

Abstract
The EFSA Panel on Food Additives and Flavourings (FAF) was requested to evaluate the safety of 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one [FL‐no: 16.134] as a new flavouring substance, in accordance with Regulation (EC) No 1331/2008. The substance has not been reported to occur naturally and is chemically synthesised. In food, it is intended to be used as a flavouring substance only in chewing gum. The chronic dietary exposure to [FL‐no: 16.134] was estimated to be 45 μg/person per day for a 60‐kg adult and 28.4 μg/person per day for a 15‐kg 3‐year‐old child. [FL‐no: 16.134] did not show genotoxicity in a bacterial reverse mutation test and an in vitro mammalian cell micronucleus assay. Based on the submitted toxicokinetic and metabolism data, it can be predicted that the flavouring substance is metabolised to innocuous products only. The Panel derived a lower confidence limit of the benchmark dose (BMDL) of 0.71 mg/kg bw per day for a 20% increase in the relative thyroid (including parathyroid) weight observed in a 90‐day toxicity study in rats. Based on this BMDL, adequate margins of exposure of 887 and 374 could be calculated for adults and children, respectively. The Panel concluded that there is no safety concern for [FL‐no: 16.134], when used as a flavouring substance at the estimated level of dietary exposure, based on the intended use and use levels as specified in Appendix B. The Panel further concluded that the combined exposure to [FL‐no: 16.134] from its use as a food flavouring substance and from its presence in toothpaste and mouthwash is also not of safety concern.
Keywords: 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one, FGE.419, FL‐no: 16.134
1. INTRODUCTION
The present scientific opinion deals with the safety assessment of 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one [FL‐no: 16.134] to be used as a new flavouring substance in food.
1.1. Background and Terms of Reference as provided by the requestor
1.1.1. Background
Only food flavourings included in the Union list may be placed on the market as such and used in foods, in accordance with Article 10 of Regulation (EC) No 1334/2008 on food flavourings.
On 27 October 2021, a new application has been introduced by the applicant “Givaudan International SA” for the authorisation of the food flavouring 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one.
1.1.2. Terms of Reference
The European Commission requests the European Food Safety Authority to carry out the safety assessment and the assessment of possible confidentiality requests of the following food flavouring: 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one, in accordance with Regulation (EC) No 1331/2008 establishing a common authorisation procedure for food additives, food enzymes and food flavourings. 1
1.2. Existing authorisations and evaluations
The use of 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one as flavouring has been evaluated by the Expert Panel of the Flavour and Extract Manufactures Association (FEMA) of the United States and considered as ‘Generally Regarded As Safe’ (GRAS) (FEMA No. 4970).
In addition, the Panel noted that a dossier on the substance has been submitted to ECHA for registration purpose under REACH Regulation 2 (https://chem.echa.europa.eu/100.391.682/overview).
2. DATA AND METHODOLOGIES
2.1. Data
The present evaluation is based on data on 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one [FL‐no: 16.134] provided by the applicant in a dossier (Documentation as provided to EFSA No. 1) to support its evaluation as a food flavouring substance.
In accordance with Article 38 of the Regulation (EC) No 178/2002 3 and taking into account the protection of confidential information and of personal data in accordance with Articles 39 to 39e of the same Regulation and of the Decision of the EFSA's Executive Director laying down practical arrangements concerning transparency and confidentiality, 4 the non‐confidential version of the dossier is published on Open.EFSA. 5
According to Art. 32c(2) of Regulation (EC) No 178/2002 and to the Decision of EFSA's Executive Director laying down the practical arrangements on pre‐submission phase and public consultations, EFSA carried out a public consultation on the non‐confidential version of the application from 15 November to 6 December 2022, for which no comments were received.
Additional information was provided by the applicant during the risk assessment process on 27 January 2023 (Documentation as provided to EFSA No. 2) and on 27 September 2023 (Documentation as provided to EFSA No. 3) in response to requests from EFSA sent on 27 September 2022 and on 14 July 2023, respectively.
2.2. Methodologies
This opinion was prepared following the principles described in the EFSA Guidance of the Scientific Committee on transparency with regard to scientific aspects of risk assessment (EFSA Scientific Committee, 2009) and following the relevant existing Guidance documents from the EFSA Scientific Committee.
The current application was submitted to EFSA before the publication of the latest EFSA guidance on data required for the risk assessment of flavourings to be used in or on foods (EFSA FAF Panel, 2022). Therefore, the safety assessment of 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one [FL‐no: 16.134] was carried out in accordance with the procedure as outlined in the EFSA scientific opinion ‘Guidance on the data required for the risk assessment of flavourings to be used in or on foods’ (EFSA CEF Panel, 2010) and the EFSA technical report ‘Proposed template to be used in drafting scientific opinions on flavouring substances (explanatory notes for guidance included)’ (EFSA, 2012).
3. ASSESSMENT
3.1. Technical data
3.1.1. Identity of the substance
The chemical structure of the flavouring substance 2‐methyl 1‐(2‐(5‐(p‐tolyl)‐1H imidazole‐2‐yl)piperidin‐1‐yl)butan‐1‐one and the specification data provided by the applicant are shown in Table 1. The flavouring substance was allocated the FLAVIS number [FL‐no: 16.134].
TABLE 1.
Specification data for 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one (Documentation as provided to EFSA Nos 1 and 2). The table was compiled by the Panel.
| Chemical name |
CAS no FL‐no EC no CoE no JECFA no FEMA no |
Chemical formula MW |
Structural formula | Physical form | Solubility data | ID test | Purity | Impurities |
Boiling point a Melting point Specific gravity b Refractive index c |
|---|---|---|---|---|---|---|---|---|---|
| 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one |
2413115‐68‐9 16.134 – – – 4970 |
C20H27N3O 325.456 g/mol |
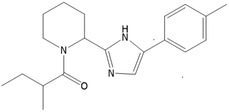
|
White crystalline solid |
Water: < 0.1% Ethanol: > 20% |
1H‐NMR 13C‐NMR GC–MS IR |
Racemic mixture of diastereomers, i.e. mixture of four components: (S)‐2 methyl‐1‐((S)‐2‐(5‐(p‐tolyl)‐1H‐imidazol 2 yl)piperidin‐1yl)butan‐1‐one; (S)‐2‐methyl‐1‐((R)‐2‐(5‐(p‐tolyl)‐1H‐imidazol 2‐yl)piperidin‐1‐yl)butan‐1‐one; (R)‐2‐methyl‐1‐((S)‐2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1yl)butan‐1‐one; (R)‐2‐methyl‐1‐((R)‐2‐(5‐(p‐tolyl)‐1H imidazole‐2‐yl)piperidin‐1‐yl)butan 1 one; Sum of stereoisomers: ≥ 99% |
Not more than 1% of the total peak area, measured through HPLC‐UV at 257 nm |
N.D. 155–157°C N.A. N.A. |
Abbreviations: GC–MS, gas chromatography‐mass spectrometry; ID, identity; IR, infrared; MW, molecular weight; N.A., not applicable; N.D., not determined; NMR, nuclear magnetic resonance.
At 1013.25 hPa, if not otherwise stated.
At 20°C, unless otherwise stated.
At 25°C, unless otherwise stated.
The identity of the flavouring substance was demonstrated by GC‐MS, 1H NMR, 13C NMR and IR analyses. Details of the analytical conditions used in each identity test were provided to EFSA (Documentation as provided to EFSA No. 1).
The flavouring substance is a racemic mixture of two diastereomers. These are present in ratios between 40/60 and 60/40. The stereoisomer ratio was investigated through HPLC‐UV using a chiral stationary phase. Details of the chromatographic method were provided by the applicant (Documentation as provided to EFSA No. 1).
The flavouring substance is obtained through chemical synthesis, with a reaction yield above 99% (final product as sum of the two diastereomers). In the original technical dossier, the applicant provided purity data for two production batches (no. VE00763902 and VE00763903) (Documentation as provided to EFSA No. 1); the analysis was performed through HPLC‐UV, and impurities were reported at concentrations below 0.4% of the total peak area. To further clarify the degree of purity of the flavouring substance, the applicant was requested to provide compositional data through mass spectrometry analyses. The applicant replied providing GC‐MS and LC‐MS data for four synthesised batches (i.e. VE00704711, VE00704712, VE00762813 and VE00762815) (Documentation as provided to EFSA No. 2). The data support that the purity of the flavouring substance exceeds 99%.
3.1.2. Organoleptic characteristics
According to the applicant, the flavouring substance when dissolved in flavour oils (to be added to foods) imparts a cooling and refreshing flavour to the final product (Documentation as provided to EFSA No. 1). At concentrations higher than 100 mg/kg, the substance results in an off‐flavour. Therefore, the applicant proposed a maximum use level of 100 mg/kg of chewing gum.
3.1.3. Manufacturing process
The approach used to manufacture, through chemical synthesis, the flavouring substance is outlined in Figure 1. The applicant described the key parameters and stages of the production process, including the purification steps.
FIGURE 1.

Approach used to synthesise the flavouring substance (Documentation as provided to EFSA No. 1).
Following an additional data request from EFSA, the applicant specified the purities of the starting materials, the reagents and the solvents used to manufacture the flavouring substance (Documentation as provided to EFSA No. 2).
Regarding the toxic elements, following an additional data request from EFSA, the applicant provided analytical data on the content of arsenic, lead, cadmium and mercury in four batches (i.e. VE00704712, VE00762813, VE00762815 and VE00799502) of the flavouring substance (Documentation as provided to EFSA No. 3). The analyses were performed by an external laboratory using inductively coupled plasma–mass spectrometry (ICP‐MS); the certificates of analyses were provided. All four batches had levels of toxic elements below the limits of quantification (LOQ), which were 0.1, 0.05, 0.01 and 0.005 mg/kg for As, Pb, Cd and Hg, respectively.
Upon request from the Panel, the applicant provided data on the residual levels of the solvents in the final product, resulting from the manufacturing process (Documentation as provided to EFSA No. 2). Considering the dietary exposure estimates, the Panel considered that the detected levels of residual solvents in the final product do not pose a safety concern.
3.1.4. Solubility and particle size
The applicant provided the solubility data presented in Table 1 (Documentation as provided to EFSA No. 1), and described the test performed to establish the dissolution rates of the flavouring substance in selected essential flavour oils. The applicant employed the methodology of ‘Ultrafiltration’, as described in the EFSA ‘Guidance on technical requirements for regulated food and feed product applications to establish the presence of small particles including nanoparticles’ (EFSA Scientific Committee, 2021). Since the original tests were performed on three technical replicates, the Panel requested the applicant to perform the experiments with three different production batches. The applicant reported that 1% (w/w) solutions of the flavouring substance were prepared for two representative carrier liquids, i.e. peppermint oil (PO) and lactic acid (LA) (Documentation as provided to EFSA No. 2). Then, aliquots of these solutions were analysed by liquid chromatography with UV detection (LC‐UV) at 260 nm before and after ultracentrifugation, with 10 kDa membrane filters. The tests were performed on batches no.: VE00704711, VE00704712 and VE00762815. The average concentrations of the flavouring substance, determined before ultrafiltration, were 0.98 ± 0.04% in PO and 1.11 ± 0.08% in LA. After ultracentrifugation, concentrations of 0.97 ± 0.03% in PO and 1.11 ± 0.15% in LA were determined. Consequently, the flavouring substance is soluble in the tested matrices. Therefore, there is no concern regarding the potential exposure of consumers to small particles, including nanoparticles, under the intended conditions of use.
3.1.5. Proposed specifications
The chemical structure of the flavouring substance [FL‐no: 16.134] and the specification data provided by the applicant, in the original dossier and in response to the EFSA requests for additional information, are summarised in Table 1 (Documentation as provided to EFSA Nos 1 and 2).
3.1.6. Stability and fate in food
Stability
With regard to the storage stability, the stereoisomer distribution of the flavouring substance was determined by exposing the neat product to two different conditions: 120 days at 37°C (batch no: 6) and 24 h at 100°C (batch no: 11). The stereoisomer ratio was determined by comparing the relative peak areas with those of the reference product (i.e. product not aged, the structure of which was determined by NMR). Analyses were performed by HPLC‐UV with a chiral stationary phase at 268 nm. Details of the experimental conditions were submitted to EFSA (Documentation as provided to EFSA No. 1).
The total amount of the substance decreased by approximately 10% during the test periods (i.e. 120 days at 37°C and 1 day, 100°C) and the stereoisomer distribution of the flavouring substance remained constant under the tested conditions (Documentation as provided to EFSA No. 2). No information on the degradation products were provided.
The stability of the flavouring substance was also tested in flavour oils (i.e. white peach, citrus blast, spearmint, wintergreen and cool mint) stored at 40°C. The flavouring substance (batch VE00704712, purity 99%) was incorporated into each flavour oil at concentrations ranging from 0.42% to 0.5% (w/w). The analyses were performed through GC‐MS and an internal standard was used for quantification. Details of the experimental conditions, along with the certificates of analysis, were submitted to EFSA (Documentation as provided to EFSA No. 1). Samples were tested at 0, 2, 4, 8 and 13 weeks of storage. Except for wintergreen oil, the flavouring substance decreased by less than 10% over the 13‐week period in all the tested oils; in wintergreen oil, no loss was observed. No information on the degradation products was provided.
In addition, the applicant assessed the stability of the proposed substance in flavoured toothpaste. Details of the experimental conditions were submitted to EFSA (Documentation as provided to EFSA No. 1). Briefly, flavoured toothpaste samples containing the flavouring substance at 20 mg/kg were stored at 4, 24 and 40°C. The concentration of the proposed flavouring was determined at day 0, 7, 13, 19, 25, 61, 90 and 273. The flavouring substance was quantified in the toothpaste samples by HPLC‐UV, using an internal standard. The decrease in content at the end of the storage period, was on average less than 6%, thus indicating an adequate stability of the flavouring substance in toothpaste.
Overall, the Panel considered the flavouring substance to be sufficiently stable under the intended conditions of use.
Fate in food
The use of the flavouring substance in food is limited to chewing gum (food category 05.3). Therefore, a method for analysis in chewing gum was developed, in the context of a ‘chew‐out’ study, the aim of which was to evaluate the dietary exposure to the flavouring substance (Documentation as provided to EFSA No. 1). Briefly, two types of chewing gum (‘bubble gum’ and ‘no coating gum’) containing the flavouring substance were chewed by 10 panellists, for 15 min (‘bubble gum’) and 30 min (‘no coating gum’). Then, chewing gums were chewed out, spiked with an internal standard and after extraction analysed by GC. After 15 and 30 min of chewing, 88.6% and 86.9%, respectively, of the flavouring substance were retained in the chewing gums. The applicant concluded that a longer chewing time period would not release a higher fraction of the substance, since at least 80% of the substance was retained and remained stable in the base matrix after a chewing period of 30 minutes.
However, the Panel noted that gums could be chewed by consumers for more than 30 minutes and requested the applicant to submit additional information. In response, the applicant provided a study from the literature that surveyed the use of chewing gums in Europe (Hearty et al., 2014), which among other parameters addressed the duration of chewing (Documentation as provided to EFSA No. 2).
The Panel recognised that the composition of the matrix used by the applicant in the chewing experiments reflects the composition of commercial products. The study from Hearty et al. (2014) showed that 81% of children and 74% of adolescents/adults chew gums for up to 30 minutes. These percentages increase to 94% (children) and 88% (adolescents/adults) for a chewing period for up to 60 minutes.
The Panel considered that the assumptions of a chewing time of 30 minutes with a release of maximum 20% of the flavouring substance, from the chewing gum matrix, are sufficiently conservative. Based on the experimental data for the release of the flavouring substance from chewing gum after 15 and 30 minutes of chewing, it is reasonable to assume that only a minor proportion of the flavouring substance may be further released from the chewing gum after 30 minutes of chewing.
3.2. Structural/metabolic similarity to flavouring substances in existing FGE
No flavouring substances structurally related to the flavouring substance were identified in existing FGEs.
3.3. Exposure assessment
3.3.1. Natural occurrence in food
2‐Methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one [FL‐no: 16.134] has not been reported to occur naturally in any food or food source (volatile compounds in food (VCF) database, version 16.8). Therefore, the only occurrence levels in food arise from its use as a flavouring substance.
3.3.2. Non‐food sources of exposure
The flavouring substance [FL‐no: 16.134] is used in oral care formulations: toothpaste and mouthwash. An estimated exposure from these uses was provided by the applicant (Table 2).
TABLE 2.
Calculation of exposure to [FL‐no: 16.134] from the use of oral care formulations.
| Age group | Product type | Level of [FL‐no: 16.134] (mg/kg) | Amount applied per use day (g/person per day) | Retention factor | Exposure | |
|---|---|---|---|---|---|---|
| μg/person per day | μg/kg bw per day a | |||||
| Adults | Toothpaste | 10 | 2.75 b | 0.05 b | 1.38 | 0.023 |
| Mouthwash | 4 | 21.62 b | 0.1 b | 8.65 | 0.145 | |
| Children | Toothpaste | 10 | 2.75 b | 0.05 b | 1.38 | 0.092 |
| Mouthwash | 4 | 21.62 b | 0.1 b | 8.65 | 0.577 | |
Values expressed per kg bw per day refer to adults, considering an average body weight of 60 kg and to children, considering an average body weight of 15 kg.
Values reported in the dossier reflect the default values used by the Scientific Committee on Consumer Safety (SCCS, 2018a).
In the calculation of the exposure from the use of oral care formulations, the applicant considered the ‘amount applied per use day (g)’ for toothpaste and for mouthwash (Documentation as provided to EFSA No 1). These amounts, i.e. 2.75 g for toothpaste and 21.62 g for mouthwash, are reported in the SCCS Guidance (SCCS, 2018a). According to the same guidance, these applied amounts are connected to retention factors of 0.05 and 0.1, respectively, thus resulting in a daily exposure to toothpaste of 0.14 g per person per day and to mouthwash of 2.16 g per person per day.
To estimate the exposure to the flavouring substance from these oral care products for adults (see Table 3), the Panel applied a body weight of 60 kg to maintain consistency with the exposure estimation via food. The applicant submitted the same exposure estimates for children of undefined age. In order to match the exposure to the flavouring substance via this non‐food sources to the exposure via food, the Panel assumed for the children a body weight of 15 kg. This will likely result in an overestimation of exposure because, e.g. the SCCS recognises that use of mouthwash by children of less than 5 years old is not recommended (SCCS, 2018b).
TABLE 3.
APET – Chronic dietary exposure as provided by the applicant and confirmed by EFSA. Exposure estimates have been adjusted by a correction factor of 0.2.
| Chronic APET | Added as flavouring substance a | Other dietary sources b | Combined c | |||
|---|---|---|---|---|---|---|
| μg/kg bw per day | μg/person per day | μg/kg bw per day | μg/person per day | μg/kg bw per day | μg/person per day | |
| Adults d | 0.8 | 45.0 | 0.0 | 0.0 | 0.8 | 45.0 |
| Children e | 1.9 | 28.4 | 0.0 | 0.0 | 1.9 | 28.4 |
Abbreviations: APET, added portions exposure technique; bw, body weight.
APET Added is calculated on the basis of the amount of flavouring added to a specific food category.
APET Other Dietary Sources is calculated based on the natural occurrence of the flavouring in a specified food category.
APET Combined is calculated based on the combined amount of added flavour and naturally occurring flavouring in a specified food category.
For the adult APET calculation, a 60 kg person is considered representative.
For the child APET calculation, a 3‐year old child with 15 kg bw is considered representative.
3.3.3. Chronic dietary exposure
The exposure assessment to be used in the Procedure for the safety evaluation is the chronic added portions exposure technique (APET) estimate (EFSA CEF Panel, 2010). The chronic APET for the flavouring substance [FL‐no: 16.134] has been calculated for adults and children (see Table 3), and these values, expressed per kg body weight (bw), will be used in the Procedure (see Appendices A and B). The chronic APET calculation is based on the proposed normal use levels and the standard portion size (see Appendix B, Table B1).
The calculated exposure has been adjusted by a correction factor of 0.2, taking into account that only about 20% of the food flavouring is actually consumed while the remaining 80% is retained by the food (chewing gum; see Section 3.1.6 Stability and fate in food).
Based on the information provided by the applicant, the Panel noted that the flavouring substance is not intended to be used in food category 13.2 (foods for infants and young children).
3.3.4. Acute dietary exposure
The acute APET calculation for the flavouring substance is based on the proposed maximum use levels and large portion size (i.e. three times standard portion size) (EFSA CEF Panel, 2010). Acute exposure has been calculated by EFSA, based on the maximum use levels proposed by the applicant and a correction factor of 0.2, for the limited release of the flavouring substance from chewing gum. Results are reported in Table 4.
TABLE 4.
APET – Acute dietary exposure as calculated by EFSA. Exposure estimates have been adjusted by a correction factor of 0.2.
| Acute APET | Added as flavouring substance a | Other dietary sources b | Combined c | |||
|---|---|---|---|---|---|---|
| μg/kg bw | μg/person | μg/kg bw | μg/person | μg/kg bw | μg/person | |
| Adults d | 3.0 | 180.0 | 0.0 | 0.0 | 3.0 | 180.0 |
| Children e | 7.6 | 113.4 | 0.0 | 0.0 | 7.6 | 113.4 |
Abbreviations: APET, added portions exposure technique; bw, body weight.
APET Added is calculated on the basis of the maximum amount of flavouring added to a specific food category.
APET Other dietary sources is calculated based on the natural occurrence of the flavouring in a specified food category.
APET Combined is calculated based on the combined amount of added flavouring and naturally occurring flavouring in a specified food category.
For the adult APET, calculation a 60‐kg person is considered representative.
For the child APET, calculation a 3‐year‐old child with 15 kg bw is considered representative.
3.3.5. Cumulative dietary exposure
The Panel considered that there are no flavouring substances with structural similarity to 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one. Therefore, the calculation of the cumulative exposure is not applicable in this opinion.
3.4. Biological and toxicological data
3.4.1. Absorption, distribution, metabolism and elimination
3.4.1.1. In vivo studies
A toxicokinetic analysis was performed within a repeated dose toxicity study (described below in Section 3.4.3.1) where 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one was administered to four groups of Sprague Dawley rats at doses of 0 (vehicle only), 150, 500, 1500 mg/kg in the feed (equal to 0, 9.4, 32.5 and 96.2 mg/kg/day for male rats and 0, 11.0, 37.4 and 112.0 mg/kg/day for female rats). Plasma samples were collected from all four dosing groups on days 8, 91 (last dose) and 92 (first day of recovery period). Urine samples were collected on day 92 (time of scheduled necropsy) and only the vehicle control and the highest dose group were analysed. The concentration of the test substance and presence of eight potential metabolites were analysed by liquid chromatography coupled with high‐resolution mass spectrometry (LC‐HRMS). Parent compound and metabolites which had been previously detected in in vitro studies were quantified by reference material or estimated based on their peak areas when no reference standard was available.
The parent compound was detected at low nM concentrations in blood plasma of all rats from the mid (500 ppm) and high (1500) dosing groups on days 8 and 91 of sampling (ranging approximately from 2 nM (at the mid dose) and 20 nM (at the high dose)), yet no longer on day 92 (first day of recovery). The parent compound was detected at mid to high nM levels in urine of the high dose group on day 92 (up to 360 nM), along with phase I metabolites, the alcohol form (M1a) and the carboxylic acid acid (M3), and phase II metabolites, the glucuronide (M23) and acyl‐glucuronide (M27). These metabolites were also detected in plasma samples of rats in the mid‐ and high‐dose groups along with some other tentatively identified metabolites (see Figure C1 and Table C1 in Appendix C).
Overall, the data show that systemic exposure of the animals to the flavouring substance via food occurs. The parent compound was cleared fast from the blood, indicating no accumulation, and metabolised in a qualitatively similar manner as observed in the in vitro study with primary human hepatocytes (see Section 3.4.1.2). The major in vivo phase I metabolite was the carboxylic acid (M3) and the major phase II metabolite the acyl glucuronide (M27). Metabolites resulting from the hydrolysis of the amide bond were not detected in plasma or urine samples, in agreement with in vitro data. The Panel noted also that, in the observed or tentatively identified metabolites, the imidazole ring remained intact.
3.4.1.2. In vitro studies
2‐Methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one was tested in primary human hepatocytes to determine the in vitro clearance and to identify the major metabolites. LC‐HRMS was applied to determine the concentration of the parent compound and to identify potential metabolites (Givaudan, 2019).
The flavouring substance was incubated with primary human hepatocytes at a concentration of 10 μM for 0, 30, 60, 120 and 240 min. Negative controls included incubation of the test chemical in the medium in the absence of cells and incubation of cells in the absence of test chemicals. Cells were incubated with 7‐ethoxycoumarin (10 μM) as positive control for 0, 60 and 240 min. Both the tested compound and 7‐ethoxycoumarin were diluted in methanol. The final concentration of methanol in the incubation was 1% (v/v).
At the end of the incubation period, reactions were stopped, purified by solid‐phase extraction and analysed by LC‐HRMS in order to measure the concentration of the parent compound and to identify potential Phase I and Phase II metabolites. The parent compound decreased up to 46.6% after 4 h incubation.
The structure of the tentative metabolites (see Appendix C) was determined by their exact mass; positions of the modifications were tentatively assigned based on MS fragmentation.
The parent compound and tentative metabolites were profiled for alerts of genotoxicity and skin sensitisation using the TIMES software. 6 The observed metabolites did not trigger any alerts for genotoxicity or skin sensitisation, which was in line with an analysis by the Panel using the OECD toolbox.
The study authors concluded that the flavouring substance is moderately metabolised in human hepatocytes involving hydroxylation(s), oxidation of the primary alcohol to the corresponding carboxylic acid and conjugation with glucuronic acid. Based on the derived metabolic map, the tested compound can be cleared from the body, through Phase I and Phase II biotransformation pathways. The Panel agrees with these conclusions.
3.4.2. Genotoxicity
3.4.2.1. In silico analysis
2‐Methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one and the metabolites tentatively identified in the in vitro metabolism study (Givaudan, 2019) were analysed for alerts of genotoxicity using the TIMES software. No structural alerts for genotoxicity were identified.
3.4.2.2. In vitro genotoxicity studies
3.4.2.2.1. Bacterial reverse mutation assay
A bacterial reverse mutation assay was conducted in Salmonella Typhimurium strains TA98, TA100, TA1535, TA1537 and in Escherichia coli WP2 uvrA to assess the mutagenicity of 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one (purity 100%, sum of isomers), both in the absence and in the presence of metabolic activation by Aroclor 1254‐induced rat liver S9 fraction (S9‐mix). Two separate experiments were conducted, using the plate incorporation method (BioReliance, 2020a). The study design complied with OECD Test Guideline (TG) 471 (OECD, 1997) and with the GLP principles. Positive control chemicals and dimethyl sulfoxide (DMSO, as vehicle control) were evaluated concurrently.
In the initial toxicity‐mutation experiment, the flavouring substance was tested at eight concentrations from 1.5 to 5000 μg/plate with and without S9‐mix, in duplicate plates. Precipitate was found at 5000 μg/plate with and without S9‐mix. Toxicity was observed at 5000 μg/plate with tester strain TA1535 in the presence and absence of S9‐mix and with E. coli in the absence of S9‐mix.
In the confirmatory experiment, the flavouring substance was tested at six concentrations from 15 to 5000 μg/plate with and without S9‐mix in triplicate plates. Precipitate was found at 5000 μg/plate with and without S9‐mix. Toxicity was observed at 5000 μg/plate with tester strain TA1535 in the presence and absence of S9‐mix.
All positive control chemicals both with and without S9‐mix induced significant increases in revertant colony numbers. Both vehicle controls and positive controls were within the respective historical control ranges.
In both experiments, no increase in the mean number of revertant colonies was observed at any tested concentration in any tester strain in the absence or presence of metabolic activation (BioReliance, 2020a). The Panel considered the study reliable without restrictions and the results of high relevance (see Table D1 in Appendix D).
3.4.2.2.2. In vitro mammalian cell micronucleus test
Human peripheral blood lymphocytes from one healthy donor were treated with 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one (purity 100%, sum of isomers). The in vitro micronucleus assay was carried out according to OECD TG 487 (OECD, 2016) and GLP principles. The cytokinesis block micronucleus assay protocol was applied. Positive controls were cyclophosphamide, mitomycin C and vinblastine. DMSO was used as negative control (BioReliance, 2020b).
A range‐finder experiment was carried out with concentrations up to the limit of solubility (2000 μg/mL). Concentrations for the micronucleus experiment were selected based on the results of this experiment.
For the micronucleus experiment, lymphocytes were treated with the flavouring substance at concentrations ranging from 59 to 280 μg/mL in the 4 h treatment, in the presence of metabolic activation (S9‐mix from rats treated with Aroclor 1254), followed by 20‐h recovery period (4 h + 20 h). A further experiment was carried out with concentrations from 59 to 202 μg/mL in the 4‐h treatment in the absence of S9‐mix (4 h + 20 h), and from 15 to 59 μg/mL in the 24‐h continuous treatment in the absence of S9‐mix. Precipitation was observed at 238 μg/mL and above in the 4 h treatment, in the presence of metabolic activation.
Cytotoxicity data, based on cytokinesis‐block proliferation index, were used to select the concentrations for the micronucleus (MN) analysis.
In the treatment of 4 h + 20 h in the presence of S9‐mix, the MN analysis was conducted at 59, 105 and 146 μg/mL (cytotoxicity of 2%, 8% and 51%, respectively).
In the treatment of 4 h + 20 h in the absence of S9‐mix, the MN analysis was performed at 84, 124 and 172 μg/mL (cytotoxicity of 17%, 27% and 57%, respectively).
In the treatment of 24 h in the absence of S9‐mix, the MN analysis was performed at 21.5, 42.5 and 50 μg/mL (cytotoxicity of 5%, 39% and 55%, respectively).
The flavouring substance did not increase the frequency of micronucleated cells compared to vehicle (DMSO) controls in any of the conditions tested. The Panel considered the study reliable without restrictions and the results of high relevance (see Table D1 in Appendix D).
3.4.2.3. In vivo genotoxicity studies
No in vivo studies were performed since no genotoxicity was observed in vitro.
3.4.2.4. Conclusion on genotoxicity studies
No indications of mutagenicity were obtained from an adequate bacterial reverse mutation assay, and no indications for clastogenicity or aneugenicity were obtained from an adequate in vitro mammalian cell micronucleus test. Therefore, the Panel concluded that 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one does not raise a concern for genotoxicity.
3.4.3. Toxicity
3.4.3.1. 90‐day toxicity study in rats
2‐Methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one (batch nr. 9, purity 99.9%) was tested in a 90‐day repeated dose toxicity study in rats followed by a 28‐recovery period with GLP compliance and according to OECD guideline 408 (OECD, 2018). In addition, toxicokinetic analysis was performed, confirming systemic exposure to the substance (for further details, see Section 3.4.1.1).
2‐Methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one was administered to four groups of 8 weeks old Sprague Dawley Crl:CD(SD) rats (15/sex/group for control and 1500 mg/kg; 10/sex/group for 150 and 500 mg/kg) at doses of 0 (vehicle only), 150, 500, 1500 mg/kg in the feed. This were equal to 0, 9.4, 32.5 and 96.2 mg/kg/day for male rats and 0, 11.0, 37.4 and 112.0 mg/kg/day for female rats. The extra 10 animals (5/sex) in the control and highest dose group had a 28‐recovery period following day 91. The doses were chosen based on a 2‐week palatability and toxicity study, where decreased food consumption and body weights were noted at 3000 mg/kg.
The feed formulations were prepared approximately every 6 days. The test substance has been shown to be stable in feed under the conditions of the study.
No test substance‐related mortality, clinical observations or effects on food consumption were observed.
Lower body weights were observed in the group of male and female rats receiving a dose of the flavouring substance of 1500 mg/kg in the feed compared to the control group; however, the difference did not exceed 10% for male rats and 5% for females. Transient significant lower body weight gains were observed for males in the 1500 mg/kg group during the study duration and at recovery.
At sacrifice, serum alkaline phosphate (ALP) concentrations were statistically significantly increased for males in the low‐ and high‐dose group (36% and 74%, respectively) compared with controls. Males receiving diet with 500 mg/kg had a non‐statistically significant increase of 28% in serum ALP compared to controls. At day 120, serum of males in the high‐dose group had a non‐significant increase in ALP concentrations (32%). Exposed females had 16%–30% increases in serum ALP, but none were statistically significant. Additionally, there were no correlating changes in liver weights or microscopic changes in the liver associated with increased ALP levels.
A statistically significant dose‐related higher mean absolute thyroid (including parathyroid) gland weight (29%–45%) were observed for all dosed females. In addition, for all exposed females, relative thyroid (including parathyroid) gland weight, relative to body weight (19%–48%, statistically significant for mid‐ and high‐dose group) and brain (28%–46%, statistically significant for all three dose groups), was increased compared to controls. No difference in thyroid (including parathyroid) gland weights between the high‐dose group and controls was observed following the 28‐day recovery period. No substance‐related difference was seen for thyroid (including parathyroid) gland weight in male rats.
There was no substance‐related effects for the other parameters measured/analysed: haematological analysis including coagulation, determination of oestrous stage at necropsy, functional observational battery, motor activity, clinical chemistry, urinalysis, thyroid hormone analysis, ophthalmic measures, macroscopic and microscopic analyses.
There were no statistically significant effects on thyroid hormones and no histopathological changes in female rats. Although the Panel noted that thyroid function is more easily perturbated in rats than humans, in the absence of further information, the large dose‐dependent increases in absolute and relative thyroid (including parathyroid) gland weights were considered by the Panel to be substance‐related and adverse. Using dose–response modelling, the Panel identified a BMDL of 0.71 mg/kg bw/day (see section 3.4.3.1.2).
3.4.3.1.1. Calculation of the Point of Departure for the assessment
The data on absolute, relative (to body weight) and relative (to brain weight) thyroid (including parathyroid) weights, were submitted to dose–response modelling using the EFSA PROAST web tool, 7 in line with the EFSA SC guidance document (EFSA Scientific Committee, 2017). Instead of using the default value of 5% for the benchmark response (BMR) for continuous endpoints, the Panel employed endpoint‐specific BMRs, based on the theory developed by Slob (2017). This theory takes better account of the natural variability in the measured parameters, than the default BMRs. This results in biologically more plausible BMRs and subsequently more plausible BMDLs. The endpoint‐specific BMR for the thyroid (including parathyroid) weight, relative to body weight was calculated with the RIVM PROAST webtool 8 and amounted to 19.3%, which is close to the standard deviation of approximately 15% from historical control data for thyroid (Marty et al., 2009).
3.4.3.1.2. Results of the dose–response modelling
For the thyroid data in the males, no dose‐related trend could be identified. Therefore, dose–response modelling was performed only for the females.
The dose–response modelling was carried out based on the exposure expressed in mg/kg bw per day as group averages over the 90‐day exposure period and based on the individual exposures in mg/kg bw per day averaged over the 90‐day exposure period.
The results of the dose–response modelling, using model averaging, have been presented in Table 5. This table gives the endpoint‐specific BMRs for the three parameters investigated (all are around 20% increase) as well as the lower (BMDL) and upper (BMDU) boundaries of the 90% confidence interval around the benchmark dose (BMD). For convenience also the ratio of these two has been presented.
TABLE 5.
Dose–response modelling results for thyroid (including parathyroid) weight parameters. a
| Thyroid (including parathyroid) weight | BMR (%) | BMD‐CI (mg/kg bw and day) | BMDU/BMDL ratio | ||
|---|---|---|---|---|---|
| Exposure as | Exposure as | ||||
| Group average dose | Individual dose | Group average dose | Individual dose | ||
| Absolute | 22.6 | 0.06–59.6 | 0.01–61.6 | 946 | 6160 |
| Relative to brain weight | 21.7 | 0.15–52.2 | 0.05–53.6 | 348 | 1072 |
| Relative to body weight | 19.3 | 0.64–41.9 | 0.71–39 | 65 | 55 |
The table presents only the results for the data for the females because there was no dose‐related trend in the males.
The most reliable estimate for a BMD (i.e. the narrowest confidence interval around it) was found for thyroid (including parathyroid) weight relative to body weight, based on the mean individual exposure estimates. Since thyroid (including parathyroid) weight depends on body weight, and since using the individual exposure estimates makes use of the maximum amount of information present in the data, preference is given to the BMDL found for the thyroid (including parathyroid) weight relative to body weight, based on the individual exposure estimates, which amounted to 0.71 mg/kg bw and day.
More details of the dose–response modelling for this parameter can be found in Appendix E.
3.5. Application of the procedure
No structural/metabolic similarity of the flavouring substance to flavouring substances in an existing FGE was identified.
Since 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one [FL‐no: 16.134] does not raise a concern for genotoxicity, it is appropriate to evaluate the use of [FL‐no: 16.134] as a flavouring substance following the stepwise evaluation procedure for individual substances as outlined in the ‘Guidance on the data required for the risk assessment of flavourings to be used in or on foods’ (EFSA CEF Panel, 2010) and Appendix A.
Step 1
The flavouring substance is allocated to structural Cramer Class III (90 μg/person per day). 9
Step 2
Based on the toxicokinetic and metabolism data (see Section 3.4.1), it can be predicted that the flavouring substance is metabolised to innocuous products, only. Therefore, it is evaluated via the left (A‐)side of the procedure (see Appendix A, Figure A1).
Step A3
The conditions of use result in chronic APET dietary exposure estimates of 0.8 and 1.9 μg/kg bw per day (45 and 28.4 μg /person per day), for adults and children, respectively. These estimates are below the TTC for Cramer Class III (90 μg/person per day). Therefore, according to the procedure, no further data would be required to conclude on the safety of the substance (see Appendix A, Figure A1).
Nevertheless, the applicant tested the flavouring substance in a subchronic toxicity study (see Section 3.4.3.1). In this study, dose‐dependent increases in absolute and relative thyroid (including parathyroid) gland weights were considered by the Panel to be substance‐related and adverse. For this effect, a BMDL of 0.71 mg/kg bw per day was calculated based on a BMR of approximately 20% for the thyroid (including parathyroid) weight relative to body weight. Using this BMDL, margins of exposure (MoE) of 887 and 374 could be calculated for adults and children, respectively, when assessing the exposure based on chronic APET. These MoEs are sufficiently large and therefore the dietary exposure to the flavouring substance does not raise a safety concern.
3.6. Assessment of acute, combined and cumulative exposure
3.6.1. Acute exposure
No signs of acute toxicity were observed in a subchronic toxicity study with dose levels up to 96.2 mg/kg per day (for male rats) and 112.0 mg/kg per day (for female rats). Since these dose levels are far above the potential acute exposure in humans (i.e. 3 μg/kg bw in adults and 7.6 μg/kg bw in children (see Table 5)), there is no concern for acute toxicity.
3.6.2. Combined exposure
Since the substance does not occur naturally in food, no exposure is anticipated from that source, but additional oral exposure to the substance may occur from its use in oral care formulations, i.e. toothpaste and mouthwash. At most this would add 0.17 μg/kg bw per day in adults and 0.67 μg/kg bw per day in children to the exposure from food, resulting in combined exposure estimates of 0.97 μg/kg bw per day in adults and 2.57 μg/kg bw per day in children. When compared to the BMDL derived from the subchronic toxicity study, the resulting MoEs would be 732 for adults and 276 for children. Taking into account that the combined exposure is likely to be overestimated, these MoEs are considered sufficiently large.
3.6.3. Cumulative exposure
Because no structurally related substances were identified, a safety assessment for cumulative exposure is not included in this FGE.
4. DISCUSSION
The European Commission requested that EFSA carry out the safety assessment of the substance 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one [FL‐no: 16.134] (CAS no. 2413115–68‐9) as a new flavouring substance in accordance with Regulation (EC) No 1334/2008.
EFSA evaluated [FL‐no: 16.134] and used the procedure as described in (EFSA CEF Panel, 2010).
No other substances with sufficient structural similarity to the flavouring substance have been identified in existing FGEs. The substance is not reported to occur naturally and is manufactured through chemical synthesis.
The provided specifications, which include a 99% purity requirement, are considered consistent with the analytical data provided. The flavouring substance is a mixture of two racemic diastereomers present in ratios between 40/60 and 60/40.
The presence of impurities was investigated through HPLC‐UV and was reported to be ≤ 0.4%. In addition, the flavouring substance was analysed for the presence of toxic elements (i.e. As, Pb, Cd and Hg). The results showed all levels to be below the LOQ for their respective analytical methods.
To investigate the potential presence of nanoparticles, the flavouring substance was assessed as required in the EFSA ‘Guidance on technical requirements for regulated food and feed product applications to establish the presence of small particles including nanoparticles’ (EFSA Scientific Committee, 2021). The flavouring substance was shown to be soluble in carriers which are relevant for its intended use in chewing gums. Therefore, there is no concern regarding the potential exposure of consumers to small particles, including nanoparticles, under the intended conditions of use.
With regard to the storage stability, the flavouring substance was tested by assessing the product at (i) normal and (ii) accelerated storage conditions. The stability of the flavouring substance was also investigated in five essential oils and in toothpaste. Based on the data available (see Section 3.1.6), the Panel considered the flavouring substance to be sufficiently stable under the intended conditions of use.
Overall, the information provided on the manufacturing process, the composition and the stability of the flavouring substance was considered sufficient by the Panel.
For the use of [FL‐no: 16.134] as a flavouring substance, adequate information on uses and use levels has been provided, as specified in Appendix B. The substance is not intended to be used in food for infants and young children. The chronic dietary exposure to the candidate substance has been estimated using the APET method. The chronic APET exposure estimates are 0.8 and 1.9 μg/kg bw per day (45 and 28.4 μg/person per day) for adults and children (15‐kg bw; 3 years old), respectively. The acute APET exposure estimates are 3 and 7.6 μg/kg bw (180 and 113.4 μg/person, for adults and children, respectively).
Based on the results from a bacterial reverse mutation test (Ames test) and an in vitro MN assay in human lymphocytes, the Panel concluded that this substance does not raise a concern for genotoxicity.
Based on the toxicokinetic and metabolism data (see Section 3.4.1), it can be predicted that the flavouring substance is metabolised to innocuous products, only. Therefore, its evaluation proceeds via the left (A‐)side of the Procedure (see Appendix A, Figure A1).
The substance is allocated to structural Cramer Class III and the APET exposure estimates are below this TTC applicable for this class (i.e. 90 μg/person per day). Therefore, according to the procedure, no further data would be required to conclude on the safety of the substance.
Nevertheless, the applicant submitted a subchronic toxicity study in which the substance was fed to rats for 91 days followed by a 28‐recovery period (see Section 3.4.3.1). In this study, the large dose‐dependent increases in absolute and relative thyroid (including parathyroid) gland weights in female rats were considered by the Panel to be substance‐related and adverse. For this endpoint, a BMDL of 0.71 mg/kg bw per day was calculated based on a BMR of 19.3% for the thyroid (including parathyroid) weight relative to body weight.
With this BMDL adequate margins of exposure of 887 and 374 for adults and children, respectively, were calculated for the use of the substance [FL‐no: 16.134] as food flavouring, for the APET exposure estimates based on the proposed use and use levels as specified in Appendix B.
The exposure from the use as flavouring substance in food was combined with exposure from oral care formulations (i.e. toothpaste and mouthwash). The margin of exposure calculated for children (i.e. 276) was still considered sufficiently large by the Panel, taking into account that the additional exposure to the substance resulting from its use in oral care formulations is likely to be overestimated in children (see Section 3.3.2).
The Panel noted that no data on acute toxicity are available. Considering the dose levels tested in the repeated dose toxicity study, which did not result in acute toxicity and are far higher than the anticipated acute exposure in humans (see Section 3.3.4), there is no concern for acute toxicity.
5. CONCLUSIONS
Overall, the Panel concluded that there is no safety concern for [FL‐no: 16.134], when used as a flavouring substance at the estimated level of dietary exposure calculated using the APET approach, based on the intended use and use levels as specified in Appendix B.
The Panel further concluded that the combined exposure to [FL‐no: 16.134] from its use as a food flavouring substance and from its presence in toothpaste and mouthwash is also not of safety concern.
DOCUMENTATION AS PROVIDED TO EFSA
Technical Information Submission for a New Flavouring Substance by Givaudan International SA to the European Food Safety Authority (EFSA) according to the “Common Authorisation Procedure for the application for evaluation of a new flavouring substance” (Regulation (EC) No 1334/2008, Regulation (EC) No 1331/2008, Regulation (EU) No 234/2011). October 2021. Submitted by Givaudan International SA. 10
Additional information received on 27 January 2023, submitted by Givaudan International SA in response to a request from EFSA (27 September 2022).
Additional information received on 27 September 2023, submitted by Givaudan International SA in response to a request from EFSA (14 July 2023).
BioReliance (2020a). Bacterial Reverse Mutation Assay. BioReliance, study number AG02KR.503.BTL. May 2020. Submitted by Givaudan International SA.
BioReliance (2020b). In Vitro Mammalian Cell Micronucleus Assay in Human Peripheral Blood Lymphocytes. BioReliance, study number AG02KR.348.BTL. April 2020. Submitted by Givaudan International SA.
Givaudan (2019). In Vitro Metabolism of GR‐50‐6449 in Primary Human Hepatocytes. Givaudan, study number RCR 153′920. December 2019. Submitted by Givaudan International SA.
Abbreviations
- APET
added portions exposure technique
- BMD
benchmark dose
- BMDL
lower confidence limit of the benchmark dose
- BMDU
upper confidence limit of the benchmark dose
- BMR
benchmark response
- BW
body weight
- CAS
chemical abstract service
- DMSO
dimethyl sulfoxide
- FAO
Food and Agriculture Organization of the United Nations
- FEMA
Flavour and Extract Manufactures Association
- FGE
flavouring group evaluation
- FLAVIS (FL)
flavour information system database
- GC‐MS
gas chromatography‐mass spectrometry
- GLP
good laboratory practiced
- GRAS
generally regarded as safe
- GSFA
codex general standard for food additives
- HPLC‐UV
high performance liquid chromatography‐ultraviolet
- HS‐GC‐MS
headspace gas chromatography‐mass spectrometry
- ICP‐MS
inductively coupled plasma mass spectrometry
- IR
infra red
- JECFA
The Joint FAO/WHO Expert Committee on Food Additives
- LOQ
limit of quantification
- MW
molecular weight
- NMR
nuclear magnetic resonance
- No
number
- OECD
Organisation for Economic Co‐operation and Development
- rptwght_bw
thyroid (including parathyroid) weight relative to body weight
- SPET
single‐portion exposure technique
- WHO
World Health Organisation
CONFLICT OF INTEREST
If you wish to access the declaration of interests of any expert contributing to an EFSA scientific assessment, please contact interestmanagement@efsa.europa.eu.
REQUESTOR
European Commission
QUESTION NUMBER
EFSA‐Q‐2022‐00141
COPYRIGHT FOR NON‐EFSA CONTENT
EFSA may include images or other content for which it does not hold copyright. In such cases, EFSA indicates the copyright holder and users should seek permission to reproduce the content from the original source.
PANEL MEMBERS
Maged Younes, Gabriele Aquilina, Laurence Castle, Karl‐Heinz Engel, Paul Fowler, Maria Jose Frutos Fernandez, Peter Fürst, Ursula Gundert‐Remy, Rainer Gürtler, Trine Husøy, Melania Manco, Wim Mennes, Peter Moldeus, Sabina Passamonti, Romina Shah, Ine Waalkens‐Berendsen and Matthew Wright.
LEGAL NOTICE
The scientific output published implements EFSA's decision on the confidentiality requests submitted on specific items. As certain items have been awarded confidential status by EFSA, they are consequently withheld from public disclosure by redaction.
ACKNOWLEDGEMENTS
The Panel wishes to thank the following for the support provided to this scientific output: Borana Dino and Agnieszka Mech.
APPENDIX A. Procedure for the safety evaluation of ‘stand‐alone’ chemically defined flavouring substances
FIGURE A1.
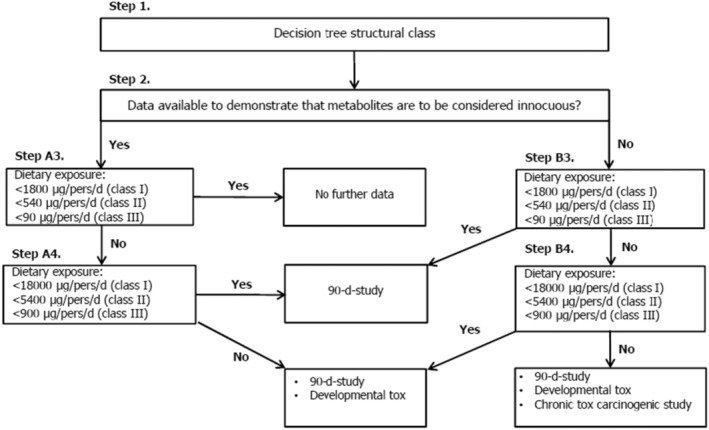
Procedure applied for the safety evaluation of 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one according to the data requirements for the risk assessment of flavourings for which no structurally related flavouring substances in existing FGEs can be identified (EFSA CEF Panel, 2010).
APPENDIX B. Food categories and use levels provided for 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one
TABLE B1.
Food categories and use levels (only these food categories are included for which use levels were provided).
| CODEX code | Food categories a | Standard portions (g) | Intended use level as flavouring substance (mg/kg) | Occurrence level from other food sources (mg/kg) | Combined occurrence level from all sources (mg/kg) | |||
|---|---|---|---|---|---|---|---|---|
| Normal | Maximum | Normal | Maximum | Normal | Maximum | |||
| 05.3 | Chewing gum | 3 (adult) | 75 | 100 | – | – | 75 | 100 |
| 1.89 (children) b | – | – | ||||||
According to Codex GSFA (General Standard for Food Additives, available at https://www.codexalimentarius.net/gsfaonline/CXS_192e.pdf) used by the JECFA in the SPET technique (FAO/WHO, 2008).
Standard portion sizes for children are obtained by multiplying the adult standard portion sizes by a factor of 0.63 (EFSA CEF Panel, 2010).
APPENDIX C. Identified and tentatively identified metabolites of 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one
FIGURE C1.
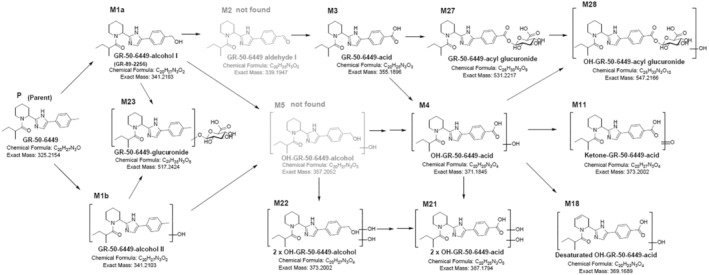
Proposed metabolic map for GR‐50‐6449 (code used by the applicant for the parent compound) with metabolites identified (M1a, M3, M27) or tentatively identified in plasma and urine samples of rats exposed to GR‐50‐6449 via the diet for 91 days. Metabolites in grey (M2 and M5) were not detected in plasma in plasma or urine samples. The square brackets indicate that position of substituents could not be determined from the mass spectra (see Documentation as provided to EFSA No. 1).
TABLE C1.
Metabolites investigated in pooled plasma and urine samples. Parent (P, GR‐50‐6449) and (tentative) metabolites in bold have been also analysed in plasma samples from individual animals, metabolites in grey‐bold were not detected in any of the samples. F, found; T, traces found; NF, not found (based on Documentation as provided to EFSA No. 1).
| ID | Name | Formula | Exact mass | Structure comment | Plasma | Urine | ||
|---|---|---|---|---|---|---|---|---|
| d8 | d91 | d92 | d92 | |||||
| P | GR‐50‐6449 | C20H27N3O | 325.2154 | Parent | F | F | T | F |
| M1a | GR‐50‐6449‐alcohol I | C20H27N3O2 | 341.2103 | Identified (reference GR‐89‐2256) | F | T | NF | F |
| M1b | GR‐50‐6449‐alcohol II | C20H27N3O2 | 341.2103 | Tentative structure (MS/MS) | T | T | NF | NF |
| M2 | GR‐50‐6449‐aldehyde | C20H25N3O2 | 339.1947 | Tentative structure | NF | NF | NF | NF |
| M3 | GR‐50‐6449‐acid | C20H25N3O3 | 355.1896 | Indicated by reference a | F | F | F | F |
| M4 | OH‐GR‐50‐6449‐acid | C20H25N3O4 | 371.1845 | Tentative structure (MS/MS); multiple peaks | F | F | F | F |
| M5 | OH‐GR‐50‐6449‐alcohol | C20H27N3O3 | 357.2052 | Not found | NF | NF | NF | NF |
| M23 | GR‐50‐6449‐glucuronide | C26H35N3O8 | 517.2424 | Tentative structure (MS/MS) | F | F | T | F |
| M27 | GR‐50‐6449‐acyl glucuronide | C26H33N3O9 | 531.2217 | Indicated by MS/MS and strucutre of M3 | F | F | F | F |
| M28 | OH‐GR‐50‐6449‐acyl glucuronide | C26H33N3O10 | 547.2166 | Tentative structure (MS/MS) | T | T | T | F |
| M6 | Amide hydrolysis metabolite | C15H19N3 | 241.1579 | < LOD (verified by reference GR‐89‐0675) | NF | NF | NF | NF |
| M7 | OH‐amide hydrolysis metabolite | C15H19N3O | 257.1528 | < LOD (verified by reference GR‐89‐2118) | NF | NF | NF | NF |
| M18 | Desaturated OH‐GR‐50‐6449 acid | C20H23N3O4 | 369.1689 | Tentative structure (2 peaks with same mass) | NF | NF | NF | F |
| M21 | 2 × OH‐GR‐50‐6449‐acid | C20H25N3O5 | 387.1794 | Tentative structure (exact mass; MS/MS) | T | T | T | F b |
| M22/M11 c | 2 × OH GR‐50‐6449‐alcohol/Ketone‐GR‐50‐6449‐acid | C20H27N3O4 | 373.2002 | Tentative structure (exact mass; MS/MS) | T | T | T | F |
The structure of M3 was indirectly confirmed due to a similar retention time and MS/MS spectrum compared to synthetic M3 and the starting material M1a.
Male rats produced more M21 than female rats.
Tentative M22 and M11 have the same exact mass and chemical formula.
APPENDIX D. Genotoxicity studies on 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one
TABLE D1.
Summary of in vitro genotoxicity data on 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one.
| Chemical name [FL‐no] | Test system in vitro | Test object | Concentrations of substance and test conditions | Result | Reliability/comments | Relevance of test system/relevance of the result | Reference |
|---|---|---|---|---|---|---|---|
| 2‐Methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one [FL‐no : 16.134] | Bacterial Reverse Mutation test |
S. typhimurium TA98, TA100, TA1535, TA1537 E. coli WP2 uvrA |
Experiment 1 (plate incorporation, +S9, −S9): 1.5–5000 μg/plate Experiment 2 (plate incorporation, +S9, −S9): 15–5000 μg/plate |
Negative |
Reliable without restrictions. Study performed according to OECD TG 471 and in compliance with GLP. |
High/High | BioReliance (2020a) |
| Micronucleus assay | Human peripheral blood lymphocytes |
59,105, 146 μg/mL (4 h + 20 h, +S9) 84, 124, 172 μg/mL (4 h + 20 h, ‐S9) 21.5, 42.5, 50 μg/mL (24 h + 0 h, ‐S9) |
Negative |
Reliable without restrictions. Study performed according to OECD TG 487 and in compliance with GLP. The given concentrations are those for the cultures that were scored for micronuclei. |
High/High | BioReliance (2020b) |
Abbreviations: GLP, Good Laboratory Practice; OECD, Organisation for Economic Co‐operation and Development.
APPENDIX E. Benchmark dose response modelling for thyroid (including parathyroid) weight relative to body weight
E.1. Data description
The endpoint to be analysed is thyroid (including parathyroid) weight, relative to body weight (rptwght_bw). Only data for females have been analysed, because no dose‐related trend was observed in the males. Exposure (‘dose’) is represented as the individual dose averaged over the exposure period (90 days).
Data used for analysis of the four dose groups are shown below:
| 0 mg/kg feed | 150 mg/kg feed | 500 mg/kg feed | 1500 mg/kg feed | ||||
|---|---|---|---|---|---|---|---|
| Dose | rptwght_bw | Dose | rptwght_bw | Dose | rptwght_bw | Dose | rptwght_bw |
| 0.0 | 83.673 | 9.7 | 53.333 | 32.5 | 76.408 | 92.5 | 80.453 |
| 0.0 | 63.091 | 9.7 | 74.754 | 33.3 | 109.059 | 95.1 | 82.943 |
| 0.0 | 50.943 | 10.3 | 67.429 | 34.1 | 73.700 | 107.6 | 106.084 |
| 0.0 | 55.804 | 10.7 | 49.291 | 34.8 | 75.633 | 110.4 | 101.829 |
| 0.0 | 59.770 | 10.8 | 68.944 | 36.2 | 86.392 | 110.9 | 69.841 |
| 0.0 | 57.338 | 11.1 | 78.286 | 37.1 | 90.253 | 113.8 | 94.141 |
| 0.0 | 61.290 | 11.5 | 65.152 | 37.3 | 80.634 | 122.0 | 86.638 |
| 0.0 | 53.640 | 11.7 | 105.556 | 40.6 | 67.033 | 132.4 | 107.759 |
| 0.0 | 49.346 | 12.0 | 77.974 | 42.9 | 81.102 | 133.1 | 58.167 |
| 0.0 | 66.238 | 12.4 | 71.613 | 45.1 | 77.959 | 143.1 | 99.588 |
E.2. Selection of the BMR
The BMR (benchmark response) is a change in mean response compared to the controls. The BMD (benchmark dose) is the dose corresponding with the BMR of interest. A two‐sided 90% confidence interval around the BMD will be estimated, the lower bound is reported by BMDL and the upper bound by BMDU.
For the current assessment, an endpoint‐specific BMR of 19.3% will be used, which is calculated from the within group variance of the rptwght_bw parameter across the dose groups, considering log‐normality. The derivation is based on a theoretical consideration by Slob (2017). For further clarification, see also the ANS Panel opinion on the re‐evaluation of sodium and potassium nitrite (EFSA ANS Panel, 2017).
E.3. Software Used
The endpoint‐specific BMR for rptwght_bw was estimated using the RIVM PROAST webtool. 11
Results of the dose–response modelling are obtained using the EFSA web tool for BMD analysis, which uses the R‐package PROAST, version 70.0, for the underlying calculations.E1
E.4. Specification of Deviations from Default Assumptions
General assumptions
None, except for the use of an endpoint‐specific BMR, rather than the default BMR of 5% for continuous endpoints (see EFSA Scientific Committee, 2017).
Dose–response models
Default set of fitted models:
| Model | Number of parameters | Formula | |
|---|---|---|---|
| Null | 1 |
|
|
| Full | No. of groups |
|
|
| Exp model 3 | 3 |
|
|
| Exp model 4 | 4 |
|
|
| Hill model 3 | 3 |
|
|
| Hill model 4 | 4 |
|
|
| Inverse exponential | 4 |
|
|
| Log‐normal family | 4 |
|
Procedure for selection of BMDL
FIGURE E1.
Flow chart for selection of BMDL
E.5. Results
E.5.1. Response variable: rptwght_bw
E.5.1.1. Fitted models
| Model | Converged | loglik | npar | AIC |
|---|---|---|---|---|
| Full | NA | NA | NA | NA |
| null model | Yes | 3.27 | 2 | −2.54 |
| Expon. m3‐ | Yes | 14.41 | 4 | −20.82 |
| Expon. m5‐ | Yes | 14.86 | 5 | −19.72 |
| Hill m3‐ | Yes | 14.41 | 4 | −20.82 |
| Hill m5‐ | Yes | 14.96 | 5 | −19.92 |
| Inv.Expon. m3‐ | Yes | 14.47 | 4 | −20.94 |
| Inv.Expon. m5‐ | Yes | 14.80 | 5 | −19.60 |
| LN m3‐ | Yes | 14.44 | 4 | −20.88 |
| LN m5‐ | Yes | 14.95 | 5 | −19.90 |
E.5.1.2. Estimated model parameters
EXP
Estimate for var‐: 0.02849
Estimate for a‐: 59.27
Estimate for BMD‐: 8.657
Estimate for d‐: 0.3154
HILL
Estimate for var‐: 0.02849
Estimate for a‐: 59.27
Estimate for BMD‐: 8.667
Estimate for d‐: 0.3166
INVEXP
Estimate for var‐: 0.0284
Estimate for a‐: 59.3
Estimate for BMD‐: 9.329
Estimate for d‐: 0.06425
LOGN
Estimate for var‐: 0.02844
Estimate for a‐: 59.29
Estimate for BMD‐: 9.048
Estimate for d‐: 0.1138
E.5.1.3. Weights for model averaging
| EXP | HILL | INVEXP | LOGN |
|---|---|---|---|
| 0.24 | 0.24 | 0.26 | 0.25 |
E.5.1.4. Final BMD values
| Endpoint | Subgroup | BMDL | BMDU |
|---|---|---|---|
| rptwght_bw | F | 0.71 | 39 |
Confidence intervals for the BMD are based on 1000 bootstrap data sets.
E.5.1.5. Visualisation
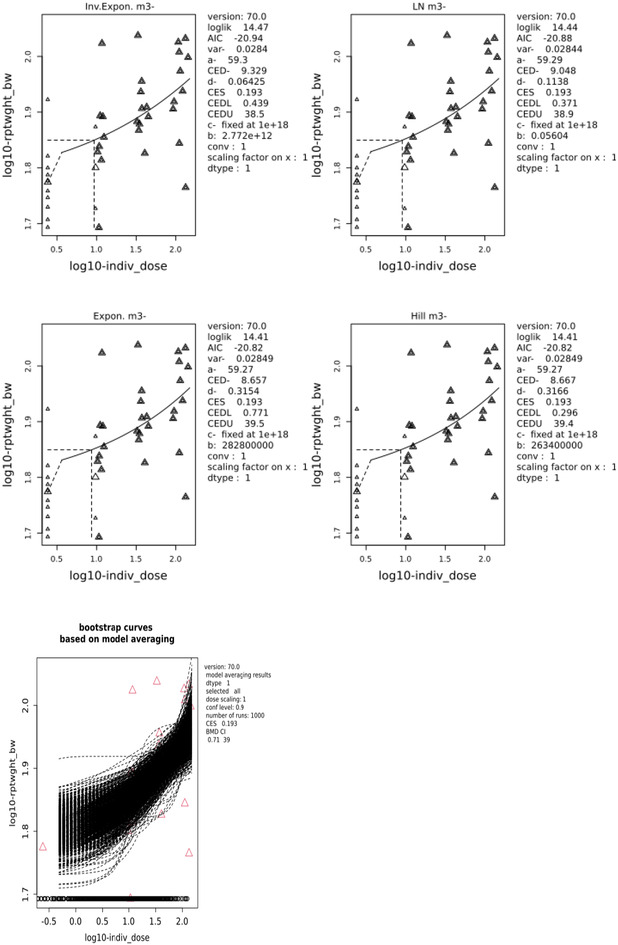
E.6. Conclusions
The dose–response analysis for statistically significant and dose‐related increase in thyroid (including parathyroid) weight, relative to body weight (rptwght_bw) resulted in a BMDL of 0.71 mg/kg bw and day, based on an endpoint‐specific BMR of 19.3% increase. This BMDL is approximately a factor of 10 below the lowest dose level used in the study, owing to the highly variable nature of the data, resulting in a rather wide confidence interval around the BMD.
EFSA FAF Panel (EFSA Panel on Food Additives and Flavourings) , Younes, M. , Aquilina, G. , Castle, L. , Degen, G. , Engel, K.‐H. , Fowler, P. J. , Frutos Fernandez, M. J. , Fürst, P. , Gundert‐Remy, U. , Gürtler, R. , Husøy, T. , Manco, M. , Moldeus, P. , Passamonti, S. , Shah, R. , Waalkens‐Berendsen, I. , Wright, M. , Benigni, R. , … Mennes, W. (2024). Flavouring group evaluation 419 (FGE.419): 2‐methyl‐1‐(2‐(5‐(p‐tolyl)‐1H‐imidazol‐2‐yl)piperidin‐1‐yl)butan‐1‐one. EFSA Journal, 22(5), e8750. 10.2903/j.efsa.2024.8750
Adopted: 21 March 2024
Notes
Regulation (EC) No 1334/2008 of the European Parliament and of the Council of 16 December 2008 on flavourings and certain food ingredients with flavouring properties for use in and on foods and amending Council Regulation (EEC) No 1601/91, Regulations (EC) No 2232/96 and (EC) No 110/2008 and Directive 2000/13/EC. OJ L 354, 31.12.2008, pp. 34–50.
Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC. OJ L 396, 30.12.2006, p. 1.
Regulation (EC) No 178/2002 of the European Parliament and of the Council of 28 January 2002 laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety. OJ L 31, 1.2.2002, p. 1.
Decision available online https://www.efsa.europa.eu/en/corporate‐pubs/transparency‐regulation‐practical‐arrangements.
The non‐confidential version of the dossier, following EFSA's assessment of the applicant's confidentiality requests, is published on Open.EFSA and is available at the following link: https://open.efsa.europa.eu/dossier/FFL‐2021‐0361.
TIMES software version 2.29.1.88, Burgas University, Bulgaria.
Determined with OECD QSAR Toolbox (version 4.5) available at https://qsartoolbox.org/.
The non‐confidential version of the dossier, following EFSA's assessment of the applicant's confidentiality requests, is published on Open.EFSA and is available at the following link: https://open.efsa.europa.eu/dossier/FFL‐2021‐0361.
REFERENCES
- EFSA (European Food Safety Authority) . (2012). Proposed template to be used in drafting scientific opinion on flavouring substances (explanatory notes for guidance included). EFSA Journal, 9(1), 45. 10.2903/sp.efsa.2012.EN-218 [DOI] [Google Scholar]
- EFSA ANS Panel (EFSA Panel on Food Additives and Nutrient Sources added to Food) . (2017). Scientific opinion on the re‐evaluation of potassium nitrite (E 249) and sodium nitrite (E 250) as food additives. EFSA Journal, 15(6), 157. 10.2903/j.efsa.2017.4786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA CEF Panel (EFSA Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids) . (2010). Guidance on the data required for the risk assessment of flavourings to be used in or on foods. EFSA Journal, 8(6), 38. 10.2903/j.efsa.2010.1623 [DOI] [Google Scholar]
- EFSA FAF Panel (EFSA Panel on Food Additives and Flavourings) . (2022). Scientific guidance on the data required for the risk assessment of flavourings to be used in or on foods. EFSA Journal, 20(12), 7673. 10.2903/j.efsa.2022.7673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee . (2009). Guidance of the scientific committee on transparency in the scientific aspects of risk assessments carried out by EFSA. Part 2: General principles. EFSA Journal, 7(7), 22. 10.2903/j.efsa.2009.1051 [DOI] [Google Scholar]
- EFSA Scientific Committee . (2017). Update: Use of the benchmark dose approach in risk assessment. EFSA Journal, 15(1), 41. 10.2903/j.efsa.2017.4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee . (2021). Guidance on technical requirements for regulated food and feed product applications to establish the presence of small particles including nanoparticles. EFSA Journal, 19(8), 6769. 10.2903/j.efsa.2021.6769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- FAO/WHO , 2008. Joint FAO/WHO Expert Committee on Food Additives. Sixty‐ninth meeting, Rome, Italy, 17–26 June 2008. Summary and conclusions. JECFA/69/SC, 21 pp. Issued 4 July 2008. Available online: https://www.fao.org/documents/card/en/c/c1dfe308‐c04e‐444d‐9885‐e2b20ef6bb07/
- Hearty, A. , Lau, A. , & Roberts, A. (2014). Chewing gum intake in Europe: A survey of intakes in France, Germany, Italy, Spain and the UK. Food Additives & Contaminants: Part A, 31(7), 1147–1157. 10.1080/19440049.2014.913104 [DOI] [PubMed] [Google Scholar]
- Marty, M. S. , Allen, B. , Chapin, R. E. , Cooper, R. , Daston, G. P. , Flaws, J. A. , Foster, P. M. D. , Makris, S. L. , Mylchreest, E. , Sandler, D. , & Tyl, R. W. (2009). Inter‐laboratory control data for reproductive endpoints required in the OPPTS 870.3800/OECD 416 reproduction and fertility test. Birth Defects Research (Part B), 86, 470–489. 10.1002/bdrb.20208 [DOI] [PubMed] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (1997). Test no. 471: Bacterial reverse mutation test. OECD guidelines for the testing of chemicals, section 4. 10.1787/9789264071247-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development) . (2016). Test no. 487: In vitro mammalian cell micronucleus test. OECD guidelines for the testing of chemicals, section 4. 10.1787/9789264264861-en [DOI]
- OECD (Organisation for Economic Co‐operation and Development) . (2018). Test no. 408: Repeated dose 90‐day oral toxicity study in rodents (OECD TG 408), in revised guidance document 150 on standardised test guidelines for evaluating chemicals for endocrine disruption. OECD Publishing. 10.1787/9789264304741-en [DOI] [Google Scholar]
- SCCS (Scientific Committee on Consumer Safety) . (2018a). SCCS notes of guidance for the testing of cosmetic ingredients and their safety evaluation 10th revision, 24–25 October 2018, SCCS/1602/18 . https://health.ec.europa.eu/system/files/2019‐02/sccs_o_224_0.pdf
- SCCS (Scientific Committee on Consumer Safety) . (2018b). Opinion on water‐soluble zinc salts used in oral hygiene products – Submission I, preliminary version adopted on 7 March 2017, final version adopted on 21–22 June 2018, SCCS/1586/17 . https://health.ec.europa.eu/system/files/2021‐08/sccs_o_207_0.pdf
- Slob, W. (2017). A general theory of effect size, and its consequences for defining the benchmark response (BMR) for continuous endpoints. Critical Reviews in Toxicology, 47(4), 342–351. 10.1080/10408444.2016.1241756 [DOI] [PubMed] [Google Scholar]