

Abstract
Biogenic monoamines—vital transmitters orchestrating neurological, endocrinal and immunological functions1–5—are stored in secretory vesicles by vesicular monoamine transporters (VMATs) for controlled quantal release6,7. Harnessing proton antiport, VMATs enrich monoamines around 10,000-fold and sequester neurotoxicants to protect neurons8–10. VMATs are targeted by an arsenal of therapeutic drugs and imaging agents to treat and monitor neurodegenerative disorders, hypertension and drug addiction1,8,11–16. However, the structural mechanisms underlying these actions remain unclear. Here we report eight cryo-electron microscopy structures of human VMAT1 in unbound form and in complex with four monoamines (dopamine, noradrenaline, serotonin and histamine), the Parkinsonism-inducing MPP+, the psychostimulant amphetamine and the antihypertensive drug reserpine. Reserpine binding captures a cytoplasmic-open conformation, whereas the other structures show a lumenal-open conformation stabilized by extensive gating interactions. The favoured transition to this lumenal-open state contributes to monoamine accumulation, while protonation facilitates the cytoplasmic-open transition and concurrently prevents monoamine binding to avoid unintended depletion. Monoamines and neurotoxicants share a binding pocket that possesses polar sites for specificity and a wrist-and-fist shape for versatility. Variations in this pocket explain substrate preferences across the SLC18 family. Overall, these structural insights and supporting functional studies elucidate the mechanism of vesicular monoamine transport and provide the basis to develop therapeutics for neurodegenerative diseases and substance abuse.
Biogenic monoamines, including catecholamines (dopamine, adrenaline and noradrenaline), histamine and serotonin, are essential neurotransmitters in the central and peripheral nervous systems, impacting motor control, cognition, memory, mood and sleep1,2. They also act as local transmitters and hormones in the endocrine and immune systems, regulating gastrointestinal movement3, blood pressure4 and immune responses5. Monoamines must be stored in secretory vesicles to maintain precise control of their quantal release into extracellular spaces, such as synaptic clefts, and to prevent their premature degradation or potential damage to neurons (Fig. 1a). VMATs uptake monoamines from the cytoplasm into storage vesicles6,7 and sequester neurotoxicants, such as 1-methyl-4-phenylpyridinium (MPP+), which is commonly used for modelling Parkinson’s disease10. VMATs have two isoforms that exhibit high sequence similarity but distinct tissue distributions: VMAT1 is mainly found in neuroendocrine cells in humans, whereas neurons and immune cells primarily express VMAT215,17. VMATs belong to the SLC18 family of human solute carriers, which contains a third member, the vesicular acetylcholine transporter (VAChT)18. These SLC18 proton antiporters accumulate a variety of single positively charged amines to high concentrations (up to 0.5 M) within the secretory vesicles, the acidic environment of which creates an outward proton gradient to drive the amine intake8,9. The mechanism of this remarkable storage ability remains unclear.
Fig. 1 |. Cryo-EM structures of the VMAT1 dimer in the ligand-free and reserpine-bound states.
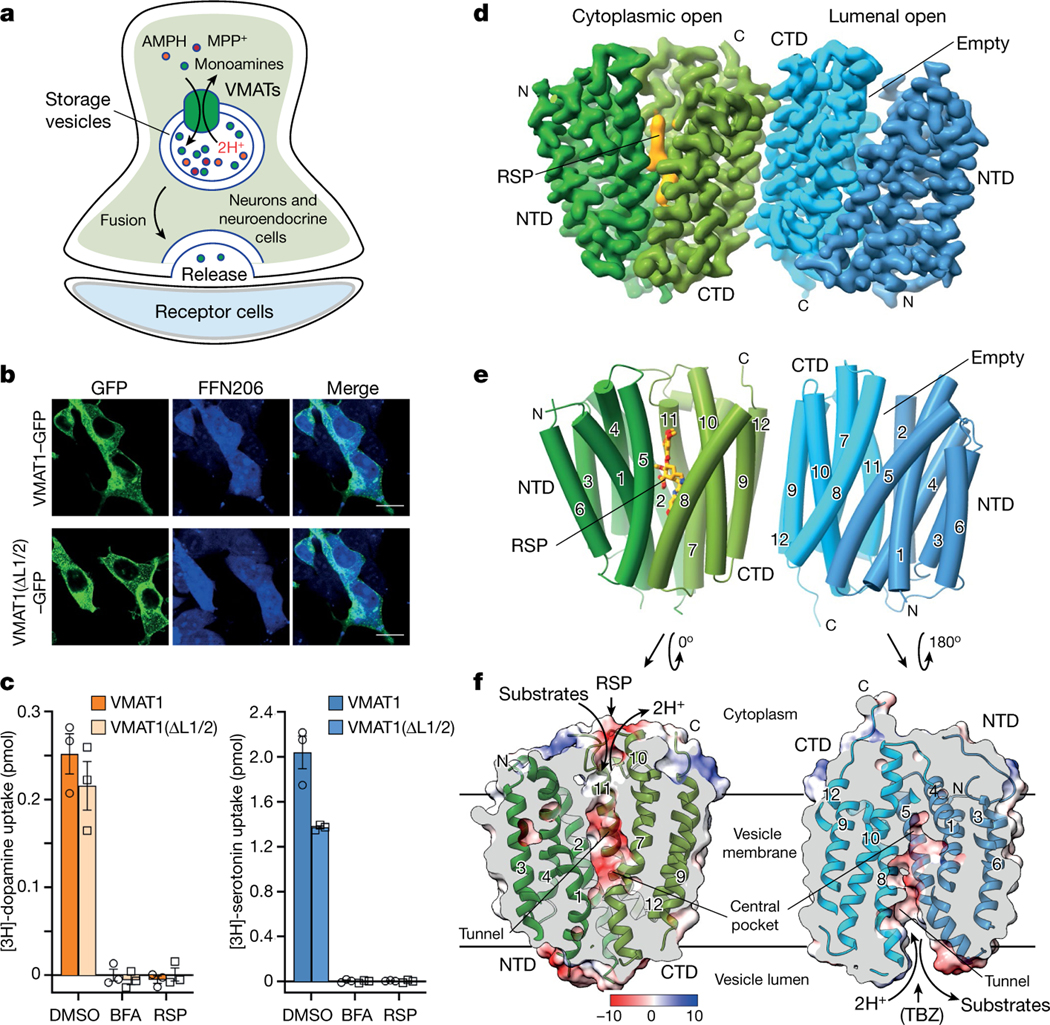
a, The cellular process of monoamine storage and release. VMATs accumulate various amine-containing compounds (coloured spheres) in secretory vesicles through proton (2H+) antiport. AMPH, amphetamine. MPP+, 1-methyl-4-phenylpyridinium. b, The subcellular location and FFN206 uptake of full-length VMAT1 and VMAT1(ΔL1/2). The experiments were repeated independently three times with similar results. Scale bars, 20 μm. c, Vesicular uptake of [3H]dopamine (left) and [3H]serotonin (right) with 5 × 105 cells. The activities of VMAT1 and VMAT1(ΔL1/2) are inhibitable by bafilomycin A (BFA), which collapses the proton gradient, and reserpine (RSP). Data are mean ± s.e.m. from tthree biological replicates. DMSO, dimethylsulfoxide. d, Cryo-EM density map of the VMAT1 dimer, coloured by the NTD and CTD in each monomer. Map contour level = 0.12 in ChimeraX. The expected orientation (cytoplasmic-open or lumenal-open) of each monomer in storage vesicles is indicated. e, The structure of the VMAT1 dimer (in the same orientation as shown in d). f, Electrostatic surface representation of the monomers, reoriented as in the vesicular membrane. The transition from the cytoplasmic-open (reserpine omitted) to the lumenal-open conformation is associated with proton export and monoamine import. TBZ, tetrabenazine.
The VMATs and their specific inhibitors have long-standing clinical importance in regulating and investigating neuronal and endocrine responses8. Reserpine is an antihypertensive and antipsychotic drug targeting both VMAT1 and VMAT2. The introduction of reserpine in the 1950s spurred the discovery of the roles of monoamines in the central nervous system and Parkinson’s disease1, “bridging the gap between biochemistry, psychiatry and neurology”11. Reserpine also marked the modern era of drug treatment for schizophrenia12, to which VMAT1 exhibits a strong genetic linkage19. Tetrabenazine derivatives, targeting VMAT2, are the only uS Food and Drug Administration (FDA)-approved treatments for Huntington’s chorea, tardive dyskinesia and Tourette’s syndrome13. In vivo imaging using radiolabelled tetrabenazine and vesamicol (targeting VAChT) has enabled the early detection and tracking of disease progression in Parkinson’s disease, Alzheimer’s disease, schizophrenia and drug addiction14. Notably, amphetamines can efficiently enter storage vesicles15 and displace stored catecholamines, prompting their synaptic release and inducing intense psychostimulation16. Despite the substantial clinical interest in treating drug addiction and neurodegenerative diseases, how these illicit drugs and therapeutic medications interact with VMATs remains unclear.
Here we report a series of cryo-electron microscopy (cryo-EM) structures of human VMAT1 in cytoplasmic-open and lumenal-open conformations, in unbound form and in complex with four monoamine neurotransmitters, the antihypertensive reserpine, the psychostimulant amphetamine and the neurotoxin MPP+. The structures and functional studies reveal the mechanisms of monoamine storage and pharmacological interactions, providing a framework for understanding disease associations and inspiring future development of targeted medications.
Structure determination
Human VMAT1, when expressed in HEK293 cells or Pichia pastoris, generates low levels of detergent-extracted protein or exhibits inhomogeneous elution profiles during fluorescence detection size-exclusion chromatography (FSEC) (Extended Data Fig. 1a). We overcame this issue by removing the poorly conserved glycosylated loop (amino acids 69–123) between the transmembrane helix 1 (TM1) and TM2 (hereafter L1/2 and so forth) of VMAT1. This deletion construct, VMAT1(ΔL1/2), shows a monodispersed FSEC peak and a considerable increase in protein yield (Extended Data Fig. 1a), enabling subsequent protein purification and structure determination. VMAT1(ΔL1/2) has a similar subcellular location to full-length VMAT1 (Fig. 1b) and largely retains the activities of importing [3H]dopamine, [3H]serotonin and a fluorescent false neurotransmitter, FFN206, into vesicle-like compartments of HEK293 cells20 (Fig. 1b,c and Extended Data Fig. 1b–e). Furthermore, VMAT1(ΔL1/2) transport requires a proton gradient and is inhibitable by reserpine, indicating preservation of VMAT1 functionality (Fig. 1c and Extended Data Fig. 1b). Consistently, L1/2 is deemed to be non-essential for VMAT function21 and most of it is absent in the related vesicular transporters SLC18B1, SLC17 and SLC32 that need to perform similar functions.
Cryo-EM analysis of VMAT1(ΔL1/2) protein particles exhibited high-resolution features during the two-dimensional (2D) classification stage (Extended Data Fig. 2a). However, extensive attempts at three-dimensional (3D) reconstruction produced only low-resolution maps (around 6 Å) owing to the small 52 kDa monomeric state and the absence of extramembrane domains that could help to resolve the signal ambiguities. Notably, with the classical inhibitor reserpine added before cell membrane disruption, VMAT1(ΔL1/2) appeared with a higher molecular mass on size-exclusion chromatography (Extended Data Fig. 2b,c). Subsequent 2D classifications revealed the formation of dimeric species (Extended Data Fig. 2a), and the best 3D class yielded a 3.5-Å-resolution map (Fig. 1d, Extended Data Figs. 3 and 4 and Extended Data Table 1). In this map, the densities of almost all regions and most side chains are unambiguous (Extended Data Fig. 3d,e). The reserpine density is clearly resolved at the centre of one monomer, whereas the second monomer remains unoccupied and, together, they form an antiparallel dimer with respect to the membrane plane (Fig. 1d–f). This dimeric assembly, possibly generated during membrane solubilization (Supplementary Discussion), is unlikely to be physiological as VMAT1 typically adopts a single orientation with both termini at the cytoplasmic side of storage vesicles7. Nevertheless, the reserpine-bound monomer exhibits an open conformation towards the cytoplasmic side (Fig. 1e,f, left), in agreement with previous studies showing that high-affinity binding of reserpine requires a cytoplasm-facing conformation of VMATs induced by the proton efflux, a process occurring also during monoamine import into the storage vesicles9,22. By contrast, without a proton gradient, VMATs adopt a conformation open to the lumen of storage vesicles9; this resting state is represented by the lumenal-open conformation of the second, unoccupied monomer (Fig. 1e,f, right). Moreover, in another high-resolution 3D class, reserpine is bound by both monomers in the cytoplasmic-open conformation (Extended Data Fig. 5a,b), indicating that the reserpine-induced dimerization can include alternative conformations (an explanation is provided in the Supplementary Discussion). Fortuitously, the antiparallel dimer enables simultaneous capture of the ligand-free and reserpine-bound conformations of VMAT1.
Alternative gating and reserpine binding
VMAT1 adopts a canonical major facilitator superfamily (MFS) fold with 12 transmembrane helices divided into the N-terminal (NTD) and C-terminal (CTD) domains, each containing two inverted repeats of three helices23. In the absence of ligands, the first two transmembrane helices of each repeat splay apart at the lumenal side of VMAT1, creating a narrow entry tunnel to a deep cavity that extends below the midpoint of the membrane (Fig. 1f). The bottom part of this cavity is negatively charged (Fig. 1f), reflecting the function of VMAT1 in transporting positively charged monoamines. This lumenal-open conformation, representing the state after monoamine release or before protonation-induced antiport (Fig. 2a), is stabilized by extensive interactions between the NTD and CTD (Fig. 2b and Supplementary Table 1), namely the ‘gating interactions’ that regulate conformational changes during the transport cycle23. The cytoplasmic end of TM5 engages with those of TM8 and TM10 and their following loops, establishing several polar interactions, including salt bridges, Asp222–Arg365 and Asp419–Arg225 (hydrogen bonded to Tyr426), that are often crucial for the gating of MFS transporters24. Additional gating residues near the central cavity, including Met229 and Tyr430, form non-polar interactions along the NTD–CTD interface. Consistently, VMAT2 studies show that mutating Arg225, Met229, Tyr426 and Tyr430 (as numbered in human VMAT1) induces the transporter towards the cytoplasmic-open state25. The NTD–CTD association is further stabilized by interactions from the connecting L6/7, which wraps around the L2/3 and binds into a groove formed between L2/3 and L10/11 (Fig. 2b). Collectively, these interactions are unique to the lumenal-open conformation and confer high stability of this state.
Fig. 2 |. Alternative conformations stabilized by gating interactions and reserpine binding.
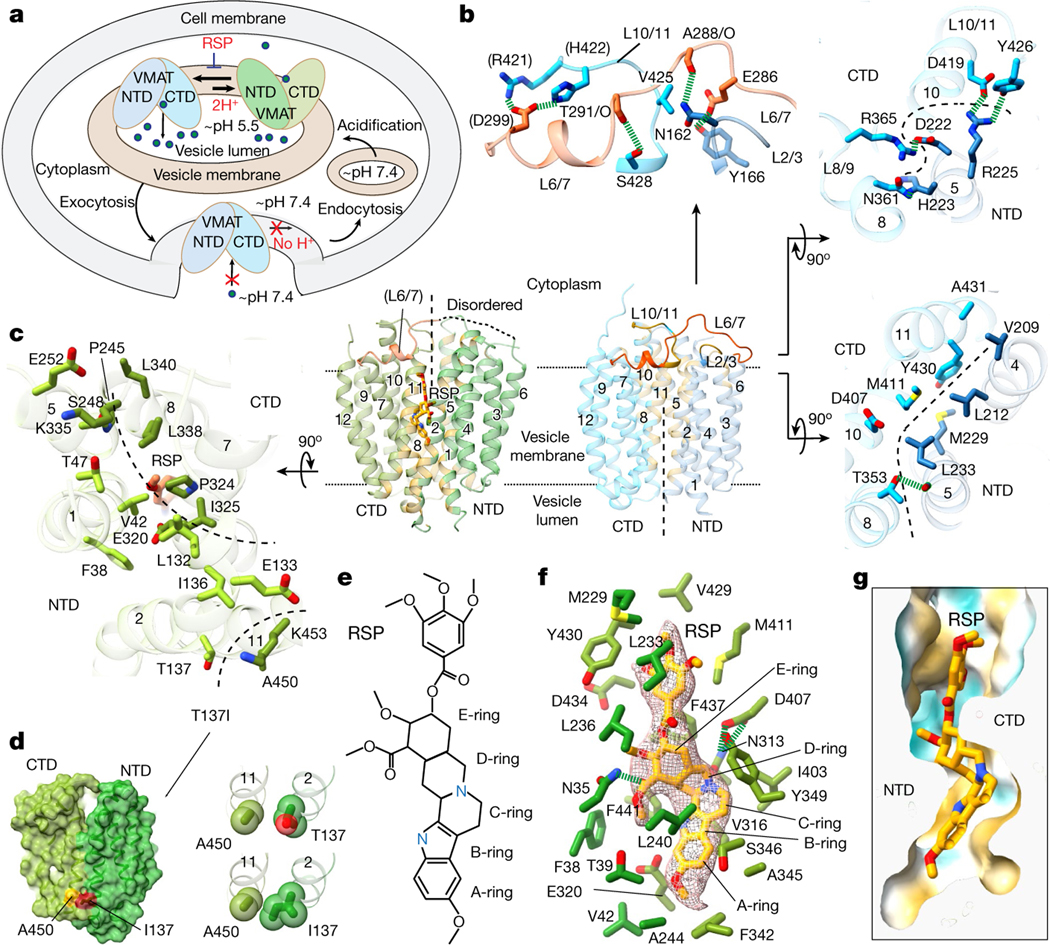
a, Cellular regulation by the alternative states. VMATs preferably adopt a lumenal-open conformation. Protonation in the acidic vesicular lumen facilities the rate-limiting transition to the cytoplasmic-open state, which binds to reserpine. After exocytosis and vesicle fusion with the plasma membrane, VMATs are trapped in an outward-open state and deactivated in the absence of a proton gradient. Endocytosis and acidification generate mature storage vesicles. b, Extensive gating interactions in the lumenal-open conformation. Interactions are formed by the loop between TM6 and TM7 (L6/7) (top), at the cytoplasmic end (top right) and near the central pocket (bottom right). The transmembrane helices and residues in the NTD and CTD are coloured as in Fig. 1, with L6/7 in red. Residues in parentheses indicate non-unique interactions to this conformation. The dashed curves indicate the NTD–CTD interface. In the overall structures, NTD–CTD-contacting regions are coloured in yellow. c, Gating interactions in the cytoplasmic-open conformation. d, The Ile137–Ala450 interaction in the Thr137Ile variant potentially favours NTD–CTD association in the cytoplasmic-open state (left). Right, the impact of this substitution is illustrated with van der Waals spheres. e, The chemical structure of reserpine. f, Densities and binding interactions of reserpine. Map contour level = 0.45 in ChimeraX. g, Complementarity of shape and hydrophobicity between reserpine and the central pocket. The surface is coloured by hydrophobicity; yellow and cyan indicate the most hydrophobic and hydrophilic regions, respectively.
In the reserpine-bound monomer, these extensive gating interactions are lost, including the stabilization by L6/7, as its N-terminal portion becomes disordered (Fig. 2b,c). The pore-lining transmembrane helices rotate to open at the cytoplasmic side, while, at the lumenal side, these transmembrane helices form alternative gating interactions between the NTD and CTD (Fig. 2b,c and Supplementary Table 1). Among the lumenal gating residues, Thr137Ile is a common missense variant of VMAT1 that has been linked to increased risks of schizophrenia, bipolar and anxiety disorders26. Brain activities in affected individuals suggest enhanced monoamine signalling27, consistent with the hyperactivity of monoamine transport of the Thr137Ile variant28. Structurally, the substitution of Thr to a non-polar and larger Ile side chain may promote an interaction with Ala450 (Fig. 2d). This interaction may enhance NTD–CTD association in the cytoplasmic-open state and facilitate the rate-limiting transition9 to this state (Supplementary Discussion), thereby increasing the transport activity. Notably, the cytoplasmic-open conformation has only about half as many gating interactions compared with the lumenal-open conformation (Supplementary Table 1). This disparity maintains VMAT1 preferably in the lumenal-open state9—a key control mechanism ensuring that the rocker-switch transition of VMAT1 transport occurs only within the membrane of storage vesicles possessing a proton gradient (Fig. 2a). Conversely, when storage vesicles fuse with the cell membrane to release monoamines, the lack of a proton gradient across the cell membrane deactivates VMAT1 to prevent unwanted recovery of monoamines.
Despite relatively weak gating interactions, the cytoplasmic-open conformation interacts extensively with the sizable reserpine molecule. Reserpine is a complex indole alkaloid featuring a pentacyclic core that consists of a methoxyindole (rings A and B) fused with a tetrahydropyridine (ring C) and a perhydroisoquinol-3-one (rings D and E), with this core further linked through an ester to a trimethoxybenzene (Fig. 2e). The methoxyindole group of reserpine reaches to the bottom of the central cavity, with the methoxy group interacting with Phe342 and the indole ring surrounded by both non-polar and polar residues, including Leu240, Val316 and Glu320 (Fig. 2f). Notably, Glu320 is surrounded by non-polar groups and is possibly protonated to form stable interactions. Rings C–E are situated further up the cavity, surrounded by several aromatic and aliphatic residues, along with the hydrogen-bonded Asn313–Asp407. Moreover, Asn35 binds to the ester group on the E ring. At the upper region, the trimethoxybenzene is surrounded by Tyr430, Leu233 and other aliphatic residues. Reserpine binding buries a 630 Å2 area and occupies the narrow gap along the central cavity (Fig. 2g). Both the NTD and CTD sides of this gap contain hydrophobic patches that match with the molecular shape of reserpine (Extended Data Fig. 5c), resulting in a substantial hydrophobic effect because the central cavity is otherwise aqueously exposed. Combining hydrophobic interactions and shape complementarity, reserpine acts as a tight-binding inhibitor29,30 for the cytoplasmic-open conformation.
Conformational changes between the reserpine-bound state and the apo state are primarily relative rigid-body rotations of the NTD and CTD (Extended Data Fig. 5d–g). However, TM11 is bent at its C-terminal in the lumenal-open conformation, whereas it transitions into a straight helix and rotates by 25° in the cytoplasmic-open conformation. Similarly, the N-terminal region of TM5 slightly rotates and its C-terminal region partly loses its helical structure. TM5 and TM11 are repeat helices that both contain multiple conserved Gly residues (Extended Data Fig. 6), and their bending or rotation is commonly observed during the rocker-switch transition in drug/antiporter family number 1 (DHA-1), an MFS subfamily that includes SLC18 members31. Thus, the reserpine-bound conformation may resemble a cytoplasmic-open state during the monoamine transport.
Recognition of monoamines
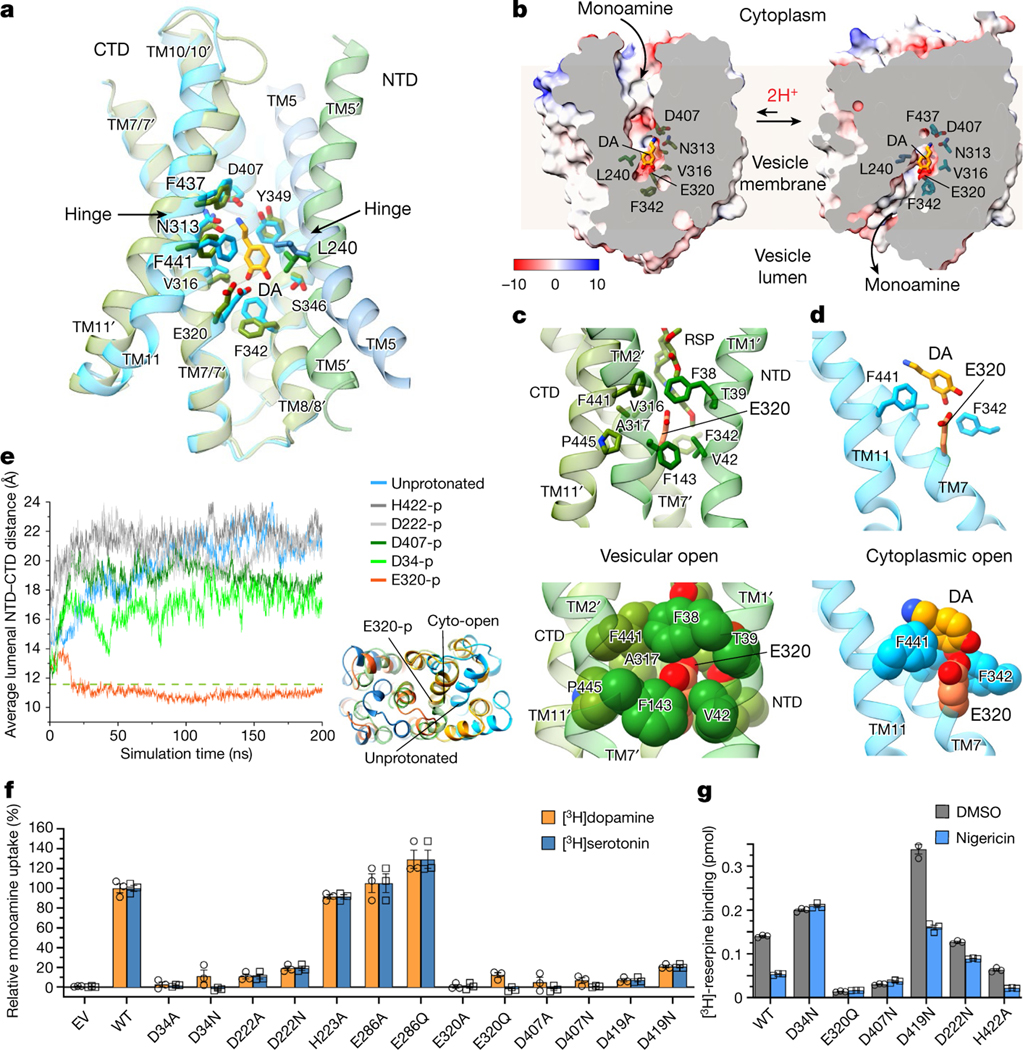
To understand the recognition and transport mechanisms of different monoamines, we determined a series of cryo-EM structures of VMAT1(ΔL1/2) complexed with four monoamines: dopamine, noradrenaline, serotonin and histamine (Extended Data Fig. 4c–f and Extended Data Table 1). These structures represent a state before substrate release, as the monoamines are bound to the lumenal-open monomer, which adopts essentially the same conformation as the unbound form (Extended Data Fig. 7a,b). The densities of these monoamines are clearly resolved (Fig. 3a–f), showing their binding within the same pocket that is situated on the side wall of the central cavity (Fig. 3a). This binding pocket mainly resides in the CTD, surrounded by TM7, TM8 and TM11, but maintains contact with TM5 of the NTD. Thus, this off-centre configuration conforms to the canonical MFS mechanism that substrate binding requires interactions from both domains23. As MFS transporters typically use a single substrate pocket for transport23,32, this pocket is most likely retained in the cytoplasm exposed, monoamine-import state of VMAT1.
Fig. 3 |. Recognition mechanism of monoamines.

a, The monoamine-binding site. The representative dopamine (DA) molecule is shown with a density map, and the overall structure is shown as a surface representation. b, The substrate pocket. Left, surface representation (orange) showing the wrist-and-fist shape of this pocket. Right, cartoon showing monoamine-binding residues and types of interactions. c–f, The binding interactions and densities of various monoamines (dopamine (c), noradrenaline (NE; d), serotonin (SERT; e) and histamine (HA; f)). The contour levels range from 0.06 to 0.1 in ChimeraX. Hydrogen bonds and salt bridges are depicted as green and orange dashed lines, respectively. Residues in parentheses do not interact with serotonin or histamine. Chemical structures are provided below. g, The [3H]dopamine- and [3H]serotonin-uptake activities of mutants (normalized to the wild type) of pocket-forming non-polar residues. 100% uptake corresponds to 0.187 pmol per 5 × 105 cells for dopamine and 0.402 pmol per 5 × 105 cells for serotonin. h, The activities of mutants at the two polar sites. 100% uptake corresponds to 0.123 pmol per 5 × 105 cells for dopamine and 0.444 pmol per 5 × 105 cells for serotonin. For g and h, data are mean ± s.e.m. from three biological replicates. EV, empty vector; WT, wild type.
The shape of this pocket resembles a fist and wrist (Fig. 3b), sculpted to bind to monoamines with various aromatic rings linked through a two-carbon side chain to the amine group (Fig. 3c–f). The fist region is capped below by Phe342, flanked by Glu320 and Ser346, and surrounded above by Val316 and Leu240, the latter of which is the only NTD residue involved in substrate binding. These residues together create a space that is capable of accommodating one or two aromatic rings, while permitting these rings to rotate or shift position in response to specific interactions (Extended Data Fig. 7c–e). The pocket then narrows towards the wrist region, constrained by Ile403 and the large side chains of Tyr349 and Phe441, to loosely grasp the two connecting carbons and position the terminal amine group for polar interactions. The top end of this pocket is defined by Phe437. Mutations of these non-polar residues to enlarge this pocket substantially decrease the activities of [3H]dopamine and [3H]serotonin uptake (Fig. 3g): Phe342Ala, Val316Ala and Leu240Ala at the fist region are inactive, and Ile403Ala and Phe441Ala at the wrist region exhibit around 20% activity compared with the wild type. Beyond these regions, Phe437Ala is tolerated for the transport activities. Overall, this binding pocket is optimized for the small aromatic ring and two-carbon side chain of monoamines, while excluding a wide array of other amine-containing molecules, such as alkaloids or polyamines, from being transported by VMATs into the storage vesicles.
The amine group of the monoamines forms a specific interaction network with a triad of polar residues: Asn313, Tyr349 and Asp407 (Fig. 3b–f). The positively charged amine group interacts electrostatically with Asp407 and donates hydrogen bonds to the Asn313 carbonyl, Asp407 carbonyl and/or Tyr349 hydroxyl. The two carbonyls of Asp407 accept hydrogen bonds from the Asn313 amide and Tyr349 hydroxyl. This network, by satisfying the charged groups and multiple hydrogen donors and acceptors within a hydrophobic environment, energetically favours monoamine binding. Alterations in this triad, including minor changes of Tyr349Phe, Asp407Asn and Asn313Asp, result in the loss of serotonin and dopamine uptake activities (Fig. 3h). These mutations also substantially lower the binding of FFN206 (Extended Data Fig. 7f), except that Asp407Glu is more tolerated for binding and activities. Other specific interactions with monoamines vary by their unique chemical structures. For the catecholamines, dopamine and noradrenaline, the two hydroxyl groups on their catechol rings interact with Glu320 and Ser346, while the extra hydroxyl group on the noradrenaline side chain interacts with the Asn313 carbonyl (Fig. 3c,d). The binding interactions of adrenaline are likely to be similar as it differs from noradrenaline only by an N-methyl group. For serotonin, the single hydroxyl group on its indole ring donates a hydrogen bond to Glu320, and its pyrrole nitrogen interacts with Ser346 (Fig. 3e). Consistently, the Glu320 and Ser346 mutations abolish activities of both dopamine and serotonin transport (Fig. 3h) and interfere with FFN206 binding (Extended Data Fig. 7f). However, the small imidazole ring of histamine lacks interactions with Glu320 and Ser346 (Fig. 3f), consistent with its low apparent affinity for VMAT1 compared with other monoamines15,17,33. Together, the amine group and ring hydroxyls of the monoamines are specifically recognized by the two polar sites within this binding pocket.
Neurotoxicants bind to substrate pocket
To understand how psychostimulants are taken up into storage vesicles, we determined the structure of VMAT1(ΔL1/2) with amphetamine (Extended Data Fig. 4g and Extended Data Table 1). Amphetamine binds to the same VMAT1 pocket as monoamine neurotransmitters (Fig. 4a and Extended Data Fig. 7g). Amphetamine chemically differs from dopamine only by the lack of hydroxyl substitutions on the phenyl ring, as well as having a characteristic α-methyl substitution in the side chain. As this α-methyl group interacts with Tyr349, the amine group of amphetamine rotates to form hydrogen bonds exclusively with Asn313, yet maintains electrostatic interaction with Asp407 (Fig. 4a and Extended Data Fig. 7g). The phenyl ring also rotates to fit between Phe342 and Tyr349, but does not form polar interaction with Glu320 due to the lack of ring hydroxyls. This structure provides direct evidence that VMAT1 binds to amphetamine in a similar manner to monoamines, confirming that amphetamine is an actual VMAT substrate16. Amphetamine-related psychostimulants, despite their diverse chemical structures, share a common α-methyl-phenethylamine motif (Fig. 4b). Given the substrate-binding plasticity (Fig. 3 and Extended Data Fig. 7c–e,g), we predict that VMATs can transport most of these psychostimulants, such as methamphetamine and 3,4-methylenedioxymethamphetamine (MDMA) (Fig. 4b). Once accumulated in storage vesicles, they displace the stored catecholamines to promote their synaptic release, leading to substantial psychostimulation16.
Fig. 4 |. Binding of neurotoxicants in the substrate pocket.

a, Binding interactions and density map of amphetamine. Contour level = 0.02 in ChimeraX. Hydrogen bonds and electrostatic interactions are shown as green and red dashed lines, respectively. Residues shown in parentheses do not interact with amphetamine. b, The chemical structures of representative amphetamine-related illicit and clinical drugs transportable by VMATs. Methamphetamine (METH) and amphetamine are traditionally abused substances. Ephedrine, used clinically to treat significant hypotension, is a precursor in the illicit synthesis of methamphetamine. Ring-substituted amphetamines are represented by MDMA (also known as ecstasy). c, Binding interactions and density map of MPP+. Contour level = 0.08 in ChimeraX.
As a substrate of VMATs, MPP+ is the active metabolite of the neurotoxin N-methyl-4-phenyltetrahydropyridine (MPTP), causing symptoms closely mimicking Parkinson’s disease10,15. The structure of VMAT1(ΔL1/2) with MPP+ reveals that it binds to the same pocket as monoamines (Fig. 4c, Extended Data Figs. 4h and 7h and Extended Data Table 1). However, in contrast to the monoamines, the amine group of MPP+ is permanently charged and situated within the pyridinium ring. This quaternary amine cannot serve as a hydrogen donor but retains electrostatic interaction with Asp407, which preserves hydrogen bonding with Asn313 and Tyr349. The pyridinium ring shifts towards the top of the pocket, snugly fitting between Tyr349 and Phe441 in the wrist region (Fig. 4c and Extended Data Fig. 7h). The 1-methyl side group of MPP+ interacts with Phe437, and its 4-phenyl group binds at the fist region to Phe342 and Ala345. These altered interactions highlight the structural adaptability of VMATs to transport amine-containing neurotoxins and environmental toxicants, sequestering them in storage vesicles to protect against neurotoxicity and degeneration10.
Monoamine storage by proton antiport
Notably, the substrate-binding pocket observed in the lumenal-open conformation is preserved in the cytoplasmic-open conformation bound by reserpine (Fig. 5a). Thus, this cytoplasmic-open conformation may resemble a conformation for substrate import34, which is expected to be cytoplasmic-open and maintain the same substrate pocket23,32. Consistently, biochemical data show that reserpine recognizes a protonation-induced, cytoplasmic-open conformation, as presented during the transport cycle9,22. When the monoamine-bound and reserpine-bound structures are superimposed by the CTD, the TM7, TM8 and TM10 forming this pocket, as well as the substrate-binding residues on these transmembrane helices, retain their conformations across the alternative states (Fig. 5a). Notably, although TM11 rotates between the two conformations, the rotation hinges around Phe437 and Phe441. Thus, the positions of these residues are effectively maintained, thereby preserving the integrity of the substrate pocket. Similarly, the hinge point of TM5 rotation is at Leu240, the only CTD residue involved in substrate binding. The two polar sites, the Asn313–Tyr349–Asp407 triad and Glu320–Ser346, maintain their respective interactions to the amine group and ring hydroxyls of the superimposed monoamines, ensuring the same monoamine binding orientation during the rocker-switch transition (Fig. 5b).
Fig. 5 |. Monoamine and proton antiport mechanism.
a, Conservation of the monoamine-binding pocket across the alternative conformations. The lumenal-open structure (coloured in blue) and cytoplasmic-open structure (green; transmembrane helices labelled with a prime) are superimposed by the CTD. The dopamine molecule is shown as presented in the lumenal-open state. b, Alternative opening of the pocket for monoamine transport. The superimposed structures (surface representation) and dopamine are same as in a. c, Glu320 protonation is favoured for interactions in the cytoplasmic-open state. Top, residues and transmembrane helices surrounding Glu320. Bottom, Glu320 and interacting residues shown in van der Waals spheres. d, In the lumenal-open state, deprotonated Glu320 interacts with monoamine and is exposed to water. e, Molecular dynamics simulation (initiated from the lumenal-open state) reveals lumenal NTD–CTD association induced by Glu320 protonation. The average NTD–CTD distances at the lumenal side were compared for candidate protonation sites (-p) and with that of the cytoplasmic-open (reserpine-bound) state (dashed line). Inset: representative conformations from the molecular dynamics simulation showing the similarity of the lumenal NTD–CTD association between Glu320-p (orange) and the cytoplasmic-open state (green), and the difference compared with the unprotonated state (blue). All of the molecular dynamics simulations were repeated at least three times. f, The [3H]dopamine- and [3H]serotonin-uptake activities of mutants at potential protonation sites. 100% uptake corresponds to 0.129 pmol per 5 × 105 cells for dopamine and 0.434 pmol per 5 × 105 cells for serotonin. g, Reserpine binding and proton-gradient dependency (collapsed by nigericin) of neutralizing mutants at candidate protonation sites. For f and g, data are mean ± s.e.m. from three biological replicates.
The transport of one monoamine molecule by VMATs is driven by the binding and release of two protons, an antiport mechanism enabling approximately 10,000-fold enrichment of the monoamines in storage vesicles9. A comparison of the two VMAT1 conformations revealed a substantial change in the local environment of Glu320; this critical residue is involved in both substrate and reserpine binding (Figs. 2 and 3 and Extended Data Fig. 7i) and its mutations abolish VMAT1 activity (Figs. 3h and 5f). In the cytoplasmic-open state, Glu320 (TM7) may require protonation to form knob-and-hole interactions with Phe38 and Val42 in TM1, Phe143 in TM2 and Phe441 in TM11, thereby favouring the NTD–CTD association between these transmembrane helices (Fig. 5c). Conversely, Glu320 in the lumenal-open state needs to be deprotonated to bind to the ring hydroxyls of monoamines (Fig. 3c–f). When monoamines lack these ring hydroxyls or are not bound, Glu320 is exposed to water (Fig. 5d). Thus, Glu320 does not contribute to NTD–CTD association in the lumenal-open state (Extended Data Fig. 8a). This alteration of transmembrane interactions suggests that Glu320 protonation may facilitate the rocker-switch transition during monoamine transport. Consistently, molecular dynamics simulation, initiated from the lumenal-open conformation, shows that Glu320 protonation induces NTD–CTD association at the lumenal side (Fig. 5e, Extended Data Fig. 8c and Supplementary Video 1). This lumenal NTD–CTD association is similar to that observed in the cytoplasmic-open state (Fig. 5e), and may therefore serve as an initial step for the cytoplasmic-open transition.
Previous studies have suggested not only Glu32025,35,36 but also Asp34 (refs. 25,35), His422 (ref. 37) or Asp407 (ref. 38) as potential protonation sites. For example, the D407E mutation in rat VMAT1 exhibits a steep and shifted pH profile38 for monoamine transport, indicating an altered pKa 9. Moreover, the VMAT1 structures reveal several protonatable gating residues as potential candidates (Fig. 2b), among which mutants of Asp222 and Asp419 are nearly inactive (Fig. 5f). However, the cryo-EM structures show that most of these residues, including Asp419, Asp222 and His422, are located at the cytoplasmic surface and not exposed to the vesicle lumen at both conformational states (Extended Data Fig. 8a), and therefore cannot independently translocate a proton. Importantly, only Asp34, Glu320 and Asp407 are situated at alternatively exposed hinge regions. Molecular dynamics simulations show that Asp34 protonation induces relatively close luminal NTD–CTD association, whereas protonation of other residues shows minor differences compared with unprotonated VMAT1 (Fig. 5e). Notably, Asp34 does not directly participate in gating interactions, but interacts exclusively within the NTD and with different surrounding residues between alternative conformations (Extended Data Fig. 8b). To further investigate these candidate sites, we assessed whether neutralizing mutants mimicking protonation could induce the cytoplasmic-open state recognizable by reserpine25 (Fig. 5g). We found that the D34N mutation in VMAT1 enables reserpine binding independent of a proton gradient, similar to the observation for VMAT225. By contrast, the reserpine binding of the D419N, D222N and H422A mutants changes with the proton gradient. However, this experiment cannot assess Glu320 and Asp407, because their mutations disrupt reserpine binding even in the presence of a proton gradient. Taken together, these experimental and computational data suggest that Asp34 and Glu320, both conserved in eukaryotic SLC18 homologues (Extended Data Fig. 6), may serve as the two protonation sites in VMAT1.
On the basis of the structural and biochemical evidence, we propose the following proton antiport mechanism (Supplementary Video 2). The lumenal-open state, stabilized by extensive gating interactions (Fig. 2b), is an energetically favoured state. However, protonation at vesicular acidic pH can destabilize this state, triggering alternative transmembrane associations and facilitating the transition to the cytoplasmic-open state. After exposure to neutral pH, the deprotonation and substrate binding triggers a return to the lumenal-open state, while the monoamines remain bound within the same pocket (Fig. 5b). Subsequent protonation releases the monoamine and induces the cytoplasmic-open transition once again. Notably, the two putative protonation sites, Asp34 and Glu320, may have different roles during this process, as only Glu320 is directly involved in the substrate binding (Extended Data Fig. 8a). In this antiport model, protonation and substrate binding are mutually exclusive events, consistent with the general mechanism of MFS proton antiporters39. As the transition to the cytoplasmic-open state requires protonation, monoamines cannot be bound and exported. Even when monoamine is bound in the deprotonated state of the lumenal-open state, export is disfavoured due to the relatively high stability of this state. Thus, VMATs import monoamines nearly unidirectionally, resulting in their substantial accumulation within storage vesicles.
Discussion
The dominant monoamine import and the protonation-precluded export constitute an effective mechanism for monoamine enrichment within storage vesicles (Supplementary Video 2). The import process proceeds through a favoured transition from the cytoplasmic-open to the lumenal-open state (Supplementary Discussion), governed by the disparity of conserved gating interactions (Fig. 2b,c and Supplementary Table 1) and substrate-binding energy. Conversely, unintended export during transition to the cytoplasmic-open state is prevented by the protonation40 that facilitates the transition but disrupts the substrate-binding site39. This disruption is also crucial to substrate release because the high monoamine concentration in storage vesicles necessitates a high dissociation constant9,41. Even with protonation, cytoplasmic-open transition remains the rate-limiting step throughout the transport process—the substrate-transport Michaelis constant (Km) is 10–100 times lower than the substrate-binding dissociation constant (Kd), indicating that the substrate-occupied import (Km) is much faster than the reappearance of an empty site (Kd) through the transition9. The proton gradient across the vesicular membrane is established by the V-type H+-ATPase that acidifies the vesicle lumen and builds up a positive electric potential inside. Although monoamine transport by VMATs is driven by the proton gradient rather than the charge gradient21, the exchange of two H+ for one monoamine molecule, carrying a single positive charge, is energetically favourable as this process dissipates the charge buildup within vesicles. Eventually, an electrochemical equilibrium may be reached, thereby setting a limit to the monoamine concentration within the vesicles. Structurally, a notable aspect of the rocker-switch transition is the strategic use of critical substrate-binding residues as hinge points for TM5 and TM11 rotation, preserving the structural integrity of the binding pocket (Fig. 5a,b). This proton-antiport mechanism is probably shared by other SLC18 family members, VMAT2 and VAChT, as they have identical proton-substrate stoichiometries, highly similar pKa values at protonation sites and consistent transport kinetics9.
The first VMAT1 structures reveal how vesicular transporters balance specificity and promiscuity in recognizing positively charged amines of various chemical structures. The binding-pocket architecture in VMAT1 is tailored to monoamines, with the wrist-and-fist shape accommodating diverse aromatic rings, invariant two-carbon side chains, and polar sites recognizing ring hydroxyls and the amine group (Fig. 3). This architecture also enables the transport of non-natural substrates including neurotoxins and psychostimulants. Variations of this pocket may explain the acetylcholine specificity of VAChT, which shares 52% sequence similarity with VMAT1. Homology modelling of VAChT shows that residues at the lower portion of the VMAT1 substrate pocket, Val316, Phe342 and Ser346, are replaced by Leu305, Trp331 and Phe335 in VAChT, respectively (Extended Data Fig. 9). These larger side chains may narrow the fist region for VAChT to grasp the aliphatic chain of acetylcholine, in contrast to the larger fist of VMATs designated for the aromatic ring of monoamines (Fig. 3). Consistent with this model, systematic mutagenesis of VAChT identified Trp331, Phe335, and the nearby Ala334 and Cys391, as substrate-binding residues41,42. As to VMAT2, this isoform shares 78% sequence similarity to VMAT1 and is specifically inhibited by tetrabenazine. Conformationally sensitive mutations in VMAT2 lead to tetrabenazine resistance43, whereas chimeric constructs of VMAT1 possessing TM5–TM8 and TM9–TM12 from VMAT2 exhibit tetrabenazine sensitivity44. Thus, the difference in the structural flexibility of these transmembrane regions, along with variations in the substrate-binding pocket45, may account for the distinct tetrabenazine sensitivity of these VMAT isoforms.
Structural elucidation of the SLC18 family provides the foundation to innovate pharmaceutical agents for neurodegenerative conditions, optimize neurological imaging and combat psychostimulant abuse. Unique intervention opportunities arise from dual targeting of the alternative conformations of VMATs by specific inhibitors with distinct pharmacokinetic properties—a rare situation among transporters32. Reserpine competitively inhibits both VMAT isoforms46 by binding at their cytoplasmic-open state with near-irreversible affinity, resulting in prolonged pharmaceutical effects47. To achieve such high affinities, future drug development should consider designing sizable molecules that align with the hydrophobic patches within the narrow gap of the central cavity (Fig. 2g and Extended Data Fig. 5c). Moreover, compounds with reduced side effects may be developed to specifically target each VMAT isoform based on their distribution of hydrophobic patches and unique hydrophilic interactions. By contrast, tetrabenazine non-competitively inhibits VMAT2 by binding on the vesicular lumen side through allosteric modulation25, but its rapid metabolism necessitates the ongoing pursuit of long-acting alternatives48. The structures may also facilitate the development of lobeline-like compounds that inhibit VMATs to decrease amphetamine-evoked dopamine release49, offering the potential to treat psychostimulant abuse. Moreover, leveraging alternative transitions, as seen with the Thr137Ile variant, could lead to the long-desired discovery of positive VMAT modulators that enhance dopamine storage, thereby treating Parkinson’s disease and other neurological conditions that are characterized by dopamine decline50. Overall, owing to the critical roles of the SLC18 family in neurotransmission and endocrine regulation, our comprehensive structural understanding of this family opens routes for therapeutic interventions.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-024-07290-7.
Methods
Constructs
The coding sequence of human VMAT1 (UniProt: P54219) contained in a pDONR-221 plasmid was acquired from DNASU and was used as the template for subcloning. For expression in P. pastoris, VMAT1 was cloned into a modified pPICZ-B vector (Invitrogen) under a methanol-inducible promoter. The expression construct contains a C-terminal PreScission protease cleavage site, followed by green fluorescent protein (GFP) and a Flag tag. For expression in HEK293T cells, VMAT1 was cloned into a modified pBudCE4.1 vector also comprising the C-terminal PreScission, GFP and Flag tag. To enhance protein stability, a series of deletion constructs at the N-terminal, C-terminal and loops between transmembrane helices were generated using standard PCR-based methods. VMAT1 mutations for uptake and binding assays were generated similarly. All nucleotide sequences were verified by DNA sequencing.
Protein expression and FSEC analysis
The plasmids were linearized by PmeI and transformed into P. pastoris by electroporation. Transformants were selected by Zeocin resistance on yeast extract peptone dextrose medium with sorbitol (YPDS) agar plates. The expression levels and elution profiles of resistant clones were compared through FSEC analysis. In brief, the Pichia cells were disrupted using a mixer mill and the cell membrane was dissolved in 2% n-dodecyl-β-D-maltoside (DDM, Anatrace) with 150 mM NaCl, 1 mM EDTA, 50 mM Tris-HCl, pH 8.0 for 1 h. After removing cell debris, the supernatant was injected into a Superose 6 increase 10/300 GL size-exclusion column (24 ml bed volume) pre-equilibrated in a buffer containing 0.05% DDM, 150 mM NaCl and 50 mM Tris-HCl, pH 8.0. The elution profile was monitored on the basis of GFP fluorescence (480 nm excitation and 520 nm emission) using the Shimadzu FSEC system. Constructs with monodispersed elution profiles and high expression levels were stored at −80 °C.
For large-scale protein expression, Pichia cells were grown in the buffered minimal glycerol medium (1.2% glycerol, 0.34% yeast nitrogen base, 1% ammonium sulfate, 0.4 mg ml−1 biotin and 100 mM potassium phosphate, pH 6.0) at 30 °C for 24 h. The growth medium was replaced with buffered minimal methanol medium (0.34% yeast nitrogen base, 1% ammonium sulfate, 0.4 mg ml−1 biotin and 200 mM potassium phosphate, pH 6.0), and protein expression was induced with 0.7% methanol. After 2 days at 25 °C, the cells were collected by centrifugation and flash-frozen in liquid nitrogen.
Protein purification
Frozen Pichia cells (70 g) were milled (Retsch, PM100) to disrupt the cell wall and resuspended in a buffer containing 0.1 mM reserpine (Sigma-Aldrich), 150 mM NaCl, 20 mM Tris-HCl, pH 8.0, and protease inhibitor cocktail (Sigma-Aldrich, 1:1,000). After incubation with reserpine for 10 min, the cells were lysed by sonication in a water–ice bath, and centrifuged at 4,000g for 15 min. The supernatant was centrifuged at 195,000g for 30 min at 4 °C. The pelleted membranes were resuspended in 50 ml buffer containing 0.4 mM reserpine, 150 mM NaCl, 20 mM Tris-HCl, pH 8.0, and subjected to Dounce homogenization on ice. Subsequently, DDM was added to a final concentration of 2% (w/v) to solubilize the membranes by stirring for 2 h at 4 °C. After centrifugation at 195,000g for 30 min, the resultant supernatant was incubated with 3 ml of anti-Flag M2 resins (Sigma-Aldrich) for 1 h at 4 °C. The resins were then washed with 50 ml wash buffer containing 0.08% (w/v) lauryl maltose neopentyl glycol (LMNG, Anatrace), 150 mM NaCl and 20 mM Tris-HCl, pH 8.0. The protein was released from the resin by cleavage with 200 μg ml−1 PreScission protease in 15 ml buffer containing 0.02% (w/v) LMNG, 150 mM NaCl and 20 mM Tris-HCl, pH 8.0. The protein was concentrated using the Amicon Ultra-15 centrifugal filter unit (100 kDa cut-off), and further purified by size-exclusion chromatography on the Superose 6 Increase 10/300 column (GE Healthcare) in a buffer containing 0.01% (w/v) LMNG, 0.0033% (w/v) glyco-diosgenin (Anatrace), 150 mM NaCl and 20 mM Tris-HCl, pH 8.0. The peak fractions were collected and concentrated for cryo-EM experiments.
Cryo-EM sample preparation and data acquisition
For monoamine-bound samples, the purified, reserpine stabilized VMAT1 dimers at around 5 mg ml−1 were incubated with 5 mM dopamine (Sigma-Aldrich), 3 mM serotonin (Sigma-Aldrich), 8 mM noradrenaline (Sigma-Aldrich) or 10 mM histamine (Sigma-Aldrich) for 1 h on ice. For amphetamine- and MPP+-bound samples, the purified VMAT1 dimers were concentrated to about 7 mg ml−1 and incubated with 1 mM amphet amine (Sigma-Aldrich) or 10 mMMPP+ (MCE) for 1 h on ice. Subsequently, 3 μl of the samples was added to glow-discharged holey carbon, 300 mesh R1.2/1.3 Au grids (Quantifoil). The grids were incubated for 20 s and blotted for 3 s at 8 °C and 90% humidity, and then plunge-frozen into liquid ethane using an EM GP2 automatic plunge freezer (Leica).
For unbound/reserpine and serotonin-bound/reserpine samples, the grids were loaded onto the Glacios (FEI) electron microscope operating at 200 kV equipped with a Falcon IV detector (Thermo Fisher Scientific). Videos were recorded using the EPU software (Thermo Fisher Scientific) with a pixel size of 0.94 Å and a nominal defocus value of −1.0 μm to −2.2 μm. The datasets of the unbound/reserpine sample were collected with a dose of 5.12 e− Å−2 s−1, and each video was recorded by 46 frames (209 ms per frame) for a 9.36 s exposure. The datasets of the serotonin-bound/reserpine sample were collected with a dose of 4.63 e− Å−2 s−1, and each video was recorded by 48 frames (204 ms per frame) for a 9.77 s exposure.
For the rest of the samples, the grids were loaded onto the Titan Krios (FEI) electron microscope operating at 300 kV, equipped with a K3 detector (Gatan) in CDS mode with a BioQuantum GIF energy filter (slit width of 20 eV) at the Hormel Institute, University of Minnesota. The videos were recorded using the EPU software (Thermo Fisher Scientific) with a pixel size of 0.664 Å (magnification of ×130,000) and a nominal defocus value of −1.0 μm to −2.0 μm. Each video consists of 40 dose-framed fractions and was recorded with a dose rate of 25 e− Å−2 s−1 and a total dose of 50 e− Å−2. Cryo-EM data collection statistics are summarized in Extended Data Table 1.
Cryo-EM data processing
A total of 4,301 videos was collected for the unbound/reserpine VMAT1 dimer and processed for patch-based motion correction using Motion-Cor251 and CTF estimation with CTFFIND (v.4.1.13)52 in cryoSPARC (v.4)53. Images with the defocus values outside of −0.6 to −2.8 μm or CTF fit resolutions worse than 5 Å were excluded from the further steps. The particles were then automatically picked from a small set of videos using Blob picker with a 320 pixel box size. Two rounds of 2D classification were performed to remove ice, contaminants and aggregates, yielding 190,189 particles. These particles were processed for 2D classification, and the classes from 72,139 particles corresponding to intact VMAT1 dimer were used as templates for further template-based particle picking. The selected particles were processed for 2D classification, resulting in 874,826 particles corresponding to the VMAT1 dimer. These particles were used to generate an ab initio model of five maps with C1 symmetry. The initial models were set as the starting references for heterogeneous refinement. The selected 3D classes were then processed for further homogeneous, non-uniform and CTF refinements, generating the final map at 3.5 Å resolution, as determined by the gold-standard measure of Fourier shell correlation using a cut-off of 0.143. Dopamine-bound (6,847 videos; 150,374 particles), noradrenaline-bound (5,601 videos; 255,378 particles), serotonin-bound (4,680 videos; 151,472 particles), histamine-bound (6,166 videos; 110,835 particles), amphetamine-bound (7,074 videos; 131,394 particles) and MPP+-bound (6,112 videos; 197,772 particles) datasets were processed using a similar procedure and yielded reconstructions at 3.4 Å, 3.4 Å, 3.3 Å, 3.7 Å, 3.5 Å and 3.6 Å, respectively, as estimated by gold-standard Fourier shell correlation. Local resolution variations were estimated from the two half-maps in cryoSPARC. The maps were sharpened by B-factor or using DeepEMhancer54.
Model building and refinement
The VMAT1 structure predicted by AlphaFold55 (AF-P54219-F1) was used as the initial model for the ligand-free monomer (vesicular open) and docked into the cryo-EM map using UCSF Chimera56. The initial model of the reserpine-bound monomer (cytoplasmic open) was generated by adjusting the transmembrane helices. The resulting dimer model was manually rebuilt in COOT57 and further refined by real space refinement in PHENIX58. This refined model was used as the initial model for VMAT1 structures with bound substrates, which were fitted into the density map using COOT. The resulting models were then manually rebuilt in COOT and further refined by real space refinement in PHENIX. The model stereochemistry was evaluated using the comprehensive validation (cryo-EM) utility with MolProbity in PHENIX. Structural figures were generated using ChimeraX59.
Fluorometric assay of FFN206 uptake
The fluorometric assay of VMAT1 activity was performed according to a previously described method with the following modifications60. HEK293T cells (ATCC, no further authentication, mycoplasma tested), cultured in DMEM and FBS, were plated onto poly-L-lysine-coated coverslips (Epredia) in six-well dishes at a density of 1.5–2.0 × 105 cells per well. After reaching 80–90% confluence, the cells were transfected with 2 μg plasmid of VMAT1 constructs with a GFP tag. Then, 2 days after transfection, the cells were washed with PBS and incubated in 1 ml Leibovitz’s L-15 Medium (Thermo) with 5 μM FFN206 for 2 h at 37 °C, 5% CO2. Subsequently, the coverslips were washed with PBS and mounted onto glass slides with an antifade mounting medium (VectorLabs). To inhibit VMAT1 activity, the cells were treated with 4 μM reserpine (Sigma-Aldrich) for 1 h or 1 μM bafilomycin A1 (BFA, Sigma-Aldrich) for 5 h, while controls were treated with DMSO. BFA specifically inhibits the V-type H+-ATPase activity and collapses the proton gradient across the vesicular membrane, which is required for the monoamine uptake activity of VMAT1. Fluorescence images were acquired using a Zeiss LSM 880 laser-scanning confocal microscope. The cell images were captured using ×40/1.4 NA oil objective with pinhole setting to 1 Airy unit. Image contrast and brightness were adjusted uniformly, and the mean fluorescence intensity in each cell area was analysed using ImageJ. The mean intensity of GFP and FFN206 fluorescence was measured for 60 cells per sample. The normalized fluorescence of FFN206 uptake was calculated by dividing the FFN206 intensity by GFP intensity. Statistical analyses were performed using one-way analysis of variance followed by a Tukey post hoc test. These experiments were independently repeated three times.
Western blot analysis
HEK293T cells were grown in six-well plates to 70–80% confluence and transfected with 1.5 μg of plasmid DNA coding for VMAT1 constructs using the polyethylenimine (PEI) transfection reagent (Polysciences). After 24 h transfection, the cells were collected and resuspended in cold RIPA buffer (1% Triton X-100, 150 mM NaCl, 50 mM Tris-HCl, pH 7.5, 1 mM PMSF, and 0.1% protease inhibitor cocktail). The samples were separated by SDS–PAGE and detected by immunoblotting on polyvinylidene fluoride membranes. The immunoblot used rabbit monoclonal antibody against β-actin (D6A8, Cell Signaling Technology, 1:1,000) or rabbit monoclonal antibody against Flag (D6W5B, Cell Signaling Technology, 1:1,000) as the primary antibody, and HRP-conjugated goat anti-rabbit antibody as the secondary antibody (0706S, Cell Signaling Technology, 1:3,000), followed by exposure to autoradiography film.
Vesicular uptake of [3H]dopamine and [3H]serotonin
The monoamine uptake and storage assays were adapted from a previous report36 with the following modifications. In brief, HEK293T cells in six-well plates were transfected with 1.5 μg plasmids encoding the VMAT1 constructs. After 24 h of transfection, cells were trypsin-detached and an aliquot was taken for the western blot estimation of protein expression level. The remaining cells were aliquoted to MultiScreenHTS 96-Well Filter Plates (Sigma-Aldrich) at 5 × 105 cells per well, and washed once with uptake buffer containing 110 mM Na-K-tartrate, 5 mM glucose, 0.2% (w/v) BSA, 5 mM MgCl2 and 20 mM K-HEPES, pH 7.4. Cells were then permeabilized in the uptake buffer with 10 mM digitonin for 10 min at 37 °C. This buffer was then replaced with the uptake buffer supplemented with 5 mM ATP, 1 mM ascorbic acid, ~20 nM [3H]serotonin (40.7 Ci mmol−1) (Perkin-Elmer) or [3H]dopamine (45.6 Ci mmol−1). Non-specific accumulation of [3H]serotonin or [3H]dopamine was measured in the presence of 4 μM reserpine. After incubation for 25 min, the reaction was terminated by removing the reaction buffer, and the cells were washed twice with ice-cold uptake buffer containing 2 mM MgSO4 (without MgCl2). The filter paper containing cells from each well was transferred to a microbeta vial and dissolved in a biodegradable scintillation cocktail (Research Products International). Radioactivity was quantified using a liquid scintillation counter (Hitachi Aloka, LSC8000). As all of the mutants express to a similar level (Supplementary Fig. 1), their relative activities presented in the figures (generated using GraphPad Prism) were not adjusted for expression levels.
Fluorescence polarization binding assay
Purified VMAT1 protein at varying concentrations (0.1–100 μM) was used to titrate 0.2 μM FFN206 in a buffer containing 0.01% (w/v) LMNG, 150 mM NaCl and 20 mM Tris-HCl, pH 8.0. After incubation for 1 h at 37 °C, the binding of FFN206 to VMAT1 was measured by fluorescence polarization, carried out in a 96-well fluorescence plate (Corning) with a SpectraMax M5 plate reader (excitation 360 nm, emission 460 nm). The Kd and Bmax values of the binding curves were fitted with a classical one-site specific-binding model using GraphPad Prism.
[3H]reserpine binding assay
The [3H]reserpine binding assay was adapted from a previous report25 with the following modifications. In brief, HEK293T cells in 15-cm-well plates were transfected with 20 μg plasmids encoding the VMAT1 constructs. After 24 h of transfection, HEK293T cells were treated 10 μM nigericin that collapses the proton gradient or with the same volume of DMSO for 2 h. Subsequently, the cells were trypsin-detached and an aliquot was taken for the FSEC estimation of the protein expression level. The remaining cells were resuspended with 5 ml wash buffer (140 mM KCl, 5 mM glucose, 5 mM MgCl2, and 20 mM HEPES, pH 7.4) supplemented with 5 mM ATP and permeabilized with 100 μM digitonin for 5 min. The binding reaction was initiated by the addition of 5 nM [3H]reserpine (ViTrax Radiochemicals; 20 Ci mmol−1), and cells were then incubated for 15 min at 37 °C. The reactions were stopped by quickly diluting the cells with ice-cold binding buffer and collected by brief centrifugation (12,700g for 1 min). The cells were then solubilized in the wash buffer containing 2% (w/v) DDM, 15 μg ml−1 DNase I (Sigma-Aldrich) and protease inhibitor mixture (Sigma-Aldrich). After 1 h of shaking at 4 °C, cells were centrifuged for 10 min at 18,900g and incubated with Ni-Magbeads (GeneScript) for 1 h at 4 °C. The beads were washed twice with wash buffer containing 0.08% DDM, and incubated with 650 μl of the same buffer with 450 mM imidazole at room temperature for 20 min. After the elution, 200 μl aliquots were measured for radioactivity using liquid scintillation.
Molecular dynamics simulation
The starting model for molecular dynamics simulation used the structure of VMAT1 in the lumenal-open state. To improve accuracy, the highest-resolution structure (amphetamine bound) was selected and this ligand was removed because the protonation-induced cytoplasmic-open transition occurs in absence of a substrate. The model was oriented in vacuole membrane using PPM (v.3.0)61. Subsequently, protonation of each candidate site25,35,37,38 was generated by using CHARMM-GUI62. The models with and without protonation were inserted into a POPC bilayer with surrounding water molecules. For each model, molecular dynamics simulations (200 ns) were repeated at least three times with the GROMACS molecular dynamics simulation package63 and CHARMM36m force field64. The distance analyses targeted the disparities between alternative conformations at the lumenal side, where NTD–CTD association may initiate the protonation-induced transition. Specifically, the average distance was measured between the lumenal ends of pore-forming transmembrane helices TM1, TM2 and TM5 in the NTD, and their repeat helices, TM7, TM8 and TM11 in the CTD. The trajectory files during the molecular dynamics simulation were generated by GROMACS and morphing motions were simulated by ChimeraX.
Homology modelling and molecular docking
The initial homology models of human VAChT were generated with Phyre265 by applying one-on-one threading with the VMAT1 structure as the template. In these initial models, the backbone conformation of VAChT was nearly identical to that of VMAT1. Side-chain conformations at the VAChT active site were adopted from the AlphaFold model (AF-Q16572-F1)55. The docking model of acetylcholine was generated by HADDOCK66, with the ‘active’ residues (directly involved in the interaction) defined approximately by those equivalent to the dopamine-interacting residues in VMAT1. Docking models with the top z scores were evaluated and the optimal model was selected, in which the amine and acetyl group of acetylcholine interact with the Tyr349-Asp407-Asn313 triad and the narrowed fist region, respectively. This docking model was further optimized with the geometry minimization program of PHENIX58.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. The VMAT1(ΔL1/2) construct enables the protein purification and is active in monoamine uptake.

a, FSEC profile comparison of full-length VMAT1 and VMAT1(ΔL1/2). These proteins are tagged with a C-terminal GFP, expressed in HEK293 or Pichia pastoris cells, and extracted in DDM. b, Vesicular uptake of FFN206 by full-length VMAT1 and VMAT1(ΔL1/2). Both can transport this false fluorescence neurotransmitter into the vesicle-like compartments of HEK293 cells (shown in single cells). The activities of full-length VMAT1 and VMAT1(ΔL1/2) are both contingent upon the proton gradient (collapsed by bafilomycin A1, BFA) and inhibitable by reserpine (RSP). c, View of multiple cells for fluorescence quantification of FFN206 transport. Experiments in b and c were repeated independently three times with similar results. d, Dose-dependent FFN206 transport curves. The FFN206 fluorescence is normalized to GFP fluorescence to account for variations in protein expression level. e, Time course of [3H]-dopamine and [3H]-serotonin uptake assays. Data in d and e are shown as mean ± s.e.m. from n = 3 biological replicates.
Extended Data Fig. 2 |. Reserpine treatment during protein purification induces the dimer formation of human VMAT1.

a, 2D classification showing that VMAT1 protein forms monomers without reserpine treatment (top) and forms dimers with reserpine treatment (bottom). The monomer particles were extracted at box size of 320 pixels (0.664 Å/pixel), and VAMT1 dimer at box size of 320 pixels (0.885 Å/pixel). b, Size-exclusion chromatography (Superose 6) shows a shift to higher molecular weight after reserpine treatment, indicative of dimer formation. The elution profiles are from size-exclusion chromatography after affinity purification (left) and a subsequent rerun (right). c, SDS-PAGE of eluted fractions from the size-exclusion rerun. Uncropped gel is shown in Supplementary Fig. 2. Purifications were repeated independently at least three times with similar results.
Extended Data Fig. 3 |. Cryo-EM data processing flowchart and density maps of individual regions in VMAT1(ΔL1/2) structure.

The dataset and 3.5 Å density map of VMAT1(ΔL1/2) dimer with amphetamine and reserpine are shown as a representative. Other datasets are similarly processed, and density maps are similar. a, A representative raw cryo-EM image. Similar images are present in the 6,930 movies selected (see c). b, Representative 2D classes. c, The data processing procedure. Three rounds of 2D classification and two rounds of heterogenous refinements generated 3.54 Å and 3.49 Å final maps for two different classes. The maps were sharpened by DeepEMhancer for model building and analysis. d-e, Densities of helices and loops in the luminal-open (d) and cytoplasmic-open (e) monomer. Contour level = 0.12 in ChimeraX.
Extended Data Fig. 4 |. Quality of cryo-EM maps.
a-h, VMAT1(ΔL1/2) dimer in unbound form and with reserpine (a); both with reserpine (b); with dopamine (DA) and reserpine (c); with noradrenaline (NE) and reserpine (d); with serotonin (SERT) and reserpine (e); with histamine (HA) and reserpine (f); with amphetamine (AMPH) and reserpine (g); and with MPP+ and reserpine (h). Left to right, Local resolution illustrations of the cryo-EM maps, angular distribution plots, half-map FSC curves, and model-to-map FSCs.
Extended Data Fig. 5 |. VMAT1 structures with bound reserpine and in unbound form.

a, Cryo-EM density map of the VMAT1 dimer, in which both monomers bind reserpine (density coloured in orange). Map contour level = 0.22 in ChimeraX. b, Helical tube representation of the dual reserpine-bound dimer structure. c, Hydrophobic effect of reserpine binding. Top, molecular structure of reserpine fits into a narrow gap at the bottom of the central cavity. Middle and bottom, The NTD and CTD sides of this gap both contain hydrophobic patches (yellow) that match well with the molecular shape of reserpine. d, Relative rigid-body rotation of the NTD and CTD between the cytoplasmic-open and lumenal-open conformations, and bending of TM5 and TM11. The NTD (left) and CTD (right) from each conformation are superimposed and shown in a side view. e, Top view of the superimposition. f, The NTD and CTD in the lumenal-open state are closely related by a 2-fold rigid body rotation. Left, the overall structure of the lumenal-open state. Right, superimposition of the NTD and CTD. TM5 and TM11, shown in dark colours, are replicated helices related by +6 numbering (as for other NTD and CTD helices). g, The NTD and CTD in the cytoplasmic-open state. Left, overall structure of the cytoplasmic-open state. Right, superimposition of the NTD and CTD.
Extended Data Fig. 6 |. Sequence alignment of SLC18 transporters.

The sequence alignment of human (h) and rat (r) SCL18 members was generated by ClustalW. The asterisks below the aligned sequences indicate conserved residues, and single and double dots represent varying levels of sequence similarity. Residues participating in substrate and reserpine binding are highlighted with orange shading and green spheres below, respectively. Unique gating residues in the cytoplasmic-open or lumenal-open conformations are indicated by green and blue bars, respectively. Interaction analysis was performed using the CONTACT program from the CCP4i suite. The potential protonation sites are indicated by red triangles. The protein folding topology (cyto: cytoplasmic side, lum: lumenal side), based on the VMAT1 structure, is denoted above the sequences. The sequence logo, produced by WebLogo, depicts the residue conservation level at each position. Initial identification of eukaryotic SLC18 homologues was achieved through the PSI-BLAST program within the Max-Planck Institute Bioinformatics Toolkit, using VMAT1 as the query sequence against a nonredundant (70%) sequence database, eukaryotes NR70. The top 500 proteins were selected, and incomplete sequences were manually removed. The multiple sequence alignment was generated by ClustalΩ. The motifs identified in the DHA-1 drug antiporter family are displayed above the sequence.
Extended Data Fig. 7 |. Substrate and inhibitor binding.

a-b, Structural superimposition of VMAT1 in unbound form and with bound dopamine showing the overall structures (a) and the substrate-binding pocket (b). c-e, Left, surface representation of the wrist-and-fist binding pocket with bound noradrenaline (c), serotonin (d), and histamine (e). Right, comparison of the binding of these monoamines (orange) with dopamine (dark grey). f, Mutations at the substrate-binding pocket reduces the binding affinity of FFN206. The binding of VMAT1 mutants is assessed by the fluorescence polarization of FFN206. Because most mutants do not achieve saturation in FFN206 binding even at very high protein concentration (100 μM), their relative affinities are estimated through binding potential (Bmax/Kd). Data are shown as mean ± s.e.m. from n = 3 biological replicates. g-h, Left, Amphetamine and MPP+ in the substrate binding pocket (surface representation). Right, comparison of their binding (orange) with dopamine (dark grey). i, Reserpine occupies the monoamine binding pocket. Left, the dopamine (dark grey) bound structure is superimposed onto the reserpine (orange) bound structure by the CTD. The protein sidechains illustrated are from the fixed reserpine-bound structure. Right, a 90-degree rotated view with the binding pocket (from the fixed reserpine-bound structure) shown in surface representation.
Extended Data Fig. 8 |. Candidate protonation sites.

a, Overall structures of the lumenal-open (left) and cytoplasmic-open conformations (right) with all candidate protonation sites. The lumenal unexposed regions in both conformations are highlighted by the orange circle. b, Local environment of D34 (zoomed view of dashed box in a) in the alternate conformations of VMAT1. c, A representative conformation from the molecular dynamics (MD) simulation of protonated E320 (E320-p, as in Fig. 5e) showing a similar NTD-CTD association as those in the cryo-EM structures. Left, Structure of the E320-p MD conformation (different orange colours). Middle, E320-p conformation is similar to that of the lumenal-open state (different blue colours) at the cytoplasmic side (orange circle). Right, E320-p conformation is also similar to that of the cytoplasmic-open state (different green colours) at the lumenal side (orange circle).
Extended Data Fig. 9 |. Homology model of VAChT.

Changes in interactions at the altered substrate-binding pocket may enable the recognition of acetylcholine (ACh). Residues in VMAT1 are in parentheses, with altered residues in VAChT highlighted in bold. Surface representation uses the altered binding pocket.
Extended Data Table 1 |.
Cryo-EM data collection, refinement and validation statistics
| Unbound/RSP (EMD-41238) (PDB 8TGJ) | RSP/RSP (EMD-41241) (PDB 8TGM) | DA/RSP (EMD-41237) (PDB 8TGI) | NE/RSP (EMD-41240) (PDB 8TGL) | SERT/RSP (EMD-41242) (PDB 8TGN) | HA/RSP (EMD-41239) (PDB 8TGK) | AMPH/RSP (EMD-41236) (PDB 8TGH) | MPP7RSP (EMD-41235) (PDB 8TGG) | |
|---|---|---|---|---|---|---|---|---|
| Data collection and processing | ||||||||
| Magnification | 150,000 | 130,000 | 130,000 | 130,000 | 150,000 | 130,000 | 130,000 | 130,000 |
| Voltage (kV) | 200 | 300 | 300 | 300 | 200 | 300 | 300 | 300 |
| Electron exposure (e-/Å2) | 47.88 | 50.00 | 50.00 | 50.00 | 45.22 | 50.00 | 50.00 | 50.00 |
| Defocus range (μm) | 1.0–2.2 | 1.0–2.0 | 1.0–2.0 | 1.0–2.0 | 1.0–2.2 | 1.0–2.0 | 1.0–2.0 | 1.0–2.0 |
| Pixel size (Å) | 0.94 | 0.664 | 0.664 | 0.664 | 0.94 | 0.664 | 0.664 | 0.664 |
| Symmetry imposed | Cl | Cl | Cl | Cl | Cl | Cl | Cl | Cl |
| Initial particle images (no.) | 874,862 | 567,815 | 567,815 | 641,349 | 629,796 | 633,820 | 893,558 | 699,136 |
| Final particle images (no.) | 148,629 | 113,342 | 150,374 | 255,378 | 151,472 | 110,835 | 131,394 | 197,772 |
| Map resolution (Å) | 3.5 | 3.5 | 3.4 | 3.4 | 3.3 | 3.7 | 3.5 | 3.6 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 3.1–5.1 | 3.1–5.5 | 3.1–4.7 | 3.0–4.6 | 3.0–5.4 | 3.2–4.8 | 3.0–4.6 | 3.2–4.8 |
| Refinement | ||||||||
| Model resolution (Å) | 3.98 | 3.44 | 3.74 | 3.71 | 3.68 | 3.68 | 3.27 | 3.70 |
| FSC threshold | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| Map sharpening B factor (Å2) | 133.4 | 116.1 | 118.0 | 120.3 | 130.2 | 129.7 | 118.1 | 136.8 |
| Model composition | ||||||||
| Non-hydrogen atoms | 5761 | 5901 | 5837 | 5849 | 5856 | 5839 | 5818 | 5856 |
| Protein residues | 761 | 767 | 759 | 769 | 770 | 770 | 767 | 770 |
| Ligands | 1 | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| B factors (Å2) | ||||||||
| Protein | 80.47 | 72.39 | 75.60 | 47.43 | 84.56 | 74.77 | 60.52 | 84.86 |
| Ligand | 27.53 | 74.14 | 93.80 | 40.62 | 68.43 | 84.63 | 67.28 | 70.92 |
| R.m.s. deviations | ||||||||
| Bond lengths (Å) | 0.003 | 0.005 | 0.743 | 0.679 | 0.004 | 0.004 | 0.005 | 0.008 |
| Bond angles (°) | 0.794 | 0.876 | 0.003 | 0.004 | 0.880 | 0.757 | 0.749 | 0.856 |
| Validation | ||||||||
| MolProbity score | 1.89 | 1.82 | 1.84 | 1.93 | 1.94 | 1.79 | 1.75 | 1.84 |
| Clashscore | 25.23 | 14.60 | 22.06 | 30.11 | 28.56 | 19.76 | 15.65 | 22.48 |
| Poor rotamers (%) | 0.49 | 0.33 | 0.65 | 0.49 | 0.16 | 0.32 | 0.33 | 0.49 |
| Ramachandran plot | ||||||||
| Favored (%) | 98.40 | 97.21 | 99.08 | 98.16 | 98.81 | 98.55 | 97.75 | 98.16 |
| Allowed (%) | 1.60 | 2.79 | 0.92 | 1.84 | 1.19 | 1.45 | 2.25 | 1.84 |
| Disallowed (%) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
The statistics are reported for VMAT1(ΔL1/2) dimer in unbound form and with reserpine (Unbound/RSP); both with reserpine (RSP/RSP); with dopamine and reserpine (DA/RSP); with noradrenaline and reserpine (NE/RSP); with serotonin and reserpine (SERT/RSP); with histamine and reserpine (HA/RSP); with amphetamine and reserpine (AMPH/RSP); and with MPP+ and reserpine (MPP+/RSP).
Supplementary Material
Acknowledgements
We thank Andrzej Krezel for comments. W.L. is supported by the Vagelos Endowed Chair, the Established Investigator Award and Collaborative Sciences Award from American Heart Association, the Forefront of Science Award from the W. M. Keck Foundation, the National Heart, Lung and Blood Institute (R01 HL121718), the Children’s Discovery Institute (MCII 2020-854), the National Institute of Allergy and Infectious Diseases (R01 AI158500) and the National Institute of General Medical Sciences (R01 GM131008). B.L. is supported by the Hormel Institute, University of Minnesota. K.W. is supported by Lundbeck foundation (R324-2019-1855). J.X. is supported by National Institute of Neurological Disorders and Stroke (R01 NS092865).
Footnotes
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-024-07290-7.
Reprints and permissions information is available at http://www.nature.com/reprints.
Competing interests The authors declare no competing interests.
Data availability
The cryo-EM maps have been deposited at the Electron Microscopy Data Bank (EMDB) under accession numbers EMD-41238 (unbound/reserpine), EMD-41241 (reserpine/reserpine), EMD-41237 (dopamine/reserpine), EMD-41240 (noradrenaline/reserpine), EMD-41242 (serotonin/reserpine), EMD-41239 (histamine/reserpine), EMD-41236 (amphetamine/reserpine) and EMD-41235 (MPP+/reserpine). The coordinates have been deposited at the Protein Data Bank (PDB) under accession numbers 8TGJ (unbound/reserpine), 8TGM (reserpine/reserpine), 8TGI (dopamine/reserpine), 8TGL (noradrenaline/reserpine), 8TGN (serotonin/reserpine), 8TGK (histamine/reserpine), 8TGH (amphetamine/reserpine) and 8TGG (MPP+/reserpine). Source data are provided with this paper.
References
- 1.Carlsson A. A half-century of neurotransmitter research: impact on neurology and psychiatry. Biosci. Rep. 21, 691–710 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Sara SJ The locus coeruleus and noradrenergic modulation of cognition. Nat. Rev. Neurosci. 10, 211–223 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Mawe GM & Hoffman JM Serotonin signalling in the gut-functions, dysfunctions and therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 10, 473–486 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carey RM Renal dopamine system: paracrine regulator of sodium homeostasis and blood pressure. Hypertension 38, 297–302 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Stone KD, Prussin C. & Metcalfe DD IgE, mast cells, basophils, and eosinophils. J. Allergy Clin. Immunol. 125, S73–S80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erickson JD & Eiden LE Functional identification and molecular cloning of a human brain vesicle monoamine transporter. J. Neurochem. 61, 2314–2317 (1993). [DOI] [PubMed] [Google Scholar]
- 7.Erickson JD, Eiden LE & Hoffman BJ Expression cloning of a reserpine-sensitive vesicular monoamine transporter. Proc. Natl Acad. Sci. USA 89, 10993–10997 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eiden LE, Schafer MK, Weihe E. & Schutz B. The vesicular amine transporter family (SLC18): amine/proton antiporters required for vesicular accumulation and regulated exocytotic secretion of monoamines and acetylcholine. Pflugers Arch. 447, 636–640 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Parsons SM Transport mechanisms in acetylcholine and monoamine storage. FASEB J. 14, 2423–2434 (2000). [DOI] [PubMed] [Google Scholar]
- 10.Liu Y. et al. A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell 70, 539–551 (1992). [DOI] [PubMed] [Google Scholar]
- 11.Carlsson A. & Healy D. in The Psychopharmacologists (ed. Healy D) Vol. 1, 51–80 (CRC Press, 1996). [Google Scholar]
- 12.Baumeister AA The chlorpromazine enigma. J. Hist. Neurosci. 22, 14–29 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Kenney C. & Jankovic J. Tetrabenazine in the treatment of hyperkinetic movement disorders. Expert Rev. Neurother. 6, 7–17 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Efange SM In vivo imaging of the vesicular acetylcholine transporter and the vesicular monoamine transporter. FASEB J. 14, 2401–2413 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Erickson JD, Schafer MK, Bonner TI, Eiden LE & Weihe E. Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc. Natl Acad. Sci. USA 93, 5166–5171 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sulzer D, Sonders MS, Poulsen NW & Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog. Neurobiol. 75, 406–433 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Peter D, Jimenez J, Liu Y, Kim J. & Edwards RH The chromaffin granule and synaptic vesicle amine transporters differ in substrate recognition and sensitivity to inhibitors. J. Biol. Chem. 269, 7231–7237 (1994). [PubMed] [Google Scholar]
- 18.Erickson JD et al. Functional identification of a vesicular acetylcholine transporter and its expression from a “cholinergic” gene locus. J. Biol. Chem. 269, 21929–21932 (1994). [PubMed] [Google Scholar]
- 19.Lohoff FW et al. Association between polymorphisms in the vesicular monoamine transporter 1 gene (VMAT1/SLC18A1) on chromosome 8p and schizophrenia. Neuropsychobiology 57, 55–60 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Hu G. et al. New fluorescent substrate enables quantitative and high-throughput examination of vesicular monoamine transporter 2 (VMAT2). ACS Chem. Biol. 8, 1947–1954 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anne C. & Gasnier B. Vesicular neurotransmitter transporters: mechanistic aspects. Curr. Top. Membr. 73, 149–174 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Darchen F, Scherman D. & Henry JP Reserpine binding to chromaffin granules suggests the existence of two conformations of the monoamine transporter. Biochemistry 28, 1692–1697 (1989). [DOI] [PubMed] [Google Scholar]
- 23.Quistgaard EM, Low C, Guettou F. & Nordlund P. Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat. Rev. Mol. Cell Biol. 17, 123–132 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Law CJ, Maloney PC & Wang DN Ins and outs of major facilitator superfamily antiporters. Annu. Rev. Microbiol. 62, 289–305 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yaffe D, Vergara-Jaque A, Forrest LR & Schuldiner S. Emulating proton-induced conformational changes in the vesicular monoamine transporter VMAT2 by mutagenesis. Proc. Natl Acad. Sci. USA 113, E7390–E7398 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lohoff FW Genetic variants in the vesicular monoamine transporter 1 (VMAT1/SLC18A1) and neuropsychiatric disorders. Methods Mol. Biol. 637, 165–180 (2010). [DOI] [PubMed] [Google Scholar]
- 27.Lohoff FW et al. Functional genetic variants in the vesicular monoamine transporter 1 modulate emotion processing. Mol. Psychiatry 19, 129–139 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sulzer D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron 69, 628–649 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deupree JD & Weaver JA Identification and characterization of the catecholamine transporter in bovine chromaffin granules using [3H]reserpine. J. Biol. Chem. 259, 10907–10912 (1984). [PubMed] [Google Scholar]
- 30.Scherman D. & Henry JP Reserpine binding to bovine chromaffin granule membranes. Characterization and comparison with dihydrotetrabenazine binding. Mol. Pharmacol. 25, 113–122 (1984). [PubMed] [Google Scholar]
- 31.Vardy E, Steiner-Mordoch S. & Schuldiner S. Characterization of bacterial drug antiporters homologous to mammalian neurotransmitter transporters. J. Bacteriol. 187, 7518–7525 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yaffe D, Forrest LR & Schuldiner S. The ins and outs of vesicular monoamine transporters. J. Gen. Physiol. 150, 671–682 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wimalasena K. Vesicular monoamine transporters: structure-function, pharmacology, and medicinal chemistry. Med. Res. Rev. 31, 483–519 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pidathala S. et al. Mechanisms of neurotransmitter transport and drug inhibition in human VMAT2. Nature 623, 1086–1092 (2023). [DOI] [PubMed] [Google Scholar]
- 35.Yaffe D. et al. Functionally important carboxyls in a bacterial homologue of the vesicular monoamine transporter (VMAT). J. Biol. Chem. 289, 34229–34240 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yaffe D, Radestock S, Shuster Y, Forrest LR & Schuldiner S. Identification of molecular hinge points mediating alternating access in the vesicular monoamine transporter VMAT2. Proc. Natl Acad. Sci. USA 110, E1332–E1341 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shirvan A, Laskar O, Steiner-Mordoch S. & Schuldiner S. Histidine-419 plays a role in energy coupling in the vesicular monoamine transporter from rat. FEBS Lett. 356, 145–150 (1994). [DOI] [PubMed] [Google Scholar]
- 38.Steiner-Mordoch S, Shirvan A. & Schuldiner S. Modification of the pH profile and tetrabenazine sensitivity of rat VMAT1 by replacement of aspartate 404 with glutamate. J. Biol. Chem. 271, 13048–13054 (1996). [DOI] [PubMed] [Google Scholar]
- 39.Schuldiner S. Competition as a way of life for H+-coupled antiporters. J. Mol. Biol. 426, 2539–2546 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maron R, Stern Y, Kanner BI & Schuldiner S. Functional asymmetry of the amine transporter from chromaffin granules. J. Biol. Chem. 258, 11476–11481 (1983). [PubMed] [Google Scholar]
- 41.Khare P, Mulakaluri A. & Parsons SM Search for the acetylcholine and vesamicol binding sites in vesicular acetylcholine transporter: the region around the lumenal end of the transport channel. J. Neurochem. 115, 984–993 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ojeda AM, Kolmakova NG & Parsons SM Acetylcholine binding site in the vesicular acetylcholine transporter. Biochemistry 43, 11163–11174 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Ugolev Y, Segal T, Yaffe D, Gros Y. & Schuldiner S. Identification of conformationally sensitive residues essential for inhibition of vesicular monoamine transport by the noncompetitive inhibitor tetrabenazine. J. Biol. Chem. 288, 32160–32171 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peter D, Vu T. & Edwards RH Chimeric vesicular monoamine transporters identify structural domains that influence substrate affinity and sensitivity to tetrabenazine. J. Biol. Chem. 271, 2979–2986 (1996). [DOI] [PubMed] [Google Scholar]
- 45.Finn JP 3rd & Edwards RH Multiple residues contribute independently to differences in ligand recognition between vesicular monoamine transporters 1 and 2. J. Biol. Chem. 273, 3943–3947 (1998). [DOI] [PubMed] [Google Scholar]
- 46.Kanner BI, Fishkes H, Maron R, Sharon I. & Schuldiner S. Reserpine as a competitive and reversible inhibitor of the catecholamine transporter of bovine chromaffin granules. FEBS Lett. 100, 175–178 (1979). [DOI] [PubMed] [Google Scholar]
- 47.Schuldiner S, Liu Y. & Edwards RH Reserpine binding to a vesicular amine transporter expressed in Chinese hamster ovary fibroblasts. J. Biol. Chem. 268, 29–34 (1993). [PubMed] [Google Scholar]
- 48.Kilbourn MR, Lee LC, Heeg MJ & Jewett DM Absolute configuration of (+)-α-dihydrotetrabenazine, an active metabolite of tetrabenazine. Chirality 9, 59–62 (1997). [DOI] [PubMed] [Google Scholar]
- 49.Dwoskin LP & Crooks PA A novel mechanism of action and potential use for lobeline as a treatment for psychostimulant abuse. Biochem. Pharmacol. 63, 89–98 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Lohr KM & Miller GW VMAT2 and Parkinson’s disease: harnessing the dopamine vesicle. Expert Rev. Neurother. 14, 1115–1117 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rubinstein JL & Brubaker MA Alignment of cryo-EM movies of individual particles by optimization of image translations. J. Struct. Biol. 192, 188–195 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Rohou A. & Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Sanchez-Garcia R. et al. DeepEMhancer: a deep learning solution for cryo-EM volume post-processing. Commun. Biol. 4, 874 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jumper J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pettersen EF et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Emsley P, Lohkamp B, Scott WG & Cowtan KD Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Afonine PV et al. Towards automated crystallographic structure refinement with phenix. refine. Acta Crystallogr. D 68, 352–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pettersen EF et al. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Black CA et al. Assessing vesicular monoamine transport and toxicity using fluorescent false neurotransmitters. Chem. Res. Toxicol. 34, 1256–1264 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lomize AL, Todd SC & Pogozheva ID Spatial arrangement of proteins in planar and curved membranes by PPM 3.0. Protein Sci. 31, 209–220 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vanommeslaeghe K. et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van Der Spoel D. et al. GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Huang J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kelley LA, Mezulis S, Yates CM, Wass MN & Sternberg MJE The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Van Zundert GCP et al. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 428, 720–725 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM maps have been deposited at the Electron Microscopy Data Bank (EMDB) under accession numbers EMD-41238 (unbound/reserpine), EMD-41241 (reserpine/reserpine), EMD-41237 (dopamine/reserpine), EMD-41240 (noradrenaline/reserpine), EMD-41242 (serotonin/reserpine), EMD-41239 (histamine/reserpine), EMD-41236 (amphetamine/reserpine) and EMD-41235 (MPP+/reserpine). The coordinates have been deposited at the Protein Data Bank (PDB) under accession numbers 8TGJ (unbound/reserpine), 8TGM (reserpine/reserpine), 8TGI (dopamine/reserpine), 8TGL (noradrenaline/reserpine), 8TGN (serotonin/reserpine), 8TGK (histamine/reserpine), 8TGH (amphetamine/reserpine) and 8TGG (MPP+/reserpine). Source data are provided with this paper.