

Abstract
High levels of transcription from the amyloid precursor protein promoter are dependent on the binding of CTCF to the APBβ core recognition sequence located between positions –82 and –93 upstream from the transcriptional start site. CTCF comprises 727 amino acids and contains 11 zinc finger motifs arranged in tandem that are flanked by 267 amino acids on the N-terminal side and 150 amino acids on the C-terminal side. Deletion of either the N- or the C-terminal regions outside of the zinc finger domain had no detrimental effect on the binding of CTCF to APBβ. However, internal deletions of zinc fingers 5–7 completely abolished binding. The binding of full-length CTCF generated a DNase I protected domain extending from position –78 to –116, which was interrupted by a hypersensitive site at position –99. Selective deletions from the N- and C-terminal sides of the zinc finger domain showed that the N-terminal end of the zinc finger domain was aligned toward the transcriptional start site. Furthermore, deletions of zinc fingers peripheral to the essential zinc fingers 5–7 decreased the stability of the binding complex by interrupting sequence-specific interactions.
INTRODUCTION
The extracellular deposition of amyloid β-protein (Aβ) is a characteristic neuropathological manifestation in Alzheimer’s disease and Down syndrome (1–3). The Aβ originates from a group of proteins designated amyloid β-protein precursors (APP), which are derived from the same gene by differential splicing (4). The APP gene is expressed at varying levels in all major tissues, including brain (5,6). Increased levels of APP gene transcript have been observed in Down syndrome and in certain areas of the brain in Alzheimer’s disease (6–9). Overexpression of APP also leads to amyloid deposition in transplanted murine hippocampal tissue with trisomy 16, the mouse equivalent of Down syndrome (10). These observations suggest that overexpression of APP may be one of several contributing factors in the formation of amyloid depositions and in the neuropathology associated with Alzheimer disease.
The promoter of the APP gene is a necessary element in regulating APP transcription and it has been shown to confer cell-type specific expression in transgenic mice (11,12). The proximal APP promoter is devoid of CCAAT and TATA boxes but contains a prominent initiator element associated with the main transcriptional start site (+1). The integrity of this initiator element is essential for both start site selection and optimal transcriptional activity (13). In addition, an intact nuclear factor binding site designated APBβ is essential for effective transcription from the APP promoter (13,14). The core recognition sequence for this binding site is located between positions –82 and –93 and its elimination reduces transcriptional activity by ~70–90% (13,14). The nuclear factor that activates transcription from APBβ was identified as CTCF (15), a nuclear regulatory protein comprising 727 amino acids (16). It contains a centrally located DNA binding domain with 11 zinc finger motifs that are flanked by 267 amino acids on the N-terminal side and 150 amino acids on the C-terminal side. This protein was first identified as a factor that binds to the chicken c-myc promoter (17) and to the silencer element of the chicken lysozyme gene (18,19). CTCF was also shown to bind to the chicken β-globin insulator and other vertebrate enhancer blocking elements (20). CTCF binds to diverse sequences by utilizing different combinations of essential zinc fingers (16,19). A functional role for CTCF in both positive and negative transcriptional regulation has been documented (14–16,20–22). The mechanism by which CTCF exerts its diverse regulatory effects remains unclear. However, it is likely to involve interactions with specific secondary factors and to depend on the position and orientation of the binding site relative to the transcriptional unit. We have therefore analyzed the binding characteristics of CTCF as a step toward elucidating its role in transcriptional activation.
MATERIALS AND METHODS
Expression of recombinant CTCF constructs
Using sequence information provided elsewhere (16), the cDNA encoding human CTCF was amplified by the polymerase chain reaction (PCR) from a library derived from the human retinoblastoma cell line Y79 (Clontech, Palo Alto, CA). The PCR products were assembled into vector pCR3 (Invitrogen, Carlsbad, CA) and subsequently, the full-length cDNA was excised and subcloned into plasmid pGEM 7Zf(-) (Promega, Madision, WI). The cDNA insert was sequenced and Taq polymerase reading errors were corrected by site-directed mutagenesis (23). Deletions from the 5′ end were introduced at restriction sites AccI (position 965), Bsu36I (position 1216) and KpnI (position 1553). Deletions from the 3′ end were introduced at restriction sites BglII (position 2142) and PstI (position 1867) (16). Individual internal zinc finger deletions (Fig. 1) were obtained by site-directed mutagenesis (23).
Figure 1.

(A) Amino acid sequence of human CTCF. The first methionine is designated as M1. All other amino acids delineating selected deletions are indicated by the specific amino acid followed by its position in the sequence. The putative zinc fingers are indicated by brackets. Amino acids removed in individual zinc finger deletions are underlined. (B) Schematic representation of the CTCF molecule indicating its N-, C-terminal and zinc finger (Zn) domain. The approximate relative positions of N- and C-terminal deletions are indicated by arrows. (C) Sequence of the three oligonucleotides APBβ[WT], APBβ[–94] (derived from the APP promoter) and C-MYC used in their double-stranded form for mobility shift electrophoresis. Native APP and c-myc promoter sequences are written in capital letters and non-promoter sequences are written in lower case. The positions of nucleotides –125, –94 and –64 within the APP promoter are delineated in the APBβ oligonucleotides. In oligonucleotide APBβ[–94] the sequence upstream of position –94 is underlined, representing the exact reproduction of the sequence as it exists in expression vector APP[–94], which is derived from plasmid CAT2bGAL (26).
Coupled in vitro transcription/translation was performed using the TNT Reticulocyte Lysate System (Promega) according to the manufacturer’s instructions. Specifically, 2 µg of template plasmid pGEM 7Zf(-) was used per 25 µl reaction mixture containing [35S]methionine. Aliquots (10 µl) of the reaction products were either analyzed by mobility shift electrophoresis (see below) or by electrophoresis in 8% SDS–polyacrylamide gels.
Expression of CTCF and its deletion constructs in yeast was accomplished with the Pichia Expression Kit (Invitrogen) according to the manufacturer’s instructions. Briefly, full-length cDNA and specific deletions encoding various CTCF constructs were excised from plasmid pGEM 7Zf(-), blunt ended, ligated with EcoRI linkers, and cloned into the EcoRI site of plasmid pHIL-D2, a vector that directs intracellular recombinant protein expression in Pichia pastoris. The vectors containing the CTCF cDNA constructs in the correct orientation were transformed into Pichia strain KM71 by electroporation. Positive clones were amplified, induced for protein expression and screened for the presence of CTCF by mobility shift electrophoresis.
Purification of CTCF
Pichia cells (5–10 g) were mixed with 25 ml of 0.5 mm glass beads, adjusted to a total volume of 50 ml with buffer E (40 mM HEPES, pH 7.6, 2 mM MgCl2,1 mM EDTA and 100 µM ZnSO4), and homogenized with the Bead Beater apparatus (Biospec Products, Inc., Bartensville, OK). Cell debris was pelleted at 5000 g for 10 min and the supernatant was supplemented with 3 M KCl to a final concentration of 200 mM. The lysate was further clarified by centrifugation at 100 000 g for 30 min.
CTCF was purified from recombinant Pichia extracts or HeLa cell nuclear extract (15) by single step SP cation exchange chromatography. Briefly, a 1 ml HiTrap SP Sepharose column (Pharmacia) was equilibrated with buffer E containing 200 mM KCl. Cleared extract was loaded on the column and proteins were eluted with a linear concentration gradient of KCl (0.2–1 M) in buffer E. Fractions of 0.5 ml were collected and CTCF binding activity was monitored by mobility shift electrophoresis. Fractions containing the peak activity were used for mobility shift electrophoresis and DNase I footprinting.
Binding reactions, mobility shift electrophoresis, DNase I footprinting and sequencing
Double-stranded oligonucleotides were 5′-end-labeled with [γ-32P]ATP using T4 polynucleotide kinase (24). The binding reaction was assembled by first mixing CTCF-containing extract with binding buffer (25 mM HEPES, pH 7.6, 100 mM KCl, 2 mM dithiothreitol and 10% glycerol) supplemented with 10 µg of yeast tRNA, 2 µg of poly(dI–dC).poly(dI–dC) and adjusted to a final concentration of 2.5% CHAPS, and 3% fetal calf serum (FCS) in a total volume of 28 µl. Extracts containing CTCF binding activity consisted either of whole (13,25) or partially purified (15) HeLa cell nuclear extract (0.1–1 µl), CTCF and its deletion constructs purified from P.pastoris (0.1–2 µl), or CTCF translated in vitro (10 µl). The mixture was preincubated for 5 min at 25°C followed by the addition of 2 µl of labeled double-stranded oligonucleotide (1 ng per binding reaction, 50 000–200 000 c.p.m.) in binding buffer. The final binding reaction was incubated for 1 h at 25°C and then electrophoresed in 2% agarose gels containing 0.5× TBE (24) at 100–150 V constant voltage.
DNase I footprinting was performed on DNA fragments extending from position –193 to +100 of plasmids APP[–488] and APP[–94] (14). The fragments were amplified by PCR using the 32P-end-labeled direct primers 5′-CGGAGGGGGCGCGTGGGGTGCAGGC and 5′-TTATGCTTCCGGCTCGTATGTTGT for synthesis of fragments APP[WT] and APP[–94], respectively. Oligonucleotide 5′-CGCCGCCACCGCCGCCGTCTCCCG was used as the reverse primer in both cases. PCR was carried out in the presence of 7% formamide in order to overcome strong secondary structure of the promoter sequence. The PCR cycle comprised a denaturing step at 95°C for 1 min, an annealing step at 47°C for 1 min, and an elongation step at 72°C for 2 min. Following PCR the fragments were purified by PAGE. The binding reactions of the radiolabeled fragments (50 000 c.p.m.) were assembled as described above with the purified CTCF proteins except that no FCS was added to the binding buffer.
The boundaries of the DNase I protected domains were determined by dideoxy sequencing (Amersham Life Sciences, Arlington Heights, IL) with the same 32P-end-labeled primers used for PCR amplification of fragments APP[WT] and APP[–94].
Mobility shift competition and dissociation rate determination
The binding reaction was assembled as described above. However, for the purpose of mobility shift competition the radiolabeled oligonucleotide was premixed with an increasing molar excess of unlabeled oligonucleotide as described in Figure 6. After electrophoresis in 2% agarose, the amount of bound and free oligonucleotide was quantitated with a GS-250 phosphor imager (Bio-Rad, Hercules, CA).
Figure 6.

Binding of recombinant CTCF constructs to APP[WT] and APP[–94]. (A) DNase I footprinting of fragments extending from position –193 to +100 in the wild-type APP promoter (lanes 1 and 2) or of fragments in which the region between positions –94 and –193 had been replaced with upstream vector sequences in plasmid APP[–94] (lanes 3–7). Fragments were 5′-end-labeled at position –193 and digested with DNase I in the absence of CTCF (lanes 1, 3 and 7), with full-length recombinant CTCF (lanes 2 and 4), or with internal zinc finger deletions Zn4 (lane 5) and Zn9 (lane 6). The protected domain of full-length CTCF bound to the wild-type APP sequence (FL CTCF[WT]) is indicated by a bracket between positions –78 and –116. Position –94, representing the point of divergence between the wild-type APP sequence (WT) and APP[–94] is indicated by an arrowhead. The protected domains of full-length CTCF and N-terminal zinc finger deletions bound to the APP[–94] sequence are indicated by brackets from position –78 to –98 (FL[–94])and –83 to –98 (ΔN-Zn), respectively. (B) DNase I footprinting of fragments extending from position –193 to +100 in plasmid APP[–94] in the absence of CTCF (lane 1) or with bound full-length CTCF (lane 2). The DNase I footprinting reactions were co-electrophoresed with a dideoxy sequencing ladder obtained with the same 32P-labeled oligonucletide used for PCR amplification (lanes 3–6). The sequence within the bracketed region is provided together with the outlines of DNase I protected domains obtained with full-length CTCF (FL[–94]) and internal deletion Zn4 (ΔN-Zn), The sequence of the APBβ binding sequence is boxed. The boundaries of the footprints are defined by the terminal nucleotides protected from DNase I digestion and their position should be considered accurate within 1 bp.
To determine the dissociation rate of the CTCF/APBβ binding complex (Fig. 5), all binding reactions were assembled with labeled oligonucleotide APBβ[WT] (Fig. 1C) as described above. However, the incubation times of the reactions were extended to at least 2 h to ensure that the diverse CTCF constructs had all reached binding equilibrium with the labeled oligonucleotide. Thereafter, unlabeled oligonucleotide in 2 µl of binding buffer was added at a 500-fold molar excess. Following incubation for the time periods specified in Figure 5, the reaction products were separated by mobility shift electrophoresis. As a control, labeled oligonucleotide was premixed with a 500-fold excess of unlabeled oligonucleotide before addition to the CTCF containing binding buffer. This control reaction represented the theoretical final binding equilibrium between labeled and unlabeled oligonucleotide. After mobility shift electrophoresis, gels were dried and the amount of free and bound radiolabeled oligonucleotide was determined on a GS-250 phosphor imager (Bio-Rad). Under the applied assay conditions no binding to the labeled oligonucleotide was detectable in the control reaction.
Figure 5.

Dissociation of CTCF constructs bound to double-stranded oligonucleotide APBβ[WT]. (A) Deletions M249 (filled triangles), M285 (squares), C525 (filled diamonds), D617 (inverted triangles), Zn4 (circles), Zn9 (open triangles) and recombinant full-length CTCF (FL) (open diamonds) were bound to 5′-end-labeled APBβ[WT] and subsequently a 500-fold molar excess of unlabeled APBβ[WT] was added (time zero). The decreasing amount of radiolabeled binding complex as a function of time was monitored by mobility shift electrophoresis and the data were fitted to exponential decay curves. The relative amount of radiolabeled binding complex at time zero was assigned the value of 100% and all subsequent values are expressed as a fraction thereof. (B) From the exponential decay curves in (A), the half-lives of the CTCF binding complexes were calculated. The data points represent the averages of 2–4 independent determinations and error bars indicate the standard deviation.
The dissociation rate was determined by plotting the relative amount of bound oligonucleotide as a function of time. The half-lives of the binding complexes were determined by fitting the data to single-phase exponential decay curves using Graph Pad Prism version 3.00 for Windows (Graph Pad Software, San Diego, CA)
RESULTS
N-terminal deletion of zinc fingers 1–6 abolishes binding to APBβ
To investigate protein domains that could potentially affect the binding of CTCF to the APP promoter target sequence, deletions from the 5′ end of the CTCF cDNA (16) were generated at available restriction sites (Materials and Methods). These deletions shifted the translational start sites to the next available methionine residues at amino acid positions M249, M285 and M422 (Fig. 1A). In terms of amino acid sequence, deletion M249 removed the N-terminal portion of the protein while leaving the zinc finger domain intact, deletion M285 extended into the first zinc finger, and deletion M422 eliminated the first six zinc fingers. A single 3′ deletion truncated the native protein at the position of amino acid D617, which removed the majority of the C-terminal end of the protein (Fig. 1A and B).
The predominant translation product derived from full-length cDNA and 3′ deletion D617 was CTCF initiated at the first methionine M1 of the open reading frame. However, numerous shorter translation products were observed that had been initiated at other methionine residues downstream from M1 (Fig. 2A, lanes 1 and 5). A similar pattern was seen with deletions M249 and M422, where the first available methionine provided the primary translational start site (Fig. 2A, lanes 2 and 4). In contrast, the most abundant translation product of deletion M285 was initiated further downstream at a site tentatively identified as M329, which extends into zinc finger 3 (Fig. 2A, lane 3).
Figure 2.
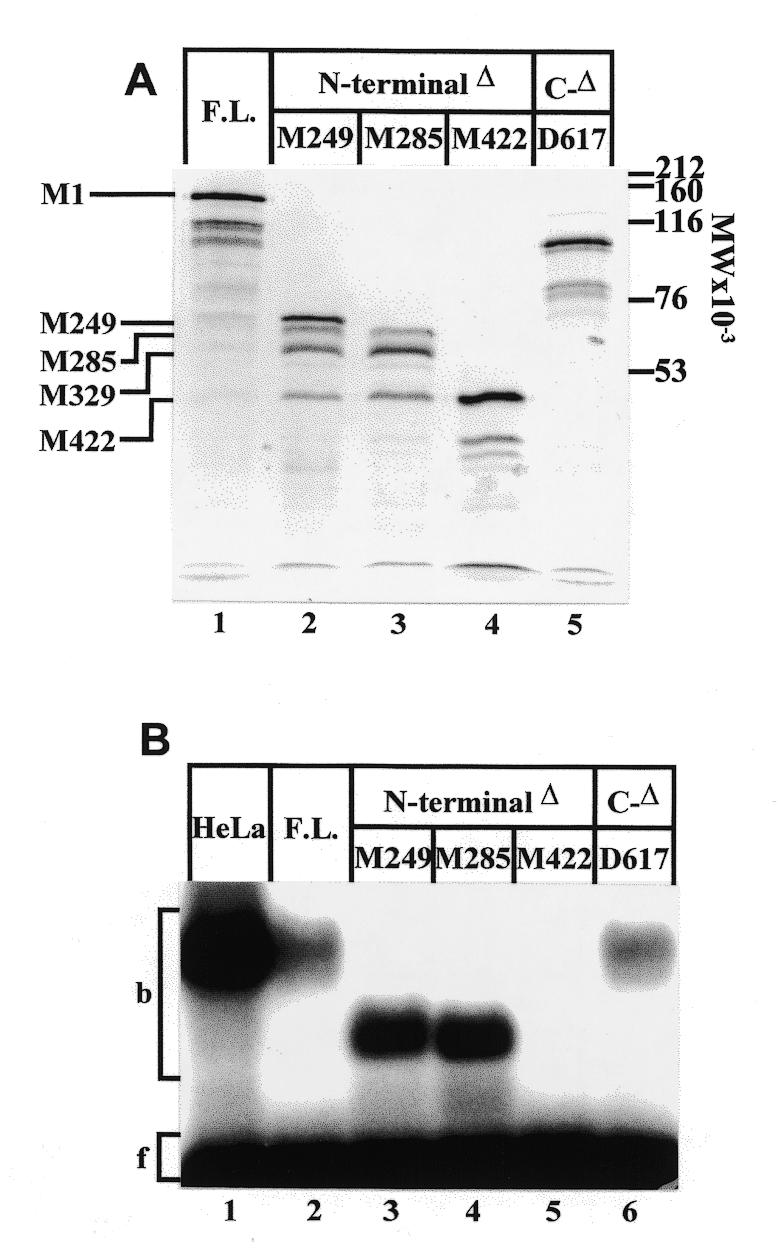
Analysis of N- and C-terminal deletions of CTCF. (A) Autoradiogram of [35S]methionine-labeled CTCF constructs. Full length CTCF (lane 1), successive N-terminal deletions at positions M249 (lane 2), M285 (lane 3), M422 (lane 4), and a C-terminal deletion at amino acid residue D617 (lane 5) were synthesized by in vitro transcription/translation and separated on a 7% SDS–polyacrylamide gel. The positions of methionine residues used as translational start sites are indicated. (B) Mobility shift electrophoresis of CTCF constructs bound to 32P-end-labeled oligonucleotide APBβ[WT] (Fig. 1C). In addition to CTCF partially purified from HeLa cell nuclear extract (lane 1), the same protein fractions as shown in (A), lanes 1–5, were used as binding factors (lanes 2–6).
The translation products were analyzed by mobility shift electrophoresis for their ability to bind to the APBβ domain (Fig. 2B). Binding was observed with the full-length translation product as well as with 5′ deletions M249 and M285 (Fig. 2B, lanes 2–4). Similarly, eliminating a large part of the C-terminal domain did not abolish CTCF binding (Fig. 2B, lane 6). However, binding was completely abolished with the translation product starting at M422, which eliminated the N-terminal domain up to zinc finger 6 (Fig. 2B, lane 5). This shows that the N-terminal region, including zinc finger 1 and probably zinc finger 3, was not required for the binding of CTCF to APBβ. The results also indicate that the apparent binding activities of N-terminal CTCF deletions M249 and M285 were consistently higher than those of the full-length molecule or C-terminal deletion D617 (Fig. 2B). Whether this was due to differences in concentration or in actual binding affinity was not systematically examined. It is conceivable that the in vitro translated product lacked correct folding of the N-terminal domain, which in turn could adversely affect binding affinity.
Zinc fingers 5–7 are essential for binding of CTCF to the APBβ domain
CTCF binds to diverse sequences by utilizing different combinations of essential zinc fingers (16,19). In order to determine which zinc fingers are indispensable for binding to the APBβ domain, each zinc finger was individually deleted from the CTCF molecule. Since the N-terminal deletion at M249 consistently displayed more pronounced binding activity to the APBβ domain (Fig. 2B), the subsequent internal zinc finger deletions were introduced into this construct (Fig. 3A).
Figure 3.
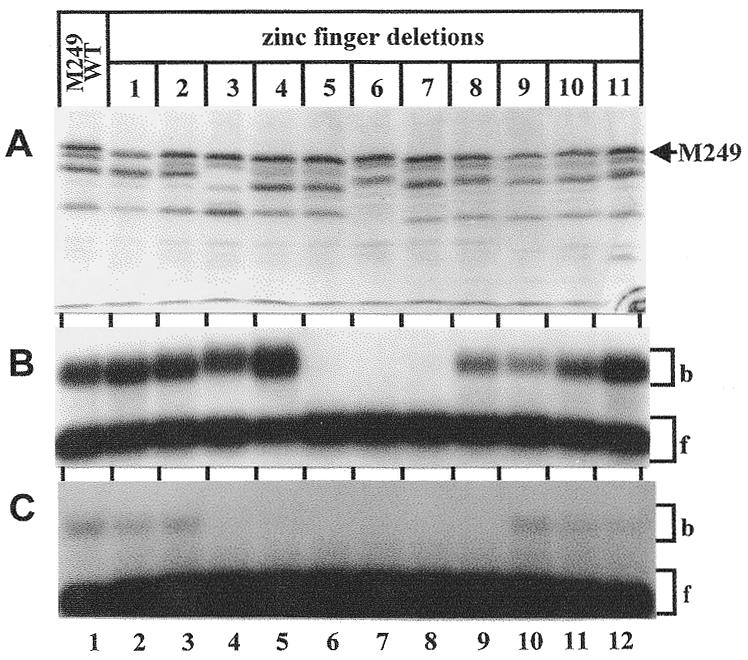
Analysis of internal zinc finger deletions in CTCF. (A) Autoradiogram of [35S]methionine-labeled wild-type CTCF (lane 1) and successive zinc finger deletions (lanes 2–12). All zinc finger deletions were introduced into N-terminal deletion M249 by site-directed mutagenesis. Proteins were synthesized by in vitro transcription/translation and separated on a 7% SDS–polyacrylamide gel. The position of the protein initiating at methionine residue 249 (M249) is indicated. (B) Wild-type N-terminal deletion M249 (lane 1) and individual zinc finger deletions introduced into this construct (lanes 2–12) were bound to 32P-end-labeled APBβ[WT] (Fig. 1C) and separated by mobility shift electrophoresis. The same constructs as presented in (A) were used for the binding reaction. The bound (b) and free (f) oligonucleotides are indicated by brackets. (C) Same as in (B), except that the C-MYC oligonucleotide (Fig. 1C) was used as a labeled probe.
Deletion of individual zinc fingers 5–7 completely abolished DNA binding activity (Fig. 3B, lanes 6–8). All other zinc finger deletions supported target binding although some variation was observed in the relative binding activities of the different zinc finger deletions. This might reflect differences in binding affinity of individual zinc finger deletions or differences in the availability of functional CTCF.
CTCF also binds to the chicken c-myc promoter and the zinc fingers that are essential for binding to this site have been extensively characterized with constructs containing successive 5′ and 3′ deletions (16). In this study we used internal zinc finger deletions for a consistent comparison of the CTCF binding sites in the chicken c-myc and human APP promoters. Under the same reaction conditions, the apparent binding activity to the c-myc promoter site was substantially lower than to the APBβ site. Furthermore, deletion of zinc fingers 3–8 abolished binding to the c-myc promoter site (Fig. 3C, lanes 4–9).
The N-terminal end of the CTCF zinc finger domain is aligned toward the transcriptional start site
The binding of CTCF to APBβ gives rise to a defined DNase I protected domain (13). Since deletions within the CTCF molecule may extend into the DNase I protected domain and modify it, they could be employed to determine the orientation of the CTCF molecule on the target site. For such an analysis it is beneficial to have available a reasonable amount of purified protein that is also appropriately folded and post-translationally modified. We therefore chose to express the protein constructs intracellularly in the yeast strain P.pastoris. This was followed by cellular breakage under non-denaturing conditions and purification by cation exchange chromatography. The purified proteins displayed a high degree of homogeneity, the majority of which were translated in their respective full-length form (Fig. 4A). The total yield of purified protein was ~0.5 mg/l of yeast culture.
Figure 4.
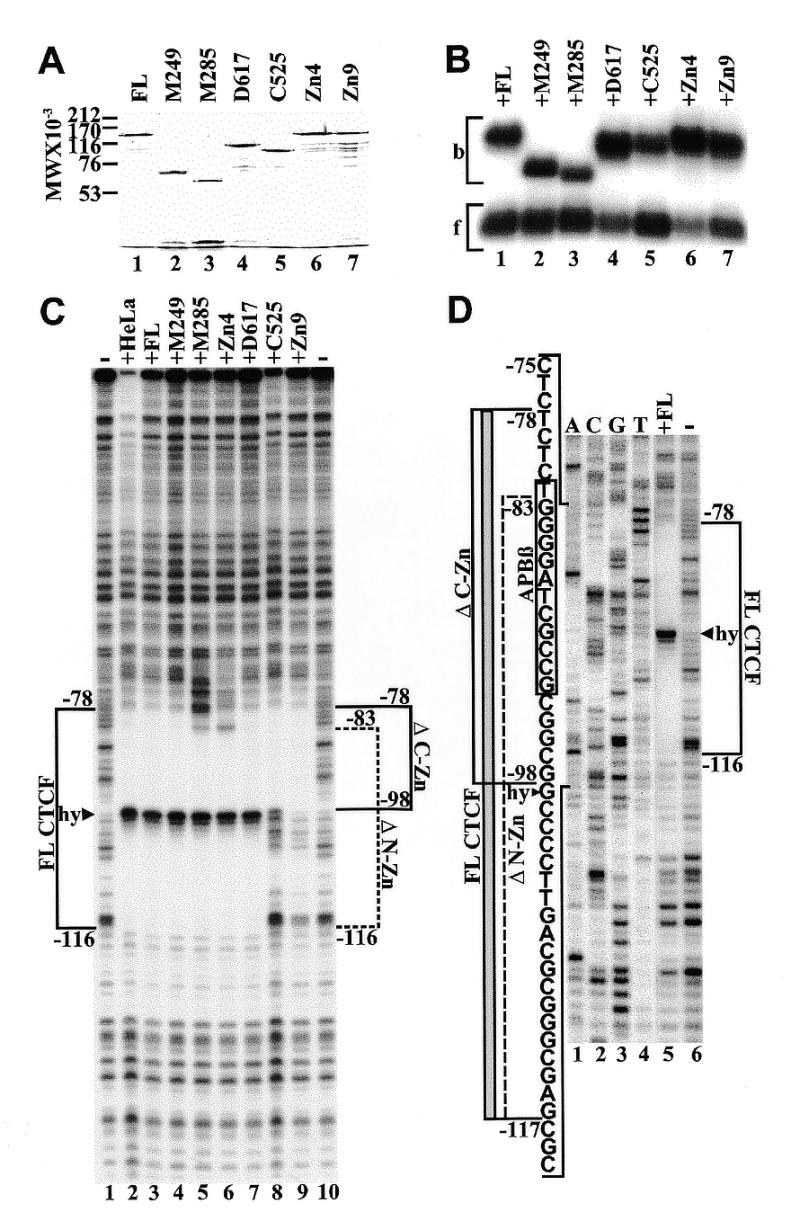
Binding of purified recombinant CTCF constructs to APP[WT]. (A) Coomassie blue stained SDS–polyacrylamide gel of full-length CTCF (lane 1), N-terminal deletions M249 (lane 2), M285 (lane 3), C-terminal deletions D617 (lane 4), C525 (lane 5), and internal zinc finger deletions Zn4 (lane 6) and Zn9 (lane 7). (B) Mobility shift electrophoresis of the same CTCF constructs shown in (A). All CTCF constructs were bound to radiolabeled oligonucleotide APBβ[WT] (Fig. 1C). Bound (b) and free (f) oligoncleotides are indicated by brackets. (C) DNase I footprinting of purified CTCF constructs bound to 5′-end-labeled wild-type APP fragment extending from position –193 to +100. Fragments were 5′-end-labeled at position –193 and digested with DNase I either in the absence of CTCF (lanes 1 and 10), or with full-length CTCF purified from HeLa cell nuclear extract (lane 2), full-length recombinant CTCF from P.pastoris (lane 3), N-terminal deletions M249 (lane 4) and M285 (lane 5), internal zinc finger deletion Zn4 (lane 6), C-terminal deletions D617 (lane 7) and C525 (lane 8), and internal zinc finger deletion Zn9 (lane 9). The DNase I footprint of full-length CTCF (FL CTCF) is delineated by a bracket from position –78 to –116 interrupted by a hypersensitive site (hy, arrowhead). Brackets also indicate the DNase I footprints generated by N-terminal (ΔN-Zn, position –83 to –116) and C-terminal (ΔC-Zn, position –98 to –78) zinc finger deletions. (D) DNase I footprinting of 5′-end-labeled wild-type APP fragment without CTCF (lane 1) or with bound full-length CTCF (lane 2). The DNase I footprinting reactions were co-electrophoresed with a dideoxy sequencing reaction obtained with the same 32P-labeled oligonucletide used for PCR amplification (lanes 3–6). The sequence within the bracketed region is provided together with the outlines of DNase I protected domains obtained with FL CTCF (gray bar). Brackets also indicate the outlines of the DNase I protected domains obtained with N- (ΔN-Zn) and C-terminal (ΔC-Zn) deletions of CTCF. The sequence of the APBβ binding sequence is boxed. The boundaries of the footprints are defined by the terminal nucleotides protected from DNase I digestion and their position should be considered accurate within 1 bp.
Full-length CTCF, N-terminal deletions M285, M249 and C-terminal deletion D617 (Fig. 1A and B) were selected for expression in P.pastoris. In addition, a new C-terminal deletion was created that terminated at position C525, which removed zinc fingers 10 and 11 (Fig. 1A). Two individual zinc finger deletions, ΔZn4 and ΔZn9, representing non-essential zinc fingers on both sides of the essential zinc fingers 5–7, were also included to examine their effect on DNase I footprinting. However, in order to assess the potential influence of an intact N-terminal region on the DNase I protected domain, zinc finger deletions ΔZn4 and ΔZn9 were analyzed within the full-length CTCF construct instead of N-terminal deletion M249, which had been used for in vitro transcription/translation (Fig. 3).
Mobility shift electrophoresis of full-length recombinant CTCF and its truncated versions resulted in a migration pattern similar to that obtained with the in vitro translated proteins (Figs 2B and 4B). However, In contrast to the in vitro translated constructs (Fig. 2B), there was no apparent difference in binding activity between full-length CTCF and N-terminal deletion M249 expressed in Pichia (Fig. 4B, lanes 1 and 2). It is notable that the elimination of 249 amino acids on the N-terminal side of CTCF (M249) caused a pronounced increase in the electrophoretic mobility of the binding complex (Fig. 4B, lanes 1 and 2). In contrast, a comparable change in mobility was not observed when ~203 amino acids (C525) were removed from the C-terminal side (Fig. 4B, lanes 1 and 5). Indeed, the mobility shift pattern of the various CTCF constructs largely paralleled their electrophoretic migration under denaturing conditions (Fig. 4A and B).
DNase I footprinting with full-length CTCF purified from HeLa cell nuclear extract resulted in a protected domain between positions –78 and –116, interrupted by a hypersensitive site at position –99 (Fig. 4C). An identical footprint was observed with full-length recombinant CTCF produced in P.pastoris, and with CTCF truncated on the N-terminal side at position M249 (Fig. 4C, lanes 1–4). This indicates that the N-terminal region of CTCF that is located outside of the zinc finger domain did not contribute to DNA binding. However, as zinc finger 1 was removed (M285), the 3′ boundary of the DNase I protected domain shifted from position –78 to –83. A similar shift was observed with the selective deletion of zinc finger 4 (Fig. 4C, lanes 5 and 6). Furthermore, when a large portion of the C-terminal domain was deleted from CTCF (D617), no change in the protection pattern was observed. However, when zinc fingers 10 and 11 were removed (C525), the 5′ boundary of the DNase I protected domain shifted from position –116 to –98. A similar result was obtained with the internal elimination of zinc finger 9 (Fig. 4C, lanes 7–9). These results, which are summarized in Figure 4D, indicate that the N-terminal end of the zinc finger domain is aligned toward the transcriptional start site of the APP promoter. Furthermore, although the zinc fingers peripheral to zinc fingers 5–7 are not absolutely essential for binding, they contribute to the integrity of the DNase I protected sequence.
Deletion of peripheral zinc fingers from CTCF alters the stability of the binding complex
To address whether these peripheral zinc fingers also contribute to the stability of the binding complex, the dissociation rate of the CTCF binding complex was measured (Fig. 5A) and the half-lives of the binding complexes were determined by fitting the data to single-phase exponential decay curves (Fig. 5A and B). The half-life of the full-length CTCF/APBβ binding complex was ~22 h and remained essentially unchanged with deletions M249 and D617 (18.6 and 23.8 h, respectively). However, when deletions were extended into the zinc finger domain, the half-life of the binding complex decreased to ~7 h for construct M285 and to 2.4 h for construct C525. With deletions of individual zinc fingers 4 and 9, the half-life decreased even more dramatically to 1.9 and 0.34 h, respectively (Fig. 5B). These results indicate that peripheral zinc fingers contribute to CTCF/APBβ binding complex stability. While zinc fingers 5–7 are absolutely required for binding, disruption of the peripheral zinc fingers increases the rate of CTCF dissociation from the binding complex.
Changing the DNA sequence upstream of the core APBβ recognition sequence alters the DNase I footprint
The involvement of peripheral zinc fingers in maintaining an integral DNase I protected domain suggests a sequence-specific interaction with the target site. We have previously reported that an APP promoter construct terminating at position –94 displays a similar promoter activity as constructs terminating further upstream, both in vivo and in vitro (13,14,26). However, in cultured rat embryonic hippocampal neurons we observed that APP[–94] displayed a somewhat lower promoter activity than constructs extending further upstream (27). Since the DNase I protected domain generated by the bound CTCF extends in the 5′ direction to position –116, it is conceivable that replacing the endogenous upstream APP promoter domain with a heterologous vector sequence could alter the binding properties of CTCF to the APBβ element. We therefore examined the DNase I footprint resulting from the binding of CTCF to APBβ in the context of the APP[–94] construct. When full-length CTCF was bound to the APBβ site of the APP[–94] sequence, a DNase I protected domain was again observed (Fig. 6). However, in contrast to the wild-type sequence, the DNase I protected domain in construct APP[–94] was truncated at a point that would be equivalent to position –98 of the wild-type sequence (Fig. 6A, lanes 2 and 4 ). This is also the position of the hypersensitive site observed in the protected domain of the wild-type sequence (Fig. 6A, lane 2). As in the wild-type sequence, deletion of zinc finger 4 shortened the DNase I protected domain in the 5′ direction from position –78 to –83 (Fig. 6A, lane 5). However, deletion of zinc finger 9 did not change the DNase I protected domain in the 3′ direction (Fig. 6A, lane 6). These results show that altering the wild type sequence upstream of position –94 produces a DNase I protected domain with full-length CTCF which is similar to that obtained by binding C-terminal deletion C525 or internal deletion ZN9 to the wild-type sequence. This indicates that the N-terminal zinc finger interaction is sequence specific.
Changing the APP promoter sequence upstream of position –94 reduces binding affinity of CTCF
To investigate the contribution of upstream promoter sequences to the relative binding affinity of CTCF to APBβ, we constructed two 80mer oligonucleotides (Fig. 1C). Oligonucleotide APBβ[WT] contained the endogenous wild-type APP promoter sequence between position –64 and –125, whereas in oligonucleotide APBβ[–94] the domain between position –94 and –125 exactly reproduced the sequence of the expression vector APP[–94] (Fig. 1C). Mobility shift competition shows that CTCF had a 3–4-fold lower binding affinity for oligonucleotide APBβ[–94] than for oligonucleotide APBβ[WT] (Fig. 7, lanes 1–8). This confirms that the integrity of the wild–type APP promoter sequence between position –125 and –94 is essential for optimal binding of CTCF and that the interaction of the C-terminal zinc fingers with this domain is sequence specific.
Figure 7.
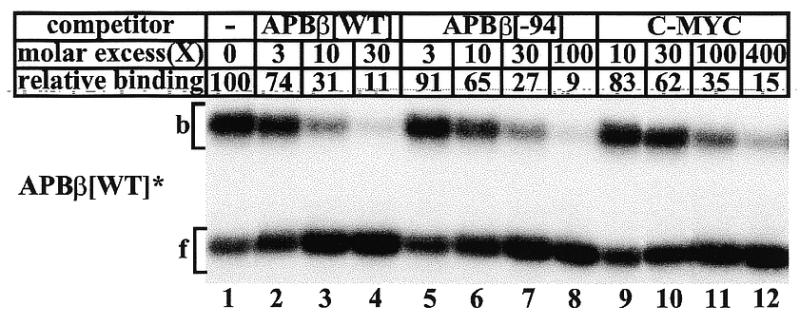
Mobility shift competition of CTCF binding to double-stranded oligonucleotide APBβ[WT]. Full-length recombinant CTCF was bound to 5′-end-labeled APBβ[WT] either without competitor (lane 1), or with a 3- (lane 2), 10- (lane 3), and 30-fold (lane 4) molar excess of unlabeled APBβ[WT], a 3- (lane 5), 10- (lane 6), 30- (lane 7), and 100-fold (lane 8) excess of unlabeled APBβ[–94], or a 10- (lane 9), 30- (lane 10) 100- (lane 11) and 400-fold (lane 12) excess of C-MYC (Fig. 4C). The amount of CTCF bound without competitor was assigned the value of 100, and all other binding activities are expressed as a fraction thereof.
Using in vitro translated CTCF constructs as binding factors, we observed that binding to the c-myc promoter was considerably lower than to the APBβ sequence (Fig. 3C). To verify that observation with CTCF obtained from HeLa cells, we included the C-MYC oligonucleotide (Fig. 1C) as a competitor. Consistent with the results obtained with in vitro translated CTCF constructs (Fig. 3B and C), these data confirm that the apparent binding affinity of CTCF for the c-myc sequence is ~10–15-fold lower than for the APBβ[WT] sequence (Fig. 7).
DISCUSSION
Accumulating evidence indicates that nuclear protein CTCF is a multifunctional regulator of numerous genes that can promote both activation and suppression of transcription as well as play a key role in transcriptional insulation (19–22,27–29). The DNA binding sites of CTCF are also diverse, and often bear no apparent similarity to each other. CTCF contains 11 zinc finger domains that are arranged in tandem. The expression of internal deletions by in vitro transcription/translation demonstrated that CTCF binding to the APP promoter required the presence of zinc fingers 5–7, whereas zinc fingers 3–8 were essential for binding to the chicken c-myc promoter (Fig. 2). In addition, a comparison between CTCF binding to the c-myc and the APP promoters under identical reaction conditions showed that the avidity for the APP promoter sequence is substantially higher (Figs 3 and 7). In principle, this result is consistent with enhancer blocking activity experiments performed on the FII insulator sequence of the chicken β-globin gene (20). In that study, the level of insulation obtained with the APBβ sequence was found to be substantially higher than with the chicken c-myc and lysozyme sequences but in the same range as the endogenous FII sequence of the chicken β-globin insulator.
CTCF is a protein with an approximate molecular mass of 82 kDa but its electrophoretic migration in SDS–PAGE suggests a much larger molecular mass in the range of 130–140 kDa. This aberrant electrophoretic mobility could to a large extent be traced to the N-terminal region of the protein (Fig. 4A) (30). We here extend that observation to non-denatured CTCF, bound to APBβ in mobility shift assays (Fig. 4A and B). The cause for this effect of the N-terminal end on electrophoretic mobility has not been determined.
DNase I footprinting of purified recombinant and native CTCF revealed a defined nuclease protected domain extending from position –78 to –116 (Fig. 4). Although from the results presented here, it is not possible to precisely assign specific contact points for each zinc finger, the zinc fingers may conceptually be organized into three groups, accounting for three distinct regions of the protected DNA (Fig. 4D). Specifically, removing zinc finger 1 (M285) and zinc finger 4 resulted in a shift in the DNase I footprinting domain from position –78 to –83, while deletions on the C-terminal part of the zinc finger domain resulted in a contraction of the footprint from position –116 to –98 (Fig. 4C and D). It is therefore reasonable to surmise that the contact points for the essential zinc fingers 5–7 are located between positions –83 and –98 of the APP promoter, which overlaps with the previously identified core recognition sequence of APBβ between positions –82 and –93 (14) (Fig. 4D). Indeed, long before CTCF was identified as the factor that binds to the APBβ domain, the DNA contact points of the binding factor were preliminarily established (14). Specifically, methylation of any G residue within the coding strand of the APBβ domain was associated with reduced DNA binding. Comparison of binding sequence, DNA contact points, and the position of the hypersensitive site with other CTCF binding sites (19) shows that a certain similarity exists between the APBβ site and the chicken lysozyme silencer element. Any similarity with the human and chicken c-myc binding sites is elusive.
The DNase I footprints further suggest that most if not all of the 11 zinc fingers contribute to the DNase I protected domain of the full-length CTCF. Disruption of peripheral zinc fingers from both the N- and C-terminal side reduced the half-life of the binding complex, although this reduction was more pronounced in deletions from the C-terminal side (Fig. 5). It is also noteworthy that individual zinc finger deletions reduced the half-life of the binding complex more than N- and C-terminal deletions while the DNase I protected domains remained unchanged (Figs 4 and 5). The shorter half-lives of the internal deletions may be explained by the continued presence of the entire N- and C-terminal regions, which are removed by progressive deletions from either side. When zinc fingers are disrupted, the presence of these regions may further destabilize binding.
The contraction of the DNase I protected domain and the associated destabilization of the binding complex as a result of peripheral zinc finger disruption suggested that the interaction of these zinc fingers with their target was sequence specific. This was further corroborated by mobility shift competition, which demonstrated that the CTCF binding activity to a sequence corresponding to APP[–94] was 3–4-fold lower than to the wild-type sequence. These observations are of particular significance since we had previously reported that plasmid APP[–94] conferred full promoter activity in a number of cell backgrounds both in vivo and in vitro (13,14,26). However, we have also recently observed lower levels of expression from the APP[–94] construct during differentiation of rat primary hippocampal neurons (27). It is conceivable that if the availability of CTCF is limiting, the difference in binding affinity between the wild-type and the APP[–94] sequences can result in lower levels of expression from transfected APP[–94] constructs during differentiation of embryonic rat hippocampal neurons (27).
DNase I footprinting with CTCF containing peripheral zinc finger deletions immediately suggests that CTCF is bound to APBβ with the N-terminal end of the zinc finger domain aligned toward the transcriptional start site (Fig. 4). This may have functional implications since CTCF can act as transcriptional activator or repressor in different genes or cell backgrounds. For example, CTCF binds to the chicken lysozyme silencer 2.4 kb upstream from the transcriptional start site. Here it synergistically represses transcription in conjunction with the thyroid hormone receptor or the retinoic acid receptor (22,28). This function may be due to differential DNA bending, which is dependent on the composition of the binding complexes on the silencer element (31). Another example of synergistic repression is provided by the coordinate action of CTCF and the thyroid hormone receptor on the TRE-containing rat genomic element 144 (29,32). Furthermore, CTCF has been found to directionally block enhancer activation by binding to the insulator element at the 5′ end of the chicken β-globin gene locus (20). Similar CTCF binding sequences were identified in a variety of insulators from diverse vertebrate species, suggesting a widespread role for CTCF in the regulation of enhancer activated genes (20). CTCF also binds to the promoter of the chicken c-myc gene where it acts as transcriptional activator or repressor, depending on the cell background (17,21,30). In the human and mouse c-myc genes CTCF binds to divergent sequences that coincide with RNA polymerase pausing sites within the transcribed region of the genes (16). Finally, in the human APP promoter CTCF has been associated with transcriptional activation (13,14,15,27).
These examples show that the function of CTCF may be influenced by a variety of factors such as the position of the binding site relative to the transcriptional unit, the sequence of the binding site, the properties of different CTCF isoforms (30), post-translational processing (33), or the binding of specific cofactors. One possible regulatory mechanism has recently been provided by the finding that the co-repressor SIN3A was found to bind to the zinc finger domain of CTCF. Histone deacetylases may then be recruited to the complex via binding to SIN3A (34). Alternatively, phosphorylation of CTCF that is dependent on specific differentiation pathways of human myeloid cells may contribute to the selection of specific DNA binding sites or regulate the binding of cofactors (33).
Acknowledgments
ACKNOWLEDGEMENT
This work was supported by National Institutes of Health Grant NS30994 (W.Q).
REFERENCES
- 1.Glenner G.G. and Wong,C.W. (1984) Biochem. Biophys. Res. Commun., 122, 1131–1135. [DOI] [PubMed] [Google Scholar]
- 2.Mann D.M., Jones,D., Prinja,D. and Purkiss,M.S. (1990) Acta Neuropathol., 80, 318–327. [DOI] [PubMed] [Google Scholar]
- 3.Masters C.L., Simms,G., Weinman,N.A., Multhaup,G., McDonald,B.L. and Beyreuther,K. (1985) Proc. Natl Acad. Sci. USA, 82, 4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang J., Lemaire,H.G., Unterbeck,A., Salbaum,J.M., Masters,C.L., Grzeschik,K.H., Multhaup,G., Beyreuther,K. and Müller-Hill,B. (1987) Nature, 325, 733–736. [DOI] [PubMed] [Google Scholar]
- 5.Golde T.E., Estus,S., Usiak,M., Younkin,L.H. and Younkin,S.G. (1990) Neuron, 4, 253–267. [DOI] [PubMed] [Google Scholar]
- 6.Neve R.L., Finch,E.A. and Dawes,L.R. (1988) Neuron, 1, 669–677. [DOI] [PubMed] [Google Scholar]
- 7.Cohen M.L., Golde,T.E., Usiak,M.F., Younkin,L.H. and Younkin,S.G. (1988) Proc. Natl Acad. Sci., USA, 85, 1227–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson S.A., McNeill,T., Cordell,B. and Finch,C.E. (1990) Science, 248, 854–857. [DOI] [PubMed] [Google Scholar]
- 9.Schmechel D.E., Goldgaber,D., Burkhart,D.S., Gilbert,J.R., Gajdusek,D.C. and Roses,A.D. (1988) Alzheimer Dis. Assoc. Disord., 2, 96–111. [DOI] [PubMed] [Google Scholar]
- 10.Richards S.-J., Waters,J.J., Beyreuthers,K., Masters,C.L., Wischik,C.M., Sparkman,D.R., White,C.L.,III, Abraham,C.R. and Dunnet,S.B. (1991) EMBO J., 10, 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wirak D.O., Bayney,R., Kundel,C.A., Lee,A., Scangos,G.A., Trapp,B.D. and Unterbeck,A.J. (1991) EMBO J., 10, 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fox N.W., Johnstone,E.M., Ward,K.E., Schrementi,J. and Little,S.P. (1997) Biochem. Biophys. Res. Commun., 240, 759–762. [DOI] [PubMed] [Google Scholar]
- 13.Quitschke W.W., Matthews,J.P., Kraus,R.J. and Vostrov,A.A. (1996) J. Biol. Chem., 271, 22231–22239 [DOI] [PubMed] [Google Scholar]
- 14.Quitschke W.W. (1994) J. Biol. Chem., 269, 21229–21233. [PubMed] [Google Scholar]
- 15.Vostrov A.A. and Quitschke,W.W. (1997) J. Biol. Chem., 272, 33353–33359. [DOI] [PubMed] [Google Scholar]
- 16.Filippova G.N., Fagerlie,S., Klenova,E.M., Myers,C., Dehner,Y., Goodwin,G., Neiman,P.E., Collins,S.J. and Lobanenkov,V.V. (1996) Mol. Cell. Biol., 16, 2802–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lobanenkov V.V., Nicolas,R.H., Adler,V.V., Paterson,H., Klenova,E.M., Polotskaja,A.V. and Goodwin,G.H. (1990) Oncogene, 5, 1743–1753. [PubMed] [Google Scholar]
- 18.Baniahmad A., Steiner,C., Köhne,A.C. and Renkawitz,R. (1990) Cell, 61, 505–514. [DOI] [PubMed] [Google Scholar]
- 19.Burcin M., Arnold,R., Lutz,M., Kaiser,B., Runge,D.F., Lottspeich,F., Filippova,G.N., Lobanenkov,V.V. and Renkawitz,R. (1997) Mol. Cell. Biol., 17, 1281–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bell A.C., West,A.G. and Felsenfeld,G. (1999) Cell, 98, 387–396. [DOI] [PubMed] [Google Scholar]
- 21.Klenova E.M., Nicolas,R.H., Paterson,H.F., Carne,A.F., Heath,C.M., Goodwin,G.H., Neiman,P.E. and Lobanenkov,V.V. (1993) Mol. Cell. Biol., 13, 7612–7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Köhne A.C., Baniahmad,A. and Renkawitz,R. (1993) J. Mol. Biol., 232, 747–755. [DOI] [PubMed] [Google Scholar]
- 23.Kunkel T.A. (1985) Proc. Natl Acad. Sci. USA, 82, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maniatis T., Fritsch,E.F. and Sambrook,J. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 25.Heberlein U. and Tjian,R. (1988) Nature, 331, 410–415. [DOI] [PubMed] [Google Scholar]
- 26.Quitschke W.W. and Goldgaber,D. (1992) J. Biol. Chem., 267, 17362–17368. [PubMed] [Google Scholar]
- 27.Yang Y., Quitschke,W.W., Vostrov,A.A. and Brewer,G.J. (1999) J. Neurochem., 73, 2286–2298. [DOI] [PubMed] [Google Scholar]
- 28.Baniahmad A., Steiner,C., Köhne,A.C. and Renkawitz,R. (1990) Cell, 61, 505–514. [DOI] [PubMed] [Google Scholar]
- 29.Awad T.A., Bigler,J., Ulmer,J.E., Hu,Y.J., Moore,J.M., Lutz,M., Neiman,P.E., Collins,S.J., Renkawitz,R., Lobanenkov,V.V. and Filippova,G.N. (1999) J. Biol. Chem., 274, 27092–27098. [DOI] [PubMed] [Google Scholar]
- 30.Klenova E.M., Nicolas,R.H., U,S., Carne,A.F., Lee,R.E., Lobanenkov,V.V. and Goodwin,G.H. (1997) Nucleic Acids Res., 25, 466–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnold R., Burcin,M., Kaiser,B., Muller,M. and Renkawitz,R. (1996) Nucleic Acids Res., 24, 2640–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bigler J. and Eisenman,R.N. (1995) EMBO J., 14, 5710–5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delgado M.D., Chernukhin,I.V., Bigas,A., Klenova,E.M. and Leon,J. (1999) FEBS Lett., 444, 5–10. [DOI] [PubMed] [Google Scholar]
- 34.Lutz M., Burke,L.J., Barreto,G., Goeman,F., Greb,H., Arnold,R., Schultheiß,H., Brehm,A., Kouzarides,T., Lobanenkov,V. and Renkawitz,R. (2000) Nucleic Acids Res., 28, 1707–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]