomes. Mol. Microbiol., 32, 907–912. [DOI] [PubMed] [Google Scholar]
- 8.Tomb J.-F., White,O., Kerlavage,A.R., Clayton,R.A., Sutton,G.G., Fleischmann,R.D., Ketchum,K.A., Klenk,H.P., Gill,S., Dougherty,B.A. et al. (1997) The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature, 388, 539–547. [DOI] [PubMed] [Google Scholar]
- 9.Hancock R.E., Alm,R., Bina,J. and Trust,T. (1998) Helicobacter pylori: a surprisingly conserved bacterium. Nat. Biotechnol., 16, 216–217. [DOI] [PubMed] [Google Scholar]
- 10.Howard K.A., Card,C., Benner,J.S., Callahan,H.L., Maunus,R., Silber,K., Wilson,G. and Brooks,J.E. (1986) Cloning the DdeI restriction-modification system using a two-step method. Nucleic Acids Res., 14, 7939–7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dubendorff J.W. and Studier,F.W. (1991) Controlling basal expression in an inducible T7 expression system by blocking the target T7 promoter with lac repressor. J. Mol. Biol., 219, 45–59. [DOI] [PubMed] [Google Scholar]
- 12.O’Connor C.D. and Timmis,K.N. (1987) Highly repressible expression system for cloning genes that specify potentially toxic proteins. J. Bacteriol., 169, 4457–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elledge S.J. and Davis,R.W. (1988) A family of versatile centromeric vectors designed for use in the sectoring-shuffle mutagenesis assay in Saccharomyces cerevisiae. Gene, 30, 303–312. [DOI] [PubMed] [Google Scholar]
- 14.Elledge S.J. and Davis,R.W. (1989) Position and density effects on repression by stationary and mobile DNA-binding proteins. Genes Dev., 3, 185–197. [DOI] [PubMed] [Google Scholar]
- 15.Elledge S.J., Sugiono,P., Guarente,L. and Davis,R.W. (1989) Genetic selection for genes encoding sequence-specific DNA-binding proteins. Proc. Natl Acad. Sci. USA, 86, 3689–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dorner L.F. and Schildkraut,I. (1994) Direct selection of binding proficient/catalytic deficient variants of BamHI endonuclease. Nucleic Acids Res., 25, 1068–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolivar F., Betlach,M.C., Heyneker,H.L., Shine,J., Rodriguez,R.L. and Boyer,H.W. (1997) Origin of replication of pBR345 plasmid DNA. Proc. Natl Acad. Sci. USA, 74, 5265–5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horiuchi T. and Inokuchi,H. (1967) Temperature-sensitive regulation system of prophage lambda induction. J. Mol. Biol., 23, 217–224. [DOI] [PubMed] [Google Scholar]
- 19.Palmer B.R. and Marinus,M.G. (1994) The dam and dcm strains of Escherichia coli—a review. Gene, 143, 1–12. [DOI] [PubMed] [Google Scholar]
- 20.Benson D.A., Boguski,D.A., Lipman,D.J., Ostell,J., Ouellette,B.F., Rapp,B.A. and Wheeler,D.L. (1999) GenBank. Nucleic Acids Res., 27, 12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boguski M.S., Lowe,T.M. and Tolstoshev,C.M. (1999) dbEST–database for ‘expressed sequence tags’. Nature Genet., 4, 332–333. [DOI] [PubMed] [Google Scholar]
- 22.Bairoch A. and Apweiler,R. (2000) The SWISS-PROT protein database and its supplement TrEMBL in 2000. Nucleic Acids Res., 28, 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barker W.C., Garavelli,J.S., Huang,H., McGarvey,P.B., Orcutt,B., Srinivasarao,G.Y., Xiao,C., Yeh,L.S., Ledley,R.S., Janda,J.F. et al. (2000) The Protein Information Resource (PIR). Nucleic Acids Res., 28, 41–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Altschul S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 25.Ruan H., Lunnen,K.D., Scott,M.E., Moran,L.S., Slatko,B.E., Pelletier,J.J., Hess,E.J., Benner,J., Wilson,G.G. and Xu,S.-Y. (1996) Cloning and sequence comparison of AvaI and BsoBI restriction-modification systems. Mol. Gen. Genet., 252, 695–699. [PubMed] [Google Scholar]
- 26.Xu Q., Peek,R.M. Jr., Miller,G.G. and Blaser,M.J. (1997) The Helicobacter pylori genome is modified at CATG by the product of HpyIM. J. Bacteriol., 179, 6807–6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morgan R.D., Camp,R.R., Wilson,G.G. and Xu,S.-Y. (1996) Molecular cloning and expression of NlaIII restriction-modification system in E. coli. Gene, 183, 215–218. [DOI] [PubMed] [Google Scholar]
- 28.Anton B.P., Heiter,D.F., Benner,J.S., Hess,E.J., Greenough,L., Moran,L.S., Slatko,B.E. and Brooks,J.E. (1997) Cloning and characterization of the BglII restriction-modification system reveals a possible evolutionary footprint. Gene, 187, 19–27. [DOI] [PubMed] [Google Scholar]
- 29.Suerbaum S., Smith,J.M., Bapumia,K., Morelli,G., Smith,N.H., Kunstmann,E., Dyrek,I. and Achtman,M. (1998) Free recombination within Helicobacter pylori. Proc. Natl Acad. Sci. USA, 95, 12619–12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heintschel von Heinegg E., Nalik,H.P. and Schmid,E.N. (1993) Characterisation of a Helicobacter pylori phage (HP1). J. Med. Microbiol., 38, 245–249. [DOI] [PubMed] [Google Scholar]
- 31.Hood D.W., Deadman,M.E., Jennings,M.P., Bisercic,M., Fleischmann,R.D., Venter,J.C. and Moxon,E.R. (1996) DNA repeats identify novel virulence genes in Haemophilus influenzae. Proc. Natl Acad. Sci. USA, 93, 11121–11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saunders N.J., Peden,J.F., Hood,D.W. and Moxon,E.R. (1998) Simple sequence repeats in the Helicobacter pylori genome. Mol. Microbiol., 27, 1091–1098. [DOI] [PubMed] [Google Scholar]
- 33.Alm R.A., Ling,L.S., Moir,D.T., King,B.L., Brown,E.D., Doig,P.C., Smith,D.R., Noonan,B., Guild,B.C., de Jonge,B.L. et al. (1999) Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature, 397, 176–180. [DOI] [PubMed] [Google Scholar]
- 34.Kong H. (1998) Analyzing the functional organization of a novel restriction modification system, the BcgI system. J. Mol. Biol., 279, 823–832. [DOI] [PubMed] [Google Scholar]