

Highlights
-
•
High expression of NAT10 in ESCC tumor samples is associated with macrophage polarization and the effect of PD-1 therapy.
-
•
METTL3-induced m6A modification leads to the upregulation of NAT10 in ESCC cells.
-
•
Silencing of NAT10 in ESCC cells can promote macrophage reprogram in a co-culture system.
Keywords: NAT10, FASN, Macrophage M2 polarization, ESCC, m6A modification
Abstract
N-acetyltransferase 10 (NAT10) is acknowledged as a tumor promoter in various cancers due to its role as a regulator of acetylation modification. Tumor-associated macrophages (TAMs) play a pivotal role in the tumor microenvironment (TME). However, the intercellular communication between esophageal squamous cell carcinoma (ESCC) cells and TAMs involving NAT10 remains poorly understood. This study aimed to elucidate the regulatory mechanism of NAT10 in modulating macrophage lipid metabolism and polarization. Experimental evidence was derived from in vitro and in vivo analyses. We explored the association between upregulated NAT10 in ESCC tissues, macrophage polarization, and the therapeutic efficacy of PD-1. Furthermore, we investigated the impact of methyltransferase 3 (METTL3)-induced m6A modification on the increased expression of NAT10 in ESCC cells. Additionally, we examined the role of exosomal NAT10 in stabilizing the expression of fatty acid synthase (FASN) and promoting macrophage M2 polarization through mediating the ac4C modification of FASN. Results indicated that NAT10, packaged by exosomes derived from ESCC cells, promotes macrophage M2 polarization by facilitating lipid metabolism. In vivo animal studies demonstrated that targeting NAT10 could enhance the therapeutic effect of PD-1 on ESCC by mediating macrophage reprogramming. Our findings offer novel insights into improving ESCC treatment through NAT10 targeting.
Graphical abstract

Introduction
Esophageal squamous cell carcinoma (ESCC), a predominant histological subtype of esophageal cancer, stands as one of the most formidable malignancies globally [1,2]. While surgery remains the primary therapeutic approach for ESCC, its applicability is limited for numerous patients who are unsuitable for direct esophagectomy [3], [4], [5]. Recent investigations have demonstrated the substantial efficacy of combined treatment involving an anti-programmed cell death protein 1 (PD-1) agent and chemotherapy, exhibiting prolonged survival in ESCC patients compared to chemotherapy alone [6], [7], [8], [9], [10]. Consequently, a comprehensive exploration of molecular mechanisms influencing the therapeutic outcomes of PD-1 therapy in ESCC patients becomes imperative.
Tumor-associated macrophages (TAMs) constitute vital components of the tumor microenvironment, displaying polarization into either M1 or M2 macrophages [11]. Emerging evidence underscores that the transition of M1 to M2 macrophages can reprogram the macrophage landscape, activating malignant processes within cancer cells by shaping the tumor microenvironment. For instance, exosomal FGD5-AS1 has been shown to stimulate cell proliferation and migration in pancreatic cancer by promoting M2 polarization of TAMs [12], while exosome-delivered CXCL14 induces M2 polarization of TAMs, fostering prostate cancer progression [13]. Previous studies have established that exosomes mediate the transfer of RNA or proteins to adjacent cells or tissues, orchestrating alterations in the tumor microenvironment and driving tumor progression [14,15]. Despite these insights, the exosome-mediated molecular mechanisms influencing intercellular communication between TAMs and ESCC cells remain elusive. This study endeavors to investigate the role of exosome-mediated intercellular communication between TAMs and ESCC cells in shaping the efficacy of PD-1 therapy.
N-acetyltransferase 10 (NAT10) emerges as a pivotal regulator of mRNA stability and translation, primarily through RNA N4-acetylcytidine (ac4C) modification [16]. Serving as a catalytic enzyme, NAT10 modifies the acetylation of tRNA, rRNA, and mRNA, exhibiting implications in various cancer types [17], [18], [19], [20], [21], [22], [23]. However, the specific effects of NAT10 on the TAM polarization remain obscure. The current study elucidates the biological impact of NAT10 on macrophage reprogramming, accompanied by an exploration of the underlying molecular mechanisms.
N6-methyladenosine (m6A) modifications exert control over RNA fate by modulating stability, translation, and pre-mRNA splicing through the actions of “writers”, “erasers”, and “readers” [24], [25], [26], [27]. Methyltransferase-like 3 (METTL3), a m6A methyltransferase, regulates critical biological processes, including cell differentiation and proliferation [28], [29], [30]. In this context, we investigate whether NAT10 is upregulated by METTL3-mediated m6A modification.
In summary, the primary objectives of this study are to unravel the role of METTL3-mediated m6A modification of NAT10 in regulating macrophage reprogramming and to delineate the impact of PD-1 therapy on ESCC.
Materials and methods
Clinical samples
Fresh tumor samples and corresponding non-tumor tissues were collected from 42 patients diagnosed with ESCC following microscopic examination by two licensed pathologists at Changhai Hospital, Second Military Medical University (Naval Medical University). The tumor samples were categorized into two groups based on the patients' response to PD-1 therapy: 20 samples from patients classified as sensitive and 22 samples from those classified as resistant. Informed consent was obtained from all patients before sample collection, and the study received approval from the Ethics Committee of Changhai Hospital, Second Military Medical University (Naval Medical University). The collected samples were promptly snap-frozen in liquid nitrogen upon isolation and stored at −80 °C.
Immunohistochemistry (IHC) staining
In brief, paraffin-embedded tissue slices were deparaffinized in xylene and subsequently rehydrated in ethanol. Antigen retrieval was performed by autoclaving, and endogenous peroxidase activity was inactivated with methanol. Tissue slices were then incubated with primary antibodies (NAT10, CD163, and CD86) at 4 °C for 12 h, followed by incubation with the secondary antibody at room temperature for 45 min. The tissues were stained with 3,3-diaminobenzidine (DAB) for 10 min. Images were captured from five random fields using a light microscope (20×).
Cell culture and treatment
Human ESCC cell lines (Eca-109, TE-5, KYSE-410, KYSE-510, and TE-1) and human normal esophageal epithelial cells (HEEC) were procured from the Chinese Academy of Sciences cell library (Beijing, China). The human monocyte cell line (THP-1) was obtained from ATCC (USA). All cell lines were cultured in DMEM (Solarbio, Guangzhou, China) supplemented with 10 % FBS and 100 units of penicillin-streptomycin (Thermo Fisher Scientific, USA) at 37 °C with a 5 % CO2 atmosphere in a humidified incubator. THP-1 cell culture was conducted in RPMI1640 medium (Corning Cellgro, Manassas, VA, USA) under the same culture conditions. THP-1 cells-derived macrophages (THP-1-DM) were induced by treating THP-1 cells with 320 nM Phorbol 12-myristate 13-acetate.
For mRNA stability measurement, cells were treated with 100 nM Actinomycin D (ActD) and incubated for various time intervals.
Cell transfections
ShRNAs against METTL3 (sh-METTL3#1, sh-METTL3#2, sh-METTL3#3), NAT10 (sh-NAT10#1, sh-NAT10#2, sh-NAT10#3), and their negative controls (sh-NC) were provided by Shanghai GenePharma Company (China). NAT10 was overexpressed by subcloning its entire sequence into the pcDNA3.1 vector to construct the overexpression vector (pcDNA3.1-NAT10), with the pcDNA3.1 empty vector used as a control. TE-5 and Eca-109 cells seeded in eight-well plates were transfected with 80 nM of synthesized shRNAs using Lipofectamine 3000 (Invitrogen, USA) and incubated for 48 h.
RT-qPCR
RNA extraction was performed using Trizol reagent. Reverse transcription was carried out with ABScript II RT Mix for qPCR (ABclonal, China). qRT-PCR was performed using TB Green® Premix Ex Taq™ II (TaKaRa, Japan). GAPDH gene was employed as an internal control. The relative gene expression was calculated using 2−ΔΔCt method.
RIP
RNA-protein interactions were validated using the Magna RIP Kit (Millipore, USA) and analyzed by RT-qPCR. Briefly, cells were lysed with RIP lysis buffer containing a protease inhibitor cocktail and RNase inhibitors. Subsequently, cell lysates and magnetic beads conjugated with specific antibodies (anti-m6A, anti-METTL3, anti-NAT10, and anti-ac4C) or IgG were incubated in RIP immunoprecipitation buffer at 4 °C for 3 h with rotation. After digestion with protease K, the immunoprecipitated RNA samples were purified by phenol/chloroform/isoamyl alcohol and 100 % ethanol, and finally detected by RT-qPCR.
Co-culture system
A co-culture system was established using a Transwell chamber (4 µm pore inserts). ESCC cells (Eca-109/TE-5) were cultured in the lower chamber, while macrophages were cultured in the upper chamber.
Exosome extraction and identification
ESCC cells were cultured in medium with 10 % exosome-free FBS. After 72 h, cell culture medium was collected, and exosomes were isolated from the supernatant by differential centrifugation at 300 g for 10 min, 2000 g for 15 min, and 12,000 g for 30 min to remove floating cells and cellular debris. The supernatant was then filtered through a 0.22-μm membrane (Millipore, USA). The resulting supernatant was passed through the membrane by centrifugal filtration at 4 × 103 g for 1 h at 4 °C with the ultrafiltration device (UFC900396, Millipore, USA). Finally, the EXO Quick-TC™ Exosome Isolation Reagent (EXOTC50A-1, System Biosciences, USA) was used for exosome isolation, as previously mentioned [31,32]. As described [33], the exosomes were observed by Transmission electron microscopy (TEM, JEM-2100, Jeol, Japan). The size distribution and concentration of exosomes were determined by Nanoparticle Tracking Analysis (NTA) according to a previous protocol [34]. Results were analyzed using the Zetasizer software (Malvern Instruments).
Cells were fixed in 4 % paraformaldehyde and permeabilized with 0.25 % Triton X-100. Next, cells were blocked with goat serum for 30 min at room temperature, incubated with anti-PKH26 overnight at 4 °C. The next day, the cells were washed and then incubated with the secondary antibody for 1 h at room temperature. THP-1-DM were stained with DAPI. Finally, the cells were randomly observed and photographed under a fluorescence microscope.
Western blot
Proteins were extracted from cell lines using RIPA buffer (Thermo) containing a protease inhibitor. Protein samples were loaded onto PAGE gels (Epizyme Biomedical Technology, Shanghai, China) and transferred to 0.2 µm immobilon PVDF membranes (Millipore Sigma). Subsequently, membranes were incubated with primary antibodies (anti-CD63, anti-TSG101, anti-NAT10, and the loading control anti-β-actin) at appropriate dilutions at 4 °C overnight. After washing, the membranes underwent incubation with secondary antibodies at a 1:5000 dilution at room temperature for 1 h. Finally, blots were visualized using an ECL system (Share-bio, Shanghai, China).
Flow cytometry
THP-1-DM were harvested and collected using cold PBS. Cells were then resuspended in flow cytometry buffer (1 × PBS buffer containing 1 % BSA), stained with anti-CD163 antibody (1:100 dilution, BioLegend) and anti-CD86 antibody (1:100 dilution, BioLegend) for 30 min on ice, and subsequently analyzed using flow cytometry (C500, Beckman, USA). The data was analyzed using FlowJo software.
TG (triglyceride) levels
Cellular TG levels and cellular fatty acid levels were separately measured using the Infinity TGs Reagent (Thermo Scientific) and the ELISA kit (Jiancheng Bio., Nanjing, China).
Fatty acid oxidation (FAO)
The FAO content of cells was measured using the FAO Assay Kit (AmyJet Scientific, catalog number BR00001). Cells were lysed and added to the components of the assay kit, following the instructions provided in the kit's manual.
Oil red staining
Cells in the active state were prepared for cell culture and subjected to formaldehyde fixation. After fixation, Oil Red dye was added for staining, followed by washing. Then, hematoxylin was used to stain the cell nuclei. The stained cells were visualized under a microscope, and images were captured to examine the staining results.
Animal experiments
Before all animal studies, ethics approval was obtained from the Animal Care Committee of Changhai Hospital, Second Military Medical University (Naval Medical University). For xenograft experiments, a total of 20 male C57/BL6 mice were purchased from the Model Animal Research Center (Nanjing, China). All mice were randomly and evenly divided into four groups. AKR cells stably transfected with sh-NC or sh-NAT10#1 were inoculated subcutaneously on the right dorsal side of mice at a density of 2 × 106 cells per mouse. Two groups of mice underwent PD-1 treatment at 7 days after injection. Tumor volume was recorded and calculated every four days according to the formula (length × width2/2). At day 28, mice were sacrificed, and each tumor was isolated and weighed. Tumor samples from the four different groups were collected for IHC staining.
For IHC staining, after the sections were deparaffinized and rehydrated, the specimens were incubated in EDTA buffer (1 mM, PH 8.0) for antigen retrieval using a high-pressure method. Then, tissue sections were incubated overnight at 4 °C with primary antibodies, including anti-NAT10, anti-CD163, and anti-CD86. DAB solution (ZSGB-BIO, Beijing, China) was used to detect target proteins, which were conjugated with a peroxidase enzyme to form a brown precipitate.
Statistical analysis
Statistical analysis of two comparison groups and multiple comparison groups was performed using unpaired Student's t-tests and one-way ANOVA in SPSS v26.0 software. Data obtained from three independent experiments are shown as mean ± SEM using GraphPad v8.0. A P-value less than 0.05 was defined as the threshold of statistical significance.
Results
High NAT10 expression in ESCC tumor samples correlates with macrophage polarization and impacts PD-1 therapy efficacy
The expression profile of NAT10 in esophageal carcinoma (ESCA) was investigated using two online databases, namely starbase v2.0 (http://starbase.sysu.edu.cn/) and ualcan (https://ualcan.path.uab.edu/analysis.html). Notably, NAT10 demonstrated a significantly elevated expression in ESCA tumor tissues when compared to normal esophageal tissues, which served as the control group (Fig. 1A, B). To further validate these findings, NAT10 expression was measured in paired tumor tissues and parental non-tumor tissues collected from 42 ESCC patients. Consistent with the database results, NAT10 expression was found to be significantly higher in ESCC tumor tissues (Fig. 1C).
Fig. 1.

High NAT10 expression in ESCC tumor samples correlates with macrophage polarization and impacts PD-1 therapy efficacy A-B. NAT10 expression in ESCA tumor tissues compared to normal esophageal tissues as the control group. Results were obtained from two online databases, including starbase v2.0 and ualcan. C. NAT10 expression in paired tumor tissues and parental non-tumor tissues obtained from 42 ESCC patients was measured using RT-qPCR. D, E. NAT10 expression in tumor tissues collected from ESCC patients, categorized as resistant or sensitive to PD-1 therapy, was measured by RT-qPCR and IHC staining. F, G. The positivity of M1 marker (CD86) and M2 marker (CD163) was assessed in tumor tissues collected from ESCC patients, categorized as resistant or sensitive to PD-1 therapy, using IHC staining. ⁎⁎P < 0.01.
Immunotherapy, particularly involving immune checkpoint inhibitors like anti-PD-1 or anti-PD-L1 antibodies, has exhibited considerable efficacy in some esophageal cancer patients. Nevertheless, the precise indications, efficacy, and underlying molecular mechanisms of immunotherapy necessitate further investigation and evaluation. We explored the potential association between NAT10 expression and the response to PD-1 therapy. RT-qPCR (Fig. 1D) and IHC (Fig. 1E) analyses revealed heightened NAT10 expression in tumor tissues from ESCC patients resistant to PD-1 therapy.
Subsequent IHC staining was conducted to assess M1 and M2 macrophage markers in tumor tissues from ESCC patients classified as either sensitive or resistant to PD-1 therapy (Fig. 1F, G). The results illustrate that the positivity of the M1 marker (CD86) was lower in the resistant groups, whereas the M2 marker (CD163) exhibited an opposing trend. These findings collectively suggest that NAT10 is upregulated in ESCC tumor samples and is associated with macrophage polarization, thereby influencing the response to PD-1 therapy.
METTL3-mediated m6A modification drives the upregulation of NAT10 in ESCC cells
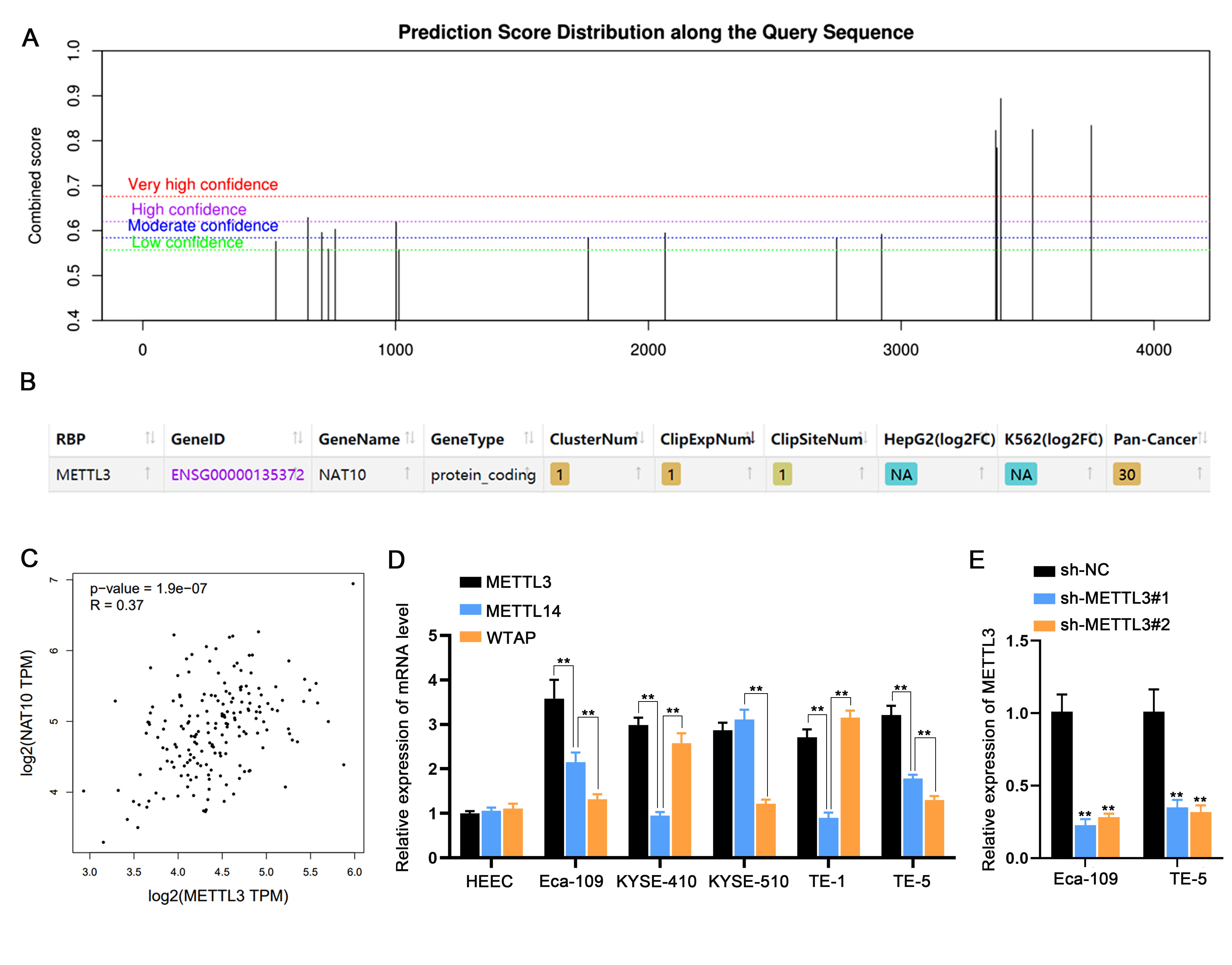
We investigated the cellular expression of NAT10 in ESCC cells, employing RT-qPCR and Western blot analyses. Comparative assessments with the normal HEEC cell line demonstrated substantial upregulation of NAT10 in five ESCC cell lines, particularly in Eca-109 and TE-5 cells, at both mRNA and protein levels (Fig. 2A). Subsequently, we delved into the mechanisms underlying NAT10 upregulation in ESCC cells. Analysis on the SRAMP database (http://www.cuilab.cn/sramp) revealed multiple m6A modification sites in NAT10 3′UTR (Fig. S1A). Further exploration on the starbase website indicated the binding of the m6A methyltransferase METTL3 to NAT10 3′UTR (Fig. S1B). Additionally, a positive correlation between METTL3 and NAT10 expression was observed in ESCA tumor tissues based on starbase pan-cancer data (Fig. S1C). Consistent with NAT10, METTL3 exhibited significant overexpression in ESCC cells (Fig. S1D).
Fig. 2.

METTL3-mediated m6A modification drives the upregulation of NAT10 in ESCC cells A. NAT10 expression in five ESCC cells was assessed using RT-qPCR and Western blot, comparing them with the normal cell line HEEC. B, C. The enrichment of NAT10 3′UTR in immunoprecipitates obtained from m6A or METTL3 antibody-incubated complexes was verified through m6A-RIP assay. D. NAT10 expression in two ESCC cells transfected with METTL3-specific shRNA or sh-NC was determined. E. The binding of NAT10 3′UTR with m6A was demonstrated by M3RIP-qPCR and agarose gel electrophoresis. F. The enrichment of NAT10 3′UTR in immunoprecipitates obtained from m6A antibody-incubated complexes was reduced after knockdown of METTL3. G. A luciferase reporter vector containing NAT10 3′UTR or NAT10 3′UTR with mutant m6A sites was constructed for luciferase reporter assay. H. The luciferase activity of the vector containing the original NAT10 3′UTR (WT) or NAT10 3′UTR with mutant m6A sites was measured in macrophages after co-transfecting METTL3-specific shRNAs. I. NAT10 mRNA lifetime profiling was performed in ESCC cells with METTL3 silencing incubated with the transcription inhibitor (ActD). ⁎⁎P < 0.01.
To investigate whether METTL3 regulates NAT10 expression through m6A modification, we performed m6A-RIP assays, confirming the enrichment of NAT10 3′UTR in immunoprecipitates from m6A or METTL3 antibody-incubated complexes (Fig. 2B, C), indicating potential m6A modification mediated by METTL3. Silencing METTL3 using shRNA in two ESCC cell lines (Fig. S1E) resulted in the anticipated downregulation of NAT10 (Fig. 2D). The binding of NAT10 3′UTR with m6A was further supported by MeRIP-qPCR and agarose gel electrophoresis (Fig. 2E). Importantly, the enrichment of NAT10 3′UTR in immunoprecipitates from m6A antibody-incubated complexes was reduced after METTL3 knockdown (Fig. 2F). We constructed a luciferase reporter vector containing NAT10 3′UTR or NAT10 3′UTR with mutated m6A sites for luciferase reporter assays (Fig. 2G). Experimental results showed reduced luciferase activity of the vector containing the original NAT10 3′UTR (WT) after co-transfection with METTL3-specific shRNAs, a tendency abolished after m6A sites were mutated in NAT10 3′UTR (Fig. 2H).
Finally, ESCC cells with METTL3 silencing were treated with the transcription inhibitor (ActD) for NAT10 mRNA lifetime profiling. Indeed, the lifetimes of NAT10 mRNA were shortened in METTL3-silenced ESCC cells (Fig. 2I). In conclusion, our findings collectively demonstrate that METTL3-induced m6A modification drives the upregulation of NAT10 in ESCC cells.
Silencing NAT10 in ESCC cells enhances macrophage reprogramming in a co-culture system
Previous studies have established that exosomal RNAs derived from tumor cells play a pivotal role in modulating the tumor microenvironment by inducing macrophage reprogramming [35], [36], [37]. In this study, we aimed to elucidate the influence of NAT10 in mediating the interaction between ESCC cells and macrophages. To address this, we established a co-culture system involving ESCC cells and THP-1-DM cells (Fig. S2A). Additionally, we silenced NAT10 expression in two specified ESCC cell lines for subsequent analysis (Fig. S2B).
Subsequent investigation involved the assessment of M2 marker expression in THP-1-DM cells co-cultured under different conditions, all four M2 markers exhibited an increased expression when co-cultured with ESCC cells (Fig. S2C). However, this augmented trend was abrogated when ESCC cells were treated with GW4869 (an exosome inhibitor) or transfected with NAT10-specific shRNA (sh#1). Similarly, the percentage of M2 macrophages (CD163 positive macrophages) increased during co-culture with ESCC cells, but this elevation was nullified by GW4869 treatment or NAT10-specific shRNA transfection (Fig. S2D). Conversely, the percentage of M1 macrophages (CD86 positive macrophages) displayed opposing trends to M2 macrophages (Fig. S2E).
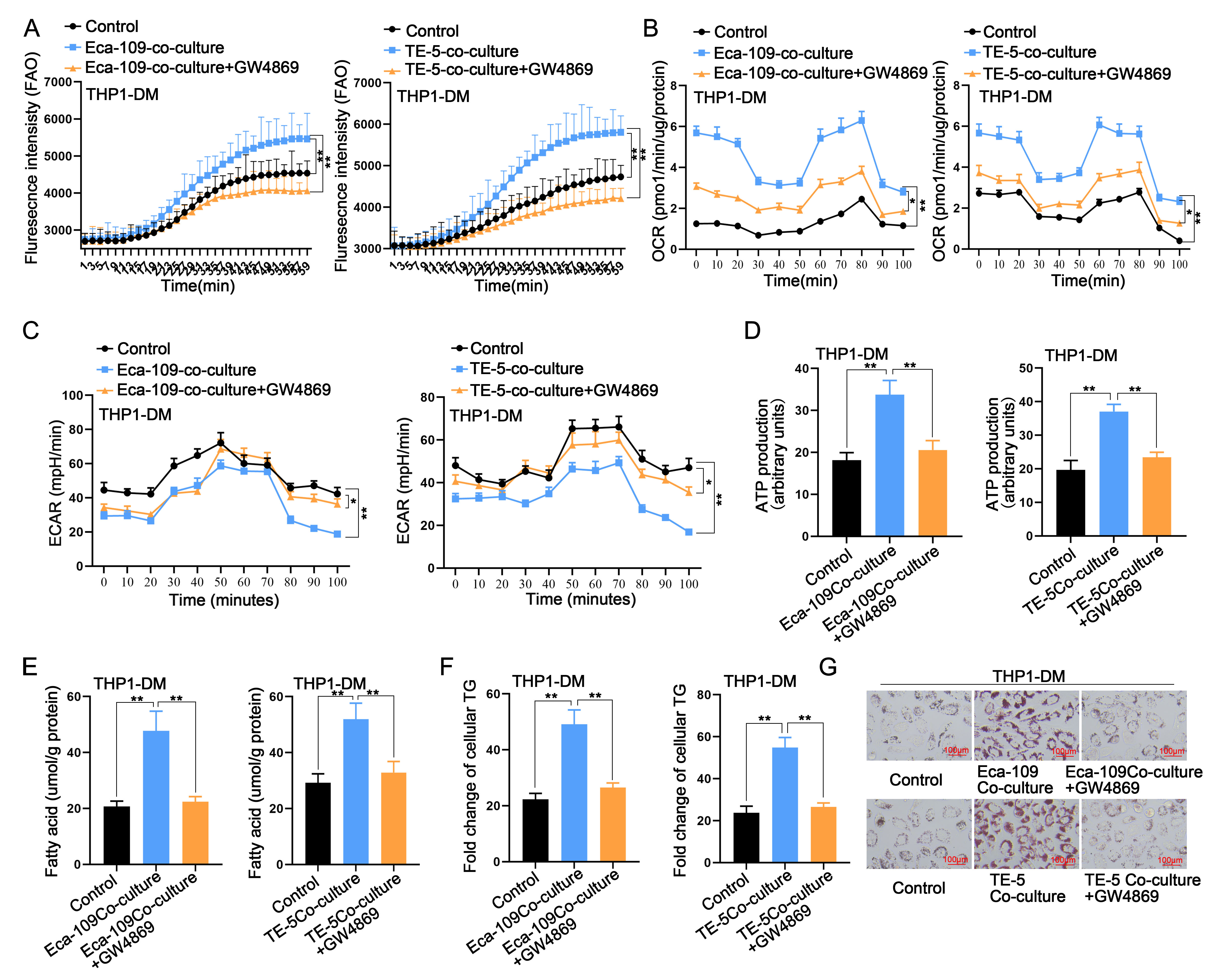
Given the crucial role of lipid metabolism in regulating macrophage functions and polarization [38,39], we investigated whether ESCC could modulate the lipid metabolism of macrophages. Initially, we analyzed FAO levels in macrophages post co-culture with ESCCs. Our findings indicated that esophageal cancer cells increased fatty acid oxidation in macrophages, a process reversible by GW4869 (Fig. S3A). Subsequently, analysis of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) revealed that esophageal cancer cells elevated OCR and decreased ECAR, signifying a shift away from glycolysis as the primary energy pathway for macrophages (Fig. S3B, C). ATP measurements further demonstrated that esophageal cancer cells enhanced the energy production rate of macrophages, an effect reversible by GW4869 (Fig. S3D). Additionally, analysis of fatty acids, TGs, and lipid droplets consistently revealed that esophageal cancer cells increased their levels in macrophages, with this effect being reversible by GW4869 (Fig. S3E–G).
In conclusion, our findings collectively suggest that tumor cells can promote the fatty acid metabolism of macrophages, with exosomes potentially participating in this process.
ESCC cell-derived exosomes promote macrophage M2 polarization by transferring NAT10
The preceding data underscored the involvement of exosomes in the interplay between ESCC cells and macrophages. To further investigate whether ESCC cell-derived exosomes exerted an influence on macrophages through NAT10, we initially isolated exosomes from ESCC cells and characterized them using TEM (Fig. 3A). Additionally, the presence of exosome surface markers (CD63 and TSG101) was confirmed through Western blot analysis (Fig. 3B). Furthermore, the size and concentration of exosomes were determined using NTA (Fig. 3C), and exosomes were labeled with PKH26 for tracing purposes (Fig. 3D).
Fig. 3.

ESCC cell-derived exosomes promote macrophage M2 polarization by transferring NAT10 A. Exosomes extracted from ESCC cells were observed using TME. B. The abundant expression of exosome surface markers (CD63 and TSG101) was assessed through Western blot. C. The diameter and concentrations of exosomes were analyzed using NTA. D. Exosomes were labeled with PKH26 for tracing. E. Changes in NAT10 protein levels in ESCC cells-derived exosomes (Eca-109-Exos or TE-5-Exos) were detected through Western blot after NAT10 overexpression or interference. F. The impact of ESCC cells-derived exosomes (Eca-109-Exos or TE-5-Exos) on NAT10 expression in macrophages was measured by Western blot. G. NAT10 expression was measured in macrophages treated with exosomes derived from ESCC cells or NAT10-silenced ESCC cells (Eca-109/sh-NAT10#1-Exos or TE-5/sh-NAT10#1-Exos). H. Expression levels of all four M2 markers were assessed in macrophages treated with ESCC cells-derived exosomes or NAT10-silenced ESCC cells-derived exosomes. I. The percentage of M2 macrophages was determined by flow cytometry analysis after treatment with the four exosomes. J. The percentage of M1 macrophages was assessed by flow cytometry analysis after treatment with the four exosomes. K. The correlation between NAT10 and M1 marker CD86, as well as NAT10 and M2 marker CD163, was evaluated in tissue expression. ⁎⁎P < 0.01.
Subsequent Western blot results demonstrated that in ESCC cells with NAT10 knockout or overexpression, the levels of NAT10 in the secreted exosomes were altered. Specifically, overexpression of NAT10 in ESCC cells led to an elevation of NAT10 content in the exosomes, while interference with NAT10 resulted in a reduction of NAT10 levels in the exosomes (Fig. 3E). Following this, we assessed the impact of ESCC cell-derived exosomes (Eca-109-Exos or TE-5-Exos) on NAT10 expression in macrophages. Our findings revealed that compared to control macrophages, NAT10 expression was heightened in macrophages treated with Eca-109-Exos or TE-5-Exos (Fig. 3F). Conversely, NAT10 expression returned to baseline levels in macrophages treated with exosomes derived from NAT10-silenced ESCC cells (Eca-109/sh-NAT10#1-Exos or TE-5/sh-NAT10#1-Exos) (Fig. 3G).
Subsequent evaluation of macrophage polarization revealed dysregulation of all four M2 markers in macrophages treated with Eca-109-Exos or TE-5-Exos, while no significant changes were observed in macrophages treated with Eca-109/sh-NAT10#1-Exos or TE-5/sh-NAT10#1-Exos (Fig. 3H). Similarly, the percentage of M2 macrophages increased upon treatment with Eca-109-Exos or TE-5-Exos but remained unchanged with Eca-109/sh-NAT10#1-Exos or TE-5/sh-NAT10#1-Exos (Fig. 3I). Conversely, the percentage of M1 macrophages exhibited opposite trends to M2 macrophages (Fig. 3J).
Finally, a correlation analysis between NAT10 expression and macrophage polarization was conducted, revealing a negative correlation between high NAT10 expression and M1 polarization, and a positive correlation with M2 polarization at the clinical specimen tissue level (Fig. 3K). In conclusion, our findings substantiate that ESCC cell-derived exosomal NAT10 promotes macrophage M2 polarization.
ESCC cells-secreted exosomal NAT10 mediates macrophage M2 polarization by facilitating lipid metabolism
Given the alterations observed in macrophage lipid metabolism in response to tumor cell co-culture conditions, we hypothesized that NAT10 might play a pivotal role in the remodeling of macrophage lipid metabolism. Consequently, we conducted a more in-depth investigation into the impact of exosomal NAT10 on mediating the lipid metabolism of macrophages. Initially, ELISA results revealed an augmentation of cellular TG in macrophages treated with Eca-109-Exos or TE-5-Exos, while no significant change was observed in macrophages treated with Eca-109/sh-NAT10#1-Exos or TE-5/sh-NAT10#1-Exos (Fig. 4A). Concurrently, lipid droplets were increased in macrophages treated with Eca-109-Exos or TE-5-Exos, whereas there was no discernible change in macrophages treated with Eca-109/sh-NAT10#1-Exos or TE-5/sh-NAT10#1-Exos (Fig. 4B). Further analyses of FAO levels in macrophages demonstrated a significant increase after treatment with ESCC cell-derived exosomes, while treatment with exosomes from NAT10-silenced ESCC cells yielded no change (Fig. 4C). Additionally, it was observed that exosomes derived from esophageal cancer cells increased the OCR of macrophages (Fig. 4D), decreased the ECAR (Fig. 4E), enhanced ATP production (Fig. 4F), and increased fatty acid content (Fig. 4G). Importantly, these effects were nullified upon interference with NAT10.
Fig. 4.

ESCC cells-secreted exosomal NAT10 mediates macrophage M2 polarization by facilitating lipid metabolism A. The cellular TG level in macrophages was measured by ELISA after treatment with ESCC cells-derived exosomes or NAT10-silenced ESCC cells-derived exosomes. B. The measurement of lipid droplets in macrophages treated with ESCC cells-derived exosomes or NAT10-silenced ESCC cells-derived exosomes. C–G. The FAO (C), OCR (D), ECAR (E), ATP (F), and fatty acid (G) levels in macrophages treated with ESCC cells-derived exosomes or NAT10-silenced ESCC cells-derived exosomes were detected using isotope experiment. *P < 0.05, ⁎⁎P < 0.01.
In summary, our findings suggest that ESCC cells-secreted exosomal NAT10 plays a pivotal role in mediating macrophage M2 polarization by modulating lipid metabolism. The observed alterations in cellular triglycerides, lipid droplets, FAO levels, and metabolic parameters highlight the crucial influence of NAT10 in orchestrating these metabolic changes, thereby providing insights into the intricate interplay between tumor cells and macrophages in the context of lipid metabolism.
NAT10 promotes M2 polarization in THP-1 cells through enhancing lipid metabolism
To unravel the effects and underlying mechanisms of NAT10 expression changes on THP-1 cell polarization, a comprehensive set of experiments was conducted in this study. The efficiency of NAT10 overexpression was meticulously evaluated through RT-qPCR and Western blot analyses (Fig. 5A). Initially, ESCC cells were transfected with pcDNA3.1 plasmid or the NAT10 overexpression plasmid pcDNA3.1-NAT10, followed by co-culturing with THP-1-DM. Subsequent RT-qPCR analysis revealed that NAT10 overexpression in ESCCs significantly promoted M2 polarization in co-cultured macrophages, as evidenced by the aberrant expression of M2 markers (Fig. 5B).
Fig. 5.

NAT10 promotes M2 polarization in THP-1 cells through enhancing lipid metabolism A. NAT10 was overexpressed in macrophages. B. The expression changes in M2 markers in THP-1-DM co-cultured with ESCC cells after NAT10 overexpression. C–I. The effects of NAT10 overexpression on cellular TG levels (C), fatty acid content (D), FAO levels (E), OCR (F), ECAR (G), ATP levels (H), and lipid droplets (I) in macrophages. J, K. The effects of NAT10 overexpression on the promotion of M2 macrophages (J) and the reduction of M1 macrophages. (K). ⁎⁎P < 0.01.
Subsequently, pcDNA3.1 plasmid or NAT10 overexpression plasmid pcDNA3.1-NAT10 were transfected into THP-1 cells, designating the control and experimental groups, respectively. According to the experimental results, NAT10 overexpression led to a substantial increase in cellular TG levels and fatty acid levels in THP-1 cells (Fig. 5C, D). This suggested that NAT10 overexpression harbored the potential to promote lipid metabolism and glycolysis in THP-1 cells. Consequently, additional experiments were conducted, revealing that NAT10 overexpression significantly elevated FAO levels and the OCR (Fig. 5E, F), while markedly inhibiting the ECAR of THP-1 cells (Fig. 5G). These findings indicate that NAT10 exerts a positive effect on lipid metabolism in THP-1 cells while concurrently exhibiting a negative effect on glycolysis.
Moreover, NAT10 overexpression was observed to promote ATP production (Fig. 5H), and oil red O staining experiments demonstrated a significantly higher number of lipid droplets in the NAT10 overexpression group compared to the control group (Fig. 5I). This further confirmed that NAT10 enhances lipid metabolism in THP-1 cells. Finally, an examination of the numbers of CD163-positive macrophages and CD86-positive macrophages in the control and experimental groups revealed that overexpression of NAT10 promoted an increase in the number of CD163-positive macrophages (Fig. 5J) and inhibited an increase in the number of CD86-positive macrophages (Fig. 5K). This signifies that NAT10 promotes the polarization of THP-1 cells towards the M2 phenotype. In culmination of all the aforementioned experiments, we can conclusively affirm that NAT10 promotes THP-1 cell M2 polarization through the augmentation of cellular lipid metabolism and concurrent inhibition of glycolysis.
NAT10 stabilizes FASN through ac4C modification
Subsequently, we conducted an in-depth analysis of the mechanisms mediated by NAT10 in macrophages. To identify NAT10 targets, we screened the GSE102113 dataset obtained from the GEO database (https://www.ncbi.nlm.nih.gov/gds/), revealing four genes correlated with lipid metabolism according to PathCards (https://pathcards.genecards.org/) (Fig. 6A). Subsequently, we examined the expression changes of these four mRNAs in macrophages following treatment with two types of exosomes. Intriguingly, only ACLY and FASN were upregulated by ESCC cell-derived exosomes (Fig. 6B). Furthermore, the overexpression of NAT10 efficiently elevated FASN expression exclusively (Fig. 6C).
Fig. 6.

NAT10 stabilizes FASN through ac4C modification A. NAT10 targets were screened from the GSE102113 dataset, and four of them were found to be correlated with lipid metabolism in PathCards. B. The expression of four targets in macrophages after treatment with two types of exosomes was measured by RT-qPCR. C. The expression of four targets in macrophages was measured by RT-qPCR after the overexpression of NAT10. D. Acetylation sites in FASN were predicted using PACES. E. The binding of NAT10 protein to FASN mRNA was demonstrated through RIP assay. F. The effect of NAT10 overexpression on the binding of NAT10 protein to FASN mRNA was demonstrated through RIP assay. G. The lifetime detection of FASN mRNA was performed in macrophages treated with or without NAT10 overexpression. H. The enrichment level of FASN in ac4C was determined by RIP assay in macrophages treated with ESCC cells-derived exosomes or NAT10-silenced ESCC cells-derived exosomes. I. The lifetime of FASN mRNA in macrophages treated with ESCC cells-derived exosomes or NAT10-silenced ESCC cells-derived exosomes was measured by RT-qPCR. J. The expression level of FASN in macrophages treated with ESCC cells-derived exosomes or NAT10-silenced ESCC cells-derived exosomes was measured by RT-qPCR. ⁎⁎P < 0.01.
Given the established role of NAT10 as an acetylation regulator, we predicted the acetylation sites in FASN (Fig. 6D). Subsequent to this, we demonstrated the binding of NAT10 protein to FASN mRNA through a RIP assay (Fig. 6E). Moreover, the overexpression of NAT10 strengthened the binding of NAT10 to FASN (Figs. 6F, S4A). To assess the stability of FASN mRNA, we performed a lifetime detection assay by treating macrophages with or without NAT10 overexpression with ActD, revealing a prolonged mRNA lifetime in NAT10-overexpressed cells (Fig. 6G). Importantly, the enrichment of FASN in ac4C was heightened in macrophages treated with Eca-109-Exos or TE-5-Exos, while remaining unchanged in macrophages treated with Eca-109/sh-NAT10#1-Exos or TE-5/sh-NAT10#1-Exos (Fig. 6H). Similarly, the ESCC cell-mediated NAT10 overexpression in co-cultured macrophages also significantly increased the ac4C modification of FASN (Fig. S4B). The FASN mRNA lifetime was similarly prolonged in macrophages treated with Eca-109-Exos or TE-5-Exos, while remaining unchanged in macrophages treated with Eca-109/sh-NAT10#1-Exos or TE-5/sh-NAT10#1-Exos (Fig. 6I). Finally, the expression level of FASN was elevated in macrophages treated with Eca-109-Exos or TE-5-Exos, while it remained unchanged in macrophages treated with Eca-109/sh-NAT10#1-Exos or TE-5/sh-NAT10#1-Exos (Fig. 6J). In conclusion, NAT10-mediated ac4C modification enhances the RNA stability of FASN.
ESCC cells-derived exosomal NAT10 regulates macrophage M2 polarization in an FASN-dependent manner
Rescue assays were conducted to definitively demonstrate the role of NAT10-regulated FASN in macrophage polarization. Prior to this, FASN was knocked out in macrophages (Fig. 7A). Using RT-qPCR and flow cytometry, we verified that the M2 polarization of macrophages promoted by NAT10 overexpression was effectively reversed by FASN knockout (Fig. 7B, C). Conversely, the M1 polarization of macrophages suppressed by NAT10 overexpression was reinstated by FASN knockout (Fig. 7D).
Fig. 7.

ESCC cells-derived exosomal NAT10 regulates macrophage M2 polarization in an FASN-dependent manner A. FASN was knocked out in macrophages. B, C. The M2 polarization of macrophages, promoted by NAT10 overexpression, was reversed by FASN knockout, as determined by RT-qPCR and flow cytometry. D. The M1 polarized macrophages suppressed by NAT10 overexpression were restored by FASN knockout, as determined by flow cytometry. E, F. The effects of FASN knockout on ESCC cells-derived exosomes-mediated macrophage M2 polarization were assessed by RT-qPCR and flow cytometry. G. The effects of FASN knockout on ESCC cells-derived exosomes-mediated macrophage M1 polarization were examined by flow cytometry. ⁎⁎P < 0.01.
Subsequently, we investigated whether FASN knockout influenced ESCC cells-derived exosomes-mediated macrophage polarization. Our findings revealed that the M2 polarization of macrophages induced by ESCC cells-derived exosomes was mitigated upon FASN knockout (Fig. 7E, F). Conversely, the M1 polarization of macrophages suppressed by ESCC cells-derived exosomes was restored by FASN knockout (Fig. 7G). These results collectively indicate that exosomal NAT10 regulates macrophage M2 polarization through an FASN-dependent mechanism.
Targeting NAT10 in ESCC cells enhances the efficacy of PD-1 therapy by modulating macrophage reprogramming
An animal study was conducted to elucidate the impact of NAT10 on in vivo tumor growth and the effectiveness of PD-1 therapy. Animal models were established by subcutaneously inoculating AKR cells stably transfected with sh-NC or sh-NAT10#1 on the right dorsal side of four groups of mice, with two of these groups undergoing PD-1 therapy. Tumors were resected from all four groups, revealing a reduction in tumor size and weight following NAT10 silencing. Notably, the most significant inhibition of tumor growth was observed in the combined NAT10 silencing and PD-1 treatment group (Fig. 8A, B).
Fig. 8.

Targeting NAT10 in ESCC cells enhances the efficacy of PD-1 therapy by modulating macrophage reprogramming Animal models were established by subcutaneously inoculating AKR cells stably transfected with sh-NC or sh-NAT10#1 on the right dorsal side of four groups of mice. Two of these groups underwent PD-1 therapy. A, B. Tumor size and weight were observed and measured in the four groups of mice. C–E. Tumor tissues isolated from the four groups were subjected to IHC staining to measure the expression of NAT10, CD163, and CD86. *P < 0.05, ⁎⁎P < 0.01.
Subsequently, IHC staining was performed on isolated tumor tissues from the four groups to assess the expression of NAT10, CD163, and CD86. It was observed that the positive expression of NAT10 and CD163 decreased with NAT10 silencing, while that of CD86 increased. The changes in the PD-1 treatment group were not significant; however, the alterations were more pronounced in the combined NAT10 silencing and PD-1 treatment group compared to the PD-1 treatment or NAT10 silencing group alone (Fig. 8C–E). These findings underscore the role of NAT10 in influencing the efficacy of PD-1 therapy through the mediation of macrophage reprogramming.
Discussion
Accumulating evidence suggests a correlation between dysregulated RNA or protein molecules in tumor samples and the polarization of TAMs [40], [41], [42], as well as the response to PD-1 therapy [43,44]. In this study, we conducted a comprehensive analysis of data from two online databases, identifying NAT10 as a significant target due to its markedly elevated expression in ESCA tumor tissues. Subsequently, we collected additional tumor samples from ESCA patients to validate NAT10 expression, confirming its higher expression in ESCC tumor tissues. PD-1 therapy, a common approach for ESCC treatment [45,46], was found to be less effective in patients with high NAT10 expression, suggesting a potential association between NAT10 expression and PD-1 therapy outcomes. Furthermore, we observed a decrease in the positivity of the M1 marker (CD86) and an increase in the M2 marker (CD163) in NAT10-overexpressing ESCC patients who were resistant to PD-1 therapy. M2-TAMs, known to inhibit the functions of CD8+ T cells and NK cells, contribute to immune escape, PD-1 monotherapy resistance, and adverse prognosis [47,48]. Combining our results, we propose that NAT10 is upregulated in ESCC tumor samples, influencing macrophage polarization, and affecting the efficacy of PD-1 therapy.
Mechanistically, we predicted abundant m6A sites in NAT10′s 3′UTR, suggesting that the upregulation of NAT10 might be induced by m6A modification. Further analysis indicated that the m6A methyltransferase METTL3 could bind to NAT10′s 3′UTR. METTL3 has been verified to be involved in the progression of various human cancers as an m6A methyltransferase [49], [50], [51]. In this study, we determined that METTL3 was highly expressed in ESCA tumor tissues and ESCC cell lines, consistent with NAT10. Further mechanistic experiments demonstrated that METTL3 enhances NAT10 expression through m6A modification at NAT10′s 3′UTR to stabilize NAT10 mRNA.
Studies have shown that tumor cell-derived exosomal RNAs can affect the tumor microenvironment by mediating macrophage reprogramming [35], [36], [37]. Our present study constructed a co-culture system for ESCC cells and THP-1-DM cells. Through a series of experiments, we determined that the expression of M2 markers and CD163-positive macrophages were all increased in THP-1-DM co-cultured with ESCC cells. Additionally, CD86-positive macrophages decreased under the same conditions. However, these changes disappeared when the co-culture system was supplemented with the exosome inhibitor GW4869 or transfected with NAT10-specific shRNA. These data suggest that NAT10 might affect macrophage reprogramming through exosomes. After isolating and identifying ESCC cell-derived exosomes, we further examined the alteration of M1/M2 polarization of macrophages. The experimental results indicated that ESCC cell-derived exosomal NAT10 promotes macrophage M2 polarization. Our study is the first to unveil the role of NAT10 in macrophage reprogramming through exosome-mediated intercellular communication.
Lipid metabolism can regulate macrophage functions and polarization [38,39]. Previous studies revealed the involvement of NAT10 in mitochondrial lipid metabolism of cancer cells [52] and fatty acid metabolism of cancer cells [53,54]. To date, whether NAT10 regulates the lipid metabolism of TAMs remains unknown. Here, we found that ESCC cell-derived exosomes or NAT10 overexpression can increase the cellular triglyceride level, the fatty acid content, and the lipid synthesis level. Therefore, we confirmed that exosomal NAT10 derived from ESCC cells promotes macrophage M2 polarization through mediating lipid metabolism.
NAT10 is a regulator of ac4C modification, exerting functions in various tumors by modifying the acetylation of tRNA, rRNA, and mRNA [17], [18], [19], [20], [21], [22], [23]. According to data analysis and bioinformatics prediction, we determined that NAT10 could target FASN, which is a lipid metabolism-related gene [55,56]. Through further investigation, we identified that NAT10 could stabilize FASN expression to induce macrophage M2 polarization by mediating the ac4C modification of FASN.
In conclusion, our study demonstrated that NAT10 from ESCC cell-derived exosomes promotes macrophage reprogramming by mediating lipid metabolism. Mechanically, NAT10 is upregulated by METTL3-induced m6A modification; NAT10 enhances FASN expression through ac4C modification. Finally, in vivo data prove that targeting NAT10 in ESCC cells can enhance the effect of PD-1 therapy by mediating macrophage reprogramming. Our findings may be conducive to finding novel targets for ESCC treatment.
Studies in humans and animals
The Ethics Committee of Changhai Hospital, Second Military Medical University (Naval Medical University) granted this study.
Informed consent and patient details
The informed consent was obtained from all patients before sample collection.
Funding
This study was supported by Medical Basic Research Project of the First Affiliated Hospital of Naval Medical University – Youth Cultivation Project, no. 2021JCQN12.
CRediT authorship contribution statement
Chun Jin: Supervision, Project administration, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Jian Gao: Methodology, Investigation, Formal analysis, Data curation. Ji Zhu: Formal analysis, Data curation. Yongqiang Ao: Formal analysis, Data curation. Bowen Shi: Writing – review & editing, Writing – original draft, Project administration. Xin Li: Writing – review & editing, Writing – original draft, Resources, Funding acquisition.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Acknowledgments
Thanks a lot to all individuals or teams that were involved in our research.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.tranon.2024.101934.
Contributor Information
Bowen Shi, Email: shihun6473230164@163.com.
Xin Li, Email: xinbilan5@163.com.
Appendix. Supplementary materials
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Kashyap M.K., Abdel-Rahman O. Expression, regulation and targeting of receptor tyrosine kinases in esophageal squamous cell carcinoma. Mol. Cancer. 2018;17(1):54. doi: 10.1186/s12943-018-0790-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 3.Kitagawa Y., Uno T., Oyama T., Kato K., Kato H., Kawakubo H., et al. Esophageal cancer practice guidelines 2017 edited by the Japan esophageal society: part 1. Esophagus. 2019;16(1):1–24. doi: 10.1007/s10388-018-0642-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang F.L., Yu S.J. Esophageal cancer: risk factors, genetic association, and treatment. Asian J. Surg. 2018;41(3):210–215. doi: 10.1016/j.asjsur.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 5.Kitagawa Y., Uno T., Oyama T., Kato K., Kato H., Kawakubo H., et al. Esophageal cancer practice guidelines 2017 edited by the Japan esophageal society: part 2. Esophagus. 2019;16(1):25–43. doi: 10.1007/s10388-018-0642-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun J.M., Shen L., Shah M.A., Enzinger P., Adenis A., Doi T., et al. Pembrolizumab plus chemotherapy versus chemotherapy alone for first-line treatment of advanced oesophageal cancer (KEYNOTE-590): a randomised, placebo-controlled, phase 3 study. Lancet. 2021;398(10302):759–771. doi: 10.1016/S0140-6736(21)01234-4. [DOI] [PubMed] [Google Scholar]
- 7.Kojima T., Shah M.A., Muro K., Francois E., Adenis A., Hsu C.H., et al. Randomized phase III KEYNOTE-181 study of pembrolizumab versus chemotherapy in advanced esophageal cancer. J. Clin. Oncol. 2020;38(35):4138–4148. doi: 10.1200/JCO.20.01888. [DOI] [PubMed] [Google Scholar]
- 8.Kato K., Cho B.C., Takahashi M., Okada M., Lin C.Y., Chin K., et al. Nivolumab versus chemotherapy in patients with advanced oesophageal squamous cell carcinoma refractory or intolerant to previous chemotherapy (ATTRACTION-3): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20(11):1506–1517. doi: 10.1016/S1470-2045(19)30626-6. [DOI] [PubMed] [Google Scholar]
- 9.Huang J., Xu J., Chen Y., Zhuang W., Zhang Y., Chen Z., et al. Camrelizumab versus investigator's choice of chemotherapy as second-line therapy for advanced or metastatic oesophageal squamous cell carcinoma (ESCORT): a multicentre, randomised, open-label, phase 3 study. Lancet Oncol. 2020;21(6):832–842. doi: 10.1016/S1470-2045(20)30110-8. [DOI] [PubMed] [Google Scholar]
- 10.Luo H., Lu J., Bai Y., Mao T., Wang J., Fan Q., et al. Effect of camrelizumab vs. placebo added to chemotherapy on survival and progression-free survival in patients with advanced or metastatic esophageal squamous cell carcinoma: the ESCORT-1st randomized clinical trial. JAMA. 2021;326(10):916–925. doi: 10.1001/jama.2021.12836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng H., Wang Z., Fu L., Xu T. Macrophage polarization in the development and progression of ovarian cancers: an overview. Front. Oncol. 2019;9:421. doi: 10.3389/fonc.2019.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He Z., Wang J., Zhu C., Xu J., Chen P., Jiang X., et al. Exosome-derived FGD5-AS1 promotes tumor-associated macrophage M2 polarization-mediated pancreatic cancer cell proliferation and metastasis. Cancer Lett. 2022;548 doi: 10.1016/j.canlet.2022.215751. [DOI] [PubMed] [Google Scholar]
- 13.Tian H.Y., Liang Q., Shi Z., Zhao H. Exosomal CXCL14 contributes to M2 macrophage polarization through NF-κB signaling in prostate cancer. Oxid. Med. Cell Longev. 2022;2022 doi: 10.1155/2022/7616696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simons M., Raposo G. Exosomes–vesicular carriers for intercellular communication. Curr. Opin. Cell Biol. 2009;21(4):575–581. doi: 10.1016/j.ceb.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 15.Azmi A.S., Bao B., Sarkar F.H. Exosomes in cancer development, metastasis, and drug resistance: a comprehensive review. Cancer Metastasis Rev. 2013;32(3–4):623–642. doi: 10.1007/s10555-013-9441-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arango D., Sturgill D., Alhusaini N., Dillman A.A., Sweet T.J., Hanson G., et al. Acetylation of cytidine in mRNA promotes translation efficiency. Cell. 2018;175(7):1872–1886. doi: 10.1016/j.cell.2018.10.030. e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang P., Hu R., Liu Z., Miao M., Jiang H., Li C. NAT10 upregulation indicates a poor prognosis in acute myeloid leukemia. Curr. Probl. Cancer. 2020;44(2) doi: 10.1016/j.currproblcancer.2019.06.006. [DOI] [PubMed] [Google Scholar]
- 18.Larrieu D., Britton S., Demir M., Rodriguez R., Jackson S.P. Chemical inhibition of NAT10 corrects defects of laminopathic cells. Science (New York, NY) 2014;344(6183):527–532. doi: 10.1126/science.1252651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito S., Horikawa S., Suzuki T., Kawauchi H., Tanaka Y., Suzuki T., et al. Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA) J. Biol. Chem. 2014;289(52):35724–35730. doi: 10.1074/jbc.C114.602698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H., Hou W., Wang H.L., Liu H.J., Jia X.Y., Zheng X.Z., et al. GSK-3β-regulated N-acetyltransferase 10 is involved in colorectal cancer invasion. Clin. Cancer Res. 2014;20(17):4717–4729. doi: 10.1158/1078-0432.CCR-13-3477. [DOI] [PubMed] [Google Scholar]
- 21.Sharma S., Langhendries J.L., Watzinger P., Kötter P., Entian K.D., Lafontaine D.L. Yeast Kre33 and human NAT10 are conserved 18S rRNA cytosine acetyltransferases that modify tRNAs assisted by the adaptor Tan1/THUMPD1. Nucleic Acids Res. 2015;43(4):2242–2258. doi: 10.1093/nar/gkv075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu X., Tan Y., Zhang C., Zhang Y., Zhang L., Ren P., et al. NAT10 regulates p53 activation through acetylating p53 at K120 and ubiquitinating Mdm2. EMBO Rep. 2016;17(3):349–366. doi: 10.15252/embr.201540505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balmus G., Larrieu D., Barros A.C., Collins C., Abrudan M., Demir M., et al. Targeting of NAT10 enhances healthspan in a mouse model of human accelerated aging syndrome. Nat. Commun. 2018;9(1):1700. doi: 10.1038/s41467-018-03770-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roundtree I.A., Evans M.E., Pan T., He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169(7):1187–1200. doi: 10.1016/j.cell.2017.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frye M., Harada B.T., Behm M., He C. RNA modifications modulate gene expression during development. Science (New York, NY) 2018;361(6409):1346–1349. doi: 10.1126/science.aau1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zaccara S., Ries R.J., Jaffrey S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019;20(10):608–624. doi: 10.1038/s41580-019-0168-5. [DOI] [PubMed] [Google Scholar]
- 27.Liu J., Harada B.T., He C. Regulation of gene expression by N(6)-methyladenosine in cancer. Trends Cell Biol. 2019;29(6):487–499. doi: 10.1016/j.tcb.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deng X., Su R., Weng H., Huang H., Li Z., Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28(5):507–517. doi: 10.1038/s41422-018-0034-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J., Yue Y., Han D., Wang X., Fu Y., Zhang L., et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014;10(2):93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin S., Choe J., Du P., Triboulet R., Gregory R.I. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell. 2016;62(3):335–345. doi: 10.1016/j.molcel.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu H., Zhou J., Mei S., Wu D., Mu Z., Chen B., et al. Circulating exosomal microRNA-96 promotes cell proliferation, migration and drug resistance by targeting LMO7. J. Cell. Mol. Med. 2017;21(6):1228–1236. doi: 10.1111/jcmm.13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J., Yao Y., Wu J., Li G. Identification and analysis of exosomes secreted from macrophages extracted by different methods. Int. J. Clin. Exp. Pathol. 2015;8(6):6135–6142. [PMC free article] [PubMed] [Google Scholar]
- 33.Lässer C., Eldh M., Lötvall J. Isolation and characterization of RNA-containing exosomes. J. Vis. Exp. 2012;(59):e3037. doi: 10.3791/3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qu Z., Wu J., Wu J., Luo D., Jiang C., Ding Y. Exosomes derived from HCC cells induce sorafenib resistance in hepatocellular carcinoma both in vivo and in vitro. J. Exp. Clin. Cancer Res. 2016;35(1):159. doi: 10.1186/s13046-016-0430-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X., Lei Y., Wu M., Li N. Regulation of macrophage activation and polarization by HCC-derived exosomal lncRNA TUC339. Int. J. Mol. Sci. 2018;19(10) doi: 10.3390/ijms19102958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang M., Chen X., Wang L., Qin L., Wang H., Sun Z., et al. Cancer-derived exosomal TRIM59 regulates macrophage NLRP3 inflammasome activation to promote lung cancer progression. J. Exp. Clin. Cancer Res. 2020;39(1):176. doi: 10.1186/s13046-020-01688-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang Y., Hu S., Li T., Qiu X. Tumor cells-derived exosomal circVCP promoted the progression of colorectal cancer by regulating macrophage M1/M2 polarization. Gene. 2023;870 doi: 10.1016/j.gene.2023.147413. [DOI] [PubMed] [Google Scholar]
- 38.Yan J., Horng T. Lipid metabolism in regulation of macrophage functions. Trends Cell Biol. 2020;30(12):979–989. doi: 10.1016/j.tcb.2020.09.006. [DOI] [PubMed] [Google Scholar]
- 39.Xiang Y., Miao H. Lipid metabolism in tumor-associated macrophages. Adv. Exp. Med. Biol. 2021;1316:87–101. doi: 10.1007/978-981-33-6785-2_6. [DOI] [PubMed] [Google Scholar]
- 40.Xiao M., Bian Q., Lao Y., Yi J., Sun X., Sun X., et al. SENP3 loss promotes M2 macrophage polarization and breast cancer progression. Mol. Oncol. 2022;16(4):1026–1044. doi: 10.1002/1878-0261.12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dan H., Liu S., Liu J., Liu D., Yin F., Wei Z., et al. RACK1 promotes cancer progression by increasing the M2/M1 macrophage ratio via the NF-κB pathway in oral squamous cell carcinoma. Mol. Oncol. 2020;14(4):795–807. doi: 10.1002/1878-0261.12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yuan Q., Gu J., Zhang J., Liu S., Wang Q., Tian T., et al. MyD88 in myofibroblasts enhances colitis-associated tumorigenesis via promoting macrophage M2 polarization. Cell Rep. 2021;34(5) doi: 10.1016/j.celrep.2021.108724. [DOI] [PubMed] [Google Scholar]
- 43.Liu C., Liu R., Wang B., Lian J., Yao Y., Sun H., et al. Blocking IL-17A enhances tumor response to anti-PD-1 immunotherapy in microsatellite stable colorectal cancer. J. Immunother. Cancer. 2021;9(1) doi: 10.1136/jitc-2020-001895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen D.L., Sheng H., Zhang D.S., Jin Y., Zhao B.T., Chen N., et al. The circular RNA circDLG1 promotes gastric cancer progression and anti-PD-1 resistance through the regulation of CXCL12 by sponging miR-141-3p. Mol. Cancer. 2021;20(1):166. doi: 10.1186/s12943-021-01475-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leone A.G., Petrelli F., Ghidini A., Raimondi A., Smyth E.C., Pietrantonio F. Efficacy and activity of PD-1 blockade in patients with advanced esophageal squamous cell carcinoma: a systematic review and meta-analysis with focus on the value of PD-L1 combined positive score. ESMO Open. 2022;7(1) doi: 10.1016/j.esmoop.2021.100380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang W., Xing X., Yeung S.J., Wang S., Chen W., Bao Y., et al. Neoadjuvant programmed cell death 1 blockade combined with chemotherapy for resectable esophageal squamous cell carcinoma. J. Immunother. Cancer. 2022;10(1) doi: 10.1136/jitc-2021-003497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muraoka D., Seo N., Hayashi T., Tahara Y., Fujii K., Tawara I., et al. Antigen delivery targeted to tumor-associated macrophages overcomes tumor immune resistance. J. Clin. Investig. 2019;129(3):1278–1294. doi: 10.1172/JCI97642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Q., Huang H., Zheng F., Liu H., Qiu F., Chen Y., et al. Resveratrol exerts antitumor effects by downregulating CD8(+)CD122(+) Tregs in murine hepatocellular carcinoma. Oncoimmunology. 2020;9(1) doi: 10.1080/2162402X.2020.1829346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taketo K., Konno M., Asai A., Koseki J., Toratani M., Satoh T., et al. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int. J. Oncol. 2018;52(2):621–629. doi: 10.3892/ijo.2017.4219. [DOI] [PubMed] [Google Scholar]
- 50.Xue L., Li J., Lin Y., Liu D., Yang Q., Jian J., et al. m(6) A transferase METTL3-induced lncRNA ABHD11-AS1 promotes the Warburg effect of non-small-cell lung cancer. J. Cell. Physiol. 2021;236(4):2649–2658. doi: 10.1002/jcp.30023. [DOI] [PubMed] [Google Scholar]
- 51.Vu L.P., Pickering B.F., Cheng Y., Zaccara S., Nguyen D., Minuesa G., et al. The N(6)-methyladenosine (m(6)A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat. Med. 2017;23(11):1369–1376. doi: 10.1038/nm.4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dalhat M.H., Mohammed M.R.S., Ahmad A., Khan M.I., Choudhry H. Remodelin, a N-acetyltransferase 10 (NAT10) inhibitor, alters mitochondrial lipid metabolism in cancer cells. J. Cell. Biochem. 2021;122(12):1936–1945. doi: 10.1002/jcb.30155. [DOI] [PubMed] [Google Scholar]
- 53.Dalhat M.H., Choudhry H., Khan M.I. NAT10, an RNA cytidine acetyltransferase, regulates ferroptosis in cancer cells. Antioxidants (Basel, Switzerland) 2023;12(5) doi: 10.3390/antiox12051116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dalhat M.H., Mohammed M.R.S., Alkhatabi H.A., Rehan M., Ahmad A., Choudhry H., et al. NAT10: an RNA cytidine transferase regulates fatty acid metabolism in cancer cells. Clin. Transl. Med. 2022;12(9):e1045. doi: 10.1002/ctm2.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu J., Luo J., He Q., Xia Y., Tian H., Zhu L., et al. Docosahexaenoic acid alters lipid metabolism processes via H3K9ac epigenetic modification in dairy goat. J. Agric. Food Chem. 2023;71(22):8527–8539. doi: 10.1021/acs.jafc.3c01606. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Q., Qian Z.Y., Zhou P.H., Zhou X.L., Zhang D.L., He N., et al. Effects of oral selenium and magnesium co-supplementation on lipid metabolism, antioxidative status, histopathological lesions, and related gene expression in rats fed a high-fat diet. Lipids Health Dis. 2018;17(1):165. doi: 10.1186/s12944-018-0815-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.