


Abstract
We have used GET Recombination, an inducible homologous recombination system for Escherichia coli, to insert one of the most common thalassaemia mutations into the intact β-globin locus in a second generation BAC vector. We first inserted a PCR fragment carrying the tetracycline resistance gene (TetR) into the β-globin gene. All recombinant clones examined contained the TetR gene at the correct target site. Next, a PCR fragment with the IVS I-110 G→A splicing mutation but no selectable marker was used to replace the TetR gene in a second round of GET Recombination. Recombinant clones were selected by plating on medium containing chlorotetracycline and fusaric acid. Although counterselection for the TetR gene is not very efficient, four recombinant colonies with the IVS I-110 mutation were identified among 480 clones screened. Analysis of the recombinant clones did not show any other modifications or rearrangements. Thus the TetR gene can be used in combination with GET Recombination to introduce point mutations and other modifications in BACs without leaving behind any operational sequences, in order to generate accurate cell and transgenic mouse models for various diseases.
INTRODUCTION
With the imminent completion of the sequencing phase of the Human Genome Project, attention is becoming focused on the functional evaluation of large genomic regions. Although new software may allow prediction of some features of each newly identified gene, it will be necessary to test such predictions in physiologically relevant systems. Understanding the mechanisms underlying the locus specificity of gene expression in various tissues and at various stages of development will also require an examination of the impact of various sequence modifications on gene expression in intact functional loci.
The genomic inserts in PAC/BAC clones are large enough to include most genes as intact functional units and thus are ideal for functional analysis in cell lines and in animal models. A major limitation in such studies has been the difficulty of introducing specific modifications in PAC/BAC clones. We have developed GET Recombination, an inducible homologous recombination system for the targeted modification of PACs and BACs in the recombination-deficient (recA–) Escherichia coli strain DH10B in which most PAC and BAC libraries are maintained (1). In this system, the ET cloning vector pBAD24 trecET (A.F.Stewart, personal communication) has been modified by insertion of the gam gene of bacteriophage λ in a polycistronic operon with the recE and recT genes, under the control of the arabinose promoter. The resulting plasmid, pGETrec (1), is superficially similar to plasmid pBAD-ETγ (2), except that in the latter plasmid only the recE gene is inducible by arabinose, while both the recT and gam genes are constitutively expressed. Constitutive expression of the gam gene from pBAD-ETγ may be too toxic to DH10B (recA–) cells under some conditions, thus limiting the efficiency of recombination (unpublished data). In contrast, tightly regulated expression of the gam gene in the pGETrec system, together with simultaneous expression of the recE and recT genes, allows efficient homologous recombination between linear DNA fragments and BACs resident in such cells.
However, the insertion of disease-causing mutations, polymorphisms and other modifications which do not include any selectable markers and which do not leave behind any operational sequences requires a two-stage homologous recombination process. In the first stage a cassette, usually consisting of a counterselection marker and an antibiotic resistance gene, is inserted in the region of interest. Recombinants are isolated by antibiotic resistance. In the second stage, the counterselection cassette is replaced by homologous recombination between its flanking regions and the two ends of a DNA fragment that contains the desired modification, without the inclusion of any operational sequences. True recombinants are identified by selection for cells that have lost the counterselection marker. Zhang et al. (2) used the sacB (3) and ccdB genes (4) as counterselection markers for the introduction of point mutations on small multicopy plasmids using the RecE system in JC9604 cells. However, the introduction of point mutations or other modifications on single copy BACs in their native E.coli DH10B strain without leaving behind any operational sequences was not demonstrated. In an alternative approach, Yang et al. used a pop-in/pop-out strategy based on expression of the recA gene in DH10B cells from a temperature-sensitive plasmid and use of the TetR gene in the presence of chlorotetracycline (cTc) and fusaric acid (FA) (5) or nickel ions (6) for counterselection. However, this approach requires the construction of a separate shuttle plasmid for each modification, while the restoration of recombination proficiency to DH10B cells via the RecA/RecBCD pathway may cause undue instability in BAC clones.
We here describe introduction of the IVS I-110 G→A splicing mutation into the β-globin locus using the TetR gene as a counterselection marker during GET Recombination. The techniques described are applicable to the targeted modification of all BAC clones in their native E.coli strain.
MATERIALS AND METHODS
Media
Bacterial cultures were grown in LB medium or on agar plates containing the following antibiotics as indicated in the text: 12.5 µg/ml chloramphenicol (Cm); 100 µg/ml ampicillin (Amp); 12.5 µg/ml tetracycline (Tet). Growth in SOC medium for 1 h was used to induce expression of antibiotic resistance after electroporation. Counterselection using the TetR gene was carried out by growing cells on FA/cTc TB plates (5) prepared as follows: Tryptone, 5 g; yeast extract 0.5 g; glucose, 0.5 g; NaCl, 4 g; 0.1 mM ZnCl2, 250 µl; 6.3 mg/ml cTc, 4 ml; agar, 7.5 g. Add water to 500 ml and autoclave for 20 min. After cooling to ∼50°C, add NaH2PO4·H2O (1 M), 36 ml; FA, (2 mg/ml, filter sterilised) 3 ml; chloramphenicol and ampicillin as required.
Plasmids
pEBAC148β, a BAC clone containing the entire β-globin locus located on a 185 kb genomic insert in a second generation PAC/BAC cloning vector has been previously described (1). pGETrec is a 6578 bp plasmid containing the E.coli recE and recT genes and the bacteriophage λ gam gene in a polycistronic operon (1). pUC18/β110 contains a 5.5 kb EcoRI fragment of the β-globin gene with the IVS I-110 G→A point mutation (Ioannou et al., unpublished results). Plasmid pBR322 was used as the template for amplification of the TetR gene.
PCR reactions
PCR primers used for preparation of the TetR cassette were obtained from Sigma (Sydney, Australia) and were additionally subjected to RPC cartridge purification. The primers TetF (5′-CAGAGAAGACTCTTGGGTTTCTGATAGGCACTGACTCTCTCTGCCTATTagtgccacctgacgtctaag-3′) and TetR (5′-CAAAGAACCTCTGGGTCCAAGGGTAGACCACCAGCAGCCTAAGGGTGGGAggagtggtgaatccgttag-3′) were used, yielding a 1499 bp product from plasmid pBR322. Upper case characters (49 bp) relate to the homology targeting arms targeting recombination into the β-globin gene, while lower case characters show the sequences used to prime amplification of the TetR gene. PCR reactions were performed in 25 µl reactions for 30 cycles (94°C, 30 s; 50°C, 30 s; 72°C, 2 min) with a high fidelity DNA polymerase, (Boehringer Mannheim, Mannheim, Germany), using the manufacturer’s specifications. The PCR product was gel purified using a commercial gel extraction kit (Qiagen, Hilden, Germany) to remove template plasmid before use for homologous recombination. PCR colony screening across the recombination junctions was performed with primers LUG1 (5′-agacaggtttaaggagacca-3′) and LUG2 (5′-gtttcccattctaaactgta-3′) in 25 µl reactions, using the same cycling conditions described above. These primers yield a PCR product of 440 bp from the normal β-globin gene, while insertion of the TetR cassette yields a 1830 bp product.
Homologous recombination
The first round of GET Recombination was routinely performed as previously described (1). For the second recombination ∼300 ng purified LUG PCR product was electroporated (Gene-Pulser II; Bio-Rad, Hercules, CA) into 30 µl of electrocompetent DH10B (pGETrec, pEBAC 148β::TetR) cells, using 1.8 kV/cm, 200 Ω, 25 µF.
Electrocompetent cells were prepared by inoculating a fresh overnight culture grown at 30°C into 250 ml of LB medium containing three antibiotics (Cm, Amp and Tet) until an OD600 of 0.5 was reached. Expression of the recE, recT and gam genes was then induced by addition of l-arabinose to a final concentration of 0.2% (w/v) for a further 40 min. The cells were harvested and made electrocompetent by a standard methodology. After electroporation, cells were incubated in 1 ml of SOC medium for 1 h and spread onto LB/agar plates containing Cm/Amp/cTc/FA. Colonies were allowed to form over a 48–60 h period. Single colonies were randomly picked, grown in 1 ml mini-cultures in LB containing Cm, and 1 µl aliquots were used directly for PCR screening.
To rescue BAC clones from the pGETrec plasmid, cells were grown on Cm plates overnight (without Amp) and replicates made on Cm and Cm/Amp plates, to identify clones growing only on Cm.
Insert analysis of BAC DNA
BAC DNA was purified using CsCl ultracentrifugation. An aliquot of 0.5 µg DNA was digested with 1–2 U EcoRI or XhoI. The products of digestion were size fractionated using CHEF under the following conditions: 1% gel in 0.5× TBE buffer at 14°C, 6 V/cm for 16 h, with a 0.1–25 s pulse time at a 120° angle for XhoI; 1% gel in 0.5× TBE at room temperature, 4 V/cm for EcoRI. The gels were stained with ethidium bromide for visualisation. Direct DNA sequencing of recombinant BAC clones was performed using the Big Dye Terminator kit (PE Applied Biosciences, Foster City, CA) using the manufacturer’s protocol.
RESULTS
The TetR gene can be used not only to confer resistance to Tet, but also as a counterselection marker on cTc/FA plates (5). We tested the usefulness of the TetR gene as a counterselection marker by inserting the IVS I-110 G→A splicing mutation into the β-globin gene of BAC pEBAC148β. TetR was amplified from pBR322 with primers TetF/TetR to yield a 1499 bp PCR fragment with 49 bp homology arms to the IVS I–exon II junction of the β-globin gene. Electroporation of this fragment into DH10B (pGETrec, pEBAC148β) cells that were induced for 40 min with l-arabinose allowed isolation of recombinant clones on Cm/Amp/Tet LB agar plates. Analysis of 10 recombinant clones by PCR with primers LUG1 and LUG2 revealed that they were all positive for correct insertion of the TetR gene at the site of interest (Fig. 1, lane 2). Sequence analysis of one of the recombinant clones showed the expected fusion of the TetR cassette into the β-globin gene without other sequence changes in the targeted region.
Figure 1.

PCR analysis of recombinant clones with primers LUG1 and LUG2. The parent clone (lane 1) gives a 440 bp product, while insertion of the TetR cassette into the β-globin gene yields a 1830 bp product (lane 2). Deletion of the TetR gene after a second round of recombination with a PCR fragment carrying only the IVS I-110 mutation in the β-globin gene restores the original PCR product size (lanes 3 and 4).
We tested the ability of cTc/FA to kill DH10B (pGETrec, pEBAC148β::TetR) cells from these clones. A 104- to 105-fold reduction in colony numbers was observed after plating of recombinant clones on cTc/FA plates. In addition, a non-specific effect of cTc/FA on colony growth rate was observed, requiring incubation for 48–60 h for the colonies to reach normal size. Thus killing of cells expressing TetR on cTc/FA medium is not very effective, with a large number of non-recombinant clones being expected to survive cTc/FA selection following electroporation and recombination to delete TetR.
The second stage of recombination was carried out with a 440 bp LUG1/LUG2 PCR product from plasmid pUC18/β110 containing only β-globin sequence with the IVS I-110 G→A splicing point mutation. Electroporation of this fragment into DH10B (pGETrec, pEBAC148β::TetR) cells yielded about 5000 colonies on cTc/FA plates. Accurate deletion of the TetR gene by recombination is expected to yield the same sized PCR product as the unmodified β-globin gene using the LUG1 and LUG2 primers. Screening of 480 colonies by PCR with primers LUG1 and LUG2 identified four clones with an apparent deletion of the TetR gene (Fig. 1, lanes 3 and 4). Sequence analysis of these clones confirmed the presence of the IVS I-110 G→A point mutation in intron I of the β-globin gene, without any other sequence changes in the targeted region (Fig. 2).
Figure 2.
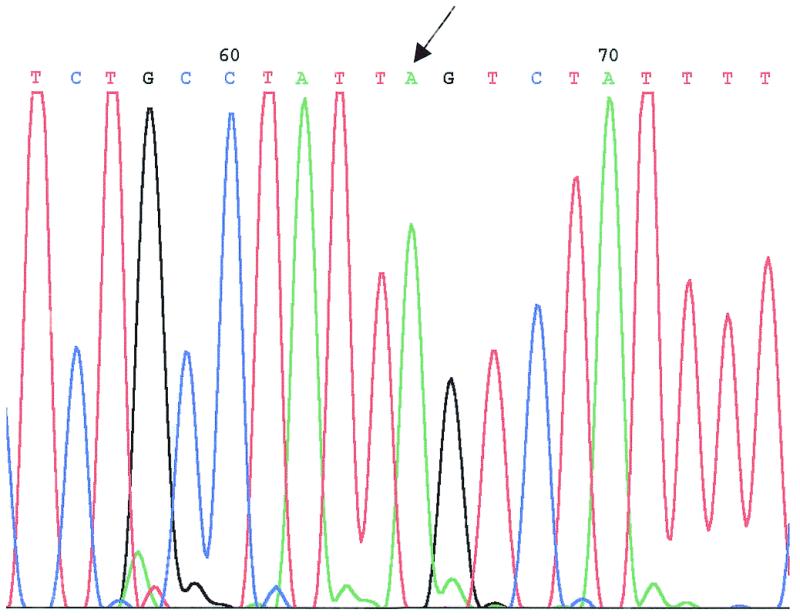
Sequencing chromatogram of one of the pEBAC148β110 clones. The arrow indicates the IVS I-110 G→A splicing mutation introduced by homologous recombination.
The overall integrity of the 185 kb genomic insert carrying the β-globin locus in the recombinant clones was evaluated by restriction enzyme digestion. There are no obvious changes in the 185 kb genomic insert after digestion with NotI or in six fragments produced after digestion with XhoI (data not shown). High resolution fingerprinting of these clones after EcoRI digestion also showed that there are no detectable differences between the recombinant clones (Fig. 3, lanes 2–5) and the parent clone (lane 1) in the 5.5 kb EcoRI fragment on which the modification took place (arrow) or on any of the other EcoRI fragments.
Figure 3.

High resolution mapping of pEBAC148β (lane 1) and four independently isolated pEBAC148β110 clones (lanes 2–5) after digestion with EcoRI. The 5.5 kb fragment on which the modification took place is indicated by an arrow.
Therefore, we were able using the TetR gene for counterselection to introduce the IVS I-110 G→A point mutation into the intact β-globin locus and demonstrate the usefulness of our two-stage GET Recombination system to perform accurate genetic manipulation of BACs.
DISCUSSION
The presence of intact functional loci in fully characterised PAC/BAC clones makes them ideal as a tool to understand the mechanisms underlying the specificity of gene expression from different loci in different tissues during development and to develop pharmacological approaches for the fine manipulation of gene expression as a means of suppressing or complementing specific genetic defects. An essential requirement to take advantage of the unique properties of PAC/BAC clones is the ability to introduce disease-causing mutations, SNPs and other genetic modifications without leaving behind any operational sequences.
In this work we demonstrate the use of GET Recombination in a two-stage approach to introduce the IVS I-110 G→A single base splicing mutation into a 185 kb genomic fragment carrying the intact β-globin locus. We have used the TetR gene both for initial selection of recombinants based on Tet resistance and for counterselection in the presence of cTc/FA medium. Although this counterselection system is not very efficient, with only a 104- to 105-fold enrichment for recombinant clones, PCR screening of pools of clones has allowed us to readily identify clones with the desired modification.
The IVS I-110 G→A mutation is the most common thalassaemia mutation in Mediterranean countries and results in ∼90% aberrant splicing of the first intron of the β-globin gene. Restoration of correct splicing may be possible either by targeting the mutant sequence on the pre-mRNA by antisense oligonucleotides (7) or by targeting the splicesome. The insertion of this mutation into the intact β-globin locus in a second generation BAC clone will greatly facilitate efforts to create a high throughput screening system for pharmacological agents that may regulate splicing specificity in a tissue-specific manner under physiologically relevant conditions. The modified β-globin gene locus is also being used to create an accurate transgenic mouse model for this mutation to facilitate testing of potential therapeutic agents.
Our approach allows the introduction of any desired modification into intact BAC clones without leaving behind operational sequences. This will greatly facilitate the coupling of knowledge and resources from the Human Genome Project to functional analysis of genomic loci as a key step in the development of targeted pharmacological approaches for the modification of gene expression.
Acknowledgments
ACKNOWLEDGEMENTS
The authors would like to acknowledge the contribution of Dr Kumaran Narayanan to some of the preliminary studies on the TetR system and Ms Evi Bashiardes of the Cyprus Institute of Neurology and Genetics for technical assistance with cloning of the IVS I-110 G→A mutation in pUC18/β110. This work was partially supported by grants from the Brockhoff Foundation, the Ronald Geoffrey Arnott Foundation and the Morris Family Trust.
REFERENCES
- 1.Narayanan K., Williamson,R., Zhang,Y., Stewart,A.F. and Ioannou,P.A. (1999) Gene Ther., 6, 442–447. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y., Buchholz,F., Muyrers,J.P.P. and Stewart,A.F. (1998) Nature Genet., 20, 123–128. [DOI] [PubMed] [Google Scholar]
- 3.Pierce J.C., Sauer,B. and Sternberg,N. (1992) Proc. Natl Acad. Sci. USA, 89, 2056–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernard P. (1996) Biotechniques, 21, 320–323. [DOI] [PubMed] [Google Scholar]
- 5.Yang X.W., Model,P. and Heitz,N. (1997) Nature Biotechnol., 15, 859–865. [DOI] [PubMed] [Google Scholar]
- 6.Podolsky T., Fong,S.T. and Lee,B.T. (1996) Plasmid, 36,112–115. [Google Scholar]
- 7.Dominski Z. and Kole,R. (1993) Proc. Natl Acad. Sci. USA, 90, 8673–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]