

Abstract
Human pluripotent stem cell-derived neural progenitor (hNP) cells are an excellent resource for understanding early neural development and neurodegenerative disorders. Given that many neurodegenerative disorders can be correlated with defects in the mitochondrial genome, optimal utilization of hNP cells requires an ability to manipulate and monitor changes in the mitochondria. Here, we describe a novel approach that uses recombinant human mitochondrial transcription factor A (rhTFAM) protein to transfect and express a pathogenic mitochondrial genome (mtDNA) carrying the G11778A mutation associated with Leber’s hereditary optic neuropathy (LHON) disease, into dideoxycytidine (ddC)-treated hNPs. Treatment with ddC reduced endogenous mtDNA and gene expression, without loss of hNP phenotypic markers. Entry of G11778A mtDNA complexed with the rhTFAM was observed in mitochondria of ddC-hNPs. Expression of the pathogenic RNA was confirmed by restriction enzyme analysis of the SfaN1-digested cDNA. On the basis of the expression of neuron-specific class III beta-tubulin, neuronal differentiation occurred. Our results show for the first time that pathogenic mtDNA can be introduced and expressed into hNPs without loss of phenotype or neuronal differentiation potential. This mitochondrial gene replacement technology allows for creation of in vitro stem cell-based models useful for understanding neuronal development and treatment of neurodegenerative disorders.
Keywords: neural progenitors, stem cells, mitochondrial DNA, TFAM, neurodegeneration
INTRODUCTION
Many human neurodegenerative disorders containing defects in the mitochondrial genome are characterized by irrevocable damage and improper functions of specialized neuronal cells or populations.1 For example, Leber’s hereditary optic neuropathy (LHON) result from point mutations in mitochondrial DNA (mtDNA) that cause retinal ganglion neuron degeneration and blindness in young adults.2,3 Presence of other large mutations/deletions in mtDNA have been linked to other neurodevelopmental and age-related disorders.4–6 Use of predictive and relevant cellular models would thus extensively benefit understanding many of these disorders, research needed to develop new drugs and novel therapeutic approaches.
The cytoplasmic hybrid cell, commonly known as cybrids, is an in vitro model in which donors’ mtDNA, obtained from platelets, is expressed against tumorigenic nuclear backgrounds in host cells depleted of their endogenous mtDNA. Studies have shown that cybrids have been used to understand mtDNA-derived bioenergetic deficiencies in neurodegenerative disorders.7–9 However, better models are needed to overcome both the glycolysis-preferring metabolism of tumor cells as hosts, and the mesodermal sources as platelets, used for cybrid creation that may not accurately reflect the genome inside the ectodermal-derived non-mitotic neural cells.
In this study, we introduced exogenous mtDNA containing the G11778A mutation, causal for LHON, into human pluripotent stem cell-derived neural progenitors (hNPs). The choice of the pathogenic mtDNA carrying the G11778A mutation associated with LHON in our study was primarily based on its ease of detection by restriction enzyme analysis after introduction into dideoxycytidine (ddC)-treated hNPs. hNPs are self-renewing stable cell lines that can be differentiated at will, along neuronal, astrocytic or oligodendroglial lineages.1,10–13 These cell lines exhibit a normal karyotype and have also exhibited potential for genomic replacement studies14 and as primary cell sources for assays of new therapeutics for neurodegenerative diseases.15,16
Human mitochondrial transcription factor A (TFAM) is a member of the high-mobility group of DNA-binding, and has been shown to be essential for mtDNA replication and transcription.17–19 Recent studies have demonstrated the potential for a recombinant human TFAM protein engineered with an N-terminal protein transduction domain (PTD), followed by a mitochondrial leader sequence (MLS) to enter rapidly into the mitochondrial compartment of cybrid cells, using its ‘mitochondrial transduction domain’ (MTD=PTD+MLS).20 These encouraging results demonstrated that the human mitochondria could be manipulated from outside the cell, and provided the impetus for our current study in assessing the potential of MTD–TFAM in delivering and expressing exogenous mtDNA into hNPs.
To our knowledge, this is the first report describing mitochondrial genetic replacement of a self-renewing neural progenitor cell type. We demonstrate the introduction and expression of exogenous mtDNA in hNPs that have had their endogenous mtDNA reduced, and their potential for being maintained and subsequently differentiated into neurons.
RESULTS
Generation and characterization of ddC-treated hNPs
Stably propagating hNPs with reduced endogenous mtDNA is a necessary first step in generating in vitro mtDNA-based models. We initially assessed the response of hNPs to 2,3 ddC treatment and treated the hNPs for a period of 6 days in medium containing 10, 20, 60 and 100 μm of 2,3 ddC (Supplementary Figures 1a and b). hNPs demonstrated a concentration-dependent loss in cell viability; compared with higher concentrations, 10 μm ddC-treated viable cells maintained neural rosette morphology (Figure 1 and Supplementary Figure 1a). Maintenance of the neural rosette structures after 10-μm ddC treatment is a good indicator that the hNP structural phenotype was not altered. Further, during early neurogenesis, once the rosettes have matured and individual cells take on a columnar morphology, they robustly express nestin and musashi1. After 6 days of ddC exposure, immunocytochemistry confirmed equivalent positive nestin expression in the ddC-treated hNPs, compared with their control-untreated counterparts (Figure 1). Quantitative PCR (qPCR) analysis (Figure 2a and Supplementary Table 1) showed that levels of Sox2, nestin, fibronectin and musashi1, genes highly expressed in hNPs,12 were the same in untreated and ddC-treated hNPs. Exposure to 10-μm ddC thus maintained viable cells without altering the representative hNP morphology or gene expression.
Figure 1.
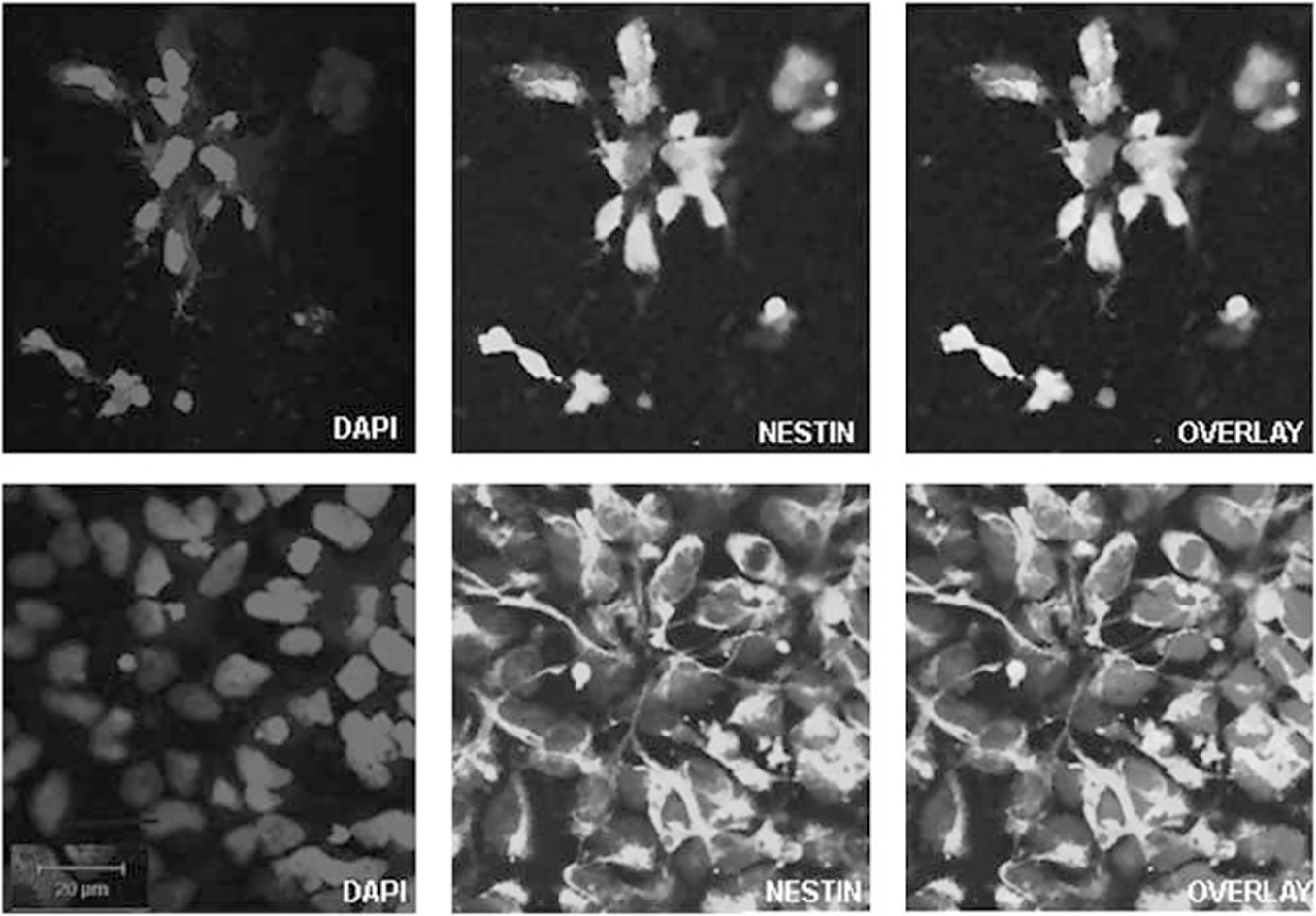
Immunocytochemical staining for nestin (green) and 4,6-diamidino-2-phenylindole (blue) in normal hNPs (bottom) and hNPs treated with 10 μm ddC for 6 days (top). Positive expression of nestin is maintained in hNPs after ddC treatment, indicating minimal alteration in characteristics of hNPs. The color reproduction of this figure is available at the Gene Therapy journal online.
Figure 2.
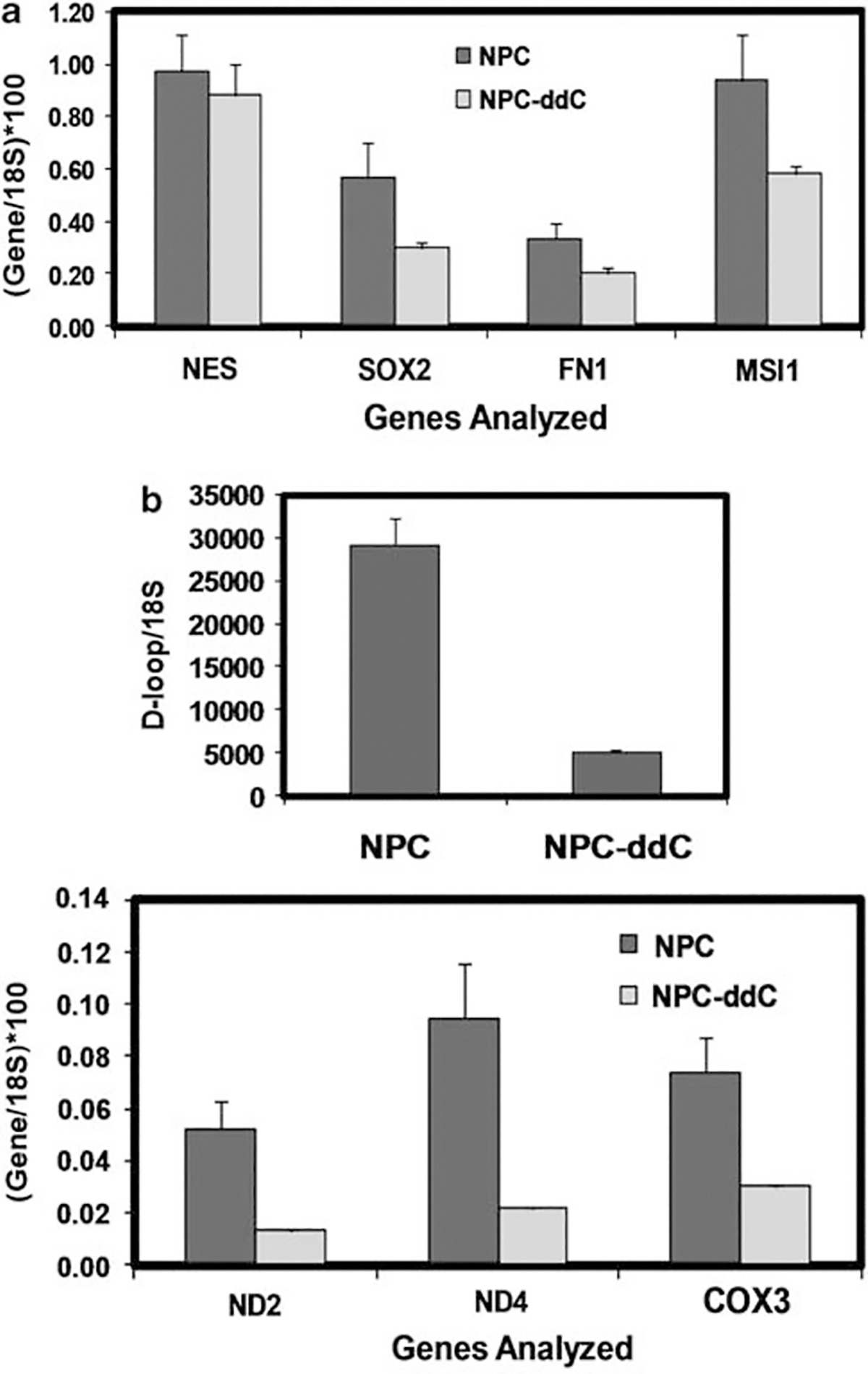
(a) Quantitative real-time PCR analysis of candidate neural progenitor markers indicates no significant change in gene expression upon ddC treatment of hNPs. RNA was reverse transcribed to cDNA, and assayed using qPCR and multiplex TaqMan probes for NESTIN (NES), SOX2, FIBRONECTIN1 (FN1) and MUSASHI1 (MSI1). Shown are 18S rRNA-normalized mean +/− s.e.m. copy numbers for NPC (normal hNP) and NPC-ddC (hNPs treated with 10 μm ddC for 6 days). One-way analysis of variance showed P>0.05 for each gene comparison. (b) Quantitative real-time PCR analysis of candidate mitochondrial markers indicates significant change in gene expression upon ddC treatment of hNPs. Extracted genomic DNA was analyzed for mtDNA copy number using qPCR and primers that amplified portions of the D-loop, ND2, ND4 and COX3 genes. Shown are 18S rRNA-normalized mean +/− s.e.m. mtcDNA copy numbers for NPC (normal hNP) and NPC-ddC (hNPs treated with 10 μm ddC for 6 days). One-way analysis of variance showed P<0.05 for D-loop and each gene comparison.
ddC inhibits mitochondrial DNA polymerase gamma’s ability to convert ddC to dideoxycytidine triphosphate21,22 and reduces mtDNA replication. We monitored the copy numbers of candidate mitochondrial genes (ND2, ND4 and COX3) in our ddC-hNP samples by qPCR analysis. Copy numbers for mtDNA-displacement loop (D-loop), and ND2, ND4 and COX3 genes (normalized to 18S rRNA) significantly decreased in ddC-hNPs (83% in D-loop, 75% in ND2, 72% in ND4, and 59% in COX3), indicating reduced endogenous mitochondrial genome content (Figure 2b). We also observed that prolonged exposure of hNPs to ddC beyond 6 days extensively reduced cell viability. Thus, for the purposes of our study and in experiments detailed below, we decided to limit ddC exposure to 6 days before introduction of the exogenous mtDNA.
Entry of pathogenic LHON mtDNA into hNPs
We then extracted mtDNA from SH-SY5Y neuroblastoma cybrid cells carrying the G11778A mutation in the mtDNA ND4 gene from a patient afflicted with LHON.2,3 These cybrids were created in a SH-SY5Y rho0 cell line, by fusion with platelets from a 42-year-old male with LHON, which contained the pathogenic mtDNA in high abundance, and were a kind gift of Dr Russell Swerdlow. The G11778A mutation eliminates an SfaN1 site present in normal mtDNA ND4 gene, facilitating its detection with restriction digestion analysis.
MTD–TFAM protein was fluorescently labeled with Alexa488 dye according to manufacturer’s instructions (Alexa Fluor 488 Protein Labeling Kit, Invitrogen, Carlsbad, CA, USA), and added to ddC-hNPs. Live cell confocal images revealed rapid entry of labeled MTD–TFAM into the mitochondrial compartment within the first 5 min (Supplementary Figure 2). Next, we purified LHON mtDNA from LHON cybrids, labeled it with Cy3 dye according to manufacturer’s instructions (Mirus, Madison, WI, USA), complexed it with unlabeled MTD–TFAM and added the mixture to ddC-hNPs. We observed slower entry of the Cy3-LHON mtDNA/MTD–TFAM complex over several hours that appears to be complete after 4–5 h (Figure 3). Our results are consistent to what has been observed earlier in cybrid models, where 100% entry of MTD–TFAM was achieved within 40 min and persisted for 6 h.20,23 Further, the entry of the mtDNA/MTD–TFAM complex was slower due to the size of the large complex, and has been observed in cybrid lines irrespective of the mtDNA used (Iyer and Bennett, unpublished data).
Figure 3.
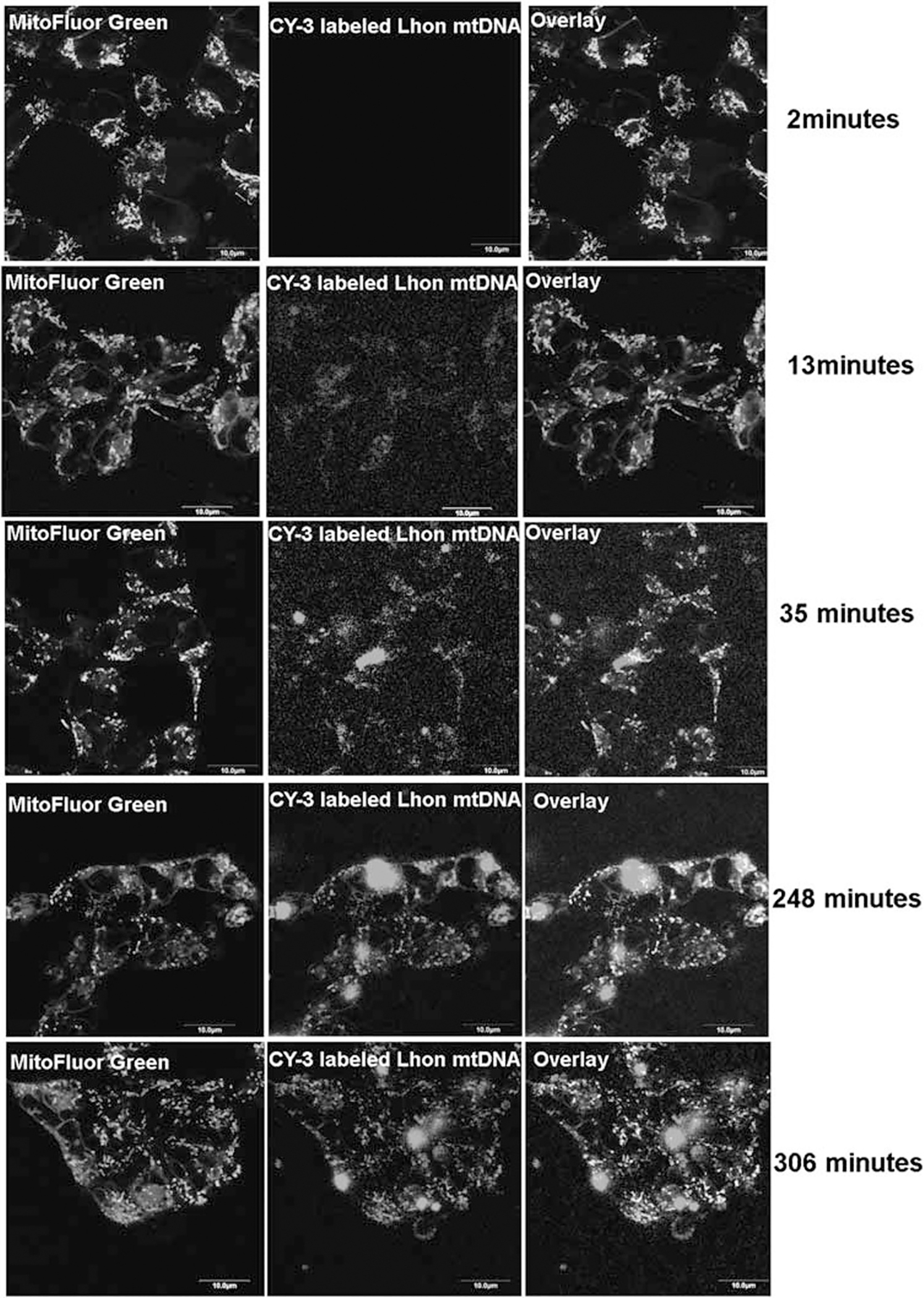
Time-lapse confocal images of entry of LHON mtDNA complexed with MTD–TFAM into ddC-treated hNPs. Entry and colocalization (yellow overlay) of Cy3-human LHON mtDNA complexed with MTD–TFAM was observed after 4 h of incubation. Mitochondria within the hNPs have been stained with MitoFluor Green. Scale bar=10 μm. The color reproduction of this figure is available at the Gene Therapy journal online.
Expression of pathogenic LHON RNA in hNPs
In independent experiments, we used restriction digestion analysis with SfaN1 to confirm the presence of LHON cDNA in transfected ddC-hNPs. As controls, we examined mtDNA from LHON cybrids and corresponding parental controls by PCR analysis, with primers designed around the ND4 region. Restriction digestion analysis revealed a ~350-bp product, confirming the presence of the LHON mtDNA (Figure 4a, lane 3). Absence of the LHON mutation in the parental SH-SY5Y cell line was confirmed by the presence of two bands at ~255 and ~108 bp (Figure 4a, lane 2), indicating complete cleavage by the SfaN1 enzyme.
Figure 4.
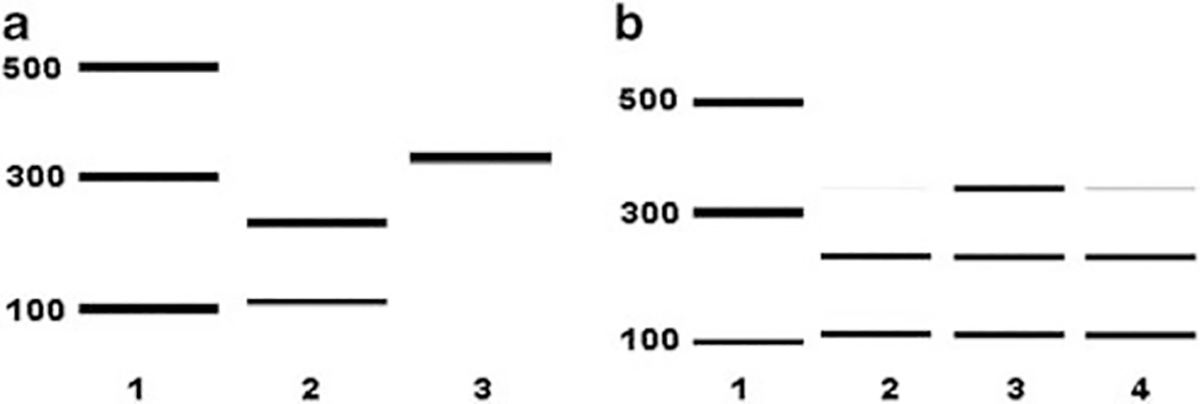
Analysis of G11778A mutation in ddC-treated hNPs treated with LHON mtDNA complexed with MTD–TFAM. (a) Electrophoretogram output corresponding to Ladder (lane 1); SfaN1 digestion pattern of ND4 PCR product from parental SY5Y cells (lane 2); and from LHON-cybrid (lane 3). (b) Electrophoretogram output corresponding to Ladder (lane 1); SfaN1 digestion pattern of ND4 PCR product from cDNA of ddC treated hNPs treated with MTD-TFAM (lane 2); with LHON- mtDNA complexed with MTD-TFAM (lane 3) and with buffer only (lane 4).
We then used SfaN1 restriction digestion analysis of cDNA to demonstrate the expression of the LHON mtDNA introduced after complexing with MTD–TFAM into ddC-hNPs. After 48 h of mtDNA introduction, we isolated total RNA from individual groups (untreated ddC-hNPs, MTD–TFAM introduced into ddC-hNPs, LHON mtDNA complexed with MTD–TFAM introduced into ddC-hNPs), reverse transcribed RNA into cDNA, and PCR amplified around the ND4 region. SfaN1 digestion analysis revealed a strong ~350-bp band, along with the existence of the ~255- and ~108-bp band in ddC-hNPs treated with LHON mtDNA complexed with MTD–TFAM (Figure 4b, lane 3). Only the ~255-bp and ~108-bp bands were present in untreated (Figure 4b, lane 2) and MTD–TFAM-treated (Figure 4b, lane 4) ddC-hNPs, respectively. The reason behind the presence of a faint band corresponding to ~350bp in the control samples (Figure 4b, lanes 2 and 4) is unclear and could be due to the presence of low-level transient polymorphisms in the residual hNP mitochondrial genome. We therefore conclude that the robust 350-bp band (Figure 4b, lane 3) is due to the introduction and expression of the exogenous LHON mtDNA. These results demonstrate that exogenous LHON mtDNA can not only be introduced into hNPs, but also maintain their expression over a 48-h period in ddC-hNPs.
Differentiation of LHON-hNPs
Following successful transfer of the LHON mtDNA into hNPs, we also investigated the differentiation potential of the manipulated hNPs. After 2 weeks of differentiation, the manipulated LHON-hNPs formed extensive neurites, exhibited neuronal morphology, and positively stained for Tuj1 (βIII tubulin, neuronal marker) (Figure 5 and Supplementary Video).
Figure 5.
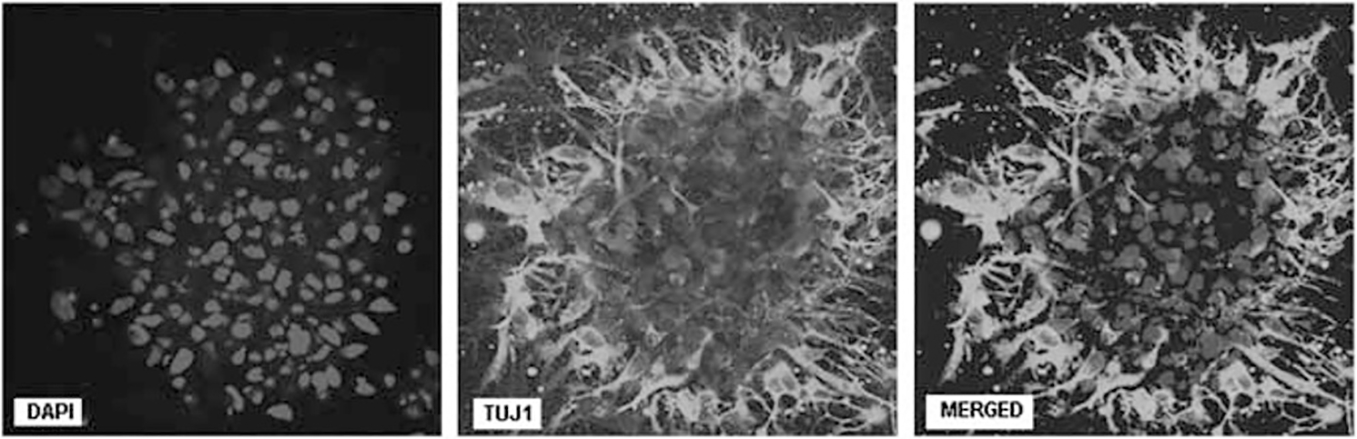
Immunocytochemical staining for Tuj1 (green) and 4,6-diamidino-2-phenylindole (blue) in 14-day differentiated neurons generated from ddC-treated hNPs, treated with LHON mtDNA complexed with MTD–TFAM. Positive expression of Tuj1 is an indication of neuronal differentiation potential of the LHON-hNPs. The color reproduction of this figure is available at the Gene Therapy journal online.
DISCUSSION
In this study, we directly introduced and expressed a LHON pathogenic mtDNA containing a G11778A mutation into a human multi-potent stem cell that possesses the genomic and anatomic properties of a neural progenitor and a developmental precursor of non-mitotic neurons. This LHON-hNP line contains and expresses LHON mtDNA and also has the capacity of differentiating into neurons. The challenging aspect of this replacement was in creating a ddC-hNP line, without compromising the viability and the integrity of the hNP marker expression. On the basis of our experimental outcomes showing reduction of endogenous mtDNA and gene expression without loss of hNP gene expression, we have demonstrated a potential approach for creating mtDNA-depleted line in hNPs treated with ddC (Figure 6).
Figure 6.
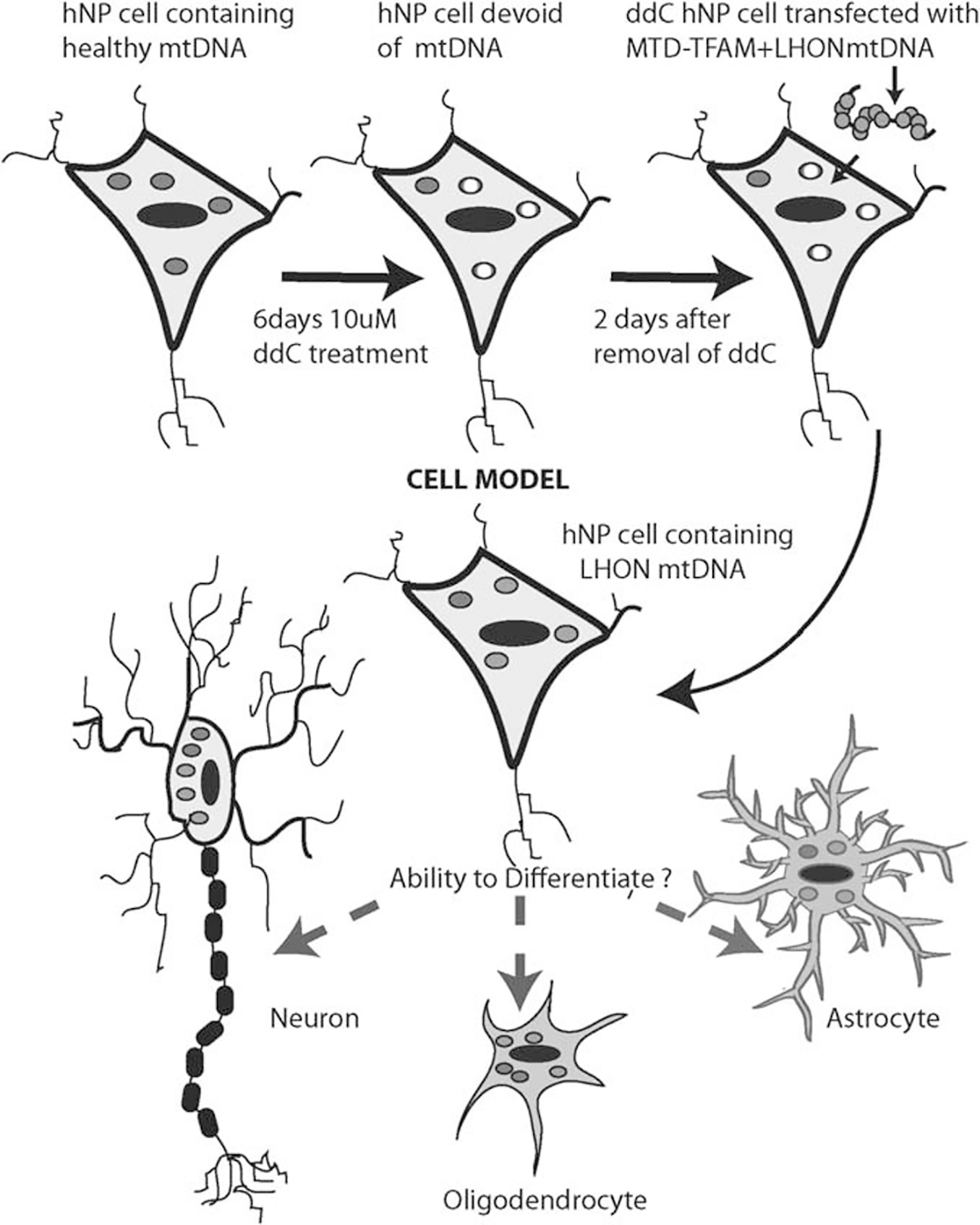
Proposed scheme outlining the steps involved in creation of neural progenitor cell model based on mitochondrial gene replacement. Red circles correspond to mitochondria containing endogenous mtDNA; white circles correspond to mitochondria depleted of their endogenous mtDNA and blue circles correspond to exogenous pathogenic mtDNA. The color reproduction of this figure is available at the Gene Therapy journal online.
Previous studies supporting the potential for mitochondrial genome replacement in stem cells have utilized different approaches in the murine and the non-human primate system.24,25 In the murine system, trans-mitochondrial embryonic stem (ES) cell lines were generated by fusion of cytoplasts carrying a variety of mtDNA mutations, into mouse ES cells that had been pretreated with rhodamine 6G, to prevent transmission of endogenous mtDNA. This study also demonstrated that neuronal differentiation could occur, although the differentiation was impaired in the stem cells carrying the mtDNA mutations that correlated with biochemical deficiencies.24 Although this study demonstrated that the mitochondrial genome could be manipulated in ES cells that could further be differentiated into neurons, the use of fibroblasts in the cybrid-based approach may not accurately reflect the genome inside non-mitotic neural cells. We believe that the advantage provided by our approach is the ability to directly introduce and create hNPs carrying pathogenic mtDNA, whereas utilizing the parental hNP protein machinery for preserving and regulating the mitochondrial function.
In the non-human primate system, mitochondrial genome replacement was achieved in mature primate oocytes by a spindlechromosomal complex transfer from one egg to an enucleated, mitochondrial-replete egg.25 The study showed that reconstructed oocytes with the mitochondrial replacement were capable of supporting normal fertilization, producing ES cell lines and healthy offspring. This approach has been proposed as an option for replacing the full complement of mitochondria in newly generated ES cell lines, but requires replacement in the primary oocyte stage. Although this approach proposed mitochondrial replacement, the end goal is an option to prevent mtDNA disease transmission in affected families. Due to the fact that this manipulation has to be carried out at the primary oocyte stage, this exciting therapeutic option might not lend itself to replacement of the mitochondrial genome in cell lines that could serve as models of complex neurodegenerative disorders, as shown in our current study.
In summary, mtDNA mutations contribute to a diverse range of neurological disorders, and generation of appropriate in vitro models that provide opportunities for mimicking specific diseases are necessary. We believe that the strength of our study lies in the simplicity of the experimental approach and in being able to manipulate the mitochondrial genome to introduce a pathogenic mtDNA into hNPs. Our novel approach provides enormous opportunities for generating relevant models that will aid in understanding and tracing the genesis of the disease as they progress forward. Defects in the mitochondrial genome contribute primarily to neuromuscular disorders, but have also been implicated in common neurodegenerative age-related disorders such as Parkinson’s disease, Alzheimer’s disease and amyotrophic lateral sclerosis.4 Studies on mtDNA mutations in the context of these neurodegenerative disorders, their role on the fate and development of neurons, and their influence on the progress of complex diseases at the cellular level will contribute to improved understanding and therapeutic possibilities.
Our novel approach also provides opportunities for creating models that will aid in understanding the fundamental questions related to homoplasmy and transmission of a particular pathogenic mtDNA as the disease progresses. In our study, it is interesting to note that both species of mtDNA (LHON and normal) replicated and transcribed within 48 h in the ddC-hNPs post-treatment with the pathogenic mtDNA complexed with MTD–TFAM (Figure 3b). What this result means in terms of continued replication and transcription of the pathogenic mtDNA in differentiated neurons, astrocytes and oligo-dendrocytes will be addressed in future studies (Figure 6). Specifically, we will determine the changes in the mutant load, when cells differentiate, as studies have shown that mitochondrial biogenesis events trigger changes in mitochondrial mass and copy number during differentiation.26 Additionally, our future studies will measure phenotypic differences (bioenergetics, oxidative stress, signaling pathway analysis) as a result of introduction of exogeneous mtDNA into hNPs and compare with cybrid-based observations. Although specific mutations like the G11778A mutation accounts for a vast majority of LHON cases in the Western world, recent identification of a different set of LHON disease-causing mutations27 provides opportunities for assessing the importance of these novel mutations in our in vitro models. Further, development of these in vitro models that carry diverse mtDNA mutations will serve as excellent components of drug-testing platforms that could contribute to alleviation of symptoms associated with many mitochondrial and neurodegenerative disorders. Hence, we propose that outcomes from this study may represent a novel reliable approach to generate in vitro models in a dish for a wide range of human neurological disorders that have mutations in the mitochondrial genome.
MATERIALS AND METHODS
hNP propagation and culture conditions
hNPs (Aruna Biomedical, Athens, GA, USA) were routinely cultured on polyornithine/laminin-coated 35-mm dishes or T-25 flasks, based on previously published protocols.10,12 Cells were propagated in neurobasal medium (Invitrogen) with B-27 supplements, leukemia inhibitory factor, basic fibroblast growth factor and antibiotics (penicillin and streptomycin). Cells were passaged at a sub-culturing ratio of 1:2 when 90–100% confluent, by scraping them off with a cell scraper.
Creation and characterization of ddC-treated hNPs
For reduction of endogenous mtDNA, hNPs were treated with 2′,3′ ddC, a potent inhibitor of mitochondrial DNA polymerase gamma for a period of 6 days. qPCR and immunocytochemical analyses were used to determine expression of mt- and hNP markers in ddC-treated hNPs. For immunocy-to-chemical analysis for detection of nestin expression, cells plated on polyornithine/laminin-coated permanox slides were washed in phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde/4% sucrose in PBS for 15min. Fixed cells were washed two times with PBS before staining. Permeabilization and blocking was carried out in blocking buffer consisting of 0.1% Triton, 3% goat serum in Tris buffer for 40 min. Cells were incubated in blocking buffer containing (rabbit anti-nestin; 1:200; Millipore, Billerica, MA, USA) for 2 h at room temperature, followed by three washes in blocking buffer, before incubation in blocking buffer containing (goat anti-rabbit conjugated Alexa 488; 1:1000; Molecular Probes Inc., Eugene, OR, USA) for 40 min at room temperature. After two washes in PBS, cells were incubated for 10 min in 4,6-diamidino-2-phenylindole (nuclear stain) and observed under the fluorescence microscope. Controls involved removal of primary antibodies from the staining protocol.
qPCR analyses on ddC-treated hNPs was conducted to determine expression of mt (D-loop, ND2, ND4, COX3) and hNP markers (NES, MSI1, FN1, SOX2) as described previously.20 Total genomic DNA and total RNA were isolated from cell pellets using All-Prep kits from Qiagen (Valencia, CA, USA) and amounts assayed with Quant-IT DNA and RNA assays (Invitrogen). Total RNA of 1 μg was reverse transcribed into cDNA using iScript (BioRad, Hercules, CA, USA). Levels of 18S rRNA, mtDNA D-loop and hNP markers were assayed using SybrGreen detection (BioRad) with Roche human genomic DNA or a full-length mtDNA PCR product, respectively, as standards.20 Copy numbers of ND2, COX3 and ND4 mitochondrial genes were assayed in a multiplex qPCR assay (BioRad Powermix) using the full-length PCR product of human mtDNA as standard. All reverse transcription qPCR was carried out in an iQ5 thermocycler (BioRad) using primers and probes designed with Beacon Designer software (PREMIER Biosoft International, Palo Alto, CA, USA). Levels of mitochondrial genes were normalized to 18S rRNA in either DNA or RNA (cDNA) samples. Data was obtained from three biological replicates, and statistical comparisons conducted using one-way analysis of variance to determine significant difference in gene expression values.
Production of MTD–TFAM, preparation of LHON mtDNA and introduction of LHON mtDNA into ddC-treated hNPs
Purification and production of recombinant human MTD–TFAM was conducted as per previously published protocols.20 Briefly, the MTD–TFAM was engineered to possess an N-terminal PTD containing 11 arginines, ligated to an SOD2 sequence for localization into the mitochondria (MLS). The final purified protein was stored in Tris-buffered 50% glycerol at −20°C. DNA-binding capacity was assayed by electrophoretic mobility shift assay, based on maximum retardation of circular DNA. For all the experiments in this study, we have used a ratio of 500 ng of circular DNA complexed with 1 μg of MTD–TFAM.
LHON cybrid cells containing a >96% homoplasmic G11778A mutation in the ND4 region of mitochondrial genome were routinely propagated in Dulbecco’s modified Eagle’s medium containing 10% FCS. The LHON mtDNA was isolated by treating genomic DNA from LHON cybrid cells to limiting amounts of exonuclease and purified.23,28
The MTD-TFAM (+/− LHON mtDNA) mix was prepared by incubating the purified MTD-TFAM to 3 μg of LHON mtDNA in PBS containing high salt Mg2+ buffer, at 37°C for 30 min. The presence of the high salt Mg2+ buffer was critical for facilitating protein binding to the DNA. hNPs were subsequently incubated in growth medium containing MTD-TFAM complexed with LHON mtDNA for two hours at 37°C. Controls involved incubating the hNPs in growth medium containing only MTD-TFAM or PBS. After the incubation period, the medium was replaced with regular growth medium and the cells subjected to different experimental analyses as outlined below.
Imaging of LHON mtDNA entry into ddC-treated hNPs
For real-time imaging of LHON mtDNA entry into hNPs, cells were propagated to ~80% confluence on laminin-coated 35mm glass bottom dishes. The Mitochondria in hNPs were stained using either Mitotraker red or Mitotracker Green (both from Mitosciences, Eugene, OR, USA) depending on whether tracking was being conducted for Alexa488-MTD-TFAM or Cy3 labeled LHON- mtDNA complexed with unlabeled MTD-TFAM respectively. MTD-TFAM was purified by dialyzing with 1×PBS buffer and labeled with Alexa 488 dye according to manufacturer’s instructions (Invitrogen). Circular mtDNA carrying the LHON G11778A mutation was labeled using Cy3 dye according to manufacturer’s instruction (Mirus, Madison, WI, USA). The labeled MTD-TFAM or the labeled LHON mtDNA complexed with MTD-TFAM was mixed in 2 ml Dulbecco’s modified Eagle’s medium and added to independent dishes of hNPs with labeled mitochondria. Real-time single plane-time lapse images were obtained every 5 min using an Olympus IX70 microscope (Olympus, Center Valley, PA, USA) with confocal capabilities.
Restriction enzyme digestion analysis
A PCR product in the ND4 gene spanning the SfaN1 site removed by the G11778A mutation was amplified from the cDNA of hNPs from independent treatments and digested with SfaN1 to check for the presence or absence of the mutation. Digested cDNA PCR products were analyzed using automated electrophoresis (Experion, BioRad).
Neuronal differentiation
The hNPs containing the LHON mtDNA were subjected to neuronal differentiation, based on published protocols.10,12 Briefly, the cells were allowed to attain 100% confluency in regular growth medium, after which the medium was replaced with medium lacking basic fibroblast growth factor and leukemia inhibitory factor for a period of 14 days. The neurons obtained were subjected to immunocytochemical analyses for beta-tubulin-III expression.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
Partial funding for this work has been provided by NIH-1K18DC009121 (Bennett/Rao), NIH- 5P50NS039788 (Bennett), Qimonda Endowment from the VCU School of Engineering (Rao) and Fellowship from the American Parkinson’s Disease Association (Iyer). Recombinant MTD–TFAM was obtained by Bennett through an MTA with Gencia Corporation, Charlottesville, VA. LHON cybrid cells were a kind gift from Dr Russell Swederlow.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Iyer S, Alsayegh K, Abraham S, Rao RR. Stem cell-based models and therapies for neurodegenerative diseases. Crit Rev Biomed Eng 2009; 37: 321–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace DC, Singh G, Lott MY, Hodge JA, Schurr TG, Lezza AM et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988; 242: 1427–1430. [DOI] [PubMed] [Google Scholar]
- 3.Yen MY, Wang AG, Wei YH. Leber’s hereditary optic neuropathy: a multifactorial disease. Prog Retin Eye Res 2006; 25: 381–396. [DOI] [PubMed] [Google Scholar]
- 4.DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci 2008; 31: 91–123. [DOI] [PubMed] [Google Scholar]
- 5.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet 2005; 39: 359–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Debray FG, Lambert M, Mitchell GA. Disorders of mitochondrial function. Curr Opin Pediatr 2008; 20: 471–482. [DOI] [PubMed] [Google Scholar]
- 7.Borland MK, Mohanakumar KP, Rubinstein JD, Keeney PM, Xie J, Capaldi R et al. Relationships among molecular genetic and respiratory properties of Parkinson’s disease cybrid cells show similarities to Parkinson’s brain tissues. Biochim Biophys Acta 2009; 1792: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghosh SS, Swerdlow RH, Miller SW, Sheeman B, Parker WD, Davis RE. Use of cytoplasmic hybrid cell lines for elucidating the role of mitochondrial dysfunction in Alzheimer’s disease and Parkinson’s disease. Ann NY Acad Sci 1999; 893: 176–191. [DOI] [PubMed] [Google Scholar]
- 9.Trimmer PA, Bennett JP Jr. The cybrid model of sporadic Parkinson’s disease. Exp Neurol 2009; 218: 320–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhara SK, Hasneen K, Machacek DW, Boyd NL, Rao RR, Stice SL. Human neural progenitor cells derived from embryonic stem cells in feeder-free cultures. Differentiation 2008; 76: 454–464. [DOI] [PubMed] [Google Scholar]
- 11.Dhara SK, Stice SL. Neural differentiation of human embryonic stem cells. J Cell Biochem 2008; 105: 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shin S et al. Long-term proliferation of human embryonic stem cell-derived neuroepithelial cells using defined adherent culture conditions. Stem Cells 2006; 24: 125–138. [DOI] [PubMed] [Google Scholar]
- 13.Young A, Assey KS, Sturkie CD, West FD, Machacek DW, Stice SL. Glial cell line-derived neurotrophic factor enhances in vitro differentiation of mid-/hindbrain neural progenitor cells to dopaminergic-like neurons. J Neurosci Res 2010; 88: 3222–3232. [DOI] [PubMed] [Google Scholar]
- 14.Dhara SK, Gerwe BA, Majumder A, Dodla MC, Boyd NL, Machacek DW et al. Genetic manipulation of neural progenitors derived from human embryonic stem cells. Tissue Eng Part A 2009; 15: 3621–3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson PG, Cherry JJ, Schwamberger S, Adams AM, Zhou J, Shin S et al. An SMA project report: neural cell-based assays derived from human embryonic stem cells. Stem Cells Dev 2007; 16: 1027–1041. [DOI] [PubMed] [Google Scholar]
- 16.Jin K, Mao X, Xie L, Galvan V, Lai B, Wang Y et al. Transplantation of human neural precursor cells in matrigel scaffolding improves outcome from focal cerebral ischemia after delayed postischemic treatment in rats. J Cereb Blood Flow Metab 2010; 30: 534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kang D, Kim SH, Hamasaki N. Mitochondrial transcription factor A (TFAM): roles in maintenance of mtDNA and cellular functions. Mitochondrion 2007; 7: 39–44. [DOI] [PubMed] [Google Scholar]
- 18.Scarpulla RC. Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 2008; 88: 611–638. [DOI] [PubMed] [Google Scholar]
- 19.Wallace DC. Animal models for mitochondrial disease. Methods Mol Biol 2002; 197: 3–54. [DOI] [PubMed] [Google Scholar]
- 20.Iyer S, Thomas RR, Portell FR, Dunham LD, Quigley CK, Bennett JP. Recombinant mitochondrial transcription factor A with N-terminal mitochondrial transduction domain increases respiration and mitochondrial gene expression. Mitochondrion 2009; 9: 196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bonawitz ND, Clayton DA, Shadel GS. Initiation and beyond: multiple functions of the human mitochondrial transcription machinery. Mol Cell 2006; 24: 813–825. [DOI] [PubMed] [Google Scholar]
- 22.Brown TA, Clayton DA. Release of replication termination controls mitochondrial DNA copy number after depletion with 2′,3′-dideoxycytidine. Nucleic Acids Res 2002; 30: 2004–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keeney PM, Quigley CK, Dunham LD, Papageorge CM, Iyer S, Thomas RR et al. Mitochondrial gene therapy augments mitochondrial physiology in a Parkinson’s disease cell model. Hum Gene Ther 2009; 20: 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirby DM, Rennie KJ, Smulders-Srinivasan TK, Acin-Perez R, Whittington M, Enriquez JA et al. Transmitochondrial embryonic stem cells containing pathogenic mtDNA mutations are compromised in neuronal differentiation. Cell Prolif 2009; 42: 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tachibana M, Sparman M, Sritanaudomchai H, Ma H, Clepper L, Woodward J et al. Mitochondrial gene replacement in primate offspring and embryonic stem cells. Nature 2009; 461: 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Facucho-Oliveira JM, St John JC. The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev 2009; 5: 140–158. [DOI] [PubMed] [Google Scholar]
- 27.Sundaresan P, Kumar SM, Thompson S, Fingert JH. Reduced frequency of known mutations in a cohort of LHON patients from India. Ophthalmic Genet 2010; 31: 196–199. [DOI] [PubMed] [Google Scholar]
- 28.McEachern MJ, Iyer S, Fulton TB, Blackburn EH. Telomere fusions caused by mutating the terminal region of telomeric DNA. Proc Natl Acad Sci USA 2000; 97: 11409–11414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.