

Abstract
Protozoan parasites represent a significant risk for public health worldwide, afflicting particularly people in more vulnerable categories and cause large morbidity and heavy economic impact. Traditional drugs are limited by their toxicity, low efficacy, route of administration, and cost, reflecting their low priority in global health management. Moreover, the drug resistance phenomenon threatens the positive therapy outcome. This scenario claims the need of addressing more adequate therapies. Among the diverse strategies implemented, the medicinal chemistry efforts have also focused their attention on the benzimidazole nucleus as a promising pharmacophore for the generation of new drug candidates. Hence, the present review provides a global insight into recent progress in benzimidazole-based derivatives drug discovery against important protozoan diseases, such as malaria, leishmaniasis and trypanosomiasis. The more relevant chemical features and structure-activity relationship studies of these molecules are discussed for the purpose of paving the way towards the development of more viable drugs for the treatment of these parasitic infections.
Keywords: Protozoan infections, antiprotozoal agents, benzimidazole-based derivatives, antimalarial compounds, antileishmanial compounds, antitrypanosomal compounds
1. INTRODUCTION
Parasitic diseases are still a plague that has a huge impact on public health and the overall social and economic well-being of affected countries, mainly developing countries. The principle human diseases caused by protozoa, including malaria, leishmaniasis and trypanosomiasis, are responsible for 790 million cases of morbidity, with an estimated 810,000 death cases annually [1]. Most parasitosis are neglected, like leishmaniasis and trypanosomiasis, since they afflict the world’s poorest populations and still do not receive the proper attention from the funding agencies, thus, research and health programs in this field remain underdeveloped [2].
The global burden of parasitic diseases is generally worsened by the absence of marketed vaccines. RTS, S/AS01 (Mosquirix™) is the first licensed malaria vaccine for P. falciparum to be widely recommended by WHO for children at risk; a two years immunization pilot program in Ghana, Kenya, and Malawi [3] has given positive results in terms of safety and efficacy, lowering the disease severity and the hospitalization rate of pediatric population with malaria infection. However, some concerns have arisen due to the short life time of immunity induced by the vaccine as well as the high production costs [4, 5]. Another vaccine candidate (R21/Matrix-M) has entered a phase 3 clinical trial (ClinicalTrials.gov Identifier: NCT04704830), and if it is approved, it would be an additional resource for the global supply. Despite this, the development of new safe and effective agents may also help manage protozoan diseases, especially for those infections where the drugs are the only weapons for prophylactic and therapeutic purposes. Most of the antiparasitic drugs are old drugs whose use for a long time has induced the emergence of parasite drug resistance [6]; the threats are indeed increasing, especially since epidemiological surveillance is coming from several countries, making the scenario more serious and complex. Hence, the identification of new drugs to treat Malaria, Leishmaniasis, and Trypanosomiasis is always challenging.
1.1. Malaria
Malaria is a life-threatening disease caused by Plasmodium parasites that are transmitted to people through the bites of infected female Anopheles mosquitoes. Five parasite species cause malaria in humans, and in particular, P. falciparum and P. vivax are the most worrisome for public health. P. falciparum is the deadliest parasite and the most prevalent throughout the African continent, while P. vivax is the most widespread outside of sub-Saharan Africa. Recent estimates suggest that 247 million clinical cases of malaria occur every year globally, with 619,000 deaths reported in 2021 [7].
The observed increase in malaria cases with respect to pre-pandemic years could be explained by the significant COVID-19 disease impact that has shifted most of the public health initiatives to contain the emergency crisis. To make matters worse, there have been long-running humanitarian crises and the repercussions of climate change on the spread of the parasitic disease [7].
The life cycle of malarial parasites starts with the growth of Plasmodium in an insect host intermediate that, during a blood meal, deposits it as sporozoite form into the bloodstream of a final host, like a human being. In the host’s liver, sporozoites evolve into merozoites by asexual development, entering the systemic circulation and invading red blood cells, causing the clinical manifestation of the disease. Characteristically, in P. vivax and ovale infections, some parasites may remain latent in the liver (hypnozoite reservoir) and then reactivate to produce blood-stage infections [8]. In erythrocytes, most parasites asexually reproduce from ring forms to trophozoites and lastly to mature schizonts. According to the different Plasmodium species, the schizonts-induced lysis of erythrocytes releases a variable number of merozoites. A group of asexual merozoites can then differentiate into male and female gametocytes that constitute the transmissible form of the parasite to an insect vector. After an infected blood-feeding, the gametocytes reach the mosquito’s midgut and form the zygotes that evolve into oocysts; they successively develop into a new progeny of sporozoites, thus restarting the infective cycle [4].
The arsenal of antimalarial drugs can be divided into three main classes, such as quinoline derivatives (chloroquine and analogues), antifolates (pyrimethamine, proguanil plus atovaquone), and artemisinin derivatives (artemether) and some distinct antibiotics [9], that are used in monotherapy or in combination chemotherapy.
Plasmodium pathogens have developed resistance to conventional therapeutics like chloroquine (CQ) and antifolates. Drug resistance phenomenon even to the artemisinin derivatives and artemisinin-based combination therapy (ACT) has been observed in several areas of Africa [7] and southeast Asia, arousing a great concern [10, 11].
The emergence of resistance has stimulated the modification of existing antimalarial scaffolds and the search for a new generation of medicines characterized by innovative mechanisms of action. Thus, many promising compounds have been tested in monotherapy or, more effectively, in combination therapies (particularly ACT) with the scope to improve efficacy and delay the onset of resistance. Different strategies are being pursued, as illustrated and discussed in some recent literature examples [12-15].
For most of the marketed antimalarials, the mechanism of action is still to be unravelled as they exhibit multitargeting behaviour. This concept is relevant in the context of multifactorial diseases, such as malaria and other vector-borne parasitosis, that are based on intricate interactions among pathogens, vectors, reservoir hosts, and environmental factors [16]. The different initiatives have not yet solved the problem of breaking the chain between the parasite reservoir in animals and infections in man. Therefore, the huge economic losses and serious damage to social well-being in endemic countries continue to be a heavy burden.
1.2. Neglected Tropical Diseases (NTDs)
NTDs represent a group of 20 different conditions widespread in 150 countries and affect about one billion people. They are caused by parasites, viruses, bacteria, fungi, and toxins [17]. Among protozoan parasites, trypanosomatids are the etiologic agents of NTDs, such as leishmaniasis, Human African Trypanosomiasis (HAT), and Chagas disease, causing significant morbidity and mortality in humans and animals.
1.2.1. Leishmaniasis
Leishmaniasis is particularly common, with an estimated 700000 to 1 million infections occurring each year [18]. It is caused by kinetoplast protozoans of the Leishmania genus, which are transmitted to mammalian hosts by the bite of the infected female phlebotomine sandflies. Over 20 Leishmania and over 90 sandfly species are known as of today. The parasites enter host cells as flagellated and motile promastigote forms which are phagocytosed by macrophages. The promastigotes differentiate into non-motile amastigote forms inside the phagosome of the host cells. Subsequent fusion with lysosomes leads to phagolysosomes in which L. parasites can proliferate and survive in the adverse environment. Macrophages are also the major effector cells able to eradicate the infection by activating a suitable immune response. Three main clinical manifestations of L. disease, depending on infecting species and human immune response, are reported: cutaneous (CL), mucocutaneous (MCL), and visceral kala-azar (VL) [19]. CL is the most frequent form and produces mainly skin ulcers that often mend spontaneously, although in some cases cause serious disability or stigma; MCL is the most deforming type affording to partial or total damage of nose, mouth and throat mucous membranes; VL represents the most dangerous form leading to death in over 95% of cases; if untreated, it results in variable fever, weight loss, enlarged liver and spleen, and anaemia [20]. The available drugs for the L. diseases include pentavalent antimonials (sodium stibogluconate and meglumine antimoniate) as conventional therapy and liposomal amphotericin B, pentamidine salts, paromomycin sulfate, and miltefosine as second-line drugs. These agents are characterized by different chemical scaffolds, which lead to several mechanisms of action. Unfortunately, the current treatments show severe side effects, drug resistance development, poor potency, and high cost. Vaccines are unavailable for human L. infections so far and only two vaccines are licensed in Europe for canine leishmaniosis (CanL) (CaniLeish® and LetiFend®) [21]. CanL is a sand fly-borne incurable pathology caused by L. infantum. It is a major zoonosis endemic in >89 countries, with an increase of imported cases in non-endemic zones, resulting in a serious public health issue. Unfortunately, remission is hardly achieved. The recurrences are common, and above all, the infected dogs represent the major reservoir of the protozoan for humans [22]. Although 50% of the infected dogs are asymptomatic, CanL is a chronic and multisystemic disease able to affect all organs. The above-mentioned vaccines offer a low protective effect. Beyond the use of topical insecticides, the first-line treatment is the combined use of human anti-leishmanial agents (pentavalent antimonials or miltefosine) with allopurinol. The development of new effective anti-leishmanial compounds is complex because the efficacy varies depending on parasite species and clinical manifestations. Besides, they have to pass three membranes to kill the intracellular amastigotes, leaving the macrophage hosts unaffected. In the last years, drug discovery in the leishmaniasis field has significantly improved due to the efforts of international consortia funded by governments and charities. In this scenario, the Drugs for Neglected Diseases initiative (DNDi), with its partners, published a target product profile (TPP) in which the minimal and optimal requisites for a new anti-leishmanial agent were detailed [23, 24]. In particular, a new agent for VL infections should be orally administered, show a good safety profile during pregnancy and in immune-deficient patients, reduce parasite burden by 95% in a short treatment, and be inexpensive.
1.2.2. Trypanosomiasis
Trypanosoma brucei is the causative agent of HAT or “sleeping sickness,” and Trypanosoma cruzi causes Chagas Disease. In particular, HAT is caused by two subspecies of T. brucei, which are morphologically indistinguishable but produce different disease courses in infected hosts: T. b. rhodesiense is endemic in eastern and southern Africa and leads to an acute disease progression, whereas T. b. gambiense is associated with a chronic form of the disease and is prevalent in Western and central Africa. To date, control efforts have drastically decreased the spread of this infection; however, sleeping sickness still represents a public health. WHO has set the target to achieve the complete eradication of HAT before 2030, seeking cooperation among multiple sectors based on the One Health approach. Indeed, diagnosis and tracking of the cases, together with the requirement of drugs and facilities to treat affected people, are strictly linked with the need for an efficient control system of vector spreading and of animal reservoirs [25]. Nowadays, there are five drugs approved for the treatment of HAT: suramin and pentamidine, administered in the first stages of the infection (hemo-lymphatic); eflornithine and melarsoprol, recommended for the second stage (meningo-encephalic). Recently, the first-line treatment options have been expanded to fexinidazole, which has been approved by the European Medicines Agency (EMA) for the treatment of T. b. gambiense infections, as a daily dose (for 10 days) against both disease’s stages [26]. However, these drugs are generally toxic and difficult to administer, and they are also prone to develop drug resistance. Thus, the research on new molecules is still intense. T. cruzi is the causative agent of Chagas disease, which is endemic in Latin America. WHO estimates that about 6-7 million people worldwide are infected with T. cruzi. Disease control is a challenging task as conventional therapy, based on benznidazole or nifurtimox, is effective only in the early phases of post-infection. The access to early diagnosis is often delayed since the acute symptoms immediately following the infection are either absent or mild and non-specific. Moreover, in endemic countries an efficient system capable of detecting and tracking the disease at onset is lacking; therefore, Chagas disease is often addressed as a “silent and silenced disease” still representing a public health concern [27].
Both T. brucei and T. cruzi are heteroxenous parasites, exploiting an insect vector to finalise their life and reproductive cycle [28]. Tsetse flies (Glossina spp) represent the intermediate hosts of T. brucei, while T. cruzi is mainly transmitted by contact with faeces/urine of infected bugs of the triatomine family. Trypanosomatids are hence transferred to the final vertebrate hosts during the insect´s blood meal [29]. Alongside the heteroxenous life cycle entailing a definitive host and an intermediate host (indirect life cycle), parasites belonging to the Trypanosomatidae family also share some peculiar structural characteristics, namely a single flagellum and a kinetoplast, an organelle containing a large massed mitochondrial DNA [28].
Trypanosomatids can exist in different forms depending on the stage of their life cycle; T. brucei differentiates into two bloodstream forms in the mammalian host: a replicative long slender trypomastigote (LSF) or a short stumpy trypomastigote form (SSF). The LSF could evade the immune system surviving inside the host, while the SSF is the tsetse-infective form. In the vector midgut, the parasites assume the epimastigote proliferative forms before evolving to the non-proliferative metacyclic trypomastigotes, which are infective for mammalian cells. Metacyclic trypomastigotes differentiate into amastigotes within the host cells and replicate; then, the intracellular amastigotes assume the form of non-replicative trypomastigotes that cause host cell lysis. Non-replicative bloodstream trypomastigotes could re-enter the triatomine vector midgut and regenerate epimastigotes [29].
2. PROMISING DRUG TARGETS
The following chapters focus on the most investigated protozoan targets and the state of the art of the related benzimidazole-based inhibitors thus far identified (Fig. 1). Noteworthy, this class of antiparasitic agents displays a wide range of biological actions, sometimes resembling those of approved antiprotozoan drugs or providing new therapeutic properties with the prospect that even more innovative drugs could be introduced in the years to follow.
Fig. (1).
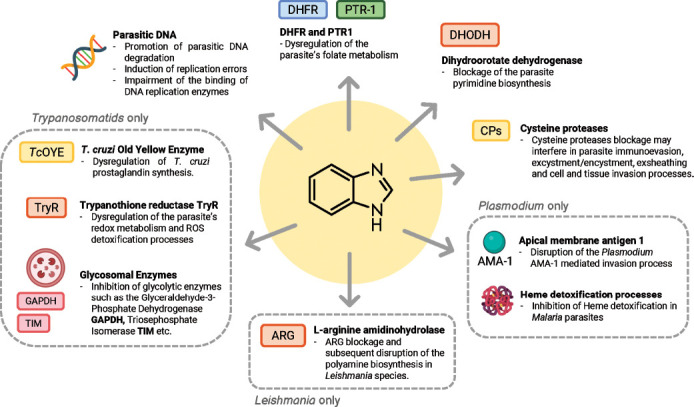
Promising drug targets in malaria, leishmaniasis and trypanosomiasis.
Several conventional drugs bear a benzimidazole ring as their main moiety or key substructure [30, 31]. Isostere of purine bases, the benzimidazole nucleus continues to draw interest in drug discovery, owing to its important biological significance in different pharmacological settings [32-34]. Concerning the antimicrobial purposes, such as antibacterial, antiviral and antiprotozoal activities, the literature reports many examples of benzimidazole derivatives [35-37].
Interestingly, the benzimidazole derivatives are often screened against diverse parasites, with a view to disclosing broad-spectrum antiprotozoal agents underlying possible common mechanisms of parasite growth inhibition. However, the study of the benzimidazole chemical space has, till now, led to the identification of hit/lead compounds, reaching only a few of them to the preclinical profiling stage in animal models of parasitic infections.
2.1. Malaria Targets
Several promising targets are emerging in the fight against malaria, such as dihydroorotate dehydrogenase (DHODH) and apical membrane antigen 1 (AMA-1). Moreover, agents binding to the minor groove of DNA and eme detoxification process inhibitors showed encouraging results (Fig. 1).
2.1.1. DNA Minor Grove
Like pentamidine, which is a bisamidinium compound at physiological pH, dicationic bisbenzamidines and structurally related heterocycles have long been known as antiparasitic DNA-targeting non-covalent agents [38, 39]. They prevalently accumulate in the mitochondrial kinetoplast of trypanosomatids rather than in the nucleus, as it occurs for Plasmodium genus [40, 41]. They bind to minor DNA grooves and cause replication errors by impairing the activity of DNA-dependent enzymes and DNA degradation, hence cell death. The protozoan DNA architecture is unusually rich in thousands of AT-repeated sequences, which are specifically recognized by diamidines when they adopt an appropriate curved shape and exhibit HBD NH groups taking contacts with N(3) of adenine and O(2) of thymine base pairs. As human DNA does not demonstrate such an arrangement, the analysis of the interspecies activity profile of this class of molecules revealed a decrease in the DNA binding capacity passing from protozoa to human cells. Therefore, they were expected to exert no toxic effects at the therapeutic concentration but only at higher doses [38, 41].
2.1.2. Dihydroorotate Dehydrogenase (DHODH)
An important enzyme implicated in DNA biosynthesis is DHODH (Fig. 1), promoting the Flavin mononucleotide-dependent formation of orotic acid, an intermediate product in the biosynthesis of pyrimidine bases for parasite survival [9, 42]. Although Plasmodium falciparum (Pf) DHODH belongs to class 2 DHODH together with human isoform, significant differences between the two enzymes were elucidated [42, 43], making the design of selective DHODH inhibitors possible. Pf DHODH inhibitors preferentially bind to a hydrophobic pocket, lined by the two N-terminal α-helices 1 and 2 and the α/β barrel domain, whose shape and flexible structure frame the main points of divergence to human ortholog, while the hydrogen-bond site (Arg265 and His185) is a conserved region across all class 2 DHODH isoforms [42]. Contrarily to parasites, humans may count on the salvage pathway as a bypass for pyrimidine biosynthesis. Thus, DHODH targeting agents may exert a limited impact on essential human functions. Therefore, this enzyme represents a validated target for addressing anti-infective agents to be applied in different pharmacological settings [42].
2.1.3. Apical Membrane Antigen 1 (AMA-1)
During the asexual blood stage, sporozoites and merozoites of Plasmodium species share a complex array of proteins to invade hepatocyte and erythrocyte host cells [44, 45], respectively, for their intracellular replication cycle. AMA-1 is an antigen on the surface of the invasive parasites, which represents the main target of naturally acquired antibodies in malaria-endemic populations, thus suggesting this protein as an important tool to set-up a vaccine [46]. Furthermore, AMA-1 is recognised as a validated target for the development of small molecules [45], enabling them to block the invasion and the growth of P. parasites' asexual blood stages (Fig. 1). Indeed, only recent studies are somehow solving the complexity of the AMA-1-mediated invasion process, pointing out that diverse pathways may occur within the Plasmodium genus [47], thus burdening the development of broad-spectrum antimalarial therapeutics.
2.1.4. Heme Detoxification
Plasmodium species spend much of their life as intracellular parasites, thus remaining safe in the host cell environment and evading the host's immune response. During infection of the human host, malaria parasites invade red blood cells and, in the digestive vacuole, metabolize a large amount of haemoglobin releasing free heme that can be toxic. Then, heme undergoes oxidation of Fe2+ to Fe3+ by molecular oxygen forming hematin (protoporphyrin IX), which finally dimerizes into non-toxic hemozoin crystals [48, 49]. The inhibition of this detoxification process (Fig. 1), based on heme catabolism, increases its accumulation in the parasite, leading to toxicity and death of the parasite. This mechanism of action is characteristic of 4-aminoquinoline-based antimalarial drugs [13], such as chloroquine, lumefantrine, quinine and mefloquine, which are usual reference compounds for biological testing.
2.1.5. Other Druggable Targets
Another critical step of the malaria parasites' life cycle is the egress event, allowing them to leave the host and promote their dissemination. A lot of parasite enzymes take part in this process, including proteases, kinases, phospholipases, and other proteins. Interestingly, all these proteins are eligible targets for the design of innovative antimalarial medicines [50-52].
2.2. Trypanosomiasis and Leishmaniasis Targets
Trypanosomatids, besides sharing the typical eukaryotic organelles, also possess some unique biological structures and hallmark enzymes that could represent promising targets for drug design.
2.2.1. ROS/RNS Detoxification
The kinetoplast is a highly specialized region of the parasite mitochondrion which contains a peculiar DNA. The kinetoplast DNA (kDNA) is organized in an intricate architecture of thousands of circular DNA molecules forming a single supramolecular structure that encodes for rRNAs and proteins of the respiratory chain or guide RNAs involved in the RNA editing processes [53]. kDNA could be subject to the attack of reactive oxygen and nitric species (ROS/RNS) produced during mitochondrial respiration by complexes I-IV. At low concentrations, these reactive species are crucial to the parasite cell physiology, involving cellular signalling or cytotoxicity. On the contrary, high levels of ROS/RNS can threaten parasite survivability [54], interfering with several biosynthetic pathways and damaging proteins and DNA. ROS release is also described as one of the defence mechanisms employed by the mammalian host immune system (macrophages) to contrast the infection. As a part of their response to reduce ROS, trypanosomatids have developed several detoxification countermeasures arranging a poll of enzymes, such as Fe- superoxide dismutases (SODs), cytochrome c peroxidase (CCP) (in Leishmania spp.), and thiol peroxidases, such as 2-Cys-peroxiredoxines, such as the tryparedoxin peroxidase (TXNPx), and the nonselenium glutathione peroxidase (nsGPXA3) [55]. TXNPx is the final enzyme of a redox cascade in which the electrons flow from NADPH to trypanothione [T(S)2/T(SH)2], a parasitic dimer derivative of glutathione in mammals. Trypanothione is the natural substrate of trypanothione reductase (TryR) (Fig. 1), a flavoprotein localised in mitochondria, glycosomes and cytosol of parasites [56]. TryR is absent in the mammalian hosts, where it is replaced by glutathione reductase (GR), showing a similar antioxidant functioning. With respect to GR, the TryR binding pocket is large, hydrophobic and exhibits a negative charge, making possible the design of selective inhibitors [57]. However, TryR undergoes a high-rate turnover. Thus, TryR activity should be inhibited by 90% to provide a significant effect on parasite viability [58].
The enzyme converts trypanothione disulfide T(S)2 into its reduced form T(SH)2, and the electron passes to another protein tryparedoxin (TXN) and finally to the tryparedoxin peroxidase (TXNPx), which catalyses hydroperoxides reduction [59]. These detoxification mechanisms sustain trypanosomatids' survival during the infection since T. cruzi TXNPx has been found to be upregulated during the infective host colonization phase. Even if the trypanosomatids thiol peroxidases redox system appears to be a potential target for the development of selective antiparasitic compounds some challenging points need to be stressed: studies on T. brucei, T. cruzi, and L. brasiliensis enlightened that the inhibition of nsGPXA3 alone is not able to produce an adequate antiparasitic effect, because of the metabolic bypass provided by TXNPx thiol peroxidases; even though they have different preferences for their substrate hydroperoxides, they could compensate each other’s work, allowing parasite survival. Conceivably, dual inhibitors could offer the attained antiparasitic effect [55].
2.2.2. Glycosomal Enzymes
The kinetoplastids are also characterised by a peculiar type of peroxisome-related organelles called glycosomes due to the presence of enzymes in the glycolytic and gluconeogenic pathways. The proteome of these organelles differs among trypanosomatids and depends on the phase of the parasite cycle; it may include glycolytic and gluconeogenesis enzymes (Fig. 1), such as hexokinase (HXK), glucokinase (GlcK), phosphoglucose isomerase (PGI), phosphofructokinase (PFK), aldolase (ALD), triosephosphate isomerase (TIM), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), phosphoglycerate kinase (PGK) etc.
TIM is a glycolytic enzyme (Fig. 1) contained in trypanosomatid glycosomes that catalyses the reversible interconversion of glyceraldehyde-3-phosphate and dihydroxyacetone phosphate, a key step of the glycolytic pathway. TIM is a homodimer that structurally adopts an α/β barrel folding; each monomer is composed of eight parallel β strands in the centre, surrounded by eight α-helices connected to each other by loops.
Glycosomes also contain enzymes involved in other metabolic pathways: ether-lipid synthesis, β-oxidation, sterol and isoprenoid synthesis, purine salvage, pyrimidine synthesis, amino acid metabolism and defence against oxidative stress [60].
2.2.3. Old Yellow Enzyme (OYE)
OYE (Fig. 1) is an oxidoreductase involved in the metabolic pathway of arachidonic acid (AA) of T. cruzi and L. parasites [61]. OYE catalyses prostaglandin F2α (PGF2α) synthesis and is also involved in the reduction of trypanocidal drugs, such as nifurtimox. PGF2α synthases play a key role in the trypanosomatids infective cycle. Thus, they are present in all the members of the trypanosomatidae family. Commonly, the parasite PGF2α synthases belong to the Aldo-Keto reductase protein family. T. cruzi diverges from this pattern since its aldo-keto reductase (TcAKT) is not capable of catalysing the PGH2 reduction to PGF2α; thus, it encodes a member of the OYEs family to carry out this reaction. OYE is a cytosolic flavoprotein NADPH oxidoreductase that can reduce nitro esters, nitroaromatics, and α,β-unsaturated compounds and is also involved in hydrogen peroxide detoxification [62].
2.2.4. L-arginine Amidinohydrolase (ARG)
In Leishmania parasites, ARG (Fig. 1) is involved in the polyamine biosynthesis by supplying L-arginine from the host. After L-arginine hydrolysis by ARG, its metabolic product, L-ornithine, is released into the cytosol and converted into putrescine by the action of ornithine decarboxylase (ODC). Finally, spermidine is obtained through the transfer of an aminopropyl group from decarboxylated S-adenosylmethionine (dsSAM) to a terminal amino group of putrescine by spermidine synthase (SPDSYN). The polyamines play an important role in the parasite survival and proliferation in the host cells. Moreover, the spermidine binds two glutathione residues, leading to trypanothione, which is implicated in the redox homeostasis of the parasite.
Interestingly, the enzyme shares 39-43% identity with human ARG [63, 64]. Instead, a 75.8% identity was observed by the alignment of ARG sequences from different L. species, with no variations concerning the active site and the metal binding regions. The variability of the compound binding affinity was attributed to amino acid differences outside the active site pocket.
In response to Th1 cytokines, the macrophages induce nitric oxide synthase (NOS) activity to produce L-citrulline and cytotoxic nitric oxide (NO) starting from L-arginine. Thus, the deprivation of this AA from the host can promote the parasite's survival and proliferation. Dissimilarity in L-arginine metabolism in cutaneous and visceral promastigote species was disclosed, reporting that L. mexicana and L. major utilize more L-arginine than L. donovani, biosynthesising a larger amount of L-ornithine and L-arginine metabolites [65]. Moreover, the L. donovani ARG was essential for ornithine and polyamine synthesis, but ODC represented the rate-limiting enzyme for polyamine production [66].
2.2.5. Cysteine Proteases (CPs)
CPs (Fig. 1) play a pivotal role in protozoa virulence, proliferation, and metabolism [67]. They are highly expressed in L. species and are structurally different from the mammalian homologous cathepsins. L. mexicana possesses three types of CPs belonging to the papain family: CPA and CPB (cathepsin L-like) and CPC (cathepsin B-like) [68]. In particular, the isoform CPB2.8 was extensively investigated as an effective antileishmanial target, and few reports described inhibitors of L. mexicana CPB2.8∆CTE, which represents the recombinant form of the CP2.8 isoform expressed without the C-terminal extension. L. mexicana CPB-deficient mutants were found to be less able to infect macrophages and induce lesions in BALB/c mice than wild-type parasites. In general, the virulence of L. mexicana was associated with the CPB capacity of stimulating the IL-4 production and the Th2 response. Interestingly, the Th2 immune response increased the susceptibility to the disease, whereas the Th1 immune response increased resistance to leishmaniasis [69]. CPB was also demonstrated to be involved in the inhibition of antigen presentation through the degradation of MHC class II molecules in the parasitophorous vacuole. Recently, other L. CPB mechanisms of action were elucidated, but their detailed description is beyond the scope of this review [70].
2.2.6. Dihydrofolate Reductase-thymidylate Synthase (DHFR-TS) and Pteridine Reductase 1 (PTR1)
As already assessed for malaria and bacterial infections, in recent years, targeting folate enzymes has increasingly been recognized as a promising strategy for the treatment of parasitic infections [71-73]. Trypanosomatids are auxotrophic for folate and other pterinic compounds, essential factors in the biosynthesis of nucleic acids and proteins. To survive, these parasites have developed a biochemical supply and reuse of these factors from the infected hosts, exploiting two NADPH-dependent key enzymes, namely the dihydrofolate reductase (DHFR) and pteridine reductase 1 (PTR1) (Fig. 1). Following the intake, pterins (such as folate or biopterin) are reduced twice, forming the active tetrahydro derivatives THF and THB, respectively. When DHFR occurs through conventional antifolates, such as MTX, the parasites overexpress PTR1, leading the enzyme to also process folate in sufficient THF levels, enabling them to survive. Keeping in mind the PTR1 metabolic bypass, the dual inhibition of both DHFR and PTR1 enzyme activities is strongly warranted for more efficient management of these parasitic diseases [72, 73].
The present review covers the last five years of literature on benzimidazole-based compounds endowed with the most promising antimalarial, antileishmanial, and antitrypanosomal profile worth of further improvements.
3. BENZIMIDAZOLE-BASED COMPOUNDS ACTIVE AGAINST MALARIA, LEISHMANIASIS, AND TRYPANOSOMIASIS
Resembling the main chemical features of the drug pentamidine, clinically used as therapy for African Trypanosomiasis and antimony-resistant leishmaniasis, Farahat et al. pursued their studies on the antiparasitic activity of bichalcophenes, developing a new series (Table 1) in which a benzimidazole or indole moiety was introduced within a three aromatic spacer to outdistance the two terminal amidine groups [74].
Table 1.
Structures and biological activity of bichalcophenes 1a-c against P. falciparum (P.f.) and T. brucei (T.b.r).

| ||||
|---|---|---|---|---|
| Compound | P.f. IC50a | SIb | T.b.r. IC50a | SIb |
| 1a: X = N, Y = O | 91.9 | 454 | 102 | 409 |
| 1b: X = CH, Y = O | 2.9 | 1000 | 15 | 1100 |
| 1c: X = CH, Y = S | 6.8 | 4400 | 6.8 | 2588 |
Note: aIC50 (nM): compound concentration that reduced the parasite load by 50%; bSI: selectivity index (IC50 L6/IC50 parasite).
Dicationic bisbenzamidines and structurally related heterocyclic analogues have long been known as parasitic DNA-targeted non-covalent agents [38].
In this regard, Farahat et al. [74] showed the benzimidazole diamidines strongly targeting the DNA minor groove by the analysis of circular dichroism (CD) spectra, and compound 1a providing nanomolar potency against P. falciparum and a high selectivity index (SI) (Table 1). Indeed, a better biological profile was exhibited by the indole isosteres 1b and 1c, for which a superior SI was noted (Table 1). In the following step, the indole diamidines were tested in vivo using a mouse model which recapitulated the acute hemolymphatic phase of African trypanosomiasis; interestingly, the 2-[5-(furan-2-yl)thiophen-2-yl]indole derivative (1c) demonstrated to cure all T.b.r. infected mice at a dose minor than that of pentamidine.
Also, the diphenyl-based bis(2-aminoimidazoline) and bisguanidine DNA binders were previously identified as potent antiprotozoal agents against Plasmodium spp and trypanosomatids (T. brucei, T. cruzi, Leishmania) [39, 75]. On the wave of these results, monocationic 5-guanidinobenzimidazoles were investigated as broad-spectrum antiprotozoal agents [76], showing to be more efficacious and safer against P. falciparum with respect to T. brucei (see cpd 2a and 2b, Table 2). No significant activity was observed against T. cruzi and L. donovani parasites. In particular, bulky aromatic rings, linked to position 2 of the 5-guanidinobenzi-midazole scaffold, were identified as a common signature for the best performance of this class of compounds, as for the rank-ordered list: biphenyl, 4-(3,4-dimethoxy-phenoxy)phenyl and bithiophene motifs. Docking studies disclosed the binding mode of compound 2a to DNA minor groove of the d(CGCGAATTCGCG)2 duplex, where the guanidinium group formed a salt bridge and HB interactions with the phosphate moiety of G22 and the NH group of benzimidazole H-bonded to T19; the biphenyl ring concurred to stabilize the complex forming weaker aromatic HBs with T7, T8, T19 and T20. Molecular dynamics revealed that the DNA-ligand (2a and 2b) interactions permitted a better stabilization of DNA conformation than its apo form, thus dampening its functional activity.
Table 2.
Structures and antiprotozoal activity of compounds 2a-c against P. falciparum (P.f.), T. brucei (T.b.r), T. cruzi and L. donovani.
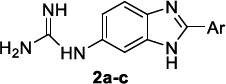
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | P.f. IC50a | SI | T.b.r. IC50a | SIb | T.c. IC50a | SIb | L.d. IC50a | SIb |
2a, Ar: 
|
0.043 | 2360 | 51 | 2 | 130 | 0.8 | 126 | 0.8 |
2b, Ar: 
|
0.089 | >1920 | 21 | >8 | 100 | >2 | 100 | >2 |
2c, Ar: 
|
0.42 | 262 | 26 | 4.2 | 103 | 1.1 | 84 | 1.3 |
Note: aIC50 (µM): compound concentration that reduced the parasite load of 50%; bSI: selectivity index (IC50 L6/IC50 parasite).
Fragment-based drug discovery (FBDD) strategies have achieved a wide consensus both in hit generation and hit-to-lead optimization process due to their potential to increase the chance of developing more targeted lead compounds that are small in size and, for this reason, highly ligand-efficient [77]. The application of this approach permitted the discovery of the benzimidazole derivatives as promising inhibitors of P. falciparum apical membrane antigen 1 (AMA-1) [45, 78], a protein responsible for the merozoite entry into the host erythrocytes. In an effort to identify novel Pf AMA-1 targeting benzimidazole agents, as a continuation of a previous study [78], R.S. Norton and P.J. Scammells identified a new series of 2-amino-1-(2-hydroxyphenyl)benzimidazoles (Table 3). The novel compound library displayed a remarkable antimalarial activity against the 3D7 laboratory reference strain, especially when the phenol moiety bore electron-donating methyl and methoxy groups. The inclusion of the phenol group in a benzofuran ring abolished the activity, while its expansion to α-naphthol was tolerated without causing any significant improvement on the activity. The compounds showed to be non-toxic, thus providing a high therapeutic window, since the ratio of CC50 HEK293 to IC50 3D7 was >103.
Table 3.
Structures and antimalarial activity of benzimidazole derivatives 3a-e.

| |||||
|---|---|---|---|---|---|
| Compound | 3D7 IC50a | Dd2 IC50a | bCam3.IIrev IC50a | cCam3.IIC580Y IC50a | Dd2-SJ Resistant IC50a |
| 3a, R = H | 77 | 97.3 | 350 | 264 | 46 |
| 3b, R = CH3 | 42 | 31.7 | 140 | 81 | 17.5 |
| 3c, R = OCH3 | 43 | - | 212 | 168 | - |
| 3d, R = 3,5-di-CH3 | 23 | - | 133 | 139 | - |
| 3e, R = 4,5-di-OCH3 | 6.4 | - | 47 | 63 | - |
Note: aIC50 (nM): compound concentration that reduced the parasite load of 50%. bCam3.II line has a K13 R539T mutation that is in Cam3.IIrev was gene-edited to wild type K13 gene. cCam3.II lines bearing the C580Y mutation in the K13 gene, which is responsible for resistance to artemisinin.
The screening of antimalarial activity was also extended to the Cambodian Cam3.IIrev and the drug-resistant Cam3.IIC580Y clinical isolates and the compounds still reached nanomolar range potencies. In the evaluation of the cross-resistance profile, compounds 3a and 3b were shown to inhibit the replication of the Dd2 chloroquine-resistant strain, harboring distinct sets of point mutations in the Pf transporter (PfCRT), such as the crucial K76T and A220S [79], and the Dd2-SJ733 strain carrying resistance to the cation-transporting ATPase 4 inhibitor SJ733 [80]. This broad spectrum of activity versus notable examples of drug-resistant strains makes the case that they could act through an innovative mechanism of action, thus deserving further in-depth analyses. The preliminary in vitro and in vivo evaluation of the pharmacokinetic (PK) profile pointed out the need for a chemical optimization for this class of 2-aminobenzimidazoles to overcome the observed low metabolic stability and moderate-low intrinsic clearance.
Bhoi et al. [81] also enlightened the good antimalarial activity of a benzimidazole set (Table 4), bearing in position 2 with a 2-phenol moiety decorated with electron-donating alkyl groups, as derived from the rearrangement of 2-formyl carvacrol. All the compounds were proven to be active against the Pf laboratory strain with IC50s ranging from 1.52-3.31 µM. The 5,6-disubstituted derivatives 4c and 4g reached the potency of the standard drug quinidine (IC50 = 0.83 µM).
Table 4.
Benzimidazole derivatives 4a-g bearing a 2-phenol moiety and their antimalarial activity.

| |
|---|---|
| Compound | 3D7 IC50a |
| 4a, R = H | 3.31 |
| 4b, R = 6-CH3 | 6.28 |
| 4c, R = 5-CH3 6-Cl | 1.52 |
| 4d, R = 6-Cl | 3.42 |
| 4e, R = 6-F | 2.71 |
| 4f, R = 6-Br | 3.19 |
| 4g, 5,6-diCl | 1.85 |
Note: aIC50 (µM): compound concentration that reduced the parasite load by 50%.
The in-silico investigation of the physicochemical properties of these compounds reported that no violation of Lipinski’s rule of five descriptors occurred and that they had a good PK profile (bioavailability score of 0.55). The analysis at molecular level of the most important features involved in the interaction of the tile compounds in complex with the Pf dihydroorotate dehydrogenase (DHODH) enzyme (PDB ID: 1TV5) showed their putative capacity of binding, albeit with some variations, to key residues of the active site, including Tyr528, Cys276, Phe278, and Ile263, as experienced by the reference compound chloroquine [81].
Singh et al. [82] developed a hybrid molecule (Table 5) where a phthalimide-based hit compound was improved by the incorporation of a benzimidazole ring and a triazole moiety as pharmacophoric elements, recurrent in several examples of natural and synthetic compounds.
Table 5.
Example of benzimidazole phthalimide hybrids 5a-e and their antimalarial activity.

| ||||||
|---|---|---|---|---|---|---|
| Compound | 3D7 IC50a | RKL-9 IC50a | SIb | |||
| - | R | R1 | R2 | - | - | - |
| 5a | CH3 | CH2CH(CH3)2 | 4-Cl-phenyl | 0.74 | 1.71 | >135 >675 |
| 5b | H | CH(CH3)CH2CH3 | 4-CF3-phenyl | 0.64 | 1.62 | >156 >781 |
| 5c | H | CH(CH3)CH2CH3 | benzyl | 0.81 | 1.88 | >123 >617 |
| 5d | CH3 | CH(CH3)CH2CH3 | 4-F-phenyl | 2.2 | 3.43 | >46 >227 |
| 5e | H | CH(CH3)2 | 4-CF3-phenyl | 2.3 | 3.92 | >43 |
Note: aIC50 (µM): compound concentration that reduced the parasite load by 50%. bSI: selectivity index (CC50/IC50). P. falciparum 3D7 sensitive and RKL-9 CQ resistant strains.
All the compounds were shown to impair the growth of the Pf3D7 strain with potencies in the submicromolar range and also of the CQ-resistant RKL-9 strain to a slightly lesser extent. Interestingly, no toxicity was observed against HepG2 cells (CC50> 100 µM), up to 500 µM against HEK2GST cells; hence, the corresponding SIs (ratio of CC50 to IC50(3D7)) were at least superior to 123 and 600 respectively (Table 5). Moreover, no haemolytic activity was observed up to 5 µM on human red blood cells, advising a preferential selectivity for infected red blood cells with respect to normal blood cells. The effect of compounds on the specific blood stage of parasite growth was evaluated after 60 hours post-treatment, and defects in invasion and egress stages of the host red blood cells were observed [50]. The biochemical study of the mechanism of action revealed that these hybrid compounds promoted the depolarization of the mitochondrial membrane potential, causing mitochondrial dysfunction and subsequent cell death; additionally, compound 5b showed to interact with the recombinant PfαI-tubulin (Kd= 26.4 µM) from E. coli strain BL21, altering microtubule dynamics. The in vivo antiparasitic activity was tested in BALB/c mice infected with P. berghei, and all derivatives demonstrated to significantly decrease the parasite load compared to an untreated control group on day 7 post-infection.
Pyrido [1,2-a]benzimidazole was advised as a promising core structure for the development of antimalarial agents [83], which were successively demonstrated to impair the heme detoxification process, leading to non-toxic hemozoin thus reducing parasite growth [48, 49, 84].
Sousa et al. [85] identified three potent pyrido [1,2-a]benzimidazole inhibitors 6a-c endowed with comparable in vitro activities against 3D7 and CQ-resistant W2 strains (Table 6). In particular, 6a was found to be the most effective, and so it was selected for addressing further studies on the mechanism of action, using some conventional drugs for the purpose of comparison. The compounds were shown to kill both ring and trophozoite stages of the parasite, similarly to CQ and AQ, thus guaranteeing a long-lasting activity, and, unlike what happens for AQ, they were not susceptible to GSH conjugation. They exhibited significantly reduced parasitaemia in mice infected by P. berghei, surpassing the effectiveness of CQ. In vitro and in vivo studies corroborated 6a binding to the cytosolic heme pool and heme-dependent toxification effects, that, however, occurred only after prolonged exposure to the compound; thus, the implication of other unexplored targets was suggested to justify the rapid parasiticidal activity. Interestingly, the compounds displayed a preferential ability to enter parasite cells more rapidly than host cells, thus exhibiting a species-selective tropism.
Table 6.
Antimalarial activity against the asexual blood stage of P. falciparum for compounds 6a-c.

| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R | R1 | X | Y | Z |
3D7
IC50a |
W2
IC50a |
| 6a |

|
CF3 | Cl | H | Cl | 60.8 | 58.8 |
| 6b |

|

|
H | H | H | 100.1 | 88.3 |
| 6c |

|
CF3 | H | Cl | Cl | 160.5 | 177.8 |
Note: aIC50 (nM): compound concentration that reduced the parasite load by 50%.
Nieto-Meneses et al. [86]. reported a series of N-benzyl-1H-benzimidazol-2-amines, among them compounds 7a and 7b (Table 7), resulted in more activity against L. mexicana cutaneous amastigotes and less cytotoxicity against murine macrophage cell line J774.2 than miltefosine, exhibiting an higher SI with respect to reference drugs (Table 7) (SI = 6.5 for amphotericin B).
Table 7.
Inhibition of several L. species proliferation for benzimidazoles 7a,b and miltefosine.

| ||||||
|---|---|---|---|---|---|---|
|
Compound
7a, R = OH, R1 = Br 7b, R = OCH3, R1 = H |
7a | 7b | Miltefosine | 7a | 7b | Miltefosine |
| Promastigotes IC50a | SIb | |||||
| L. mexicana | 2.62 | 3.21 | 15.34 | 91.76 | 317.75 | 10.23 |
| L. braziliensis | 54.05 | 127.7 | 48.37 | 4.88 | 9.61 | 3.25 |
| L. donovani | 198.47 | 223.8 | 114.68 | 1.33 | 5.48 | 1.37 |
| Amastigotes IC50a | SIb | |||||
| L. mexicana | 0.28 | 0.26 | 0.51 | 942.8 | 3939.2 | 307.8 |
| L. braziliensis | 34.22 | 33.78 | 22.67 | 7.71 | 30.32 | 6.92 |
| L. donovani | 222.23 | 218.24 | 66.74 | 1.18 | 4.69 | 2.35 |
Note: aIC50 (µM): compound concentration that reduced the parasite load of 50%; bSI: selectivity index (CC50/IC50); CC50 from J774.2 cells.
The compounds were characterized by a methyl group at N-1 of the benzimidazole ring, an oxygen atom at the C-2 position of the benzyl portion (hydroxyl or methoxyl for 7a and 7b, respectively) and no substituents in the benzenoid moiety; the modification of these main structural features led to less active analogues. Based on the encouraging results, the activity of the two compounds was also evaluated against the promastigotes and amastigotes of various Leishmania species. In detail, only benzimidazole 7a was slightly less effective than miltefosine against L. braziliensis mucocutaneous promastigotes, whereas both derivatives were less active than reference compound against L. donovani visceral promastigotes (Table 7). Regarding intracellular amastigotes, they were more active than both miltefosine and amphotericin B (IC50 0.87 µM) against L. mexicana, showing high SI values, whereas they exhibited lower activity than miltefosine against L. braziliensis and mostly L. donovani amastigotes.
To elucidate the benzimidazoles target, their ability to inhibit recombinant L. mexicana arginase (LmARG) was evaluated using the QuantiChomTM Arginase Assay Kit in the presence of Nor-NOHA (N-Hydroxy-nor-L-arginine) as positive control. Interestingly, compound 7b inhibited the activity of LmARG by 68.27%, resulting in about 3-fold more activity than 7a (24.34% inhibition) and 2-fold more activity than Nor-NOHA (32% inhibition); interestingly, 7b did not inhibit the activity of human arginase but stimulated it by 24.21%. An in silico docking study was performed, and compound 7b was the best ranked with a score of -5.5 kcal/mol with respect to -4.46 kcal/mol of its analogue 7a. Based on calculated physicochemical descriptors, both compounds fulfilled all Lipinski’s and Veber’s rules, prospectively offering a good bioavailability profile.
As result of a preliminary screening on their in-house database, De Luca et al. [87] reported four 1,2-disubstituted-1H-benzo[d]imidazole derivatives 8a-d (Table 8) as effective inhibitors (97.4-92.0% inhibition at 20µM) of L. mexicana CPB2.8∆CTE without inducing toxicity to similar human cysteine proteases cathepsin-B (no inhibition) and cathepsin-L (maximum of 20% inhibition).
Table 8.
Structures, enzymatic and cellular activity of benzimidazoles 8a-d.

| ||||||
|---|---|---|---|---|---|---|
| Compound | R | R1 | X |
CPB2.8∆CTE
Inhibition (%) |
(Ki µM)a | L. infantum amastigotes IC50b |
| 8a | H | H | SO2 | 95.6 | 0.23 | 50.8 |
| 8b | H | CO2CH3 | SO2 | 97.2 | 0.18 | 20.3 |
| 8c | Cl | CO2CH3 | SO2 | 97.4 | 0.15 | 32.5 |
| 8d | Cl | CO2CH3 | CH2 | 92.0 | 0.69 | 6.8 |
Note: aAssuming compounds as competitive inhibitors; bIC50 (µM): compound concentration that reduced the parasite load by 50%.
The three compounds 8a-c, characterized by an electron-rich sulfone bridging moiety between N1 of the benzimidazole nucleus and the phenyl ring, resulted in more activity than their analogue 8d bearing a methylene group as linker at the same position. The presence of an electron-withdrawing group in the phenyl ring of the 2-thioacetamide portion, as well as the introduction of an additional substituent to the C-6 position of the benzimidazole core, positively influenced the activity (8a vs. 8b and 8b vs. 8c). The cell-based assay using L. infantum intracellular amastigotes and miltefosine as a positive control (IC50 4.5 µM) revealed an opposite behavior of the compounds against the whole organism, being 8d more active than its analogues 8a-c. Benzimidazole 8d was also the most cytotoxic (CC50 8.0 µM) in primary peritoneal mouse macrophages (PMM). The main structural determinant between compounds 8a-c and 8d is the sulfone group, which, as a strong H-bond acceptor, probably promotes the binding to the catalytic site of the target enzyme, the cysteine proteinase CPB2.8∆CTE, but negatively affects the passive diffusion by the biological barriers due to its polar nature. In silico studies by using the homology model of mature L. mexicana CPB2.8∆CTE were reported. The calculated Ki values obtained by noncovalent redocking for compounds 8a-d followed the same trend as the experimental values, whereas the ligand efficiency (LE) values showed that derivatives 8a,d (LE 0.28) were slightly more powerful than analogues 8b,c (0.26 and 0.25, respectively). The best-docked pose of benzimidazoles 8a,d pointed out their different accommodation on the enzymatic surface based on their substituents pattern. The binding with the enzyme exploited almost all H-bond interactions, although one π-H interaction for compound 8c between the imidazole and the hydrogen of Asn162 and three π-π interactions for compound 8d between the imidazole and the pyrrole of Trp185 and between the ring of the benzimidazole and the pyrrole or the phenyl of Trp185, were also reported. The compounds did not fulfill the whole Lipinski’s rule, whereas during in silico ADME studies, they exhibited a promising oral availability with a human intestinal absorption >96% but low Caco-2 permeability (48.11-11.92%) and an efficacious plasma protein binding (PPB = 100). To investigate the preclinical value of these compounds, the Authors performed Ames’s test. Except for derivative 8c, they resulted in no mutagen, and 8a and 8d were also noncarcinogenic in rats and mice.
Kumar et al. performed a virtual screening of 221 compounds from several online databases (PubMed, ChEMBI, DrugBank) by using the predicted TryR structure [88]. Fifty-five compounds that resulted from the screening were able to bind the enzyme, and among them, 7 satisfied the docking score threshold of -7.00 kcal/mol. Among them, the benzimidazole derivative 9 (Fig. 2) showed in silico good oral bioavailability and no toxicity. The compound strongly interacted with the TryR with a docking score value of -9.7 kcal/mol, and MD simulations enlightened a stable ligand-protein interaction. Enzymatic and cell-based assays on 9 revealed a TryR dose-dependent inhibition and an anti-leishmanial activity against L. major cutaneous promastigotes with an IC50 of 44.10 µM, although 3-fold less active than mitelfosine (IC50 13.92 µM), without evincing a significant effect on RAW 264.7 macrophage cells viability. The cytotoxic effect of benzimidazole 9 on L. major promastigotes led to an increase of cells in apoptosis and necrosis with respect to the control and morphological changes in the parasites, such as cell size decrease, flagellar size reduction and cell membrane bleeding. Interestingly, compound 9 induced a dose-dependent NO release in RAW 264.7 macrophage cells, and in vivo experiments caused a significant reduction of parasite load and foot pad lesion size.
Fig. (2).

Structures of benzimidazoles 9-14 endowed with anti-leishmanial activity.
Kapil et al. [89] performed two receptor ligand pharmacophore models for homology-modelled dihydrofolate reductase-thymidylate synthase (DHFR-TS) and pteridine reductase 1 (PTR1) (PDB ID: 1E7W) with the aim of designing dual acting antileishmanial inhibitors. In detail, the generated DHFR-TS model showed four main structural features: one hydrogen bond acceptor (HBA), three hydrogen bond donors (HBD), one hydrophobic (HY) and one negative ionizable (NI) with a selectivity score of 14.289, whereas the generated PTR1 model exhibited six main structural features: one HBA, two HBD, one HY, two aromatic rings (RA) with a selectivity score of 11.429. The two models were used to screen an in-house database, and 5 benzimidazole derivatives were identified as hits. Compound 10 was mapped on the two validated models, and some overlap emerged: NH of benzimidazole on HBD feature, benzyl group on RA/HY features, the benzyl substituents on HBA and NI features. Benzimidazole 10 showed some similar structural characteristics, with derivative 9 bearing both a phenyl ring at the C-2 position of the benzimidazole core and a pyridyl/phenyl ring decorated in the para position with a polar substituent. Docking studies demonstrated the binding of compound 10 with the active site of both targets and MD simulation, the formation of a stable complex between 10 and the two proteins. The low MM-GBSA binding energy for human thymidylate synthase (1JU6) versus DHFR-TS and PTR1 pointed out the valuable selectivity of compound 10. The anti-leishmanial activity of benzimidazole 10 was evaluated against L. donovani promastigotes (IC50 68 µM), resulting in about 6-fold less activity than miltefosine (IC50 12 µM). Compounds 11 and 12 (Fig. 2), bearing at N-1 of the benzimidazole core a pyridinylmethyl moiety, were reported by Patel et al. [90]. Both compounds showed antiprotozoal activity against L. mexicana (11, IC50 1.36 µM; 12, IC50 0.66 µM) and T. cruzi (11, IC50 3.38 µM; 12, IC50 3.84 µM). Interestingly, compound 11 was 2-fold more active than miltefosine (IC50 1.35 µM) against L. parasite, and both compounds were about 3-fold more active than benznidazole (IC50 11.14 µM) against T. protozoan. Benzimidazole 11 fullfilled all the Lipinski’s, Veber’s, Ghose’s and Egan’s rules, whereas compound 12 showed one violation in Ghose and Egan's filter screening. Pharmacokinetic properties for both compounds were predicted, showing a high gastrointestinal absorption. The compounds inhibited several cytochromes; 11 also exhibited blood-brain barrier (BBB) permeability and was a P-glycoprotein (Pgp) substrate. Moreover, both derivatives were not cytotoxic and hepatotoxic but carcinogenetic and mutagenetic and 12 also immunotoxic.
Sánchez-Salgado et al. [91] reported an interesting chemoinformatic approach using the ChEMBL database along with an additional literature search to recover the benzimidazole nucleus as a target scaffold. About 235 benzimidazoles were identified with activity against several L. species. Most of the compounds were active against L. major and L. donovani followed by L. infantum; a moderate number of derivatives showed activity against L. mexicana and few compounds against L. amazonensis and L. braziliensis. In detail, 54% of compounds were active in visceral (L. donovani and L. infantum), 44.3% in cutaneous (L. major, L. mexicana and L. amazonensis) and 1.7% in mucocutaneous leishmaniasis (L. braziliensis). The activity of benzimidazoles deeply varied based upon the different substitution patterns, where the 1,2,5 and 6 positions on the benzimidazole core resulted relevant for the Structure-Activity Relationship (SAR) studies. Hence, the authors screened the benzimidazole 13 (Fig. 2), previously tested against L. mexicana promastigotes (IC50 20.80 µM), versus other L. species [92]. In particular, compound 13 resulted in more activity against L. amazonensis promastigotes (IC50 4.11 µM) than miltefosine (IC50 9.56 µM) and worked similarly to miltefosine against L. infantum promastigotes (IC50 8.63 µM and 7.42 µM, respectively). Furthermore, the compound was screened against the intracellular amastigote stage of leishmania parasites, which reproduces a more physiological and relevant model of the disease in humans. The compound retained its activity but to a lesser extent than miltefosine.
Interestingly, only a few benzimidazole derivatives were reported as active against L. amazonensis in the literature so far. The investigation of the mechanism of parasite inhibition allowed to observe an alteration of the tubulin-microtubule equilibrium in L. infantum promastigotes when incubated with an anti-β-tubulin antibody and compound 13. Moreover, in in vitro studies 13 showed a strong decrease in parasite load in the infected macrophages of both species, with particular regard to L. amazonensis infections.
Recently, immunotherapy has led to interesting results in the treatment of CanL by using compounds able to sustain the host immune response against the infection. Among these agents, the benzimidazolone domperidone 14 (Fig. 2), a dopamine D2 receptor antagonist, was approved (Leisguard®) in the prevention and treatment of CanL alone or in combination with conventional anti-Leishmania chemotherapy. The compound is able to potentiate the phagocytic cell activity involved in the Th1 immune response due to a release of serotonin, which increases the prolactin blood concentrations. Prolactin is a proinflammatory lymphocyte-derived cytokine that stimulates T CD4+ lymphocytes, the release of cytokines, such as IL-2 and IL-12, IFN-γ and TNF-α, thus activating the NK cells and macrophages, and causes a decrease of CD4+ Th2 cytokines and TNF-β [93, 94]. As mentioned above, Th1 immune response enhances the resistance to leishmaniosis [69]. Very recently, Cavalera et al. reported the role of domperidone in ameliorating kidney functions in leishmaniotic dogs affected by chronic kidney disease (CKD) by maintaining stable serum creatinine and by reducing anti-L. infantum antibody titres, globulins, gamma globulins, and C-reactive protein [95]. The same authors reported the efficacy of domperidone combined with a renal diet to slow the progression of nephropathy in leishmaniotic dogs with CKD [96].
Ferreira et al. [97] carried out a hit-to-lead optimization study starting from the 2-aminobenzimidazole 15a (Table 9) emerged as a hit in a GlaxoSmithKline high-throughput screening (HTS) diversity set of 1.8 million compounds against L. donovani, T. cruzi and T. brucei by using whole-cell phenotypic assays. Compound 15a showed moderate activity against L. infantum without affecting mammalian cells and good permeability [98]. However, it exhibited high lipophilicity and low metabolic stability in mouse liver microsomes (MLM) due to oxidative reactions and subsequent glucuronidation on the propyl chain at N-1 of the benzimidazole ring. An exhaustive optimization study on hit 15a clarified the key structural features endowed with beneficial effects on the anti-leishmania profile. In detail, the presence of electron-withdrawing groups (EWGs) on the phenyl ring of the benzimidazole core increased the metabolic stability, whereas the addition of a nitrogen atom increased the cytotoxicity. Among the EWG substituents, the hydrophobic ones maintained the activity, whereas the polar ones jeopardized the activity. The bioisosteric replacement of the phenyl ring linked to the amide fragment with a pyrazole and a pyridine nucleus had a favourable effect on the potency and metabolic stability. The presence of a hydrophobic chain on the N-1 of the benzimidazole was essential for the activity and improved the metabolic stability in the case of branched and less lipophilic alkyl groups. The results of this SAR study led to the benzimidazole compounds 15b-e exhibiting a better activity, an excellent metabolic stability and a lower lipophilicity than hit 15a (Table 9).
Table 9.
Anti-leishmanial activity, metabolic stability and lipophilicity of benzimidazoles 15a-e.

| ||||||
|---|---|---|---|---|---|---|
| Compound | R | R1 | R2 | L. infantum IC50a | MLM CIint µg/min/mg | eLogD |
| 15a | H |

|

|
12 | 3094 | 4.6 |
| 15b | 6-CN |

|

|
4.1 | 17 | 3.4 |
| 15c | 6-CN |

|

|
2.2 | 20 | 2.7 |
| 15d | 6-CN |

|

|
0.53 | 28 | 3.0 |
| 15e | 6-CN |

|

|
0.84 | 27 | 3.1 |
Note: aIC50 (µM): compound concentration that reduced the parasite load by 50%.
Selected compounds 15a and 15d were then tested against the amastigotes of several VL and CL strains and showed good activity with an IC50 range of 0.10-24.82 µM.
In vitro ADME data demonstrated that all analogues 15a-e had a comparable mouse and human microsomal stability and good passive permeability as well as plasma stability and protein binding capacity. Derivatives 15d and 15e evinced poor aqueous kinetic solubility. To investigate the safety profile of this class of compounds, inhibition of hERG and cytochrome P450 isoforms was assessed. All benzimidazoles 15a-e weakly inhibited the hERG channel and the cytochrome P450 family enzymes and did not interfere with the activity of key human enzymes and receptors, such as PDE3A, PDE4B2, 5-HT1A, B2 bradykinin, as an example. Compounds 15a and 15d, showing the best PK profiles in vivo with good bioavailability and plasma half-lives, were tested in an acute VL mouse model using an L. infantum strain. Unfortunately, both compounds lost their activity, probably due to the low systemic exposure and the poor safety profile.
Tonelli et al. [99] developed two series of benzimidazole derivatives, which proved to be active against L. tropica and L. infantum (Table 10). The first series bore a long alkyl chain and an ammonium moiety in position 2 of the benzimidazole to mimic the structure of miltefosine, while the second series was characterized by 2-phenyl/2-benzyl benzimidazole scaffold substituted in position 1 with a basic chain resembling that of sitamaquine. More than 75% of the compounds in both series were dual inhibitors of L. tropica and L. infantum promastigotes. Generally, the quaternization of 2-alkylbenzimidazoles improved the activity, which fell in the low micromolar/submicromolar range; in particular, compound 16a displayed 228- and 93-fold lower IC50s than those of miltefosine against both Leishmania species (Table 10). However, the cytotoxicity linearly increased with the potency, so the SI was low versus HMEC cells but superior versus Vero76 cells. Among the compounds featured by a basic side chain, the 1-quinolizidinylmethyl derivatives were more potent than dialkylaminoalkyl ones, and compound 16b resulted in being the most potent (Table 10). Also, this compound manifested some degree of toxicity against HMEC cells, thus showing low SI values, but was not toxic against Vero76 cells (CC50> 100 µM). The hit compounds 16a and 16b were then evaluated against the intramacrophagic amastigote stage of L. infantum, demonstrating for compound 16a an IC50= 0.31 µM, about 3-fold superior to that of miltefosine, while for compound 16b an IC50= 4.76 µM. Therefore, further exploration of the chemical space around the 2-alkyl and 2-benzyl benzimidazole scaffold may be worthy of consideration.
Table 10.
In vitro data on antileishmanial activity against L. tropica and L. infantum and toxicity against the human endothelial cell line (HMEC-1) and/or monkey kidney cell (Vero76) of benzimidazole derivatives 16a,b and miltefosine.
| Compound | - | IC50a | SIb | |
|---|---|---|---|---|
| - | HMEC | Vero76 | ||

|
L. tropica promastigotes | 0.19 | 4.10 | 30.5 |
| L. infantum promastigotes | 0.34 | 2.29 | 17.1 | |
| L. infantum amastigotes | 0.31 | - | - | |

|
L. tropica promastigotes | 3.70 | 4.58 | >27 |
| L. infantum promastigotes | 4.76 | 3.61 | >21 | |
| Miltefosine | L. tropica promastigotes | 43.26 | 2.3 | - |
| L. infantum promastigotes | 31.26 | 3.2 | - | |
| L. infantum amastigotes | 1.05 | - | - | |
Note: aIC50 (µM): compound concentration that reduced the parasite load of 50%. HMEC, human mammary epithelial cells and primate Vero76 cells.
Recent studies have detected the naphtoquinone scaffold as a starting point for the development of novel antiprotozoan compounds. In particular 2-aryloxynaphtoquinone-based compounds have shown an interesting inhibitory profile towards T.cruzi [100]. Adding to the growing interest towards the benzimidazole scaffold as the main pharmacophoric unit of several molecules endowed with antiprotozoan activity, the bioisosteric substitution of the benzene fused ring of the naphtoquinone core with an imidazole ring has been explored [101].
The antiprotozoan activity of these compounds was evaluated in vitro towards the epimastigote form of T. cruzi, Dm28c strain (Table 11). The non-infective epimastigote form could be found in the midgut of triatomine and acquire the ability to infect mammalian hosts only after its conversion to metacyclic trypomastigote. All the benzimidazole-quinone compounds manifested higher antiparasitic activity than the reference compound nifurtimox. Compounds 17a-d, characterized by the phenoxy- moiety in R1, appeared to be the most active with IC50 values in the 1.42-4.07 μM range. Compounds 17a-d were subsequently tested against the trypomastigote infective form of T. cruzi (Dm28c strain), showing even better inhibitory potency, ranging from 0.65 to 1.39 μM and a promising selectivity index > 10.
Table 11.
In vitro activity, cytotoxicity and SI on epimastigote and trypomastigote forms of T. cruzi of benzimidazole derivatives 17a-d.

| ||||
|---|---|---|---|---|
| Compound | IC50a Epim. | IC50a Tryp. | IC50a Vero cell | SIb (Tryp.) |
| 17a | 1.42 ± 0.07 | 0.65 ± 0.10 | 6.89 ± 0.50 | 10.6 |
| 17b | 4.04 ± 0.30 | 0.88 ± 0.14 | 10.09 ± 0.70 | 11.5 |
| 17c | 4.07 ± 0.27 | 0.96 ± 0.13 | 10.20 ± 0.79 | 10.6 |
| 17d | 3.73 ± 0.32 | 1.39 ± 0.40 | 7.65 ± 0.66 | 5.5 |
| Nifurtimox | 21.05 ± 0.90 | 10.00 ± 0.40 | >200 | >20 |
Note: aIC50 (µM): compound concentration that reduced the parasite load of 50%; bSI: selectivity index (IC50 Vero/IC50 parasite).
In order to assess the mechanism of action of this class of compounds, the target identification was attempted by means of molecular modelling studies through induced-fit docking (IFD) methodology. T. cruzi GAPDH, TryR, and OYE were selected as putative targets due to their involvement in the mechanism of action of some naphthoquinone-based multitarget antiparasitic compounds [100, 102, 103]. Indeed, on the basis of the calculated docking scores, only TryR and OYE resulted to be probable target enzymes of this series.
The IFD of the previously studied naphthoquinones on TryR (PDB code: 1BZL) allowed the identification of the Z-site of this enzyme as the potential binding site due to the interaction of the compounds with a hydrophobic region formed by Phe396, Pro 462, and Leu399 residues. The best compound, 17a, showed high affinity to TryR with a docking score of - 7.905 kcal/mol, involving H-bond interactions between the quinone carbonyl groups and Lys62 and Ser464; the N3 of the imidazole ring with Lys62 and the oxygen atom of the phenoxy group with Asn433. Additionally, hydrophobic interactions between the phenoxy group and the residues Leu63, Leu399 and Phe396 concurred with the stabilization of the complex. The comparison between the complex of the imidazoquinone 17a and its naphthoquinone analogue pointed out the key contribution of N-3 of the imidazole ring for the inhibition of TryR and the observed in vitro activity towards T. cruzi.
Benzimidazole-quinone 17a also showed a strong binding affinity with OYE (docking score of - 7.905 kcal/mol), smoothly fitting a binding site close to the FMN prosthetic group (PDBCode: 3ATZ): both H-bond interactions by its CO groups of the quinone moiety with Tyr364, Asn198, His195 of OYE and hydrophobic, and π-π stacking interactions of the phenoxy ring with Phe71 and Tyr200 were observed. However, further studies are needed to provide experimental evidence of TryR and/or OYE as the likely molecular targets of this class of benzimidazole-quinone derivatives [100].
Several inhibitors belonging to benzothiazole [104, 105], benzoxazole [106], and benzimidazole [107] chemical classes exhibited the ability to target the TIM enzyme. In this field, Vázquez-Jiménez et al. [108] detected novel benzimidazole compounds capable of interacting with the TcTIM interface region by performing a ligand-based virtual screening (LBVS) from the ZINC15 database. From a total of 67,141 benzimidazole-containing compounds, 1604 compounds were selected because of their binding energy values between -10.6 and -8.9 Kcal/mol and adherence to Lipinski rules. These compounds were subsequently clustered into ten groups considering the types of interactions in the complex with the enzyme. Only two compounds, 18a and 18b, were thus tested against blood trypomastigotes of the NINOA and INC-5 strains of T. cruzi to assess their trypanocidal activity (Table 12).
Table 12.
Trypanocidal activity of compounds 18a,b.

| |||
|---|---|---|---|
| Compound | LC50a NINOA | LC50a INC-5 | Docking Score (Kcal/mol) |
| 18a | 155.86 ± 3.4 | 226.30 ± 15.4 | -10.4 |
| 18b | 179.55 ± 19.7 | 179.71 ± 19.0 | -10.2 |
| Nifurtimox | 70.41 ± 8.0 | 139.37 ± 3.0 | nd |
| Benznidazole | 130.72 ± 8.8 | 191.28 ± 1.8 | nd |
Note: a LC50 (µM) = half-maximal lytic concentration; nd = not determined.
In vitro determination of the half-maximal lytic concentration (LC50) confirmed that the trypanocidal ability of the compounds was comparable to that of the drugs nifurtimox and benznidazole. Molecular dynamic simulations of compounds 18a and 18b with T. cruzi triose phosphate isomerase showed that the 18b-TcTIM complex is more stable than that of the control ligand L1 (structure not shown), which was previously described as a TcTIM inhibitor [109].
Regarding the SwissADME calculated physicochemical properties, the two compounds shared drug-like characteristics, with moderate water solubility, good intestinal adsorption, and no brain-blood barrier permeability. However, they seemed to be vulnerable to rapid hepatic inactivation and also to inhibit almost all the CYP450 isoforms (except for 1A2 isoform), thus suggesting a high risk of causing drug-drug interactions. Both 18a and 18b were predicted to be P-glycoproteins (P-gp) substrates, an efflux pump that is related to the risk of developing drug resistance.
Finally, docking calculations unravelled a preferential selectivity of 18b for the trypanosomal isoform of the glycolytic enzyme, TcTIM (DGbind= -10.2 Kcal/mol), over the human isoform (HsTIM: DGbind= -5.9 Kcal/mol) [108].
Beltran-Hortelano et al. [110] screened against T. cruzi some Mannich base-type derivatives, whose activity was previously related to the inhibition of the trypanosomal superoxide dismutase, a Fe-containing metalloprotein (Fe-SOD) [111]. The library was categorised into different groups (series a-g) according to common structural features. The only benzimidazole-containing cluster was series b, composed of 17 compounds, among which 19a and 19b demonstrated good antiparasitic activity against T. cruzi CL-Lu: Neon clone epimastigotes (Table 13). However, no further studies were performed about them due to the high toxicity demonstrated against BSR mammalian cells.
Table 13.
Antitrypanosomal activity against T. cruzi epimastigotes and cytotoxicity of derivatives 19a,b.
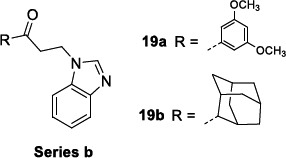
| |||
|---|---|---|---|
| Compound |
IC50a
CL-Luc: Neon Clone Epim. |
IC50a
BSR Cells |
SIb |
| 19a | 11.1 ± 3.2 | 70.1 ± 9.6 | 6 |
| 19b | 24.8 ± 5.8 | 67.6 ± 6.1 | 3 |
| Benznidazole | 3.8 ± 0.7 | 191.28 ± 1.8 | 67 |
Note: aIC50 (µM): compound concentration that reduced the parasite load of 50%; bSI: selectivity index (IC50 BSR/IC50 parasite).
As mentioned above, for the benzimidazole-based antimalarials, the pentamidine amidine motif was also explored in the search for novel DNA-binding anti-trypanosoma agents and even better broad-spectrum antiparasitic compounds. In this context, Bistrović et al. [112] synthesised a series of 5-amidino benzimidazoles as anti-bacterial and anti-protozoal compounds, characterized by a hydrophobic 1-aryl-1,2,3-triazole moiety.
These compounds were tested in vitro for the antiparasitic activity against T. brucei bloodstream form (strain 221) and toxicity against the mammal L6 cell line (Table 14). They inhibited T. brucei in the low micromolar range (IC50= 1.1 to 1.6 μM). These compounds characteristically share a p-methoxy phenyl moiety on the triazole ring and differ from each other only for the structure of the amidino motif. Notably, the best inhibitor, 20b, proved to be 4-fold more potent than the reference compound Nifurtimox. The insertion of other groups on the phenyl ring or other types of aromatic moieties negatively affected the antiparasitic activity, which reduced or even abolished. The binding affinity of the compounds to the nucleic acids, observed by UV/vis spectroscopy, was linearly correlated with the anti-microbial activity but not with the antiprotozoal one, thus reasonably assuming that other pathways and targets could also be involved in the mechanism of action.
Table 14.
Antitrypanosomal activity of compounds 20a-c against T. brucei.

| ||
|---|---|---|
| Compound | IC50a T. brucei | SIb |
| 20a | 1.5 ± 0.3 | 200 |
| 20b | 1.1 ± 0.3 | >270 |
| 20c | 1.6 ± 0.4 | 165 |
| Nifurtimox | 4.4 ± 0.7b | nd |
Note: aIC50: compound concentration that reduced the parasite load of 50%; bSI: selectivity index (IC50 L6/IC50 parasite); nd = not determined.
The class of 2-aminobenzimidazoles was identified by high-throughput screening (HTS) technology applied to a compound library of GlaxoSmithKline (GSK) in order to develop novel libraries of molecules targeting leishmaniasis [97] and trypanosomiasis [113, 114]. Subsequent hit optimization studies explored antiparasitic potency, cytotoxicity, adsorption, metabolism, and excretion as parameters for the progression of the most promising derivatives. The initial profiling of the hit compound 21a confirmed its potency towards T. cruzi and T.brucei, no observable toxicity on HepG2 and MRC-5 mammalian cells (T. cruzi strains IC50 = 0.63 and 1.26 μM; T. b. brucei IC50 = 2.0 μM; MRC-5 CC50 > 64 μM; HepG2 CC50 >100) and good permeability, but a rapid in vitro clearance in mouse liver microsomes (MLM) and a high LogD (eLogD =4.6). The hit-to-lead and lead optimization programs focused on the substitution of the benzimidazole ring in positions 5 and 6, on the variation of the N-alkyl chain and of the acyl fragment linked to the 2-amino group.
McNamara et al. observed that the p-Cl substitution of the phenyl ring was an important requisite for the activity, as its substitution with stronger electron-withdrawing CF3 or OCF3 groups only produced a negligible improvement. The replacement of the phenyl ring with biphenyl, naphthyl rings or a long aliphatic chain still provided equipotent compounds to 21a. A comparable SAR trend was observed against T. brucei, whereby the effect of the substituents in the diverse positions of the phenyl ring was similar for almost all the entries (mean IC50 3.5 μM).
The chemical space around the N1-alkyl chain of the benzimidazole ring was extensively explored, starting from the N-propyl chain of 21a. Analysing the in vitro IC50 values towards T. cruzi, the propyl chain proved to be the best choice since its shortening or removal was responsible for the loss of activity, whilst its elongation, as in the butyl and pentyl chains, led to a slight improvement of the potency but to a higher lipophilicity. Regarding T. brucei, the chain length variation did not affect the activity (IC50 3.1-4.4 μM). The variation of the nature (ureido and reverse amide groups) and the length of the amide linker on C2 of the benzimidazole ring was inspected in another series without observing any substantial improvement over the phenylacetamide moiety of the prototype 21a.
Finally, only the inclusion of halogen atoms (F, Cl and Br) in the 5 or 6 position of the benzimidazole ring allowed for more potent derivatives than 21a (Tables 15 and 16). However, the cytotoxicity parallelly increased, leading to a low therapeutic window. Analogous trend was observed in the case of T.b. brucei.
Table 15.
Antitrypanosomal activity and SI of hit 21a against two different strains of T. cruzi and T. brucei.

| ||
|---|---|---|
| Parasite | IC50a | SIe |
| T. cruzib | 0.63 | 79 |
| T. cruzic | 1.26 | 40 |
| T. bruceid | 2.00 | 50 |
Note: aIC50: compound concentration that reduced the parasite load by 50%. bT. cruzi primary growth inhibition assay performed with NIH-3T3 primary mouse embryonic fibroblast cells host cells. cT. cruzi intracellular imaging assay performed with H9c2 rat embryonic cardiomyocytes. dT. brucei primary growth inhibition assay in complete HMI9 medium. eSI: selectivity index (CC50 mammalian cell line/IC50 parasite).
Table 16.
Antitrypanosomal activity and SI of compounds 21b-f against T. cruzi and T. brucei.

| |||||
|---|---|---|---|---|---|
| Compound | R |
T. cruzi
IC50a |
SIb |
T.b. brucei
IC50 |
SIc |
| 21b | 5-Cl | 0.5 | 15.9 | 4.4 | 84% |
| 21c | 5-Br | 0.79 | >63.3 | 16 | >5.18 |
| 21d | 6-Cl | 0.5 | 12.6 | 2.4 | >34.6 |
| 21e | 6-Br | 0.4 | 32.5 | 1.5 | >55.3 |
| 21f | 6-F | 1.3 | 19.2 | 0.90 | 15.5 |
| Benznidazole | 1.58 | 20 | nd | nd | |
| Pentamidine | nd | nd | 0.0014 | nd | |
Note: aIC50: compound concentration that reduced the parasite load of 50%; bSI: selectivity index (CC50 H9c2 /IC50 T. cruzi); cSI: selectivity index (CC50 HEK-293 /IC50 T. brucei); nd = not determined.
A follow-up study was published by Rezende Júnior et al. [114], where novel benzimidazoles structurally related to hit 21a were synthesized and tested for their activity against T. cruzi, and for their ADME-Tox properties. Keeping the 1-alkyl-2-acylamino benzimidazole as the scaffold, diverse decorations in the 5/6 positions of the benzimidazole ring, 1-alkyl chains, and 2-acylamino fragments were introduced. As priority criteria for the progression of the molecules, a high potency and an adequate ADME-Tox profile were selected (IC50 < 5 μM, selectivity index (SI) > 30, MLM clearance < 30 μL/min/mg and LogD close to 3).
In order to modulate the overall lipophilicity of the compounds, the phenylacetic fragment underwent several modifications, introducing picolinic and nicotinic acid moieties, which allowed to retain good IC50s generally lower than 10 μM. Regarding the 1-alkyl moiety, the trifluoroethyl chain was identified as the best substitution to balance the potency and the metabolic stability (21g) (Table 17).
Table 17.
Antitrypanosomal activity, metabolic stability and lipophilicity of benzimidazoles 21g-j.

| |||||
|---|---|---|---|---|---|
| Compound | R | T. cruzi IC50 a Tulahuen CL2 Strain | SIb | MLM CIint µg/min/mg | eLogD |
| 21g |

|
2.6 ± 0.66 | >25 | 18 | 3.1 |
| 21h |

|
0.28 ± 0.30 | >231 | 27 | 3.1 |
| 21i |

|
0.23 ± 0.96 | 34 | 28 | 3.0 |
| 21j |

|
1.5 ± 3.19 | >42 | 25 | 2.9 |
| Benznidazole | 2.1 | nd | nd | nd | |
Note: aIC50: compound concentration that reduced the parasite load of 50%; bSI: selectivity index (CC50 MRC-5 /IC50 parasite); nd = not determined.
Unchanging the 6-CN and the 1-trifluoroethyl chain, some alternative heteroaromatic acylamines were sampled. The trifluoromethylpyridine derivative 21h and compounds 21i and 21j bearing the trifluoromethyl pyrazole rings showed improved potencies and good metabolic stability, low lipophilicity, and high selectivity indexes (Table 17).
The selected compounds 21g-j were then evaluated against T. cruzi amastigotes, which exhibited a concentration- and time-dependent antiparasitic activity. Interestingly, differing from the traditional antiparasitic agents which are considered fast killers, these compounds could behave as slow killers since they require more cycles of parasite replication to exert their antiparasitic activity. The study of physicochemical properties pointed out that these compounds possess good permeability but low kinetic solubility values at pH 7.4 and 2.0. Whilst cytotoxicity against the host cell line MRC-5 used in the first screening was generally low (mean CC50 >50 μM), it increased against HepG2 and PMM cells. From the analysis of the risk for off-target adverse events, no significant evidence of activity was observed against a panel of key human enzymes. Thus, the compounds warrant a deep investigation of their promising safety profile.
CONCLUSION
The management of protozoan diseases still represents a challenging task in WHO programmes, strengthening the need for solving the gap in innovation and more adequate investments in basic research and product development and new vector control strategies and technologies. The rapid global expansion of travel and trade, the growing interactions between humans and the animal reservoirs of pathogens and vector species, and the emerging environmental problems make the scenario very complex. Additionally, the report of natural coinfections involving diverse Plasmodium and Leishmania species poses a greater risk of morbidity and severe diseases, as the concomitant incidence of the two diseases in the same individual showed to jeopardise the host immune response [115]. Nevertheless, the impact of the COVID-19 pandemic has drastically shifted the priorities of public health. Thus, the prevention, diagnosis, and treatment strategies against protozoan diseases were unavoidably disregarded [116]. The existing drugs for human and animal vector-borne diseases are scarce and are affected by suboptimal efficacy, toxicity, and drug resistance. The development of novel antiprotozoal drugs may also take inspiration from the benzimidazole nucleus as a privileged pharmacophore in medicinal chemistry, thanks to its bioisosterism with naturally occurring purine nucleobases. The benzimidazole-based derivatives have demonstrated the ability to tackle distinct or even more multiple targets implicated in the pathogenesis of protozoan infections and have provided the potential for a broad spectrum antiparasitic activity. In this regard, the multitarget approach may be a valid tool to treat complex diseases like protozoan ones where a parasite, a vector (ectoparasite) and a host are strictly interconnected. New directions in the compounds’ design should focus on the identification of novel targets, especially those involved in the liver and transmission stages of the disease for a more meaningful blockage of the infection spreading to other human hosts. The SAR insights gathered through this survey offer the inspiration for developing more promising benzimidazole-based derivatives to treat the infections caused by Malaria, Leishmania, and Trypanosoma parasites. However, most of the proposed studies have stopped at the initial phases of drug discovery pipeline, limiting the biological evaluation at the enzymatic and cellular level, sometimes without identifying the molecular target. As only a few studies have explored the drug-likeness of these molecules, further investigations are warranted to translate them into therapeutics to handle these infectious diseases in humans and animals more effectively.
ACKNOWLEDGEMENTS
This project is based upon work from COST Action CA2111 < One Health drugs against parasitic vector-borne diseases in Europe and beyond >, supported by COST (European Cooperation in Science and Technology). Fig. (1) was created with BioRender.com.
LIST OF ABBREVIATIONS
- CL
Cutaneous
- MCL
Mucocutaneous
- VL
Visceral Kala-azar
- DNDi
Drugs for Neglected Diseases Initiative
- TPP
Target Product Profile
- EMA
European Medicines Agency
- LSF
Trypomastigote
- SSF
Short Stumpy Trypomastigote Form
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
Dr. Anna Carbone and Dr. Michele Tonelli are on the Editorial Advisory Board of the Journal Current Medicinal Chemistry.
REFERENCES
- 1.Pereira M.A., Santos-Gomes G. Parasitic infection and immunity: A special biomedicines issue. Biomedicines. 2022;10(10):2547. doi: 10.3390/biomedicines10102547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Why do neglected tropical diseases suffer low priority? Available from: https://www.afro.who.int/news/why-do-neglected-tropical-diseases-suffer-low-priority (Accessed May 1, 2023).
- 3.Q&A on RTS S malaria vaccine. Available from: https://www.who.int/news-room/questions-and-answers/item/q-a-on-rts-s-malaria-vaccine.
- 4.Nadeem A.Y., Shehzad A., Islam S.U., Al-Suhaimi E.A., Lee Y.S. Mosquirix™ RTS, S/AS01 vaccine development, immunogenicity, and efficacy. Vaccines. 2022;10(5):713. doi: 10.3390/vaccines10050713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandley P., Ranjan R., Kumar S., Rohatgi S. Host-parasite interactions during Plasmodium infection: Implications for immunotherapies. Front. Immunol. 2023;13:1091961. doi: 10.3389/fimmu.2022.1091961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Capela R., Moreira R., Lopes F. An overview of drug resistance in protozoal diseases. Int. J. Mol. Sci. 2019;20(22):5748. doi: 10.3390/ijms20225748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.World malaria report. Available from: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022 (Accessed Jan 7, 2023).
- 8.Zanghi G., Vaughan A.M. Plasmodium vivax pre-erythrocytic stages and the latent hypnozoite. Parasitol. Int. 2021;85:102447. doi: 10.1016/j.parint.2021.102447. [DOI] [PubMed] [Google Scholar]
- 9.Belete T.M. Recent progress in the development of new antimalarial drugs with novel targets. Drug Des. Devel. Ther. 2020;14:3875–3889. doi: 10.2147/DDDT.S265602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plewes K., Leopold S.J., Kingston H.W.F., Dondorp A.M. Malaria. Infect. Dis. Clin. North Am. 2019;33(1):39–60. doi: 10.1016/j.idc.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 11.Nsanzabana C. Resistance to artemisinin combination therapies (ACTs): Do not forget the partner drug! Trop. Med. Infect. Dis. 2019;4(1):26. doi: 10.3390/tropicalmed4010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones R.A., Panda S.S., Hall C.D. Quinine conjugates and quinine analogues as potential antimalarial agents. Eur. J. Med. Chem. 2015;97:335–355. doi: 10.1016/j.ejmech.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Tibon N.S., Ng C.H., Cheong S.L. Current progress in antimalarial pharmacotherapy and multi-target drug discovery. Eur. J. Med. Chem. 2020;188:111983. doi: 10.1016/j.ejmech.2019.111983. [DOI] [PubMed] [Google Scholar]
- 14.Kamchonwongpaisan S., Charoensetakul N., Srisuwannaket C., Taweechai S., Rattanajak R., Vanichtanankul J., Vitsupakorn D., Arwon U., Thongpanchang C., Tarnchompoo B., Vilaivan T., Yuthavong Y. Flexible diaminodihydrotriazine inhibitors of Plasmodium falciparum dihydrofolate reductase: Binding strengths, modes of binding and their antimalarial activities. Eur. J. Med. Chem. 2020;195:112263. doi: 10.1016/j.ejmech.2020.112263. [DOI] [PubMed] [Google Scholar]
- 15.Blasco B., Leroy D., Fidock D.A. Antimalarial drug resistance: Linking Plasmodium falciparum parasite biology to the clinic. Nat. Med. 2017;23(8):917–928. doi: 10.1038/nm.4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Antonovics J. Transmission dynamics: Critical questions and challenges. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017;372(1719):20160087. doi: 10.1098/rstb.2016.0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neglected tropical diseases - GLOBAL. Available from: https://www.who.int/health-topics/neglected-tropical-diseases (Accessed May 1, 2023).
- 18.Leishmaniasis. Available from: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (Accessed May 1, 2023).
- 19.Liu D., Uzonna J.E. The early interaction of Leishmania with macrophages and dendritic cells and its influence on the host immune response. Front. Cell. Infect. Microbiol. 2012;2:83. doi: 10.3389/fcimb.2012.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mann S., Frasca K., Scherrer S., Henao-Martínez A.F., Newman S., Ramanan P., Suarez J.A. A review of leishmaniasis: Current knowledge and future directions. Curr. Trop. Med. Rep. 2021;8(2):121–132. doi: 10.1007/s40475-021-00232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaye P.M., Mohan S., Mantel C., Malhame M., Revill P., Le Rutte E., Parkash V., Layton A.M., Lacey C.J.N., Malvolti S. Overcoming roadblocks in the development of vaccines for leishmaniasis. Expert Rev. Vaccines. 2021;20(11):1419–1430. doi: 10.1080/14760584.2021.1990043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morales-Yuste M., Martín-Sánchez J., Corpas-Lopez V. Canine leishmaniasis: Update on epidemiology, diagnosis, treatment, and prevention. Vet. Sci. 2022;9(8):387. doi: 10.3390/vetsci9080387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rao S.P.S., Barrett M.P., Dranoff G., Faraday C.J., Gimpelewicz C.R., Hailu A., Jones C.L., Kelly J.M., Lazdins-Helds J.K., Mäser P., Mengel J., Mottram J.C., Mowbray C.E., Sacks D.L., Scott P., Späth G.F., Tarleton R.L., Spector J.M., Diagana T.T. Drug discovery for kinetoplastid diseases: Future directions. ACS Infect. Dis. 2019;5(2):152–157. doi: 10.1021/acsinfecdis.8b00298. [DOI] [PubMed] [Google Scholar]
- 24.Katsuno K., Burrows J.N., Duncan K., van Huijsduijnen R.H., Kaneko T., Kita K., Mowbray C.E., Schmatz D., Warner P., Slingsby B.T. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discov. 2015;14(11):751–758. doi: 10.1038/nrd4683. [DOI] [PubMed] [Google Scholar]
- 25.Trypanosomiasis, human African (sleeping sickness). Available from: https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (Accessed May 1, 2023).
- 26.Lindner A.K., Lejon V., Chappuis F., Seixas J., Kazumba L., Barrett M.P., Mwamba E., Erphas O., Akl E.A., Villanueva G., Bergman H., Simarro P., Kadima Ebeja A., Priotto G., Franco J.R. New WHO guidelines for treatment of gambiense human African trypanosomiasis including fexinidazole: Substantial changes for clinical practice. Lancet Infect. Dis. 2020;20(2):e38–e46. doi: 10.1016/S1473-3099(19)30612-7. [DOI] [PubMed] [Google Scholar]
- 27.Chagas disease. Available from: https://www.who.int/news-room/fact-sheets/detail/chagas-disease-(american-trypanosomiasis) (Accessed May 1, 2023).
- 28.da Costa K.M., Valente R.C., Fonseca L.M., Freire-de-Lima L., Previato J.O., Mendonça-Previato L. The history of the abc proteins in human trypanosomiasis pathogens. Pathogens. 2022;11(9):988. doi: 10.3390/pathogens11090988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sandes J.M., de Figueiredo R.C.B.Q. The endoplasmic reticulum of trypanosomatids: An unrevealed road for chemotherapy. Front. Cell. Infect. 2022:12. doi: 10.3389/fcimb.2022.1057774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tahlan S., Kumar S., Narasimhan B. Pharmacological significance of heterocyclic 1H-benzimidazole scaffolds: A review. BMC Chem. 2019;13(1):101. doi: 10.1186/s13065-019-0625-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ajani O.O., Aderohunmu D.V., Ikpo C.O., Adedapo A.E., Olanrewaju I.O. Functionalized benzimidazole scaffolds: Privileged heterocycle for drug design in therapeutic medicine. Arch. Pharm. 2016;349(7):475–506. doi: 10.1002/ardp.201500464. [DOI] [PubMed] [Google Scholar]
- 32.Choudhary S., Arora M., Verma H., Kumar M., Silakari O. Benzimidazole based hybrids against complex diseases: A catalogue of the SAR profile. Eur. J. Pharmacol. 2021;899:174027. doi: 10.1016/j.ejphar.2021.174027. [DOI] [PubMed] [Google Scholar]
- 33.Yadav G., Ganguly S. Structure activity relationship (SAR) study of benzimidazole scaffold for different biological activities: A mini-review. Eur. J. Med. Chem. 2015;97:419–443. doi: 10.1016/j.ejmech.2014.11.053. [DOI] [PubMed] [Google Scholar]
- 34.Alzhrani Z.M.M., Alam M.M., Nazreen S. Recent advancements on benzimidazole: A versatile scaffold in medicinal chemistry. Mini Rev. Med. Chem. 2022;22(2):365–386. doi: 10.2174/1389557521666210331163810. [DOI] [PubMed] [Google Scholar]
- 35.Akhtar W., Khan M.F., Verma G., Shaquiquzzaman M., Rizvi M.A., Mehdi S.H., Akhter M., Alam M.M. Therapeutic evolution of benzimidazole derivatives in the last quinquennial period. Eur. J. Med. Chem. 2017;126:705–753. doi: 10.1016/j.ejmech.2016.12.010. [DOI] [PubMed] [Google Scholar]
- 36.Ebenezer O., Jordaan M.A., Carena G., Bono T., Shapi M., Tuszynski J.A. An overview of the biological evaluation of selected nitrogen-containing heterocycle medicinal chemistry compounds. Int. J. Mol. Sci. 2022;23(15):8117. doi: 10.3390/ijms23158117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cichero E., Calautti A., Francesconi V., Tonelli M., Schenone S., Fossa P. Probing in silico the benzimidazole privileged scaffold for the development of drug-like anti-rsv agents. Pharmaceuticals. 2021;14(12):1307. doi: 10.3390/ph14121307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barrett M.P., Gemmell C.G., Suckling C.J. Minor groove binders as anti-infective agents. Pharmacol. Ther. 2013;139(1):12–23. doi: 10.1016/j.pharmthera.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 39.Dardonville C., Nue Martinez J.J. Bis(2-aminoimida- zolines) and bisguanidines: Synthetic approaches, antiparasitic activity and DNA binding properties. Curr. Med. Chem. 2017;24(33):3606–3632. doi: 10.2174/0929867324666170623091522. [DOI] [PubMed] [Google Scholar]
- 40.Hamilton W.L., Claessens A., Otto T.D., Kekre M., Fairhurst R.M., Rayner J.C., Kwiatkowski D. Extreme mutation bias and high AT content in Plasmodium falciparum. Nucleic Acids Res. 2017;45(4):1889–1901. doi: 10.1093/nar/gkw1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wilson W.D., Tanious F.A., Mathis A., Tevis D., Hall J.E., Boykin D.W. Antiparasitic compounds that target DNA. Biochimie. 2008;90(7):999–1014. doi: 10.1016/j.biochi.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boschi D., Pippione A.C., Sainas S., Lolli M.L. Dihydroorotate dehydrogenase inhibitors in anti-infective drug research. Eur. J. Med. Chem. 2019;183:111681. doi: 10.1016/j.ejmech.2019.111681. [DOI] [PubMed] [Google Scholar]
- 43.Phillips M.A., Rathod P.K. Plasmodium dihydroorotate dehydrogenase: A promising target for novel anti-malarial chemotherapy. Infect. Disord. Drug Targets. 2010;10(3):226–239. doi: 10.2174/187152610791163336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fernandes P., Loubens M., Le Borgne R., Marinach C., Ardin B., Briquet S., Vincensini L., Hamada S., Hoareau-Coudert B., Verbavatz J.M., Weiner A., Silvie O. The AMA1-RON complex drives Plasmodium sporozoite invasion in the mosquito and mammalian hosts. PLoS Pathog. 2022;18(6):e1010643. doi: 10.1371/journal.ppat.1010643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Devine S.M., MacRaild C.A., Norton R.S., Scammells P.J. Antimalarial drug discovery targeting apical membrane antigen 1. MedChemComm. 2017;8(1):13–20. doi: 10.1039/C6MD00495D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Drew D.R., Wilson D.W., Elliott S.R., Cross N., Terheggen U., Hodder A.N., Siba P.M., Chelimo K., Dent A.E., Kazura J.W., Mueller I., Beeson J.G. A novel approach to identifying patterns of human invasion-inhibitory antibodies guides the design of malaria vaccines incorporating polymorphic antigens. BMC Med. 2016;14(1):144. doi: 10.1186/s12916-016-0691-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee S.K., Low L.M., Andersen J.F., Yeoh L.M., Valenzuela Leon P.C., Drew D.R., Doehl J.S.P., Calvo E., Miller L.H., Beeson J.G., Gunalan K. The direct binding of Plasmodium vivax AMA1 to erythrocytes defines a RON2-independent invasion pathway. Proc. Natl. Acad. Sci. 2023;120(1):e2215003120. doi: 10.1073/pnas.2215003120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Villiers K.A., Egan T.J. Heme detoxification in the malaria parasite: A target for antimalarial drug development. Acc. Chem. Res. 2021;54(11):2649–2659. doi: 10.1021/acs.accounts.1c00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quadros H.C., Silva M.C.B., Moreira D.R.M. The role of the iron protoporphyrins heme and hematin in the antimalarial activity of endoperoxide drugs. Pharmaceuticals. 2022;15(1):60. doi: 10.3390/ph15010060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dvorin J.D., Goldberg D.E. Plasmodium egress across the parasite life cycle. Annu. Rev. Microbiol. 2022;76(1):67–90. doi: 10.1146/annurev-micro-041320-020659. [DOI] [PubMed] [Google Scholar]
- 51.Tan M.S.Y., Blackman M.J. Malaria parasite egress at a glance. J. Cell Sci. 2021;134(5):jcs257345. doi: 10.1242/jcs.257345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramaprasad A., Burda P-C., Koussis K., Thomas J., Pietsch E., Calvani E., Howell S., MacRae J., Snijders A., Gilberger T-W., Blackman M. A malaria parasite phospholipase facilitates efficient asexual blood stage egres. bioRxiv. 2023:532312. doi: 10.1371/journal.ppat.1011449. [DOI] [PMC free article] [PubMed]
- 53.Cavalcanti D.P., de Souza W. The kinetoplast of trypanosomatids: From early studies of electron microscopy to recent advances in atomic force microscopy. Scanning. 2018;2018:1–10. doi: 10.1155/2018/9603051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Menna-Barreto R.F.S., de Castro S.L. The double-edged sword in pathogenic trypanosomatids: The pivotal role of mitochondria in oxidative stress and bioenergetics. BioMed Res. Int. 2014;2014:1–14. doi: 10.1155/2014/614014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tomás A.M., Castro H. Redox metabolism in mitochondria of trypanosomatids. Antioxid. Redox Signal. 2013;19(7):696–707. doi: 10.1089/ars.2012.4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Irigoín F., Cibils L., Comini M.A., Wilkinson S.R., Flohé L., Radi R. Insights into the redox biology of Trypanosoma cruzi: Trypanothione metabolism and oxidant detoxification. Free Radic. Biol. Med. 2008;45(6):733–742. doi: 10.1016/j.freeradbiomed.2008.05.028. [DOI] [PubMed] [Google Scholar]
- 57.Stoll V.S., Simpson S.J., Krauth-Siegel R.L., Walsh C.T., Pai E.F. Glutathione reductase turned into trypanothione reductase: Structural analysis of an engineered change in substrate specificity. Biochemistry. 1997;36(21):6437–6447. doi: 10.1021/bi963074p. [DOI] [PubMed] [Google Scholar]
- 58.Krieger S., Schwarz W., Ariyanayagam M.R., Fairlamb A.H., Krauth-Siegel R.L., Clayton C. Trypanosomes lacking trypanothione reductase are avirulent and show increased sensitivity to oxidative stress. Mol. Microbiol. 2000;35(3):542–552. doi: 10.1046/j.1365-2958.2000.01721.x. [DOI] [PubMed] [Google Scholar]
- 59.Borsari C., Quotadamo A., Ferrari S., Venturelli A., Cordeiro-da-Silva A., Santarem N., Costi M.P. Chapter Two : Scaffolds and biological targets avenue to fight against drug resistance in leishmaniasis. In: Botta M., editor. Annual reports in medicinal chemistry. Neglected Diseases: Extensive Space for Modern Drug Discovery. Vol. 51. Academic Press; 2018. pp. 39–95. [Google Scholar]
- 60.Quiñones W., Acosta H., Gonçalves C.S., Motta M.C.M., Gualdrón-López M., Michels P.A.M. Structure, properties, and function of glycosomes in Trypanosoma cruzi. Front. Cell. Infect. Microbiol. 2020;10:25. doi: 10.3389/fcimb.2020.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Veloso-Silva L.L.W., Dores-Silva P.R., Bertolino-Reis D.E., Moreno-Oliveira L.F., Libardi S.H., Borges J.C. Structural studies of old yellow enzyme of Leishmania braziliensis in solution. Arch. Biochem. Biophys. 2019;661:87–96. doi: 10.1016/j.abb.2018.11.009. [DOI] [PubMed] [Google Scholar]
- 62.Díaz-Viraqué F., Chiribao M.L., Trochine A., González-Herrera F., Castillo C., Liempi A., Kemmerling U., Maya J.D., Robello C. Old yellow enzyme from Trypanosoma cruzi exhibits in vivo prostaglandin F2α synthase activity and has a key role in parasite infection and drug susceptibility. Front. Immunol. 2018;9:456. doi: 10.3389/fimmu.2018.00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Balaña-Fouce R., Calvo-Álvarez E., Álvarez-Velilla R., Prada C.F., Pérez-Pertejo Y., Reguera R.M. Role of trypanosomatid’s arginase in polyamine biosynthesis and pathogenesis. Mol. Biochem. Parasitol. 2012;181(2):85–93. doi: 10.1016/j.molbiopara.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 64.Ilari A., Fiorillo A., Baiocco P., Poser E., Angiulli G., Colotti G. Targeting polyamine metabolism for finding new drugs against leishmaniasis: A review. Mini Rev. Med. Chem. 2015;15(3):243–252. doi: 10.2174/138955751503150312141044. [DOI] [PubMed] [Google Scholar]
- 65.Westrop G.D., Williams R.A.M., Wang L., Zhang T., Watson D.G., Silva A.M., Coombs G.H. Metabolomic analyses of leishmania reveal multiple species differences and large differences in amino acid metabolism. PLoS One. 2015;10(9):e0136891. doi: 10.1371/journal.pone.0136891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boitz J.M., Gilroy C.A., Olenyik T.D., Paradis D., Perdeh J., Dearman K., Davis M.J., Yates P.A., Li Y., Riscoe M.K., Ullman B., Roberts S.C. Arginase is essential for survival of Leishmania donovani promastigotes but not intracellular amastigotes. Infect. Immun. 2016;85(1):e00554–e16. doi: 10.1128/IAI.00554-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Siqueira-Neto J.L., Debnath A., McCall L.I., Bernatchez J.A., Ndao M., Reed S.L., Rosenthal P.J. Cysteine proteases in protozoan parasites. PLoS Negl. Trop. Dis. 2018;12(8):e0006512. doi: 10.1371/journal.pntd.0006512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mottram J.C., Coombs G.H., Alexander J. Cysteine peptidases as virulence factors of Leishmania. Curr. Opin. Microbiol. 2004;7(4):375–381. doi: 10.1016/j.mib.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 69.Alexander J., Bryson K. T helper (h)1/Th2 and: Paradox rather than paradigm. Immunol. Lett. 2005;99(1):17–23. doi: 10.1016/j.imlet.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 70.Casgrain P.A., Martel C., McMaster W.R., Mottram J.C., Olivier M., Descoteaux A. Cysteine peptidase b regulates leishmania mexicana virulence through the modulation of GP63 Expression. PLoS Pathog. 2016;12(5):e1005658. doi: 10.1371/journal.ppat.1005658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shamshad H., Bakri R., Mirza A.Z. Dihydrofolate reductase, thymidylate synthase, and serine hydroxy methyltransferase: Successful targets against some infectious diseases. Mol. Biol. Rep. 2022;49(7):6659–6691. doi: 10.1007/s11033-022-07266-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tassone G., Landi G., Linciano P., Francesconi V., Tonelli M., Tagliazucchi L., Costi M.P., Mangani S., Pozzi C. Evidence of pyrimethamine and cycloguanil analogues as dual inhibitors of Trypanosoma brucei pteridine reductase and dihydrofolate reductase. Pharmaceuticals. 2021;14(7):636. doi: 10.3390/ph14070636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pöhner I., Quotadamo A., Panecka-Hofman J., Luciani R., Santucci M., Linciano P., Landi G., Di Pisa F., Dello Iacono L., Pozzi C., Mangani S., Gul S., Witt G., Ellinger B., Kuzikov M., Santarem N., Cordeiro-da-Silva A., Costi M.P., Venturelli A., Wade R.C. Multitarget, selective compound design yields potent inhibitors of a kinetoplastid pteridine reductase 1. J. Med. Chem. 2022;65(13):9011–9033. doi: 10.1021/acs.jmedchem.2c00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Farahat A.A., Ismail M.A., Kumar A., Wenzler T., Brun R., Paul A., Wilson W.D., Boykin D.W. Indole and benzimidazole bichalcophenes: Synthesis, DNA binding and antiparasitic activity. Eur. J. Med. Chem. 2018;143:1590–1596. doi: 10.1016/j.ejmech.2017.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cardona-G W., Yepes A.F., Herrera-R A. Hybrid molecules: Promising compounds for the development of new treatments against leishmaniasis and chagas disease. Curr. Med. Chem. 2018;25(30):3637–3679. doi: 10.2174/0929867325666180309111428. [DOI] [PubMed] [Google Scholar]
- 76.Doganc F., Celik I., Eren G., Kaiser M., Brun R., Goker H. Synthesis, in vitro antiprotozoal activity, molecular docking and molecular dynamics studies of some new monocationic guanidinobenzimidazoles. Eur. J. Med. Chem. 2021;221:113545. doi: 10.1016/j.ejmech.2021.113545. [DOI] [PubMed] [Google Scholar]
- 77.Jahnke W., Erlanson D.A., de Esch I.J.P., Johnson C.N., Mortenson P.N., Ochi Y., Urushima T. Fragment-to-lead medicinal chemistry publications in 2019. J. Med. Chem. 2020;63(24):15494–15507. doi: 10.1021/acs.jmedchem.0c01608. [DOI] [PubMed] [Google Scholar]
- 78.Krishnarjuna B., Lim S.S., Devine S.M., Debono C.O., Lam R., Chandrashekaran I.R., Jaipuria G., Yagi H., Atreya H.S., Scanlon M.J., MacRaild C.A., Scammells P.J., Norton R.S. Solution NMR characterization of apical membrane antigen 1 and small molecule interactions as a basis for designing new antimalarials. J. Mol. Recognit. 2016;29(6):281–291. doi: 10.1002/jmr.2529. [DOI] [PubMed] [Google Scholar]
- 79.Kim J., Tan Y.Z., Wicht K.J., Erramilli S.K., Dhingra S.K., Okombo J., Vendome J., Hagenah L.M., Giacometti S.I., Warren A.L., Nosol K., Roepe P.D., Potter C.S., Carragher B., Kossiakoff A.A., Quick M., Fidock D.A., Mancia F. Structure and drug resistance of the Plasmodium falciparum transporter PfCRT. Nature. 2019;576(7786):315–320. doi: 10.1038/s41586-019-1795-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jiménez-Díaz M.B., Ebert D., Salinas Y., Pradhan A., Lehane A.M., Myrand-Lapierre M.E., O’Loughlin K.G., Shackleford D.M., Justino de Almeida M., Carrillo A.K., Clark J.A., Dennis A.S.M., Diep J., Deng X., Duffy S., Endsley A.N., Fedewa G., Guiguemde W.A., Gómez M.G., Holbrook G., Horst J., Kim C.C., Liu J., Lee M.C.S., Matheny A., Martínez M.S., Miller G., Rodríguez-Alejandre A., Sanz L., Sigal M., Spillman N.J., Stein P.D., Wang Z., Zhu F., Waterson D., Knapp S., Shelat A., Avery V.M., Fidock D.A., Gamo F.J., Charman S.A., Mirsalis J.C., Ma H., Ferrer S., Kirk K., Angulo-Barturen I., Kyle D.E., DeRisi J.L., Floyd D.M., Guy R.K. (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc. Natl. Acad. Sci. 2014;111(50):E5455–E5462. doi: 10.1073/pnas.1414221111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bhoi R.T., Rajput J.D., Bendre R.S. An efficient synthesis of rearranged new biologically active benzimidazoles derived from 2-formyl carvacrol. Res. Chem. Intermed. 2022;48(1):401–422. doi: 10.1007/s11164-021-04601-9. [DOI] [Google Scholar]
- 82.Singh V., Hada R.S., Jain R., Vashistha M., Kumari G., Singh S., Sharma N., Bansal M., Poonam, , Zoltner M., Caffrey C.R., Rathi B., Singh S. Designing and development of phthalimides as potent anti-tubulin hybrid molecules against malaria. Eur. J. Med. Chem. 2022;239:114534. doi: 10.1016/j.ejmech.2022.114534. [DOI] [PubMed] [Google Scholar]
- 83.Ndakala A.J., Gessner R.K., Gitari P.W., October N., White K.L., Hudson A., Fakorede F., Shackleford D.M., Kaiser M., Yeates C., Charman S.A., Chibale K. Antimalarial pyrido[1,2-a]benzimidazoles. J. Med. Chem. 2011;54(13):4581–4589. doi: 10.1021/jm200227r. [DOI] [PubMed] [Google Scholar]
- 84.Mali S.N., Pandey A. Hemozoin (beta-hematin) Formation inhibitors: Promising target for the development of new antimalarials: Current update and future prospect. Comb. Chem. High Throughput Screen. 2022;25(11):1859–1874. doi: 10.2174/1386207325666210924104036. [DOI] [PubMed] [Google Scholar]
- 85.Sousa C.C., Dziwornu G.A., Quadros H.C., Araujo-Neto J.H., Chibale K., Moreira D.R.M. Antimalarial Pyrido[1,2-a]benzimidazoles exert strong parasiticidal effects by achieving high cellular uptake and suppressing heme detoxification. ACS Infect. Dis. 2022;8(8):1700–1710. doi: 10.1021/acsinfecdis.2c00326. [DOI] [PubMed] [Google Scholar]
- 86.Nieto-Meneses R., Castillo R., Hernández-Campos A., Maldonado-Rangel A., Matius-Ruiz J.B., Trejo-Soto P.J., Nogueda-Torres B., Dea-Ayuela M.A., Bolás-Fernández F., Méndez-Cuesta C., Yépez-Mulia L. In vitro activity of new N-benzyl-1H-benzimidazol-2-amine derivatives against cutaneous, mucocutaneous and visceral Leishmania species. Exp. Parasitol. 2018;184:82–89. doi: 10.1016/j.exppara.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 87.De Luca L., Ferro S., Buemi M.R., Monforte A.M., Gitto R., Schirmeister T., Maes L., Rescifina A., Micale N. Discovery of benzimidazole-based Leishmania mexicana cysteine protease CPB2.8ΔCTE inhibitors as potential therapeutics for leishmaniasis. Chem. Biol. Drug Des. 2018;92(3):1585–1596. doi: 10.1111/cbdd.13326. [DOI] [PubMed] [Google Scholar]
- 88.Kumar A., Nimsarkar P., Singh S. Systems pharmacology aiding benzimidazole scaffold as potential lead compounds against leishmaniasis for functional therapeutics. Life Sci. 2022;308:120960. doi: 10.1016/j.lfs.2022.120960. [DOI] [PubMed] [Google Scholar]
- 89.Kapil S., Singh P.K., Kashyap A., Silakari O. Structure based designing of benzimidazole/benzoxazole derivatives as anti-leishmanial agents. SAR QSAR Environ. Res. 2019;30(12):919–933. doi: 10.1080/1062936X.2019.1684357. [DOI] [PubMed] [Google Scholar]
- 90.Patel V.M., Patel N.B., Chan-Bacab M.J., Rivera G. N -Mannich bases of benzimidazole as a potent antitubercular and antiprotozoal agents: Their synthesis and computational studies. Synth. Commun. 2020;50(6):858–878. doi: 10.1080/00397911.2020.1725057. [DOI] [Google Scholar]
- 91.Sánchez-Salgado J.C., Bilbao-Ramos P., Dea-Ayuela M.A., Hernández-Luis F., Bolás-Fernández F., Medina-Franco J.L., Rojas-Aguirre Y. Systematic search for benzimidazole compounds and derivatives with antileishmanial effects. Mol. Divers. 2018;22(4):779–790. doi: 10.1007/s11030-018-9830-7. [DOI] [PubMed] [Google Scholar]
- 92.Hernández-Luis F., Hernández-Campos A., Castillo R., Navarrete-Vázquez G., Soria-Arteche O., Hernández-Hernández M., Yépez-Mulia L. Synthesis and biological activity of 2-(trifluoromethyl)-1H-benzimidazole derivatives against some protozoa and Trichinella spiralis. Eur. J. Med. Chem. 2010;45(7):3135–3141. doi: 10.1016/j.ejmech.2010.03.050. [DOI] [PubMed] [Google Scholar]
- 93.Gómez-Ochoa P., Castillo J.A., Gascón M., Zarate J.J., Alvarez F., Couto C.G. Use of domperidone in the treatment of canine visceral leishmaniasis: A clinical trial. Vet. J. 2009;179(2):259–263. doi: 10.1016/j.tvjl.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 94.Baxarias M., Martínez-Orellana P., Baneth G., Solano-Gallego L. Immunotherapy in clinical canine leishmaniosis: A comparative update. Res. Vet. Sci. 2019;125:218–226. doi: 10.1016/j.rvsc.2019.06.009. [DOI] [PubMed] [Google Scholar]
- 95.Cavalera M.A., Gernone F., Uva A., D’Ippolito P., Roura X., Paltrinieri S., Zatelli A. Effect of domperidone (leisguard®) on antibody titers, inflammatory markers and creatinine in dogs with leishmaniosis and chronic kidney disease. Parasit. Vectors. 2021;14(1):525. doi: 10.1186/s13071-021-05030-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ratcliffe N.A., Furtado Pacheco J.P., Dyson P., Castro H.C., Gonzalez M.S., Azambuja P., Mello C.B. Overview of paratransgenesis as a strategy to control pathogen transmission by insect vectors. Parasit. Vectors. 2022;15(1):112. doi: 10.1186/s13071-021-05132-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ferreira R.A.A., Junior C.O.R., Martinez P.D.G., Koovits P.J., Soares B.M., Ferreira L.L.G., Michelan-Duarte S., Chelucci R.C., Andricopulo A.D., Galuppo M.K., Uliana S.R.B., Matheeussen A., Caljon G., Maes L., Campbell S., Kratz J.M., Mowbray C.E., Dias L.C. 2-aminobenzimidazoles for leishmaniasis: From initial hit discovery to in vivo profiling. PLoS Negl. Trop. Dis. 2021;15(2):e0009196. doi: 10.1371/journal.pntd.0009196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Peña I., Pilar Manzano M., Cantizani J., Kessler A., Alonso-Padilla J., Bardera A.I., Alvarez E., Colmenarejo G., Cotillo I., Roquero I., de Dios-Anton F., Barroso V., Rodriguez A., Gray D.W., Navarro M., Kumar V., Sherstnev A., Drewry D.H., Brown J.R., Fiandor J.M., Julio Martin J. New compound sets identified from high throughput phenotypic screening against three kinetoplastid parasites: An open resource. Sci. Rep. 2015;5(1):8771. doi: 10.1038/srep08771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tonelli M., Gabriele E., Piazza F., Basilico N., Parapini S., Tasso B., Loddo R., Sparatore F., Sparatore A. Benzimidazole derivatives endowed with potent antileishmanial activity. J. Enzyme Inhib. Med. Chem. 2018;33(1):210–226. doi: 10.1080/14756366.2017.1410480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Espinosa-Bustos C., Ortiz Pérez M., Gonzalez-Gonzalez A., Zarate A.M., Rivera G., Belmont-Díaz J.A., Saavedra E., Cuellar M.A., Vázquez K., Salas C.O. New amino naphthoquinone derivatives as anti-trypanosoma cruzi agents targeting trypanothione reductase. Pharmaceutics. 2022;14(6):1121. doi: 10.3390/pharmaceutics14061121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.López-Lira C., Tapia R.A., Herrera A., Lapier M., Maya J.D., Soto-Delgado J., Oliver A.G., Graham Lappin A., Uriarte E. New benzimidazolequinones as trypanosomicidal agents. Bioorg. Chem. 2021;111:104823. doi: 10.1016/j.bioorg.2021.104823. [DOI] [PubMed] [Google Scholar]
- 102.Bruno S., Uliassi E., Zaffagnini M., Prati F., Bergamini C., Amorati R., Paredi G., Margiotta M., Conti P., Costi M.P., Kaiser M., Cavalli A., Fato R., Bolognesi M.L. Molecular basis for covalent inhibition of glyceraldehyde-3-phosphate dehydrogenase by a 2-phenoxy-1,4-naphthoqui- none small molecule. Chem. Biol. Drug Des. 2017;90(2):225–235. doi: 10.1111/cbdd.12941. [DOI] [PubMed] [Google Scholar]
- 103.Uchiyama N., Kabututu Z., Kubata B.K., Kiuchi F., Ito M., Nakajima-Shimada J., Aoki T., Ohkubo K., Fukuzumi S., Martin S.K., Honda G., Urade Y. Antichagasic activity of komaroviquinone is due to generation of reactive oxygen species catalyzed by Trypanosoma cruzi old yellow enzyme. Antimicrob. Agents Chemother. 2005;49(12):5123–5126. doi: 10.1128/AAC.49.12.5123-5126.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Téllez-Valencia A., Ávila-Ríos S., Pérez-Montfort R., Rodríguez-Romero A., Tuena de Gómez-Puyou M., López-Calahorra F., Gómez-Puyou A. Highly specific inactivation of triosephosphate isomerase from Trypanosoma cruzi. Biochem. Biophys. Res. Commun. 2002;295(4):958–963. doi: 10.1016/S0006-291X(02)00796-9. [DOI] [PubMed] [Google Scholar]
- 105.Téllez-Valencia A., Olivares-Illana V., Hernández-Santoyo A., Pérez-Montfort R., Costas M., Rodríguez-Romero A., López-Calahorra F., Tuena de Gómez-Puyou M., Gómez-Puyou A. Inactivation of triosephosphate isomerase from Trypanosoma cruzi by an agent that perturbs its dimer interface. J. Mol. Biol. 2004;341(5):1355–1365. doi: 10.1016/j.jmb.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 106.Flores Sandoval C.A., Cuevas Hernández R.I., Correa Basurto J., Beltrán Conde H.I., Padilla Martínez I.I., Farfán García J.N., Nogueda Torres B., Trujillo Ferrara J.G. Synthesis and theoretic calculations of benzoxazoles and docking studies of their interactions with triosephosphate isomerase. Med. Chem. Res. 2013;22(6):2768–2777. doi: 10.1007/s00044-012-0264-y. [DOI] [Google Scholar]
- 107.Velázquez-López J.M., Hernández-Campos A., Yépez-Mulia L., Téllez-Valencia A., Flores-Carrillo P., Nieto-Meneses R., Castillo R. Synthesis and trypanocidal activity of novel benzimidazole derivatives. Bioorg. Med. Chem. Lett. 2016;26(17):4377–4381. doi: 10.1016/j.bmcl.2015.08.018. [DOI] [PubMed] [Google Scholar]
- 108.Vázquez-Jiménez L.K., Juárez-Saldivar A., Gómez-Escobedo R., Delgado-Maldonado T., Méndez-Álvarez D., Palos I., Bandyopadhyay D., Gaona-Lopez C., Ortiz-Pérez E., Nogueda-Torres B., Ramírez-Moreno E., Rivera G. Ligand-based virtual screening and molecular docking of benzimidazoles as potential inhibitors of triosephosphate isomerase identified new trypanocidal agents. Int. J. Mol. Sci. 2022;23(17):10047. doi: 10.3390/ijms231710047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Song D., Ma S. Recent development of benzimidazole-containing antibacterial agents. ChemMedChem. 2016;11(7):646–659. doi: 10.1002/cmdc.201600041. [DOI] [PubMed] [Google Scholar]
- 110.Beltran-Hortelano I., Atherton R.L., Rubio-Hernández M., Sanz-Serrano J., Alcolea V., Kelly J.M., Pérez-Silanes S., Olmo F. Design and synthesis of mannich base-type derivatives containing imidazole and benzimidazole as lead compounds for drug discovery in chagas disease. Eur. J. Med. Chem. 2021;223:113646. doi: 10.1016/j.ejmech.2021.113646. [DOI] [PubMed] [Google Scholar]
- 111.Beltran-Hortelano I., Perez-Silanes S., Galiano S. Trypanothione reductase and superoxide dismutase as current drug targets for Trypanosoma cruzi: An overview of compounds with activity against chagas disease. Curr. Med. Chem. 2017;24(11):1066–1138. doi: 10.2174/0929867323666161227094049. [DOI] [PubMed] [Google Scholar]
- 112.Bistrović A., Krstulović L., Harej A., Grbčić P., Sedić M., Koštrun S., Pavelić S.K., Bajić M., Raić-Malić S. Design, synthesis and biological evaluation of novel benzimidazole amidines as potent multi-target inhibitors for the treatment of non-small cell lung cancer. Eur. J. Med. Chem. 2018;143:1616–1634. doi: 10.1016/j.ejmech.2017.10.061. [DOI] [PubMed] [Google Scholar]
- 113.McNamara N., Rahmani R., Sykes M.L., Avery V.M., Baell J. Hit-to-lead optimization of novel benzimidazole phenylacetamides as broad spectrum trypanosomacides. RSC Med. Chem. 2020;11(6):685–695. doi: 10.1039/D0MD00058B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.de Oliveira Rezende Júnior C., Martinez P.D.G., Ferreira R.A.A., Koovits P.J., Miranda Soares B., Ferreira L.L.G., Michelan-Duarte S., Chelucci R.C., Andricopulo A.D., Matheeussen A., Van Pelt N., Caljon G., Maes L., Campbell S., Kratz J.M., Mowbray C.E., Dias L.C. Hit-to-lead optimization of a 2-aminobenzimidazole series as new candidates for chagas disease. Eur. J. Med. Chem. 2023;246:114925. doi: 10.1016/j.ejmech.2022.114925. [DOI] [PubMed] [Google Scholar]
- 115.Ornellas-Garcia U., Cuervo P., Ribeiro-Gomes F.L. Malaria and leishmaniasis: Updates on co-infection. Front. Immunol. 2023;14:1122411. doi: 10.3389/fimmu.2023.1122411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Formenti B., Gregori N., Crosato V., Marchese V., Tomasoni L.R., Castelli F. The impact of COVID-19 on communicable and non-communicable diseases in Africa: A narrative review. Infez. Med. 2022;30(1):30–40. doi: 10.53854/liim-3001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]