

Abstract
DNA topoisomerases and DNA site-specific recombinases are involved in a diverse set of cellular processes but both function by making transient breaks in DNA. Type IB topoisomerases and tyrosine recombinases cleave DNA by transesterification of an active site tyrosine to generate a DNA–3′-phosphotyrosyl–enzyme adduct and a free 5′-hydroxyl (5′-OH). Strand ligation results when the 5′-OH attacks the covalent complex and displaces the enzyme. We describe the synthesis of 3′-phospho-(para-nitrophenyl) oligonucleotides (3′-pNP DNAs), which mimic the natural 3′-phosphotyrosyl intermediate, and demonstrate that such pre-activated strands are substrates for DNA ligation by vaccinia topoisomerase and Cre recombinase. Ligation occurs by direct attack of a 5′-OH strand on the 3′-pNP DNA (i.e., without a covalent protein–DNA intermediate) and generates free para-nitrophenol as a product. The chromogenic DNA substrate allows ligation to be studied in real-time and in the absence of competing cleavage reactions and can be exploited for high-throughput screening of topoisomerase/recombinase inhibitors.
INTRODUCTION
DNA topoisomerases and site-specific DNA recombinases carry out cleavage and ligation reactions involving DNA phosphodiester bonds. Type IB topoisomerases and members of the λ integrase family of recombinases (also called tyrosine recombinases) have similar tertiary structures and reaction mechanisms (1–6). Strand cleavage by these enzymes is the result of transesterification of an active site tyrosine nucleophile to one strand of duplex DNA to generate a covalent DNA–(3′-phosphotyrosyl)–enzyme intermediate and a 5′-hydroxyl (5′-OH) DNA leaving group. Strand ligation is the result of a second transesterification event in which a 5′-OH attacks the covalent intermediate and displaces the enzyme (Fig. 1A). The 3′-covalent adduct is transient because the rate of cleavage (kcl) exceeds the rate of religation (krel). In a typical topoisomerase reaction, the covalently bound monomeric protein releases the 5′-OH strand and permits it to swivel about the opposing phosphodiester on the complementary strand before catalyzing religation, causing a change in DNA linking number. In the case of recombinases, the reactive 5′-OH for religation does not come from the original cleaved DNA strand but from a partner strand cleaved by a second recombinase protomer within a synaptic complex. Paired strand exchanges by recombinases result in the formation and resolution of Holliday junctions.
Figure 1.

Elementary topoisomerase reactions and rationale for suicide substrates. (A) DNA strand cleavage results from attack of an active-site tyrosine residue forming a covalent 3′-phosphotyrosine–DNA intermediate. DNA strand ligation results from attack of the displaced 5′-OH and expulsion of the active site tyrosine. (B) Pre-activated 3′-pNP DNA is illustrated on the right. Enzyme-mediated transesterification of the 5′-OH will expel para-nitrophenol. If a mutant enzyme lacking the tyrosine nucleophile is used, then competing cleavage reactions cannot occur.
These two pathways are not mutually exclusive; many tyrosine recombinases have topoisomerase activity (7–9) and type IB topoisomerases can catalyze intermolecular strand ligation and Holliday junction resolution (10–12). In addition, recent studies have shown that the catalytic repertoire of these enzymes is not limited to DNA strand exchange, but embraces transesterification and ligation reactions involving RNA as well as non-nucleic acid nucleophiles (13–20). These ‘unconventional’ reactions highlight a key theme in the evolution of phosphoryl transfer enzymes, i.e., that relatively subtle changes of the enzyme active site or the structure of the nucleic acid substrate can convert a topoisomerase/recombinase into a DNA endonuclease, an RNA endonuclease or a polynucleotide ligase.
Site-specific DNA recombinases are involved in a large number of cellular functions including integration/excision of mobile DNA elements, control of gene expression, resolution of dimeric DNA chromosomes, and the generation of genetic diversity. Topoisomerases play a major role in maintaining proper DNA topology during transcription and replication, and are targets for anti-tumor drugs such as camptothecin. Mechanistic studies of these important enzymes depend on the ability to accurately measure the forward and reverse transesterification reactions in isolation. For example, suicide cleavage substrates have been of enormous value in studying the structural basis for transesterification chemistry. One class of suicide substrates incorporates a 5′-bridging phosphorothiolate linkage at the scissile phosphate (6,21–24). This modification generates a 5′-sulfyhydryl (5′-SH) leaving strand instead of a 5′-OH following transesterification of the active site tyrosine. Because the 5′-SH is an incompetent nucleophile for the religation reactions, the enzyme–DNA covalent adduct is trapped at the nick.
Substrates that allow exclusive focus on the religation reaction are not as readily available. In some topoisomerase systems it has been possible to measure the rate of strand ligation independent of strand cleavage by assaying the ability of an exogenous oligonucleotide to attack a pre-activated enzyme–DNA covalent complex under single-turnover conditions (25). This analysis is predicated on the ability to form the covalent intermediate in high yield and, ideally, to isolate the enzyme–DNA adduct. These conditions are not readily applicable to recombinases because strand ligation occurs in a synaptic complex of four recombinase protomers and four DNA cleavage sites. In addition, very few systems are sufficiently well-defined kinetically to ensure that the reactions occur under single-turnover conditions.
An alternative approach is to chemically synthesize pre-activated DNA ligation substrates. For example, Sadowsky and colleagues (26,27) demonstrated that Flp recombinase can join DNA strands at a nick without formation of a covalent enzyme–DNA intermediate, provided that the 3′-phosphate (3′-PO4) terminus at the nick is chemically activated by esterification to a tyrosine residue. Mutation of the Flp active site Tyr343 to Phe had no apparent effect on the high efficiency with which such activated ends were sealed. The latter result demonstrated that ligation can be assayed in the absence of competing cleavage reactions, because the free tyrosine product diffuses away from the active site and the Flp Y343F mutant cannot recleave the ligated strand. Jayaram and colleagues (28,29) have since shown that Flp normally acts through a trans cleavage mechanism, whereby one molecule of Flp binds the DNA target site and catalyzes attack by the tyrosine of a second Flp monomer on the scissile phosphate. Thus, the activation of the 3′-end via a phosphodiester to a single tyrosine is essentially equivalent to the natural covalent intermediate formed during FLP-mediated strand transfer.
Cre recombinase and mammalian topoisomerase I are also capable of sealing DNA nicks with 3′-phosphotyrosine/5′-OH termini, albeit much less efficiently than Flp (26,27). Given the crystallographic evidence that Cre and mammalian topoisomerase I normally cleave DNA in cis (1,6), their low efficiency of ligation of a 3′-phosphotyrosine-activated strand may reflect steric clashes of the extra tyrosine at their respective active sites. The capacity of active site mutants of Cre and mammalian topoisomerase I to ligate 3′-phosphotyrosine-activated strands was not examined. It is therefore possible that replacement of tyrosine with a much smaller side chain might free up space to accommodate a DNA-bound tyrosine. Esterification of DNA to lower molecular weight tyrosine analogs might also circumvent steric constraints at the active site.
We have developed a post-synthetic method for attaching a relatively small tyrosine analog, para-nitrophenol, onto the 3′-end of oligonucleotides. We demonstrate here that the resulting pre-activated 3′-phospho-(para-nitrophenyl) oligonucleotides (3′-pNP) support DNA strand ligation by wild-type vaccinia topoisomerase (a prototypal type IB enzyme) and Cre recombinase and as well as by mutated versions containing Phe or Ala in lieu of the nucleophilic tyrosine on the enzyme. Hence, DNA ligation occurs in the absence of any competing DNA cleavage reactions (Fig. 1B). The 3′-pNP substrates are also useful because the ligation reaction releases para-nitrophenol, which can be assayed spectrophotometrically. This important feature allows DNA strand ligation to be assayed in real time and has implications for developing high-throughput assays for elementary topoisomerase reactions.
MATERIALS AND METHODS
Synthesis of 3′-pNP DNA
Oligonucleotides (1 µmol scale) were synthesized using 3′-PO4 CPG (Glen Research, Sterling, VA) and standard DNA phosphoramidites on an ABI 392 automated DNA synthesizer. After the last coupling step, the final 5′-dimethoxytrityl protecting group (5′-DMT) was not removed so that the final oligonucleotide contained a 3′-PO4 but lacked a free 5′-OH. The resin was incubated in concentrated ammonia at 55°C for 12 h and the resulting supernatant was dried in vacuo to a powder. The powder was dissolved in 250 µl of 2 mM MgCl2, 100 mM MES (pH 5.5) and any insoluble material was removed by centrifugation. Aliquots of 200 µl of 3 M para-nitrophenol (Aldrich, Milwaukee, WI) in acetonitrile and 0.048 g of 1-[3-(dimethylamino)propyl]-3-ethycarbodiimide hydrochloride (EDC) (Aldrich) were added sequentially. The resulting two immiscible layers where shaken vigorously in the dark at room temperature to form an emulsion. After 12–16 h, 250 µl of water was added and the aqueous layer was extracted three times with 500 µl each of ethyl acetate. The oligonucleotide was precipitated with the addition of 0.1 vol of 3 M sodium acetate and 3 vol of absolute ethanol. The resulting pellet was resuspended in 250 µl of 80% glacial acetic acid at 0°C and immediately precipitated with the sequential addition of 50 µl of 3 M sodium acetate and 900 µl of absolute ethanol.
The 3′-derivatized DNA (3′-pNP DNA) was purified by reverse phase chromatography on a Hewlett Packard 1050 HPLC using a RP-318 250 mm × 4.6 mm column (Bio-Rad, Hercules, CA) at 1 ml/min, 5→25% acetonitrile gradient (buffered with 100 mM triethylammonium acetate pH 7.0) over 30 min. Peak fractions (0.5 ml) were pooled, concentrated in vacuo, and stored in 1 mM Tris (pH 7.5), 0.1 mM EDTA at –20°C.
Purification of vaccinia topoisomerase
Wild-type (WT) vaccinia topoisomerase and mutated versions Y274F, Y274A and R223A were expressed in Escherichia coli BL21 cells by infection with bacteriophage λCE6 (30) and then purified from a soluble bacterial lysate by phosphocellulose column chromatography. The protein concentrations of the phosphocellulose preparations were determined by using the Bio-Rad dye-binding reagent with bovine serum albumin as the standard.
Topoisomerase substrate preparation
DNA oligonucleotides containing the 3′-pNP were 5′-end-labeled with [γ-32P]ATP and purified by electrophoresis through a 20% polyacrylamide gel. The labeled oligonucleotides were eluted from an excised gel slice and then hybridized to unlabeled downstream oligonucleotides and complementary oligonucleotides at a ratio of 1:4:4. Annealing reaction mixtures containing 0.2 M NaCl and oligonucleotides were heated to 70°C and then slow-cooled to 22°C. For spectrophotometric measurement of the release of para-nitrophenol, the 3′-pNP oligonucleotide was annealed to the downstream oligonucleotide and complementary oligonucleotide at a molar ratio of 1:2:2 in the absence of NaCl. The hybridized DNAs were stored at 4°C.
Purification of Cre recombinase
WT Cre recombinase and mutated versions Y324F, Y324A and Y324V were expressed in E.coli BL21(DE3) cells with the addition of 1 mM IPTG and then purified from a soluble bacterial lysate by phosphocellulose column chromatography as previously described (31).
Recombinase substrate preparation
DNA oligonucleotides containing the 3′-pNP moiety were 5′-end-labeled with [γ-32P]ATP and purified by electrophoresis through a 20% polyacrylamide gel or by passage through a Bio-gel P-10 (Bio-Rad) spin column. The labeled oligonucleotides were hybridized to unlabeled downstream oligonucleotides and complementary oligonucleotides at a ratio of 1:2:10. Annealing reaction mixtures containing 0.1 M KCl and oligonucleotides were heated to 90°C and then slow-cooled to room temperature.
RESULTS
Synthesis of 3′-pNP oligonucleotides
Previously described methods for synthesis of 3′-phosphotyrosyl DNA oligonucleotides relied upon serial 3′ to 5′ addition of standard base-protected nucleotide building blocks to a tyrosine-derivatized resin (32). This approach is limited because other tyrosine analogs cannot be easily attached to the resin and incorporation of different tyrosine analogs would require separate chemical synthesis of different resins. We have investigated the alternative approach of derivatizing oligonucleotides post-synthetically to produce 3′-modified DNA. The advantage of this approach is that a single oligonucleotide can be derivatized with different reagents so that a more diverse set of analogs can be attached to the 3′-end of the DNA.
para-Nitrophenol was chosen for several reasons. First, this analog is significantly smaller than tyrosine (139 versus 181 g/mol). As discussed above, the pre-activated substrate must be accommodated within the enzyme active site and para-nitrophenyl derivatized DNA (3′-pNP) might be expected to cause less steric clash. Second, the electron withdrawing para-nitro moiety makes this analog a better leaving group than tyrosine. Finally, free para-nitrophenol, but not 3′-pNP DNA, absorbs 400–405 nm wavelength light. This allows the ligation reaction to be monitored in real time because the ligation reaction is predicted to generate free para-nitrophenol (Fig. 1B). The 3′-pNP DNA substrate is therefore similar to para-nitrophenyl-thymidine 3′-phosphate, a substrate used to assay spleen phosphodiesterase and enzymes of similar specificity (33).
The post-synthetic approach for modifying the 3′-end of oligonucleotides detailed in Figure 2A was a modification of a protocol used to derivatize nucleotides and oligonucleotides (34). The water-soluble condensing agent, EDC, was used to specifically modify the 3′-PO4 of DNA (Fig. 2A). Using optimized conditions, ∼60% of the 3′-PO4 DNA was converted to 3′-pNP DNA after 12 h. No product was formed when a 3′-OH terminated oligonucleotide was used (data not shown). No detectable side products accumulated after 12 h, however side products were detected by reverse phase HPLC analysis (see below) after 24 h. One potential side reaction is the condensation of a 5′-OH and the 3′-PO4. In order to minimize this reaction, the 5′-DMT group was not cleaved from the oligonucleotide following the last coupling reaction. The 5′-DMT group was removed following the EDC condensation reaction and before subsequent purification steps. The 5′-DMT may also improve the solubility of the oligonucleotide in the condensation reaction.
Figure 2.

Synthesis of 3′-pNP DNA. (A) The 3′-terminal phosphate of an oligonucleotide is illustrated on the left; condensation with para-nitrophenol yields 3′-pNP DNA, illustrated on the right. (B) 3′-PO4 and 3′-pNP DNA (23mer) were resolved by reverse phase chromatography (see Materials and Methods). The resulting chromatogram (A260 versus time) is shown. (C) 5′-end-labeled 23mer containing a 3′-PO42– (lane 1), 3′-pNP (lane 2), or 3′-PO42– and 3′-OH mixture (lane 3) were resolved on a 20% acrylamide, 8 M urea, 0.5× TBE (45 mM Tris–borate, 1.25 mM EDTA) 0.4 mm sequencing gel. The resulting autoradiogram is shown.
The 3′-pNP DNA is more hydrophobic than the starting material and can be purified by reverse phase HPLC. To demonstrate this, equal amounts of 3′-PO4 and purified 3′-pNP DNA were mixed and then separated by reverse phase HPLC. A portion of the resulting chromatogram is shown in Figure 2B. When a sample of the later-eluting material (3′-pNP DNA) was incubated with spleen phosphodiesterase, the reaction product strongly absorbed 400 nm light. Incubation of the sample with venom phosphodiesterase did not generate material that absorbed 400 nm light. These results indicate that para-nitrophenol was attached to the 3′-PO4 (data not shown). We have successfully derivatized and purified oligonucleotides of different sequence composition and lengths (from 6- to 45mer). We estimate that 3′-PO4 and 3′-pNP oligonucleotides up to 50 nt in length could be separated using these methods.
Addition of para-nitrophenol to the DNA 3′-terminus would also be expected to increase the molecular weight of the resulting oligonucleotide. To test this prediction, 3′-PO4 DNA and 3′-pNP DNA (25mer) were 5′-end-labeled and analyzed by electrophoresis through a 20% polyacrylamide gel. An autoradiogram of the gel is shown in Figure 2C. As expected, the 5′-labeled 3′-pNP DNA (lane 2) migrated more slowly through the gel than 5′-labeled 3′-PO4 DNA (lane 1). In lane 3, a mixture of 5′-labeled 3′-PO4 and 3′-OH DNAs was resolved. The 3′-OH DNA migrated slower than the 3′-PO4 DNA and only slightly faster than the 3′-pNP DNA. This result demonstrates that the slower mobility of 3′-pNP versus 3′-PO4 DNA resulted from an increase in molecular weight and a decrease in charge (3′-PO4 versus 3′-phosphodiester). This pattern is very similar to polyacrylamide gel electrophoresis (PAGE) analysis of 3′-phosphotyrosine derivatized oligonucleotides (35) and is expected from the relatively small size of the para-nitrophenyl group.
Vaccinia topoisomerase catalyzes ligation of a 3′-pNP/5′-OH nick
Vaccinia topoisomerase is a prototype of the type IB topoisomerase family (36). The poxvirus topoisomerase is distinguished from the eukaryotic nuclear topoisomerase I by its compact size (314 amino acids) and its site-specificity in DNA transesterification. Vaccinia topoisomerase binds and cleaves duplex DNA at a pentapyrimidine target sequence 5′-(T/C)CCTT↓ (37). The T↓ nucleotide is linked to Tyr274 of the enzyme. The individual rate constants for cleavage and religation of DNA containing a single CCCTT target site have been measured under single-turnover and equilibrium conditions (25,38–40). Indeed, vaccinia topoisomerase is the only member of the type IB topoisomerase/tyrosine recombinase superfamily for which a detailed kinetic scheme is available.
Can vaccinia topoisomerase catalyze attack of a properly positioned 5′-OH strand on a CCCTTp target site that has been chemically pre-activated by esterification to para-nitrophenol (3′-pNP)? To test if this reaction was possible, a 5′-32P-labeled 18mer 3′-pNP oligonucleotide pCATATCCGTGTCGCCCTTpNP was prepared and hybridized to a complementary 60mer strand and a 5′-OH 18mer acceptor strand to form the nicked duplex substrate shown in Figure 3. Reaction of this molecule with a 5-fold molar excess of WT topoisomerase for 24 h at 37°C resulted in the formation of a novel radiolabeled 36mer ligation product that was well-resolved by PAGE from the input 32P-labeled 18mer strand (Fig. 3A). Digestion of the reaction products with proteinase K prior to PAGE resulted in the appearance of a doublet of labeled species migrating at ∼20 nt. This cluster corresponded to the 5′-32P-labeled 18mer strand covalently linked to Tyr274 plus one or more flanking amino acids (18). Note that the covalent adduct of full-length topoisomerase bound to 18mer DNA did not migrate into the gel and was therefore not visualized in Figure 3. We did not detect the covalent DNA–peptide adducts when the reaction mixture was adjusted to 0.5 M NaCl after the reaction with topoisomerase and immediately prior to proteinase K digestion (Fig. 3B). It is well documented that high ionic strength will elicit strand closure by topoisomerase bound to an equilibrium CCCTT substrate, by virtue of instantaneous salt-mediated dissociation of the topoisomerase from DNA as soon as it catalyzes strand religation (40,41). The apparent decay of the covalent adduct upon transient exposure to NaCl after prolonged reaction with the 3′-pNP/5′-OH substrate underscores that the enzyme remained catalytically active. This experiment showed that vaccinia topoisomerase can ligate a nick containing a 3′-pNP phosphodiester.
Figure 3.
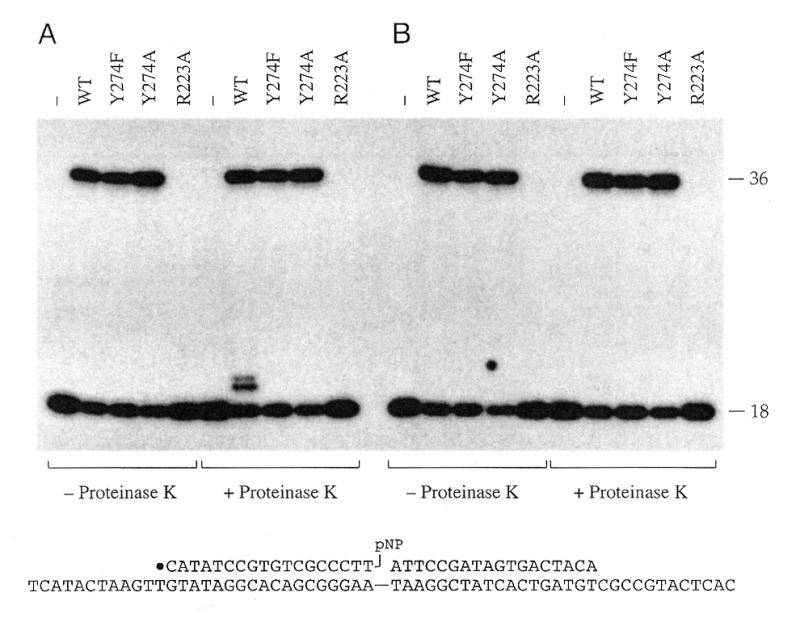
Ligation activity of vaccinia topoisomerase does not require the active site nucleophile Tyr274, but requires catalytic residue Arg223. Reaction mixtures containing (per 20 µl) 50 mM Tris–HCl (pH 7.5), 0.5 pmol of the nicked 3′-pNP/5′-OH DNA substrate (illustrated at the bottom of the figure), and 2.5 pmol of topoisomerase (WT, Y274F, Y274A or R223A) were incubated at 37°C for 24 h. Topoisomerase was omitted from control reaction mixtures (–). Duplicate aliquots (20 µl) were then withdrawn and either quenched immediately with SDS (A) or adjusted to 0.5 M NaCl and incubated at 22°C for 30 s prior to quenching with SDS (B). The samples were then digested with proteinase K where indicated. The DNA was ethanol-precipitated and then analyzed by electrophoresis through a 15% polyacrylamide gel containing 7 M urea in TBE (90 mM Tris–borate, 2.5 mM EDTA). An autoradiogram of the gel is shown. The positions of the 5′-32P-labeled 18mer 3′-pNP strand and the 36mer ligation product are indicated. Note that labeled DNA covalently linked to the full-length topoisomerase polypeptide did not enter the gel and was therefore not seen in the autoradiogram.
Ligation of a 3′-pNP/5′-OH nick does not require the active site tyrosine
The vaccinia topoisomerase mutants Y274F and Y274A bind to the CCCTT target site, but absolutely cannot transesterify to the scissile phosphodiester to form a covalent intermediate. Therefore, it was striking that the Y274F and Y274A proteins did catalyze the ligation of the nicked 3′-pNP/5′-OH substrate to form a 32P-labeled 36mer product. The extent of strand joining during a 24 h reaction was grossly comparable for WT, Y274F and Y274A proteins (Fig. 3A). The DNA–peptide adduct formed after proteolytic digestion of the WT topoisomerase reaction products was not detected when the Y274F and Y274A ligation products were treated with proteinase K (Fig. 3A), consistent with the identification of the ∼20mer cluster as a degradation product of the covalent topoisomerase–DNA complex.
At 125 nM enzyme and 25 nmol DNA, the Y274A ligation reaction displayed pseudo first-order kinetics (Fig. 4). An endpoint of 70% of the input labeled 3′-pNP strand converted to 36mer was attained at 18–24 h. The data fit well to a single exponential over three half-lives with an apparent rate constant for 3′-pNP/5′-OH ligation of 5 × 10–5 s–1. This value is at least four orders of magnitude slower than the rate constant for topoisomerase-catalyzed attack of 5′-OH DNA on the covalent DNA–(3′-phosphotyrosyl)–enzyme intermediate (krel ∼ 1 s–1). The ligation reaction of WT topoisomerase attained a similar endpoint and the apparent rate constant (3.3 × 10–5 s–1) was about half that of the Y274A mutant (Fig. 4). Thus, elimination of the bulky tyrosine side chain did not significantly enhance the rate of 3′-pNP/5′-OH ligation. We surmise that other steric or conformational factors serve to constrain the ligation reaction rate (see Discussion). The initial rate of the ligation reaction of Y274F was one-half that of WT topoisomerase and 30% that of Y274A. We surmise from this result that the hydrophobic phenyl moiety imposes a slightly less favorable environment than a polar phenol side chain.
Figure 4.

Kinetics of ligation by vaccinia topoisomerase. Reaction mixtures containing (per 20 µl) 50 mM Tris–HCl (pH 7.5), 0.5 pmol of nicked 3′-pNP/5′-OH DNA substrate and 2.5 pmol of topoisomerase (WT, Y274F or Y274A) were incubated at 37°C. The reactions were initiated by addition of topoisomerase. Aliquots were withdrawn at the times specified and incubated with NaCl to 0.5 M final concentration for 30 s. The reactions were then quenched by adding SDS to 0.5%. The samples analyzed by electrophoresis through a 15% polyacrylamide gel containing 7 M urea in TBE. The extent of ligation [36mer/(18mer + 36mer)] was quantitated by scanning the gel using a FUJIX Bio-Imaging Analyzer and plotted as a function of time.
These experiments substantiate a reaction pathway by which topoisomerase catalyzes direct attack of the 5′-OH on the 3′-pNP without forming a covalent enzyme–substrate intermediate.
Catalytic residue Arg223 is required for 3′-pNP/5′-OH ligation
In order to address if the 3′-pNP/5′-OH ligation reaction is catalyzed by the same functional groups on the enzyme that mediate the standard topoisomerase transesterification reactions (cleavage and ligation), we tested the 3′-pNP/5′ OH ligase activity of mutant protein R223A. Alanine substitution at Arg223 elicits a 10–5 decrement in the rate of attack of a 5′-OH oligonucleotide on the covalent intermediate. The R223A mutation has no effect on the non-covalent binding of topoisomerase to DNA. Activity is restored when lysine is introduced at this position (41). It was proposed, based on these mutational data, that the side chain of Arg223 of vaccinia topoisomerase makes an essential contact with one phosphate oxygen in the transition state. In the Cre–DNA cocrystal, the side chain of Arg292 (equivalent to Arg223 of vaccinia topoisomerase) makes a monodentate contact to a non-bridging phosphate oxygen in the covalent and non-covalent Cre–DNA complexes (1).
We found that the vaccinia R223A protein was inactive in polynucleotide ligation on the nicked 3′-pNP/5′-OH duplex substrate (Fig. 3). R223A did not accumulate covalent adduct either. We surmise that the mechanism of activation of the scissile phosphate for nucleophilic attack during 3′-pNP/5′-OH strand joining is the same as that of cleavage/religation.
Release of para-nitrophenol
Delineation of an enzymatic reaction requires that all products be identified. We have shown above that vaccinia topoisomerase catalyzes ligation of a nick with 3′-pNP and 5′-OH termini. The proposed reaction pathway would be expected to generate free para-nitrophenol in molar equivalency to the DNA strand transfer product. Free para-nitrophenol is a chromophore that absorbs light in the visual range. Release of para-nitrophenol during reaction of 50 µM unlabeled nicked 3′-pNP/5′-OH DNA with 100 µM WT topoisomerase was thereby monitored by an increase in A405 over time. The reaction proceeded to an extent that ∼60% of the 3′-pNP DNA was converted to free para-nitrophenol product during an 18 h reaction (Fig. 5). The single turnover kinetics of para-nitrophenol release (Fig. 5) closely paralleled the kinetics of DNA ligation (Fig. 4).
Figure 5.

Release of para-nitrophenol. Reaction mixtures containing 50 mM Tris–HCl (pH 7.5), 100 mM NaCl, 50 µM of unlabeled nicked 3′-pNP/5′-OH DNA and 100 µM of WT topoisomerase were incubated at 37°C. The reactions were initiated by addition of topoisomerase. Aliquots (10 µl) were withdrawn at the times specified and optical densities at 405 nm were measured using an ultra micro volume cell (5–7 µl working volume) (Amersham Pharmacia, Little Chalfont, UK). The yield of para-nitrophenol is plotted as a function of time.
The use of chromogenic substrates has practical implications for real-time assays of topoisomerase catalysis and high-throughput screening for candidate inhibitors. A drawback of para-nitrophenol as the chromophore is that relatively high concentrations (>10 µM) are required for spectrophotometric detection (E400nm = 18 300 M–1cm–1), which thereby mandates the use of high initial substrate and enzyme concentrations. This issue may be circumvented by synthesis and testing of additional 3′-modified chromogenic or fluorogenic DNAs that afford enhanced sensitivity in detection of the chromophore/fluorophore leaving groups.
Transesterification of topoisomerase to 3′-pNP in the absence of ligation
We have shown that 3′-pNP/5′-OH ligation need not proceed through a covalent topoisomerase–DNA intermediate. Yet, WT topoisomerase forms a covalent adduct during the ligation reaction. Does this complex represent a potential reaction intermediate in the ligation pathway (albeit not an obligate one) or is the covalent complex exclusively a by-product arising by ex post facto cleavage of the ligated duplex DNA product? In essence, the question is whether vaccinia topoisomerase can directly transesterify to the 3′-pNP DNA strand in the absence of prior strand closure. To address this scenario, we replaced the 5′-OH acceptor strand at the nick with a 5′-SH strand of identical sequence. The 5′-SH terminus is at least four orders of magnitude less effective than a 5′-OH as a nucleophile in the standard pathway of DNA strand joining by vaccinia topoisomerase (24).
Parallel reactions of 3′-pNP/5′-SH and 3′-pNP/5′-OH substrates with Y274A and Y274F showed that the 5′-thiol was completely inactive as a nucleophile in attack on the 3′-pNP strand (Fig. 6). (Note that any ligation of the 5′-SH strand would have been detectable because the mutant enzymes cannot recleave the ligation product.) WT topoisomerase again formed both a ligated product and a covalent DNA–protein adduct during its reaction with the 3′-pNP/5′-OH DNA. The instructive finding was that WT topoisomerase reacted with the 3′-pNP/5′-SH nick to yield low levels of the covalent adduct, but formed no ligation product (Fig. 6). We conclude that WT vaccinia topoisomerase can directly transesterify to the 3′-pNP end according to the reaction scheme illustrated in Figure 6. If one assumes that the free para-nitrophenol product is released from the covalent complex (a likely prospect given that the topoisomerase releases even a 5′-OH DNA leaving group unless it is annealed to the complementary non-scissile strand), then the observed lower extent of accumulation of covalent adduct on 3′-pNP/5′-SH versus 3′-pNP/5′-OH DNA implies that most of the covalent adduct generated during incubation with 3′-pNP/5′-OH DNA arises via cleavage of the ligated duplex product.
Figure 6.

Transesterification to 3′-pNP in the absence of ligation. Reaction mixtures containing (per 20 µl) 50 mM Tris–HCl (pH 7.5), 0.5 pmol of nicked 3′-pNP DNA substrates (containing either 5′-SH or 5′-OH at the nick) and 2.5 pmol of topoisomerase (WT, Y274F or Y274A) were incubated at 37°C for 24 h. The reactions were quenched with SDS and the mixtures were digested with proteinase K. Control reactions lacked topoisomerase (–). The DNA was ethanol-precipitated and analyzed by electrophoresis through a 20% polyacrylamide gel containing 7 M urea in TBE. An autoradiogram of the gel is shown. The covalent DNA–peptide adducts are indicated by the parenthesis on the right. A proposed reaction pathway is illustrated on the left.
3′-pNP/5′-OH ligation by Cre recombinase
Cre-mediated strand cleavage and ligation reactions occur at specific phosphodiester bonds within a well-defined recombination site, termed a lox site. A single lox site contains two inverted Cre binding sites; one Cre monomer directs the cleavage and ligation of the ‘top’ strand and the second Cre monomer directs the cleavage and ligation of the complementary ‘bottom’ strand (42,43). In order to test whether 3′-pNP DNA could be joined by a tyrosine recombinase, a 25mer 5′-32P-labeled oligonucleotide containing a 3′-pNP moiety was hybridized with two other oligonucleotides to form a singly nicked ‘top’ strand lox site. The resulting nick placed the 25mer 3′-pNP and a 5′-OH 22mer strand at one site of Cre-mediated catalysis (Fig. 7). The lox site was incubated in the absence (lane 1) or presence (lane 2) of CreY324A and the reaction products were resolved by PAGE. CreY324A-catalyzed displacement of the nitrophenyl group by the 5′-OH yielded a 47-nt strand transfer product (Fig. 7, lane 2). In order to verify that the ligation product was the result of strand transfer to the 5′-OH oligonucleotide at the nick, a second 3′-pNP/5′-OH lox site was constructed with a 5′-OH 27mer acceptor strand at the nick. In this instance, reaction with CreY324A yielded a larger 52mer ligation product (Fig. 7, lane 3). Reaction of CreY324A with a different nicked lox site containing 3′-PO4 and 5′-OH termini at the nick resulted in no detectable strand transfer (Fig. 7, lane 4). The fact that a Cre mutant that lacks a tyrosine nucleophile catalyzed this ligation reaction strongly argues that ligation must result from the direct attack of the 5′-OH from the acceptor oligonucleotide on the 3′-pNP.
Figure 7.
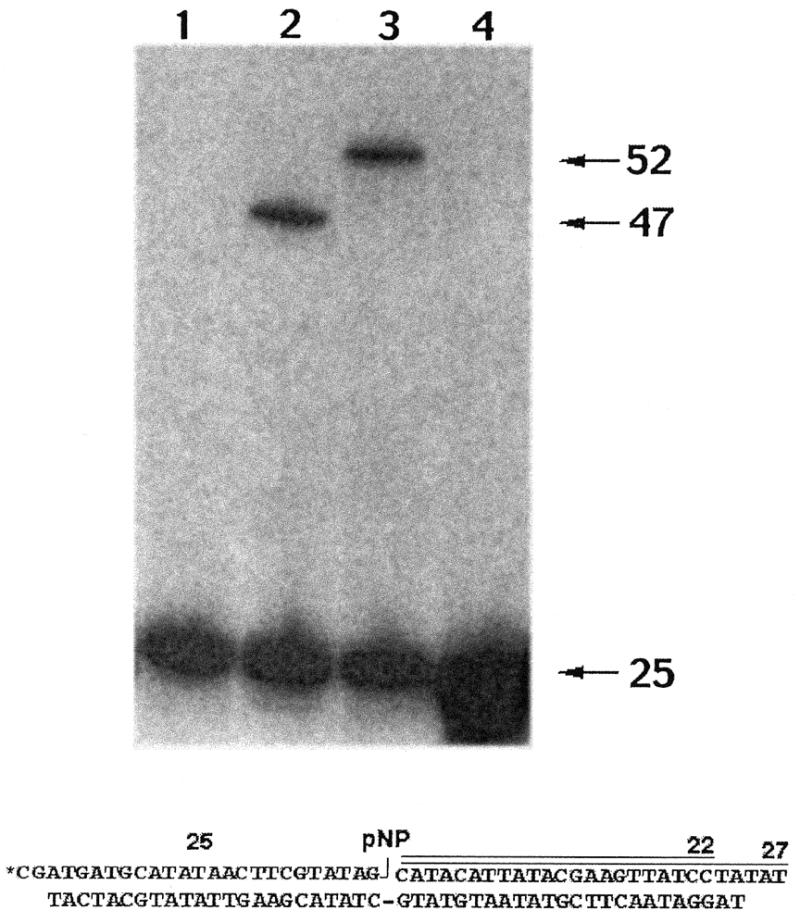
Ligation activity of Cre recombinase. Reaction mixtures containing (per 20 µl) 100 mM Tris–HCl (pH 7.5), 4 mM spermidine, 4 µg/ml BSA, 10% glycerol, 100 nM 3′-pNP/5′-OH or 3′-PO4/5′-OH DNA substrate as specified and 2.5 µM of Cre recombinase Y324A were incubated at 30°C for 2 h. Recombinase was omitted from control reaction mixtures (lane 1). Reactions analyzed in lanes 2 and 3 were identical except that different 5′-OH oligonucleotides were used to form the lox site-containing 3′-pNP/5′-OH substrates depicted at the bottom of the figure; the substrate in lane 2 was prepared with a 22-nt acceptor oligonucleotide whereas the substrate in lane 3 was formed with a 27-nt acceptor stand. Reactions were quenched and the products resolved on a 15% polyacrylamide gel containing 8 M urea in TBE. An autoradiogram of the gel is shown. The positions of the 5′-32P-labeled 25mer 3′-pNP substrate strand and the 47mer or 52mer ligation products are indicated on the right.
As described above, the chemically synthesized 3′-pNP moiety acts as a leaving group in the ligation transesterification reaction. Although the resulting para-nitrophenol presumably diffuses away from the active site, the nitrophenyl group must be accommodated within the active site of the enzyme prior to displacement. We therefore tested the ability of different active site mutants that would be predicted to create more space to accommodate the nitrophenyl group to catalyze strand ligation. 5′-End labeled oligonucleotides containing a 3′-pNP moiety were hybridized to form a nicked lox site as described above. This pre-activated lox site was then incubated with WT Cre, CreY324A, CreY324V and CreY324F and the amount of ligated product was measured as a function of time (Fig. 8). The results demonstrate that the wild-type and all three mutants catalyze ligation at the same rate. Forty percent of the substrate was converted to ligated product after an overnight incubation (data not shown). It is also important to note that covalent Cre–DNA intermediates can be easily detected using 5′-end-labeled phosphorothiolate suicide substrates (21), however no Cre–DNA adducts could be detected when WT Cre recombinase was incubated with 5′-end-labeled 3′-pNP DNA (data not shown).
Figure 8.
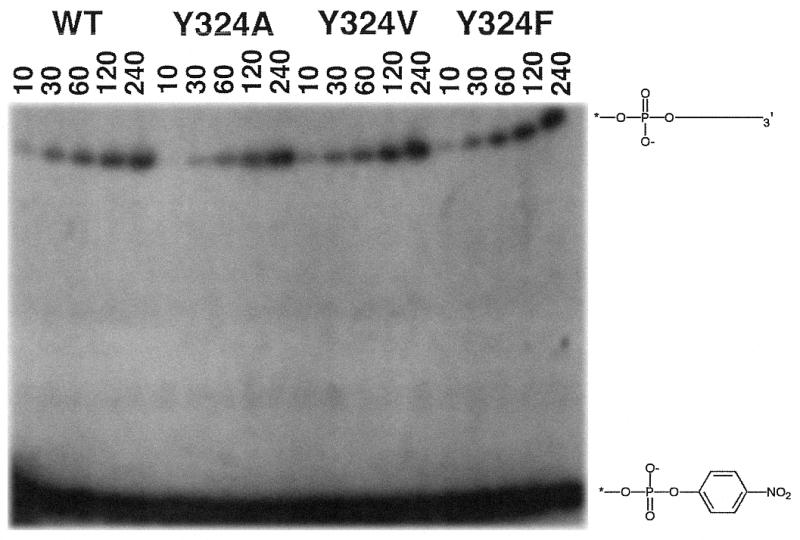
Ligation activity of Cre recombinase does not require the active site nucleophile Tyr324. Reaction mixtures containing (per 40 µl) 100 mM Tris–HCl (pH 7.5), 4 mM spermidine, 4 µg/ml BSA, 10% glycerol, 100 nM 3′-pNP/5′-OH DNA substrate and 2.5 µM Cre recombinase (WT, CreY324A, CreY324V or CreY324F) were incubated at 30°C. The reactions were initiated by addition of recombinase and aliquots (7.5 µl) were withdrawn at the times specified and quenched by the addition of an equal volume of 8 M urea. The samples were analyzed by electrophoresis through a 15% polyacrylamide gel containing 8 M urea in TBE. The resulting autoradiogram is shown. The position of 3′-pNP DNA is shown at the bottom of the autoradiogram and the position of ligated product is illustrated above.
DISCUSSION
We have described the synthesis of oligonucleotides containing a 3′-phospho-(para-nitrophenyl) moiety. These oligonucleotides (3′-pNP DNA) mimic the 3′-phosphotyrosine adducts formed when type IB topoisomerases and tyrosine recombinases cleave DNA except that the tyrosine analog (para-nitrophenyl) is not covalently attached to the enzyme. Vaccinia topoisomerase and Cre recombinase can catalyze the attack of a 5′-OH DNA on 3′-pNP DNA to displace para-nitrophenol and ligate the DNA backbone. Mutated versions of vaccinia topoisomerase and Cre that lack the tyrosine nucleophile, and therefore cannot form a covalent intermediate, still catalyze this ligation reaction with kinetics similar to the respective wild-type enzymes. These results demonstrate that 3′-pNP DNA substrates can be used as single-turnover ligation substrates because DNA strand transfer can be assayed in the absence of any competing DNA cleavage reactions.
It is possible to measure the rate of ligation by vaccinia topoisomerase because the enzyme–DNA covalent complex can be recovered in high yield. However, this is not easily accomplished for tyrosine recombinases. During a complete recombination reaction, four phosphodiester bonds are cleaved and four new DNA phosphodiester bonds are formed. These reactions occur in a complex of at least four different recombinase monomers. It is therefore very difficult to pre-form functional enzyme–DNA adducts to assay DNA strand ligation. In addition, because the cleavage/ligation transesterification reactions are isoenergenic and occur in a synaptic complex, ligation intermediates that form may not be detected because competing cleavage reactions may reform starting material, or another reaction product along the reaction pathway. 3′-pNP DNA substrates solve these problems because the substrates are chemically synthesized and can be introduced into the reaction as ‘nicked’ substrates. Most importantly, 3′-pNP/5′-OH nicked substrates allow individual ligation reaction products to accumulate because subsequent tyrosine-mediated cleavage reactions are not possible if the ligation is performed by a mutant enzyme that lacks a tyrosine nucleophile.
There are two caveats to this conclusion. First, it is possible that the released para-nitrophenol can re-attack the ligated DNA product thereby allowing subsequent cleavage reactions by regenerating an activated phosphodiester. Indeed, Lee and Jayaram have demonstrated that Flp recombinase tyrosine mutants can use para-nitrophenol as an exogenous nucleophile in DNA cleavage reactions (29). However, these reactions are inefficient and require free concentrations of 1–10 mM para-nitrophenol (29). In the experiments described above, the highest concentration of free para-nitrophenol is 0.00004 mM. It is also important to note that, unlike Cre and vaccinia topoisomerase, Flp-catalyzed DNA cleavage occurs in trans; the tyrosine residue used to cleave the phosphodiester backbone comes from an Flp protomer bound to a second partner DNA substrate. One would therefore predict that Flp, unlike Cre or vaccinia toposiomerase, would be able to use exogenous trans-cleaving nucleophiles. It has been clearly demonstrated that vaccinia topoisomerase cannot use exogenous nucleophiles for DNA cleavage reactions (20).
Second, it is theoretically possible that when a tyrosine mutant is used to catalyze ligation, a different enzyme-bound nucleophile attacks the 3′-pNP DNA or can attack the ligated DNA to form an activated phosphodiester for subsequent reactions. Many results argue against such an aberrant side reaction. For example, no enzyme–DNA adducts could be detected for the tyrosine mutants described using 5′-bridging phosphorothiolate (21) cleavage suicide substrates (data not shown). In addition, covalent adducts were easily detected when WT vaccinia topoisomerase was used but no covalent adducts could be detected when a variety of different tyrosine mutants were used (Fig. 3). The same result was even obtained when ligation was blocked by replacing the 5′-OH with a 5′-SH at the nick (Fig. 6). If a previously unidentified enzyme-bound nucleophile was attacking the 3′-pNP linkage or ligated DNA product, a covalent topoisomerase–DNA adduct would be expected to accumulate under this circumstance. In addition, the fact that vaccinia topoisomerase R223A exhibited no ligation activity (Fig. 3) argues that the 3′-pNP/5′-OH ligation reaction is catalyzed by the same functional groups on the enzyme that mediate the standard topoisomerase transesterification reactions. Finally, because the rate of Cre catalyzed 3′-pNP/5′-OH ligation is the same when wild-type or tyrosine mutants are used, it is unlikely that a previously unidentified side reaction occurs specifically in the tyrosine-deficient reaction.
It is worth emphasizing that the rate of vaccinia topoisomerase-catalyzed 3′-pNP/5′-OH ligation (5 × 10–5 s–1) is at least four orders of magnitude slower than the rate constant for topoisomerase-catalyzed attack of 5′-OH DNA on the covalent DNA-(3′-phosphotyrosyl)–enzyme intermediate (krel ∼ 1 s–1). As discussed above, it is not yet possible to make this same comparison for any tyrosine recombinase because these systems are not kinetically well-defined and recombinase–DNA complexes cannot be reliably assayed. The simplest explanation for the observed slow rate of 3′-pNP/5′-OH ligation is that steric constraints within the active site prevent the para-nitrophenyl group from achieving an optimal fit relative to the other catalytic side chains and/or an optimal apical orientation of the para-nitrophenyl leaving group relative to the attacking 5′-OH DNA nucleophile. Another possible explanation is that the enzyme bound 3′-pNP substrate must go through a slow conformational transition to place the para-nitrophenyl group in the active site (e.g., if the para-nitrophenyl ring intercalates into the nick instead of projecting out of the DNA helix). This conformational transition would not be necessary in the normal reaction because the positioning is certainly determined by the covalent attachment of the tyrosine to the enzyme.
The para-nitrophenyl group is relatively small and we entertained the hypothesis that mutations that would better accommodate 3′-pNP within the active site (topoisomerase Y274A and CreY324A) might improve the rate of 3′-pNP ligation. Clearly this is not the case. We cannot rule out the possibility that the smaller side chains in lieu of tyrosine cause unexpected distortions in the active site that inhibit catalysis per se and thereby mask salutary effects on 3′-pNP binding.
The DNA 3′-pNP strand transfer reaction described herein differs from the usual topoisomerase/recombinase-mediated transesterifications in that it occurs at a pre-existing nick. A cardinal feature of enzymatic nick-joining reactions is the covalent modification of one side of the nick to activate it for attack by the opposing hydroxyl. Whereas vaccinia topoisomerase ligated very slowly when the activating moiety at the nick is a 3′-pNP, it is much more active when the nick is activated by a 2′,3′-cyclic phosphodiester (17). For example, sealing of a 2′,3′-cyclic phosphate (2′,3′>p)/5′-OH nick proceeds to the reaction endpoint in <10 min, compared to ∼18 h for a 3′-pNP/5-OH nick. Although the active site tyrosine is not essential for ligation of a nick with a 2′,3′>p, its presence does accelerate the reaction rate. Catalysis through the covalent intermediate is ∼20-fold faster than direct attack of the 5′-OH on the 2′,3′>p (17). This is in contrast to the situation described here for 3′-pNP activated substrates, in which the tyrosine does not contribute to the observed ligation rate. A simple explanation for why the 2′,3′>p is a much better substrate for ligation by topoisomerase is that there is little or no steric clash of a cyclic phosphate in the active site.
The fact that vaccinia topoisomerase can catalyze the release of para-nitrophenol (Fig. 5) from 3′-pNP DNA has important implications for developing high throughput assays for new anti-cancer drugs. This conclusion follows from the observation that eukaryotic topoisomerases have been demonstrated to be valuable targets for anti-cancer drugs (44). A structurally diverse set of anti-cancer compounds including camptothecin, some minor groove binding ligands, some indolocarbazole derivatives and protoberberine alkaloids, appear to function by specifically blocking or slowing the rate of religation following strand cleavage by human topoisomerase I. As a result, these topoisomerase poisons stabilize the normally transient 3′-phosphotyrosyl covalent complex and turn the topoisomerase into a DNA damaging agent. Clearly, new drugs that also block religation would be expected to slow the release of para-nitrophenol from 3′-pNP DNA. Because para-nitrophenol can be assayed colorimetrically, a large number of individual compounds or mixtures can be quickly screened.
Acknowledgments
ACKNOWLEDGEMENTS
This work was supported by grants GM46330 (to S.S.) and GM58596 (to A.B.B.) from the National Institutes of Health.
REFERENCES
- 1.Guo F., Gopaul,D.N. and Van Duyne,G.D. (1997) Nature, 389, 40–46. [DOI] [PubMed] [Google Scholar]
- 2.Kwon H.J., Tirumalai,R., Landy,A. and Ellenberger,T. (1997) Science, 276, 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Subramanya H.S., Arciszewska,L.K., Baker,R.A., Bird,L.E., Sherratt,D.J. and Wigley,D.B. (1997) EMBO J., 16, 5178–5187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hickman A.B., Waninger,S., Scocca,J.J. and Dyda,F. (1997) Cell, 89, 227–237. [DOI] [PubMed] [Google Scholar]
- 5.Cheng C., Kussie,P., Pavletich,N. and Shuman,S. (1998) Cell, 92, 841–850. [DOI] [PubMed] [Google Scholar]
- 6.Redinbo M.R., Stewart,L., Kuhn,P., Champoux,J.J. and Hol,W.G.J. (1998) Science, 279, 1504–1513. [DOI] [PubMed] [Google Scholar]
- 7.Kikuchi Y. and Nash,H.A. (1979) Proc. Natl Acad. Sci. USA, 76, 3760–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abremski K., Frommer,B. and Hoess,R.H. (1986) J. Mol. Biol., 192, 17–26. [DOI] [PubMed] [Google Scholar]
- 9.Cornet F., Hallet,B. and Sherratt,D.J. (1997) J. Biol. Chem., 272, 21927–21931. [DOI] [PubMed] [Google Scholar]
- 10.Shuman S. (1992) J. Biol. Chem., 267, 8620–8627. [PubMed] [Google Scholar]
- 11.Shuman S. (1992) J. Biol. Chem., 267, 16755–16758. [PubMed] [Google Scholar]
- 12.Sekiguchi J., Seeman,N.C. and Shuman,S. (1996) Proc. Natl Acad. Sci. USA, 93, 785–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christiansen K., Knudsen,B.R. and Westergaard,O. (1994) J. Biol. Chem., 269, 11367–11373. [PubMed] [Google Scholar]
- 14.Petersen B.Ø. and Shuman,S. (1997) Nucleic Acids Res., 25, 2091–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sekiguchi J. and Shuman,S. (1997) Mol. Cell, 1, 89–97. [DOI] [PubMed] [Google Scholar]
- 16.Sekiguchi J., Cheng,C. and Shuman,S. (1997) J. Biol. Chem., 272, 15721–15728. [DOI] [PubMed] [Google Scholar]
- 17.Shuman S. (1998) Mol. Cell, 1, 741–748. [DOI] [PubMed] [Google Scholar]
- 18.Wittschieben J., Petersen,B.Ø. and Shuman,S. (1998) Nucleic Acids Res., 26, 490–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lisby M., Krogh,B.O., Boege,F., Westergaard,O. and Knudsen,B.R. (1998) Biochemistry, 37, 10815–10827. [DOI] [PubMed] [Google Scholar]
- 20.Krogh B.O. and Shuman,S. (2000) Biochemistry, 39, 6422–6432. [DOI] [PubMed] [Google Scholar]
- 21.Burgin A.B., Huizenga,B.H. and Nash,H.A. (1995) Nucleic Acids Res., 15, 2973–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burgin A.B. and Nash,H.A. (1995) Curr. Biol., 5, 1312–1321. [DOI] [PubMed] [Google Scholar]
- 23.Hwang Y., Burgin,A. and Bushman,F. (1999) J. Biol. Chem., 274, 9160–9168. [DOI] [PubMed] [Google Scholar]
- 24.Krogh B.O., Cheng,C., Burgin,A. and Shuman,S. (1999) Virology, 264, 441–451. [DOI] [PubMed] [Google Scholar]
- 25.Stivers J.T., Shuman,S. and Mildvan,A.S. (1994) Biochemistry, 33, 327–339. [DOI] [PubMed] [Google Scholar]
- 26.Pan G. and Sadowski,P.D. (1992) J. Biol. Chem., 267, 12397–12399. [PubMed] [Google Scholar]
- 27.Pan G., Leutke,K., Juby,C.D., Brousseau,R. and Sadowski,P. (1993) J. Biol. Chem., 268, 3683–3689. [PubMed] [Google Scholar]
- 28.Chen J., Lee,J. and Jayaram,M. (1992) Cell, 69, 647–658. [DOI] [PubMed] [Google Scholar]
- 29.Lee J. and Jayaram,M. (1993) J. Biol. Chem., 268, 17564–17579. [PubMed] [Google Scholar]
- 30.Shuman S., Golder,M. and Moss,B. (1988) J. Biol. Chem., 263, 16401–16407. [PubMed] [Google Scholar]
- 31.Abremski K. and Hoess,R. (1984) J. Biol. Chem., 259, 1509–1514. [PubMed] [Google Scholar]
- 32.Zhao B.P., Panigrahi,G.B., Sadowski,P.D. and Krepinsky,J.J. (1996) Tetrahedron Lett., 37, 3093–3096. [Google Scholar]
- 33.Razzell W.E. (1963) In Colowick,S.P. and Kaplan,N.O. (eds), Methods in Enzymology. Academic Press, New York and London, Vol. VI, pp. 236–258.
- 34.Shabarova Z. (1988) Biochimie, 70, 1323–1334. [DOI] [PubMed] [Google Scholar]
- 35.Yang S., Burgin,A., Huizenga,B., Yao,K. and Nash,H. (1996) Proc. Natl Acad. Sci. USA, 93, 11534–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shuman S. (1998) Biochim. Biophys. Acta, 1400, 321–337. [DOI] [PubMed] [Google Scholar]
- 37.Shuman S. and Prescott,J. (1990) J. Biol. Chem., 265, 17826–17836. [PubMed] [Google Scholar]
- 38.Stivers J.T., Shuman,S. and Mildvan,A.S. (1994) Biochemistry, 33, 15449–15458. [DOI] [PubMed] [Google Scholar]
- 39.Stivers J.T., Jagadeesh,G.J., Nawrot,B., Stec,W.J. and Shuman,S. (2000) Biochemistry, 39, 5561–5572. [DOI] [PubMed] [Google Scholar]
- 40.Wittschieben J. and Shuman,S. (1997) Nucleic Acids Res., 25, 3001–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng C., Wang,L.K., Sekiguchi,J. and Shuman,S. (1997) J. Biol. Chem., 272, 8263–8269. [DOI] [PubMed] [Google Scholar]
- 42.Landy A. (1999) Proc. Natl Acad. Sci. USA, 96, 7122–7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoess R., Wierzbicki,A. and Abremski,K. (1987) Proc. Natl Acad. Sci. USA, 84, 6840–6844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang H.K., Morris-Natsche,S.L. and Lee,K.H. (1997) Med. Res. Rev., 17, 367–425. [DOI] [PubMed] [Google Scholar]