

Abstract
Disordered lipid accumulation in the arterial wall is a hallmark of atherosclerosis. Previous studies found that the expression of triggering receptor expressed on myeloid cells 2 (TREM2), a transmembrane receptor of the immunoglobulin family, is increased in mouse atherosclerotic aortic plaques. However, it remains unknown whether TREM2 plays a role in atherosclerosis. Here we investigated the role of TREM2 in atherosclerosis using ApoE knockout (ApoE−/−) mouse models, primary vascular smooth muscle cells (SMCs), and bone marrow-derived macrophages (BMDMs). In ApoE−/− mice, the density of TREM2-positive foam cells in aortic plaques increased in a time-dependent manner after the mice were fed a high-fat diet (HFD). Compared with ApoE−/− mice, the Trem2−/−/ApoE−/− double-knockout mice showed significantly reduced atherosclerotic lesion size, foam cell number, and lipid burden degree in plaques after HFD feeding. Overexpression of TREM2 in cultured vascular SMCs and macrophages exacerbates lipid influx and foam cell formation by upregulating the expression of the scavenger receptor CD36. Mechanistically, TREM2 inhibits the phosphorylation of p38 mitogen-activated protein kinase and peroxisome proliferator activated-receptor gamma (PPARγ), thereby increasing PPARγ nuclear transcriptional activity and subsequently promoting the transcription of CD36. Our results indicate that TREM2 exacerbates atherosclerosis development by promoting SMC- and macrophage-derived foam cell formation by regulating scavenger receptor CD36 expression. Thus, TREM2 may act as a novel therapeutic target for the treatment of atherosclerosis.
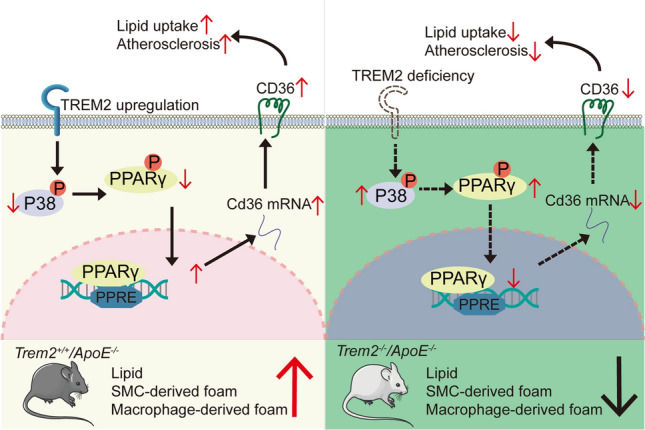
Graphical abstract
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-023-04786-9.
Keywords: TREM2, Lipid metabolism, Cholesterol uptake, Foam cell, Atherosclerosis
Introduction
Atherosclerosis is the leading cause of cardiovascular and cerebrovascular diseases [1, 2]. The accumulation of LDL-carried cholesterol in the arterial wall is one of the most striking features of atherosclerosis. Macrophages engulf these lipids and transdifferentiate into foam cells, a phenomenon of lipid metabolism disorder that is a hallmark of atherosclerosis. In addition, recent studies have found that approximately 50% of foam cells are derived from smooth muscle cells (SMCs) [3–6]. Foam cell formation is a pathological hallmark throughout the atherosclerotic diseases. Targeting foam cells is one of the current clinical approaches to prevent the progression of atherosclerosis [7, 8]. More specifically, foam cells are thought to determine plaque stability and progression. However, to date, the regulatory mechanism of foam cell formation remains unclear.
The triggering receptor expressed on myeloid cells 2 (TREM2) is a transmembrane receptor of the immunoglobulin family with a wide variety of ligands [9, 10]. TREM2 has been shown to have pathogenic effects leading to central nervous system dysfunction, diabetes, and liver-related disease by regulating lipid metabolism, inflammation, immune cell viability, and apoptosis [9, 11–15]. Furthermore, lipid metabolism also plays an important role in the occurrence and progression of atherosclerosis. Previous scRNA-seq studies of total aortic CD45-positive cells have found high expression of TREM2 in atherosclerosis-prone models [16–18]. However, the precise role of TREM2 in the atherosclerotic process remains elusive.
In the present study, we demonstrate that TREM2 plays an important role in lipid metabolism in atherosclerosis. TREM2 expression is increased in atherosclerotic-prone mouse plaques in a time-dependent manner. Conversely, TREM2 deficiency attenuates atherosclerotic plaque size and lipid deposition in ApoE−/− mice (Trem2−/−/ApoE−/−) compared with Trem2+/+/ApoE−/− mice. Furthermore, TREM2 promotes both macrophage-derived and SMC-derived foam cell formation by activating the scavenger receptor CD36. This indicates that TREM2 is a novel target for the treatment of atherosclerosis.
Materials and methods
Array data acquisition and processing
The gene expression profiles of GSE43292 were downloaded from Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE43292). We used the keywords “TREM2” and “Carotid” to search GEO datasets for related profiles. Thirty-two hypertensive patients with atheroma plaque and adjacent carotid tissue were included in GSE43292. Then the online statistical tool GEO2R was used to analyze the differentially expressed genes (DEGs) and identify TREM2 in Excel. Moreover, we analyzed log2 (fold change) and − log10 (P value) to discriminate TREM2 expression. We also utilized the GEO profile GDS5083 to obtain TREM2 values in the two groups.
Animal preparation
All animal experiments conformed to the Management Rules of the Chinese Ministry of Health and were approved by the Ethical Committee of Union Hospital affiliated with Tongji Medical School of Huazhong Technology University (Wuhan, China). All animal procedures were in accordance with the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. Animals were housed in SPF-rated (specific pathogen-free) animal centers and maintained on a 12-h light/dark cycle. Male ApoE−/− mice were purchased from Vital River Animal Center (Beijing, China). TREM2-knockout mice (Trem2−/−) on the C57BL/6N background were purchased from Cyagen (Santa Clara, CA, USA, https://www.cyagen.com/cn/zh-cn/sperm-bank-live/83433). Trem2−/− mice were crossed with ApoE−/− mice to generate Trem2−/−/ApoE−/− mice. The knockout mice were validated by genotyping and PCR. Subsequently, 8-week-old male Trem2+/+/ApoE−/− mice and male Trem2−/−/ApoE−/− mice were fed a HFD consisting of 1.25% cholesterol and 20% fat (#D12079B; Special Diet Services Ltd., Essex, UK) for 12 weeks [19, 20]. At the end of the experiments, mice were fasted for 4 h and euthanized by 100% CO2 gas for approximately 5 min in the CO2 chamber with a 30–70% fill rate followed by cervical dislocation. Then mice were perfused with 20 mL phosphate-buffered saline (PBS) before surgically obtaining tissue samples of the heart, aorta, and carotid arteries.
Analysis of atherosclerotic lesions
The entire aorta was isolated from the aortic sinus to the bifurcation of the common iliac artery and fixed with 4% paraformaldehyde solution for 24 h. Then the surrounding adipose tissue from the aorta was removed and opened longitudinally. The artery was then stained with Oil Red O (ORO) solution for 15 min at room temperature. The ORO-stained plaque area, as a percentage of the total lumen surface area, was determined using Adobe Photoshop software (Adobe Inc., San Jose, CA, USA). All morphometric analyses were performed in a double-blinded manner. For the aortic sinus, hematoxylin and eosin (H&E), Masson and ORO staining were performed on serial 6 μm cross-sections. The necrotic core was defined as the area with no nucleus. The fibrous caps were quantified by the thickness from the outer edge of the cap to the largest necrotic core boundary. Means were taken by averaging three serial cryosections from each mouse. Then we observed and photographed these sections under an Olympus light microscope (Olympus Corporation, Japan). The lesion area was measured with Adobe Photoshop software [21].
Immunofluorescence (IF) staining
Frozen sections of the aortic sinus were stained with ORO and H&E staining. For immunofluorescence staining, tissue sections were co-incubated with primary antibodies against TREM2, CD68, α-smooth muscle actin (α-SMA), or 4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene (BODIPY; #D3922; Thermo Fisher Scientific Inc., Waltham, MA, USA). The immune complexes were detected with a fluorescently labeled secondary antibody, and 4ʹ,6-diamidino-2-phenylindole (DAPI; #G1012; Wuhan Servicebio Technology Co., Ltd.) was used to mark the nuclei. The positive cells in the immunostained sections were observed and captured by a Nikon A1–Si confocal microscope (Nikon Corporation, Tokyo, Japan). The positive area and positive cell count in the plaque in each section were measured using the NIS-element software (Nikon Corporation) and ImageJ Pro Plus.
Cell culture and treatment
The experimental method for obtaining mouse primary aortic SMCs (mSMCs) was the same as described previously [22]. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; HyClone Inc., Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS; #0500; Sciencell Research Laboratories, Carlsbad, CA, USA), 100 U/mL penicillin, and 100 U/mL streptomycin and incubated at 37 °C in a humidified atmosphere with 5% CO2.
Raw264.7 cells were purchased from the Cell Resource Center of Shanghai Institute (Shanghai, China). Raw264.7 cells were cultured as described above.
Bone marrow-derived macrophages (BMDMs) were isolated from femurs of Trem2+/+ mice or Trem2−/− mice with a C57 background in RPMI-1640 containing 10% FBS. Macrophage colony-stimulating factor (M-CSF, 20 ng/mL, Peprotech) was added to the culture medium for 6 days. The cells were then used for subsequent experiments.
Cell lines stably overexpressing TREM2 were obtained after transduction with an appropriate lentiviral vector prepared by Shanghai Genechem Co., Ltd. (Shanghai, China) for follow-up experiments (LV-TREM2). Lentiviral GFP-tagged vectors were used as the control group (LV-GFP). After selection for 3 days in puromycin-containing medium (10 ng/mL), a lentivirus-transfected cell line was obtained for use in subsequent experiments. CD36 small-interfering RNAs (siRNAs) were purchased from Augctgenes CO., Ltd. (Wuhan, China). The RiboFect CP Transfection kit (#166T; RiboBio, Guangzhou, China) was used to transfect siRNA constructs. Cells were starved in serum-free medium overnight before conducting all functional studies.
DiI-oxLDL uptake analysis
The analysis of the uptake of Dil-labeled oxLDL (DiI-oxLDL; #YB-0010; Yiyuan Biotechnologies Co., Ltd., Guangzhou, China) has been described elsewhere [23]. mSMCs and Raw264.7 cells were cultured in the appropriate medium and incubated with DiI-oxLDL (10 μg/mL) for 4 h. Then we washed the cells with PBS three times and analyzed the uptake level of lipids by fluorescence microscopy. Moreover, flow cytometry analysis was used to calculate the mean fluorescence intensity (mFI).
ORO staining and BODIPY staining
Cells were fixed with 4% paraformaldehyde in PBS (#G0002; Wuhan Servicebio Technology Co., Ltd., Wuhan, China) and then washed three times with PBS, followed by ORO staining (#G1015; Wuhan Servicebio Technology Co., Ltd.) for 15 min at room temperature as described previously [24]. An Olympus optical microscope (Olympus Corporation, Tokyo, Japan) was used to assess staining, which was quantified by Image-Pro Plus 6.0 software (Media Cybernetics, Inc., Rockville, MD, USA). For BODIPY staining, cells were washed with PBS three times and incubated with BODIPY (1 μg/mL) for 1 h [25]. Cells were collected and detected by flow cytometry.
Western blot analysis
Cell lysate and aortic homogenate were prepared with RIPA lysis buffer (#PM0013B; Beyotime Biotechnology, Shanghai, China) containing complete protease inhibitors (#G2006, #G2007 and #G2008; Wuhan Servicebio Technology Co., Ltd. and mixed with loading buffer (Biosharp, Shanghai, China). Cell lysates and aortic shredded mixtures (30–50 μg) were then subjected to Western blot analysis with the following rabbit antibodies: MSR1, OLR1, CD36, ABCA1, ABCG1, total and p-p38, total and p-PPARγ. Ultimately, the immunoreactive protein bands were detected by enhanced chemiluminescence like described previously [26]. The protein GAPDH was used as an internal control. More information on antibodies is available in supplementary file Table S1.
Real-time PCR
Total RNA was extracted from mSMCs and Raw264.7 cells using TRIzol reagent (#15596026, Invitrogen, Carlsbad, CA, USA). The obtained total RNA was used to prepare cDNA, which was then amplified using a SYBR Premix Ex TaqTM Kit (#RR420A; Takara Bio Inc., Kusatsu, Shiga, Japan) on a thermocycler (Bio-Rad Laboratories, Hercules, CA, USA). The amplification program is set according to the instructions (https://www.vazyme.com/Home.html). ACTB served as the control for comparison. More information on Primers is available in supplementary file Table S2. qRT-PCR was set up with two secondary wells and performed in at least triplicate independent experiments. Data analysis was performed using the comparative CT method as previously described [27].
Statistical analysis
Continuous variables are expressed as the mean ± SEM unless otherwise specified. The two groups were compared using unpaired student's t test. Multiple groups were analyzed using analyses of variance (ANOVA) followed by Tukey’s multiple comparisons. P < 0.05 was considered statistically significant. All data were analyzed using GraphPad Prism 9.0 software (GraphPad Software Inc., San Diego, CA, USA).
Results
Upregulation of TREM2 expression in plaques during atherogenesis
Previous studies have suggested that TREM2 is involved in lipid metabolism and that the expression of TREM2 mRNA is increased in atherosclerosis [14, 16–18, 28]. We analyzed the GSE43292 dataset from the Gene Expression Omnibus (GEO) datasets (including 32 patients with atheroma plaque and adjacent carotid tissue) and found that the expression of TREM2 mRNA was increased in carotid artery plaques compared with macroscopically intact tissue (Fig. 1a, P < 0.0001, https://www.ncbi.nlm.nih.gov/geo/tools/profileGraph.cgi?ID=GDS5083:8126279). Given the results in human atherosclerotic plaques, we detected the mRNA level in mice during plaque development. The TREM2 mRNA level in the aorta of ApoE−/− mice was increased in a time-dependent manner (Fig. 1b). Furthermore, we treated Raw264.7 cells and mSMCs, the two most abundant types of cells in atherosclerotic plaques, with the atherogenic factor oxLDL (50 µg/mL) in vitro and found a time-dependent increase in TREM2 mRNA (Fig. 1c) [3, 26, 29]. Confocal fluorescence microscopy also further confirmed the trend of TREM2 expression in plaques of Trem2+/+/ApoE−/− mice (Fig. 1d, e). We validated the antibody using knockout mice to rule out nonspecific staining (Supplementary material online, Fig. S1a). These data suggest that TREM2 is increased in atherosclerotic plaques.
Fig. 1.

TREM2 expression is increased in atherosclerotic plaques. a The mRNA levels of TREM2 in human atherosclerotic plaque and non-carotid plaque vascular tissue (adjacent carotid tissue). The original data came from the GEO database GSE43292. student’s t test. b TREM2 mRNA was detected in the aorta from ApoE−/− mice fed a HFD for different periods (week; n = 5/group, one-way ANOVA test followed by a post hoc Tukey’s test). c Graph shows the trend of TREM2 mRNA expression after stimulation with oxLDL (50 µg/mL) for different times (n = 4/group, one-way ANOVA followed by a post hoc Tukey’s test). d, e Representative confocal images of TREM2 (red) in the atherosclerotic plaques of the aortic sinus from ApoE−/− mice in the indicated groups (n = 6/group). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; blue). Scale bar: 50 µm (upper panel), 25 µm (lower panel). One-way ANOVA test followed by a post hoc Tukey’s test. All data are presented as the mean ± SEM. *P < 0.05, **P < 0.01, ****P < 0.0001; ns, P > 0.05. AS atherosclerosis, H&E staining hematoxylin–eosin staining
Knockout of TREM2 attenuates atherosclerosis progression
To ascertain the causal role of TREM2 in atherogenesis, we knocked out TREM2 in ApoE−/− mice and generated Trem2−/−/ApoE−/− mice. The genotype of the mice was confirmed by RT-PCR (Supplementary material online, Fig. S1b). We fed Trem2+/+/ApoE−/− mice and Trem2−/−/ApoE−/− mice a HFD for 12 weeks to induce atherosclerosis and examined changes in aortic plaques. ORO staining showed that the whole aortic lesion area was decreased in Trem2−/−/ApoE−/− mice compared with Trem2+/+/ApoE−/− mice (Fig. 2a). In addition, the aortic arch lesion size and necrosis core of aortic tissue were decreased, while the thickness of fibrous caps was increased in the Trem2−/−/ApoE−/− group compared with the Trem2+/+/ApoE−/− group (Fig. 2a–c and Supplementary material online Fig. S1e). Consistent with the reduced whole aortic lesion area and aortic arch lesion size by ORO staining, lipid staining also decreased at the level of the aortic sinus by ORO staining in Trem2−/−/ApoE−/− mice (Fig. 2b, c). However, TREM2 deficiency had no effect on body weight, lipoprotein profiles, or collagen content (Supplementary material online, Fig. S1c–e). Then we used BODIPY to observe the lipid burden and foam cell number in the aortic sinus. BODIPY staining showed the same tendency as ORO-positive area statistics in the aortic sinus (Fig. 2b, c). These data suggest that knockout of TREM2 reduces plaque progression and lipid accumulation in the plaque and that TREM2 is involved in lipid metabolism and has an effect on the thickness of the fibrous cap.
Fig. 2.

Deletion of TREM2 ameliorates plaque formation and lipid deposition. a Trem2+/+/ApoE−/− and Trem2−/−/ApoE−/− mice were fed a HFD for 12 weeks. The percentage of whole aorta and aortic arch area that was occupied by the ORO-positive area was assessed (n = 6–7/group, student’s t test). Scale bar: 0.5 cm. b Images of H&E, ORO, and BODIPY-stained tissues showing the aortic sinus level obtained from mice. The red area shows plaque lipid in the aorta. Representative images of the aortic sinus were obtained from mice in the Trem2+/+/ApoE−/− and Trem2−/−/ApoE−/− groups. For H&E staining, the necrotic core was marked by a yellow line and zoomed in locally. Scale bar: 100 μm. c Representative images showing plaque area, the percentage of the plaque area of the necrotic core and ORO-positive area (n = 6/groups, student’s t test). The foam cell number of the plaque area occupied by BODIPY is presented among the indicated groups (green; n = 6/group, student’s t test). DAPI was used to stain nuclei (blue). All data are expressed as the mean ± SEM from three to five independent experiments. *P < 0.05, **P < 0.01, ****P < 0.0001
TREM2 is expressed in foam cells derived from SMCs and macrophages in plaques
Lipid deposition is a hallmark of atherosclerosis and contributes to plaque progression and instability. We examined whether TREM2 is directly involved in lipid metabolism dysfunction in atherosclerosis. Therefore, we co-immunostained TREM2-specific antibodies with BODIPY in plaques. The results showed that BODIPY-positive cells were co-localized with TREM2 in plaques (Fig. 3). The number of BODIPY/TREM2-positive cells gradually increased with atherosclerosis progression (Fig. 3a, d). These results were consistent with our in vivo observations that TREM2 deficiency decreases the lipid burden in the aortic root. As the main sources of foam cells are SMCs and macrophages, we investigated the role of TREM2 in the foaming process of these two kinds of cells [7].
Fig. 3.

TREM2 is expressed in foam cells derived from macrophages and SMCs in plaques. a–c Representative images of aortic sinuses obtained from the Trem2+/+/ApoE−/− and Trem2−/−/ApoE−/− groups co-stained TREM2 (red) with BODIPY (a, green), CD68 (b, green), and α-SMA (c, green). For fluorescence images, nuclei were stained with DAPI (blue). d–f The number of TREM2/BODIPY- (d), TREM2/CD68-, (e) or TREM2/α-SMA-positive cells (f) per mm2 of plaques in the indicated groups is shown (n = 6–7/group, one-way ANOVA followed by a post hoc Tukey’s test). All data are expressed as the mean ± SEM from three to five independent experiments. Scale bar: 25 μm. ***P < 0.001, ****P < 0.0001
TREM2 antibodies were stained with an SMC marker (α-SMA) and macrophage marker (CD68) to perform immunofluorescence staining in mouse aortic sinuses from mice fed a HFD for different periods. The results showed that TREM2 was co-localized with α-SMA and CD68 in plaques. Clearly, the number of TREM2/α-SMA-positive and TREM2/CD68-positive cells increased with the progression of atherosclerosis (Fig. 3b, c, e, f), indicating that TREM2 is upregulated in atherosclerotic lesions and enriched in SMCs and macrophages, underpinning our hypothesis that TREM2 is involved in foaming in SMCs and macrophages during atherogenesis.
TREM2 stimulates foam cell formation through the CD36 receptor
To verify our hypothesis that TREM2 is involved in forming foam cells from macrophages and SMCs, we used lentiviral constructs to overexpress TREM2 in Raw264.7 cells and mSMCs, respectively (Supplementary material online, Fig. S2a). ORO staining analysis of the cells after stimulation for 24 h with oxLDL (50 μg/mL) revealed lipid accumulation in the two types of cells. Specifically, the results showed that the ORO-positive area per cell in the TREM2-overexpressing Raw264.7 cell and mSMC groups (LV-TREM2) was increased by approximately 2.0- and 2.8-fold, respectively (Fig. 4a, b), compared to that in the control group (LV-GFP). These results were confirmed in primary BMDMs. Flow cytometry showed that the BODIPY fluorescence intensity of Trem2−/− BMDMs was weaker than that of Trem2+/+ mouse-derived BMDMs (Supplementary material online, Fig. S2b). This result was consistent with what we observed in RAW264.7 cells in vitro. Thus, these data indicate that TREM2 promotes macrophage and SMC cholesterol accumulation and foam cell formation, which is abolished by the knockout of TREM2. All these results are consistent with previous studies showing that TREM2 promotes lipid uptake in microglial cells [30]. To elucidate the mechanism by which TREM2 promotes foam cell formation, we further examined the changes in the protein levels of lipid uptake receptors (CD36, MSR1, and OLR1) and efflux receptors (ABCG1 and ABCA1), which are major lipid receptors for lipid metabolism in atherosclerosis. The results showed that TREM2 did not significantly affect the expression of the lipid uptake receptors MSR1 and OLR1. Similarly, the expression of the lipid efflux receptors ABCG1 and ABCA1 was not affected by the expression of TREM2. However, it is worth noting that the protein level of CD36 was significantly increased in Raw264.7 cells and mSMCs of the LV-TREM2 group compared to the LV-GFP group (Fig. 4c, d). Moreover, TREM2 increased the mRNA levels of CD36 but not the transcription of other receptors (Fig. 4e, f and Supplementary material online, Fig. S2c). The mFI of CD36 in plaques also showed a substantial decrease in Trem2−/−/ApoE−/− mice (Supplementary material online, Fig. S2d). In congruence, this result was confirmed in primary BMDMs and mSMCs that the expression of CD36 was decreased during the knockout of TREM2 (Fig. 4g and Supplementary material online, Fig. S2e).
Fig. 4.

TREM2 promotes foam cell formation in vitro. Lentiviral vectors were constructed to deliver the TREM2 gene (LV-TREM2). Lentiviral GFP (LV-GFP) was used as the control group (LV-GFP). a, b Images of ORO-stained ox-LDL-loaded Raw264.7 cells and mSMCs (a) in the indicated group. Graphs (b) show the ORO area positive per cell (n = 9/group, student’s t test). Scale bar: 10 μm. c, d Representative Western blots (left) of extracts obtained from Raw264.7 cells and mSMCs among the indicated groups of LV-GFP versus LV-TREM2 (n = 6/group, student’s t test). e, f Quantitative real-time PCR analysis of the mRNA levels of CD36 in macrophages and mSMCs in the LV-GFP or LV-TREM2 group. Data are normalized to ACTB expression (n = 5–6/group, student’s t test). g, Western blotting and statistics of BMDMs and mSMCs isolated from Trem2+/+ and Trem2−/− mice after stimulation with oxLDL at 50 µg/mL (n = 6/group, student’s t test). All data are expressed as the mean ± SEM from three to five independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001
CD36 is the predominant receptor affecting lipid uptake in atherosclerosis, so we used DiI-oxLDL to assess the level of lipid uptake. Immunofluorometric analysis and flow cytometry showed that TREM2 promoted DiI-oxLDL uptake in both Raw264.7 cells and mSMCs (Fig. 5a, b), which was in line with a previous study in microglial cells [30]. Consistent with these results, BMDMs from Trem2−/− mice showed lower mFI than those from Trem2+/+ mice (Supplementary material online, Fig. S3a). To further assess the effect of TREM2 on CD36 expression, we used a siRNA to inhibit CD36 expression in cells overexpressing TREM2. The CD36 protein and mRNA levels were much lower in siCD36 group compared with negative control (NC) group (Supplementary material online, Fig. S3b and c). The results revealed that CD36 silencing inhibited the TREM2-induced increase in lipid accumulation and uptake in Raw264.7 cells and mSMCs (Fig. 5c–f), indicating the role of TREM2-CD36 signaling in atherosclerosis. This trend is consistent with the results we observed in vivo and primary cells of Trem2−/− mice (Fig. 4g and Supplementary material online, Fig. S2d-e). These data underpin the importance of TREM2 in regulating foam cell formation by promoting lipid influx regulated by CD36.
Fig. 5.

TREM2 stimulates cholesterol uptake by regulating CD36. a Representative fluorescence images of Raw264.7 cells and mSMCs labeled with DiI-oxLDL at 37 °C for 4 h. Scale bar: 10 μm. b The mean DiI-oxLDL burden detected by flow cytometry (n = 9/group, student’s t-test). c and e Optical microscopy images (top) or confocal laser scanning microscopy images (bottom) of Raw264.7 cells and mSMCs labeled by ORO or DiI-oxLDL. CD36 small-interfering RNA (siRNA)-transfected Raw264.7 cells and mSMCs in the LV-GFP and LV-TREM2 groups before stimulation with oxLDL or DiI-oxLDL. Scale bars: 10 μm. Graphs (d, f) show the area positive for ORO per cell or mean DiI-oxLDL burden (mFI, n = 6 and 9/group, respectively; two-way ANOVA test followed by a post hoc Tukey’s test). All data are expressed as the mean ± SEM from 3 to 5 independent experiments. *P < 0.05, ***P < 0.001, ****P < 0.0001. mFI mean fluorescence intensity
TREM2 regulates CD36 expression by regulating p38MAPK and PPARγ phosphorylation
The transcription factor PPARγ is the most important factor regulating the transcription of CD36 [31–33]. Western blot analysis revealed that TREM2 did not affect the protein expression level of PPARγ under oxLDL stimulation (Fig. 6a–d). However, the phosphorylation level of PPARγ at s273 was affected by TREM2. In the LV-TREM2 group, the protein level of p-PPARγ was significantly reduced, indicating that TREM2 inhibits the phosphorylation of PPARγ compared to that in the LV-GFP group. These results indicate that TREM2 regulates the transcription level of CD36 by regulating the phosphorylation level of PPARγ, which in turn regulates lipid accumulation and foam cell formation in the atherosclerosis process.
Fig. 6.

TREM2 regulates MAPK-p38 and PPARγ phosphorylation. a–d Representative Western blots and normalized ratios of p-p38:t-p38, p-JNK1/2:t-JNK1/2, p-ERK1/2:t-ERK1/2, and p-PPARγ:t-PPARγ in RAW264.7 cells or mSMCs infected with LV-GFP or LV-TREM2 group (n = 6/group, student’s t test). e, f Representative images of ORO- and DiI-oxLDL-positive areas in Raw264.7 cells or mSMCs obtained from the indicated group. Nuclei were stained with DAPI (blue). RAW264.7 cells and mSMCs were cultured with the p38 phosphorylation agonist dehydrocorydaline (Deh) before oxLDL or DiI-oxLDL stimulation, and then the ORO- or DiI-oxLDL-positive area was analyzed (n = 6–9/group, two-way ANOVA followed by a post hoc Tukey’s test). g, h Immunoblot analysis of the expression of p-p38, t-p38, p-PPARγ, t-PPARγ, and CD36 in RAW264.7 cells and mSMCs treated with dehydrocorydaline (Deh) or an equal volume of DMSO (ctr, n = 6/group, two-way ANOVA test followed by a post hoc Tukey’s test). Quantitative data represent the fold change observed after normalizing the indicated protein band intensities to GAPDH. All data are presented as the mean ± SEM from three to five independent experiments. Scale bar: 10 μm. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. mFI mean fluorescence intensity. Deh, dehydrocorydaline
The mitogen-activated protein kinase (MAPK) pathway is a classical pathway involved in the activation of transcription factors and regulates atherosclerosis and obesity, [34, 35] which is involved in both PPARγ signaling and the TREM2 signaling pathway [12, 36]. Thus, we examined the activation of MAPK signaling (including p38, JNK1/2, and ERK1/2) after overexpression of TREM2. The results showed that phosphorylated p38 (p-p38) in the LV-TREM2 group was reduced in Raw264.7 cells and mSMCs compared to the LV-GFP group. In contrast, the total p38 (t-p38) level did not change significantly. The levels of p-JNK, t-JNK, p-ERK1/2, and t-ERK1/2 were not altered by the expression of TREM2. These results suggest that p-p38 is involved in the phosphorylation of PPARγ. Next, we investigated the effects of the p38 phosphorylation agonist dehydrocorydaline (Deh, 5 μM) in vitro. Deh or dimethyl sulfoxide (DMSO, ctr) was used to stimulate cells for 12 h in the LV-TREM2 and LV-GFP groups, and then the cell foaming model was established by oxLDL. We found that increased lipid uptake and accumulation induced by TREM2 were inhibited after Deh stimulation (Fig. 6e, f), which aligns with our expectations. Furthermore, the results of Western blot analysis also showed that the activity of PPARγ and the expression of CD36 decreased after treatment with p38 phosphorylation agonists (Fig. 6g, h). These data demonstrate that TREM2 regulates the activity of PPARγ by inhibiting the phosphorylation level of p-p38, leading to the promotion of CD36 expression and lipid uptake.
We also investigated the changes in the expression levels of the aforementioned proteins in vivo. The results further confirm that TREM2 affects atherosclerotic progression through p38/PPARγ signaling and downregulation of CD36 expression (Fig. 7a, b). In addition, CD36 was decreased in Trem2−/−/ApoE−/− mice compared with Trem2+/+/ApoE−/− mice. The lipid load and foam cells in the aortic sinus were also significantly reduced through CD68-BODIPY and α-SMA-BODIPY staining (Fig. 7c–e). Taken together, these data suggest that deletion of TREM2 inhibits foam cell formation in atherosclerosis by regulating CD36 expression.
Fig. 7.

TREM2 deficiency decreases foam cell formation in vivo. a Immunoblot analysis of the expression of p-p38, t-p38, p-PPARγ, t-PPARγ, and CD36 in extracts from the aortas of mice in the indicated group. Trem2+/+/ApoE−/− mice and Trem2−/−/ApoE−/− mice were fed a HFD for 12 weeks to induce atherosclerotic lesions. Quantitative data represent the fold change observed. Protein expression levels are calculated in (b n = 6/group, student’s t test). c Fluorescence images of CD68-BODIPY or α-SMA-BODIPY at the aortic sinus from Trem2+/+/ApoE−/− mice and Trem2−/−/ApoE−/− mice. Nuclei were stained with DAPI (blue). Scale bar: 50 μm. d, e The number of CD68/BODIPY- or α-SMA/BODIPY-positive cells per mm2 of plaques in the indicated groups is presented (n = 6/group, student’s t test). All data are shown as the mean ± SEM from three to five independent experiments. *P < 0.05, **P < 0.01
Discussion
This study found that TREM2 is increased in atherosclerotic plaques and that deletion of TREM2 attenuates atherosclerotic progression in ApoE−/− mice. In vitro, overexpression of TREM2 exacerbates foam cell formation in macrophages and mSMCs by promoting lipid uptake through regulating CD36 expression. Therefore, TREM2 is a potential target for atherosclerosis treatment. Herein, we elucidated the role of TREM2 in atherosclerosis and the mechanisms regulating foam cell formation, as shown in the Graphic Abstract.
TREM2 is a transmembrane receptor belonging to the immunoglobulin family with a wide range of ligands, including lipids, lipoproteins, cellular debris, and DNA [11]. In central nervous system dysfunction, diabetes, and liver-related disease, it has been found that TREM2 dysfunction is involved in lipid metabolism and regulates pathophysiological processes [37–41]. Recent studies using scRNA-seq of total aortic CD45-positive cells also found that one of the cell populations showed high TREM2 expression in atherosclerosis-prone mouse aortae [16–18, 28, 42]. Additionally, we analyzed the GSE43292 dataset from the Gene Expression Omnibus (GEO) datasets (including 32 hypertensive patients with atheroma plaque and adjacent carotid tissue) and found increased expression of TREM2 mRNA in human carotid plaques compared to adjacent carotid tissue. However, the causal role of TREM2 in atherosclerosis has not been previously explored.
To disclose the precise role of TREM2 in atherogenesis, ApoE−/− mice were crossbred with Trem2−/− mice to generate Trem2−/−/ApoE−/− double-knockout mice. The results show that TREM2 plays an important role in the formation of atherosclerotic plaques. Root morphometry revealed that TREM2 deficiency significantly reduced plaque size, lipid burden, necrotic core area, and BODIPY-positive cells per mm2 area compared to Trem2+/+/ApoE−/− mice. In addition, Xu et al. studies suggested that geniposide inhibited lipid metabolism and atherosclerosis in HFD-fed ApoE−/− mice by regulating TREM2 expression [43]. All these data suggest that TREM2 is closely related to foam cell formation and lipid metabolism in atherosclerosis. Our results also show that the knockout of TREM2 has an effect on plaque fibrous caps. Additionally, collagen deposition showed an increasing trend, although it was not statistically significant.
Recent studies have reported that TREM2 is expressed in foam cells in atherosclerosis [14, 16–18, 28]. In addition, Do et al. reported that TREM2-positive macrophages promote foam cell formation in acne lesions [44]. We performed immunofluorescence staining to co-immunostain TREM2 with the foam cell lipid marker BODIPY and found that TREM2 is primarily located in the atherosclerotic plaque core and foam cells. Previous studies have found that macrophages and SMCs are the two main sources of foam cells [3, 45]. To trace the origin of TREM2-positive foam cells, we performed immunofluorescence staining and found that TREM2 is co-localized with SMCs (α-SMA-positive) and macrophages (CD68-positive) in plaques and that TREM2/BODIPY/α-SMA-positive and TREM2/BODIPY/CD68-positive cells were increased in a time-dependent manner. These results confirm that TREM2 increases foam cell formation in atherosclerosis.
The formation of foam cells is an important hallmark of atherosclerosis [46]. Foam cells are mainly due to lipid metabolism disorders, resulting in intracellular cholesterol load. However, the mechanism responsible for foam cell formation is not completely understood. Scavenger receptors uncontrollably uptake modified lipoprotein, and lipid efflux receptor barriers are mainly involved in lipid metabolism during atherosclerosis, [47–50] subsequently increasing the lipid load of blood vessels. Yeh et al. found that TREM2 promotes Dil-acLDL uptake and binds to APOB in microglia in Alzheimer’s disease (AD) [30]. Analogously, Linnartz-Gerlach et al. showed that microglia decrease phagocytosis capability in Trem2−/− mice [51]. These data suggest that TREM2 is involved in lipid phagocytosis, which is in line with our findings. In vitro, our data reveal that TREM2 exacerbates foam cell formation by increasing the transcript levels of scavenger receptor CD36 and promoting lipid uptake. Yet, the expression of CD36 is reduced when TREM2 is knockout. CD36 is a class B scavenger receptor that regulates excessive ox-LDL uptake, resulting in lipid metabolism disorders and foam cell formation [52, 53]. We further validated the role of CD36 and found that CD36 gene silencing abolished TREM2-induced lipid accumulation in Raw264.7 cells and mSMCs, indicating that the TREM2-induced pro-atherosclerotic effect is dependent on CD36 expression. In addition, the Dil-oxLDL uptake assay is consistent with previous findings that TREM2 promotes lipid uptake in microglial cells [30]. These data underpin the point that TREM2 regulates lipid influx and foam cell formation in atherosclerosis.
We found that TREM2 promotes CD36 expression at the transcriptional level. Hendrikx et al. recently reported that TREM2 knockout decreases CD36 transcription in nonalcoholic steatohepatitis (NASH) [54]. Similarly, a recent study by Kim et al. also reported high expression of CD36 in TREM2-positive macrophages [55]. PPARγ is an important transcription factor contained at the CD36 promoter, [56] which promotes CD36 mRNA expression and is involved in lipid metabolism in human fatty liver disease, atherosclerosis, diabetes, and obesity [36, 57–63]. Previous studies found that PPARγ is also involved in TREM2-dependent signaling [39]. Accordingly, we detected the effect of PPARγ on TREM2-CD36 signaling and indicated that TREM2 promotes the activation of PPARγ, therefore promoting CD36 transcription. To elucidate the molecular mechanism involved in TREM2 and PPARγ-CD36 signaling, we next detected changes in the MAPK pathway, which is one of the signaling pathways that regulate PPAR in metabolic disease [64–67]. MAPK includes the p38 (also known as MAPK14) pathway, extracellular-signal-regulated kinase (ERK) pathway, and JUN N-terminal kinase (JNK) pathway. Classical MAPK signaling regulates many pathophysiological effects, such as inflammation, lipid metabolism, proliferation, migration, and apoptosis [68, 69]. Previous studies showed that MAPK is regulated by TREM2 and involved in PPARγ regulation in Parkinson’s disease, Alzheimer's disease, obesity, diabetes, and atherosclerosis [57, 70–75]. However, it remains unknown whether MAPK is involved in the regulation of TREM2 and regulates PPARγ activity in foam cells. The present study suggests that TREM2 overexpression inhibits p38 phosphorylation rather than JNK1/2 or ERK1/2, subsequently activating PPARγ and CD36 expression. In addition, pharmacological activation of p38 inhibits the pro-atherosclerotic effect of TREM2 on CD36 expression. These results are consistent with previous studies showing that TREM2 inhibited p38 phosphorylation in diabetes mellitus and chronic cerebral hypoperfusion to regulate inflammation [12, 39], and subsequently increase the active form of PPARγ. Collectively, these data suggest that p38/PPARγ signaling is involved in TREM2-regulated foam cell formation and CD36 receptor upregulation.
TREM2 is a novel lipid receptor. We demonstrated that it is involved in the progression of atherosclerosis by regulating the expression of the scavenger receptor CD36 in our study. The current pharmacological prevention of atherosclerosis is mainly lipid-lowering therapy, including statins, PCSK9 inhibitors, and antiplatelet drugs [76, 77]. Previous study suggested that TREM2 regulates atherosclerosis by regulating the process of autophagy through the mTOR pathway [43]. Our previous study also demonstrated that P2Y12R receptor inhibitor clopidogrel plus statins reverse plaque progression in atherosclerosis [3]. Yet, there is an urgent need for new targets for the treatment of atherosclerosis due to the complications of statins and antiplatelet drugs in some patients and the high price of PCSK9 inhibitors [78–80]. In this article, we revealed that the regulation of TREM2 delays the progression of plaque in the hyperlipidemia state, thus providing a new idea for the clinical treatment of hyperlipidemia state.
Recent studies on the relationship between TREM2 and atherosclerosis have been increasing, suggesting that TREM2 may play an essential role in atherosclerosis. In this article, we verified the mechanism of TREM2 promoting the expression of CD36 through the P38-PPARγ pathway, thereby promoting foam cell formation. However, TREM2 and MAPK seem act as “signaling bottlenecks”. They signal a range of ligands and transmit them to effectors to elicit a complex pathological and physiological responses in vivo environment. Therefore, the regulatory mechanism of TREM2 in atherosclerosis still needs a long way to be explored. In addition, the transgenic mice with global knockout of TREM2 for the experiments in this manuscript were used, more precise conditional gene knockout mice in macrophages and SMCs may be needed for further verification.
In summary, this study elucidates the important role of TREM2 in promoting lipid metabolism and foam cell formation in atherosclerotic progression. TREM2 promotes macrophage- and SMC-derived foam cell formation by regulating the expression of CD36. This study provides a novel target for the treatment of atherosclerosis.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1Fig. S1 TREM2 has no significant effect on body weight, lipid profile or collagen deposition. a. Antibody specificity validation of TREM2 molecules (red) in Trem2−/− mice. Scale bar: 50 µm. Nuclei were stained with DAPI (blue). b, Genotyping of TREM2 gene knockout in mice. Average body weights (c) and lipid profiles (d) of Trem2+/+/ApoE−/− and Trem2−/−/ApoE−/− mice (n = 6/group, student’s t-test). e, The percentage of collagen content and the thickness of fibrous caps were assessed in Masson staining images. Scale bar: 200 μm (n = 6/group, student’s t-test). *P < 0.05; ns, P > 0.05 (PDF 4323 KB)
Supplementary file2Fig. S2 TREM2 deficiency decreases CD36 expression. a, PCR-detected lentivirus overexpressing TREM2 in RAW264.7 cells and mSMCs (n = 6-7/group, student’s t-test). b, TREM2 stimulates primary BMDM foam cell formation. BMDMs isolated from Trem2+/+ and Trem2−/− mice were stimulated with oxLDL at 50 µg/mL for 24 h. Then the cells were incubated with BODIPY in DMEM for 1 h. A flow analyzer evaluated the mFI of the two groups of cells (n = 3/group, student’s t-test). c, Quantitative real-time PCR analysis of the mRNA levels of MSR1, OLR1, ABCA1 and ABCG1 in macrophages and mSMCs in the LV-GFP or LV-TREM2 group. Data are normalized to ACTB expression (n = 6/group, student’s t-test). d, Relative expression of CD36 (green) at the aortic sinus in Trem2+/+/ApoE−/− and Trem2−/−/ApoE−/− mice by immunofluorescence (n = 4/group, student’s t-test). Scale bar: 50 µm. e, Quantitative real-time PCR analysis of the mRNA levels of CD36 in primary BMDMs and mSMCs isolated from Trem2+/+ and Trem2−/− mice (n = 4/group, student’s t-test). *P < 0.05, **P < 0.01, ****P < 0.0001; ns, P > 0.05; mFI, mean fluorescence intensity (PDF 1510 KB)
Supplementary file3Fig. S3 Knockout of the TREM2 gene inhibits lipid uptake. a, Flow analysis showed the mFI of BMDMs isolated from Trem2+/+ and Trem2−/− mice stimulated with DiI-oxLDL at 10 µg/mL for 4 h (n = 3/group, student’s t-test). b, WB and PCR levels of CD36 expression in macrophages and mSMCs transfected with the control vector (NC) or the CD36 siRNA (siCD36, n = 4-6/group, student’s t-test). *P < 0.05, **P < 0.01, ****P < 0.0001; mFI, mean fluorescence intensity (PDF 123 KB)
Acknowledgements
We thank the Central Laboratory of Union Hospital of Tongji Medical College for providing a confocal microscope and flow cytometer.
Abbreviations
- ApoE−/−
Apolipoprotein E knockout
- AS
Atherosclerosis
- BMDMs
Bone marrow-derived macrophages
- BODIPY
4,4-difluoro-1,3,5,7,8-pentamethyl-4-bora-3a,4a-diaza-s-indacene
- CD36
Cluster of differentiation 36 Deh dehydrocorydaline
- DAPI
4ʹ,6-diamidino-2-phenylindole
- DEGs
Differentially expressed genes
- Deh
Dehydrocorydaline
- DiI-oxLDL
Dil-labeled oxLDL
- GEO
Gene Expression Omnibus
- HFD
High-fat diet
- IF
Immunofluorescence
- MAPK
Mitogen-activated protein kinase
- M-CSF
Macrophage colony-stimulating factor
- Mfi
Mean fluorescence intensity
- mSMCs
Mouse primary aortic SMCs
- ORO
Oil Red O
- OxLDL
Oxidized low-density lipoprotein
- p-ERK1/2
Phosphorylated ERK1/2
- p-JNK
Phosphorylated JNK
- p-p38
Phosphorylated p38
- PPARγ
Peroxisome proliferator activated-receptor gamma
- SMCs
Vascular smooth muscle cells
- t-ERK1/2
Total ERK1/2
- t-JNK
Total JNK
- t-p38
Total p38
- TREM2
Triggering receptor expressed on myeloid cells 2
- α-SMA
α-Smooth muscle actin
Author contributions
Conceptualization: LM; methodology: XG and BL; formal analysis and investigation: XG, CW, FZ, XX, LN, and JC; writing–original draft preparation: XG and BL; writing–review and editing: LM and XG; funding acquisition: LM.
Funding
This work was supported by the National Natural Science Foundation of China (no. 81974182 and no. 82171325 to LM).
Data availability
All the data supporting the findings of this study are available within the paper. Data will be made available on reasonable request, not applicable for material.
Declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethics approval
This study was approved by the Tongji Medical School of Huazhong Technology University, China.
Consent to participate
Not applicable.
Consent for publication
All authors have been involved in writing the manuscript and consented to publication.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Xiaoqing Guo and Bowei Li contributed equally to this article.
References
- 1.Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ Res. 2016;118:535–546. doi: 10.1161/CIRCRESAHA.115.307611. [DOI] [PubMed] [Google Scholar]
- 2.Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R, et al. Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pi S, Mao L, Chen J, Shi H, Liu Y, Guo X, et al. The P2RY12 receptor promotes VSMC-derived foam cell formation by inhibiting autophagy in advanced atherosclerosis. Autophagy. 2021;17:980–1000. doi: 10.1080/15548627.2020.1741202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118:653–667. doi: 10.1161/CIRCRESAHA.115.306256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. 2014;129:1551–1559. doi: 10.1161/CIRCULATIONAHA.113.005015. [DOI] [PubMed] [Google Scholar]
- 6.Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M, et al. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res. 2014;115:662–667. doi: 10.1161/CIRCRESAHA.115.304634. [DOI] [PubMed] [Google Scholar]
- 7.Wang D, Yang Y, Lei Y, Tzvetkov NT, Liu X, Yeung AWK, et al. Targeting foam cell formation in atherosclerosis: therapeutic potential of natural products. Pharmacol Rev. 2019;71:596–670. doi: 10.1124/pr.118.017178. [DOI] [PubMed] [Google Scholar]
- 8.Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472–477. doi: 10.1126/science.aaf6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deczkowska A, Weiner A, Amit I. The physiology, pathology, and potential therapeutic applications of the TREM2 signaling pathway. Cell. 2020;181:1207–1217. doi: 10.1016/j.cell.2020.05.003. [DOI] [PubMed] [Google Scholar]
- 10.Shi Y, Holtzman DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. 2018;18:759–772. doi: 10.1038/s41577-018-0051-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kober DL, Brett TJ. TREM2–ligand interactions in health and disease. J Mol Biol. 2017;429:1607–1629. doi: 10.1016/j.jmb.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang J, Liu Y, Zheng Y, Luo Y, Du Y, Zhao Y, et al. TREM-2-p38 MAPK signaling regulates neuroinflammation during chronic cerebral hypoperfusion combined with diabetes mellitus. J Neuroinflammation. 2020;17:2. doi: 10.1186/s12974-019-1688-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, et al. Lipid-associated macrophages control metabolic homeostasis in a Trem2-dependent manner. Cell. 2019;178:686–698 e14. doi: 10.1016/j.cell.2019.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. 2020;127:402–426. doi: 10.1161/CIRCRESAHA.120.316903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Damisah EC, Rai A, Grutzendler J. TREM2: modulator of lipid metabolism in microglia. Neuron. 2020;105:759–761. doi: 10.1016/j.neuron.2020.02.008. [DOI] [PubMed] [Google Scholar]
- 16.Depuydt MAC, Prange KHM, Slenders L, Ord T, Elbersen D, Boltjes A, et al. Microanatomy of the human atherosclerotic plaque by single-cell transcriptomics. Circ Res. 2020;127:1437–1455. doi: 10.1161/CIRCRESAHA.120.316770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-cell RNA-Seq reveals the transcriptional landscape and heterogeneity of aortic macrophages in murine atherosclerosis. Circ Res. 2018;122:1661–1674. doi: 10.1161/CIRCRESAHA.117.312509. [DOI] [PubMed] [Google Scholar]
- 18.Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ Res. 2018;123:1127–1142. doi: 10.1161/CIRCRESAHA.118.312804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reddick RL, Zhang SH, Maeda N. Atherosclerosis in mice lacking apo E. Evaluation of lesional development and progression. Arterioscler Thromb. 1994;14:141–147. doi: 10.1161/01.atv.14.1.141. [DOI] [PubMed] [Google Scholar]
- 20.Jawien J, Nastalek P, Korbut R. Mouse models of experimental atherosclerosis. J Physiol Pharmacol. 2004;55:503–517. [PubMed] [Google Scholar]
- 21.Feng B, Zhang D, Kuriakose G, Devlin CM, Kockx M, Tabas I. Niemann-Pick C heterozygosity confers resistance to lesional necrosis and macrophage apoptosis in murine atherosclerosis. Proc Natl Acad Sci USA. 2003;100:10423–10428. doi: 10.1073/pnas.1732494100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niu X, Pi SL, Baral S, Xia YP, He QW, Li YN, et al. P2Y12 promotes migration of vascular smooth muscle cells through cofilin dephosphorylation during atherogenesis. Arterioscler Thromb Vasc Biol. 2017;37:515–524. doi: 10.1161/ATVBAHA.116.308725. [DOI] [PubMed] [Google Scholar]
- 23.Le Guezennec X, Brichkina A, Huang YF, Kostromina E, Han W, Bulavin DV. Wip1-dependent regulation of autophagy, obesity, and atherosclerosis. Cell Metab. 2012;16:68–80. doi: 10.1016/j.cmet.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 24.Xu S, Huang Y, Xie Y, Lan T, Le K, Chen J, et al. Evaluation of foam cell formation in cultured macrophages: an improved method with Oil Red O staining and DiI-oxLDL uptake. Cytotechnology. 2010;62:473–481. doi: 10.1007/s10616-010-9290-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Selvais CM, Davis-Lopez de Carrizosa MA, Nachit M, Versele R, Dubuisson N, Noel L, et al. AdipoRon enhances healthspan in middle-aged obese mice: striking alleviation of myosteatosis and muscle degenerative markers. J Cachexia Sarcopenia Muscle. 2023;14:464–478. doi: 10.1002/jcsm.13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niu X, Pi SL, Baral S, Xia YP, He QW, Li YN, et al. P2Y(12) promotes migration of vascular smooth muscle cells through cofilin dephosphorylation during atherogenesis. Arterioscler Thromb Vasc Biol. 2017;37:515–524. doi: 10.1161/ATVBAHA.116.308725. [DOI] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Cochain C, Saliba AE, Zernecke A. Letter by cochain et al regarding article, "Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models". Circ Res. 2018;123:e48–e49. doi: 10.1161/CIRCRESAHA.118.314120. [DOI] [PubMed] [Google Scholar]
- 29.Yang S, Xia YP, Luo XY, Chen SL, Li BW, Ye ZM, et al. Exosomal CagA derived from Helicobacter pylori-infected gastric epithelial cells induces macrophage foam cell formation and promotes atherosclerosis. J Mol Cell Cardiol. 2019;135:40–51. doi: 10.1016/j.yjmcc.2019.07.011. [DOI] [PubMed] [Google Scholar]
- 30.Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. 2016;91:328–340. doi: 10.1016/j.neuron.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 31.Wang H, Franco F, Tsui YC, Xie X, Trefny MP, Zappasodi R, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol. 2020;21:298–308. doi: 10.1038/s41590-019-0589-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang C, Luo X, Chen J, Zhou B, Yang M, Liu R, et al. Osteoprotegerin promotes liver steatosis by targeting the ERK-PPAR-gamma-CD36 pathway. Diabetes. 2019;68:1902–1914. doi: 10.2337/db18-1055. [DOI] [PubMed] [Google Scholar]
- 33.Chen CH, Leu SJ, Hsu CP, Pan CC, Shyue SK, Lee TS. Atypical antipsychotic drugs deregulate the cholesterol metabolism of macrophage-foam cells by activating NOX-ROS-PPARgamma-CD36 signaling pathway. Metabolism. 2021;123:154847. doi: 10.1016/j.metabol.2021.154847. [DOI] [PubMed] [Google Scholar]
- 34.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim EK, Choi EJ. Compromised MAPK signaling in human diseases: an update. Arch Toxicol. 2015;89:867–882. doi: 10.1007/s00204-015-1472-2. [DOI] [PubMed] [Google Scholar]
- 36.Oppi S, Nusser-Stein S, Blyszczuk P, Wang X, Jomard A, Marzolla V, et al. Macrophage NCOR1 protects from atherosclerosis by repressing a pro-atherogenic PPARgamma signature. Eur Heart J. 2020;41:995–1005. doi: 10.1093/eurheartj/ehz667. [DOI] [PubMed] [Google Scholar]
- 37.Haass C. Loss of TREM2 facilitates tau accumulation, spreading, and brain atrophy, but only in the presence of amyloid pathology. Neuron. 2021;109:1243–1245. doi: 10.1016/j.neuron.2021.03.029. [DOI] [PubMed] [Google Scholar]
- 38.Lee SH, Meilandt WJ, Xie L, Gandham VD, Ngu H, Barck KH, et al. Trem2 restrains the enhancement of tau accumulation and neurodegeneration by beta-amyloid pathology. Neuron. 2021;109:1283–1301 e6. doi: 10.1016/j.neuron.2021.02.010. [DOI] [PubMed] [Google Scholar]
- 39.Park M, Yi JW, Kim EM, Yoon IJ, Lee EH, Lee HY, et al. Triggering receptor expressed on myeloid cells 2 (TREM2) promotes adipogenesis and diet-induced obesity. Diabetes. 2015;64:117–127. doi: 10.2337/db13-1869. [DOI] [PubMed] [Google Scholar]
- 40.Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, et al. TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron. 2020;105:837–854 e9. doi: 10.1016/j.neuron.2019.12.007. [DOI] [PubMed] [Google Scholar]
- 41.Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, et al. TREM2 maintains microglial metabolic fitness in Alzheimer's disease. Cell. 2017;170:649–663 e13. doi: 10.1016/j.cell.2017.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willemsen L, de Winther MP. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J Pathol. 2020;250:705–714. doi: 10.1002/path.5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu YL, Liu XY, Cheng SB, He PK, Hong MK, Chen YY, et al. Geniposide enhances macrophage autophagy through downregulation of TREM2 in atherosclerosis. Am J Chin Med. 2020;48:1821–1840. doi: 10.1142/S0192415X20500913. [DOI] [PubMed] [Google Scholar]
- 44.Do TH, Ma F, Andrade PR, Teles R, de Andrade Silva BJ, Hu C, et al. TREM2 macrophages induced by human lipids drive inflammation in acne lesions. Sci Immunol. 2022;7:eabo2787. doi: 10.1126/sciimmunol.abo2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bartels ED, Christoffersen C, Lindholm MW, Nielsen LB. Altered metabolism of LDL in the arterial wall precedes atherosclerosis regression. Circ Res. 2015;117:933–942. doi: 10.1161/CIRCRESAHA.115.307182. [DOI] [PubMed] [Google Scholar]
- 46.Ouimet M, Marcel YL. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler Thromb Vasc Biol. 2012;32:575–581. doi: 10.1161/ATVBAHA.111.240705. [DOI] [PubMed] [Google Scholar]
- 47.Matsuo M. ABCA1 and ABCG1 as potential therapeutic targets for the prevention of atherosclerosis. J Pharmacol Sci. 2022;148:197–203. doi: 10.1016/j.jphs.2021.11.005. [DOI] [PubMed] [Google Scholar]
- 48.Yu XH, Fu YC, Zhang DW, Yin K, Tang CK. Foam cells in atherosclerosis. Clin Chim Acta. 2013;424:245–252. doi: 10.1016/j.cca.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 49.Platt N, Gordon S. Is the class A macrophage scavenger receptor (SR-A) multifunctional?—The mouse's tale. J Clin Invest. 2001;108:649–654. doi: 10.1172/JCI13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shu H, Peng Y, Hang W, Nie J, Zhou N, Wang DW. The role of CD36 in cardiovascular disease. Cardiovasc Res. 2022;118:115–129. doi: 10.1093/cvr/cvaa319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Linnartz-Gerlach B, Bodea LG, Klaus C, Ginolhac A, Halder R, Sinkkonen L, et al. TREM2 triggers microglial density and age-related neuronal loss. Glia. 2019;67:539–550. doi: 10.1002/glia.23563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Collot-Teixeira S, Martin J, McDermott-Roe C, Poston R, McGregor JL. CD36 and macrophages in atherosclerosis. Cardiovasc Res. 2007;75:468–477. doi: 10.1016/j.cardiores.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 53.Tian K, Xu Y, Sahebkar A, Xu S. CD36 in atherosclerosis: pathophysiological mechanisms and therapeutic implications. Curr Atheroscler Rep. 2020;22:59. doi: 10.1007/s11883-020-00870-8. [DOI] [PubMed] [Google Scholar]
- 54.Hendrikx T, Porsch F, Kiss MG, Rajcic D, Papac-Milicevic N, Hoebinger C, et al. Soluble TREM2 levels reflect the recruitment and expansion of TREM2(+) macrophages that localize to fibrotic areas and limit NASH. J Hepatol. 2022 doi: 10.1016/j.jhep.2022.06.004. [DOI] [PubMed] [Google Scholar]
- 55.Kim SM, Mun BR, Lee SJ, Joh Y, Lee HY, Ji KY, et al. TREM2 promotes Abeta phagocytosis by upregulating C/EBPalpha-dependent CD36 expression in microglia. Sci Rep. 2017;7:11118. doi: 10.1038/s41598-017-11634-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93:241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 57.Huang CC, Chou CA, Chen WY, Yang JL, Lee WC, Chen JB, et al. Empagliflozin ameliorates free fatty acid induced-lipotoxicity in renal proximal tubular cells via the PPARgamma/CD36 pathway in obese mice. Int J Mol Sci. 2021 doi: 10.3390/ijms222212408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kotla S, Singh NK, Rao GN. ROS via BTK-p300-STAT1-PPARgamma signaling activation mediates cholesterol crystals-induced CD36 expression and foam cell formation. Redox Biol. 2017;11:350–364. doi: 10.1016/j.redox.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu L, Zhang S, Zhang Q, Wei S, Wang G, Luo P. The molecular mechanism of hepatic lipid metabolism disorder caused by NaAsO2 through regulating the ERK/PPAR signaling pathway. Oxid Med Cell Longev. 2022;2022:6405911. doi: 10.1155/2022/6405911. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 60.Nicholson AC. Expression of CD36 in macrophages and atherosclerosis: the role of lipid regulation of PPARgamma signaling. Trends Cardiovasc Med. 2004;14:8–12. doi: 10.1016/j.tcm.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 61.Feng J, Han J, Pearce SF, Silverstein RL, Gotto AM, Jr, Hajjar DP, et al. Induction of CD36 expression by oxidized LDL and IL-4 by a common signaling pathway dependent on protein kinase C and PPAR-gamma. J Lipid Res. 2000;41:688–696. doi: 10.1016/S0022-2275(20)32377-4. [DOI] [PubMed] [Google Scholar]
- 62.Li H, Ruan XZ, Powis SH, Fernando R, Mon WY, Wheeler DC, et al. EPA and DHA reduce LPS-induced inflammation responses in HK-2 cells: evidence for a PPAR-gamma-dependent mechanism. Kidney Int. 2005;67:867–874. doi: 10.1111/j.1523-1755.2005.00151.x. [DOI] [PubMed] [Google Scholar]
- 63.Lim HJ, Lee S, Lee KS, Park JH, Jang Y, Lee EJ, et al. PPARgamma activation induces CD36 expression and stimulates foam cell like changes in rVSMCs. Prostaglandins Other Lipid Mediat. 2006;80:165–174. doi: 10.1016/j.prostaglandins.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 64.Lu Y, Zhang M, Wang S, Hong B, Wang Z, Li H, et al. p38 MAPK-inhibited dendritic cells induce superior antitumour immune responses and overcome regulatory T-cell-mediated immunosuppression. Nat Commun. 2014;5:4229. doi: 10.1038/ncomms5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Han Y, Wang J, Jin M, Jia L, Yan C, Wang Y. Shentong Zhuyu decoction inhibits inflammatory response, migration, and invasion and promotes apoptosis of rheumatoid arthritis fibroblast-like synoviocytes via the MAPK p38/PPARgamma/CTGF pathway. Biomed Res Int. 2021;2021:6187695. doi: 10.1155/2021/6187695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hua B, Liu Q, Gao S, Li W, Li H. Protective role of activating PPARgamma in advanced glycation end products-induced impairment of coronary artery vasodilation via inhibiting p38 phosphorylation and reactive oxygen species production. Biomed Pharmacother. 2022;147:112641. doi: 10.1016/j.biopha.2022.112641. [DOI] [PubMed] [Google Scholar]
- 67.Tian C, Jin X, Ye X, Wu H, Ren W, Zhang R, et al. Long term intake of 0.1% ethanol decreases serum adiponectin by suppressing PPARgamma expression via p38 MAPK pathway. Food Chem Toxicol. 2014;65:329–334. doi: 10.1016/j.fct.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 68.Liu Y, Shepherd EG, Nelin LD. MAPK phosphatases-regulating the immune response. Nat Rev Immunol. 2007;7:202–212. doi: 10.1038/nri2035. [DOI] [PubMed] [Google Scholar]
- 69.Bassi R, Heads R, Marber MS, Clark JE. Targeting p38-MAPK in the ischaemic heart: kill or cure? Curr Opin Pharmacol. 2008;8:141–146. doi: 10.1016/j.coph.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 70.Huang W, Lv Q, Xiao Y, Zhong Z, Hu B, Yan S, et al. triggering receptor expressed on myeloid cells 2 protects dopaminergic neurons by promoting autophagy in the inflammatory pathogenesis of Parkinson's disease. Front Neurosci. 2021;15:745815. doi: 10.3389/fnins.2021.745815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Su VY, Yang KY, Chiou SH, Chen NJ, Mo MH, Lin CS, et al. Induced pluripotent stem cells regulate triggering receptor expressed on myeloid cell-1 expression and the p38 mitogen-activated protein kinase pathway in endotoxin-induced acute lung injury. Stem Cells. 2019;37:631–639. doi: 10.1002/stem.2980. [DOI] [PubMed] [Google Scholar]
- 72.Ruganzu JB, Peng X, He Y, Wu X, Zheng Q, Ding B, et al. Downregulation of TREM2 expression exacerbates neuroinflammatory responses through TLR4-mediated MAPK signaling pathway in a transgenic mouse model of Alzheimer's disease. Mol Immunol. 2022;142:22–36. doi: 10.1016/j.molimm.2021.12.018. [DOI] [PubMed] [Google Scholar]
- 73.Gessi S, Fogli E, Sacchetto V, Merighi S, Varani K, Preti D, et al. Adenosine modulates HIF-1{alpha}, VEGF, IL-8, and foam cell formation in a human model of hypoxic foam cells. Arterioscler Thromb Vasc Biol. 2010;30:90–97. doi: 10.1161/ATVBAHA.109.194902. [DOI] [PubMed] [Google Scholar]
- 74.Hopkins PN. Molecular biology of atherosclerosis. Physiol Rev. 2013;93:1317–1542. doi: 10.1152/physrev.00004.2012. [DOI] [PubMed] [Google Scholar]
- 75.Ren M, Guo Y, Wei X, Yan S, Qin Y, Zhang X, et al. TREM2 overexpression attenuates neuroinflammation and protects dopaminergic neurons in experimental models of Parkinson's disease. Exp Neurol. 2018;302:205–213. doi: 10.1016/j.expneurol.2018.01.016. [DOI] [PubMed] [Google Scholar]
- 76.Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. 2019;5:56. doi: 10.1038/s41572-019-0106-z. [DOI] [PubMed] [Google Scholar]
- 77.Cholesterol Treatment Trialists C, Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–1681. doi: 10.1016/S0140-6736(10)61350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Collins R, Reith C, Emberson J, Armitage J, Baigent C, Blackwell L, et al. Interpretation of the evidence for the efficacy and safety of statin therapy. Lancet. 2016;388:2532–2561. doi: 10.1016/S0140-6736(16)31357-5. [DOI] [PubMed] [Google Scholar]
- 79.Zheng SL, Roddick AJ. Association of aspirin use for primary prevention with cardiovascular events and bleeding events: a systematic review and meta-analysis. JAMA. 2019;321:277–287. doi: 10.1001/jama.2018.20578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Antithrombotic Trialists C. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324:71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file1Fig. S1 TREM2 has no significant effect on body weight, lipid profile or collagen deposition. a. Antibody specificity validation of TREM2 molecules (red) in Trem2−/− mice. Scale bar: 50 µm. Nuclei were stained with DAPI (blue). b, Genotyping of TREM2 gene knockout in mice. Average body weights (c) and lipid profiles (d) of Trem2+/+/ApoE−/− and Trem2−/−/ApoE−/− mice (n = 6/group, student’s t-test). e, The percentage of collagen content and the thickness of fibrous caps were assessed in Masson staining images. Scale bar: 200 μm (n = 6/group, student’s t-test). *P < 0.05; ns, P > 0.05 (PDF 4323 KB)
Supplementary file2Fig. S2 TREM2 deficiency decreases CD36 expression. a, PCR-detected lentivirus overexpressing TREM2 in RAW264.7 cells and mSMCs (n = 6-7/group, student’s t-test). b, TREM2 stimulates primary BMDM foam cell formation. BMDMs isolated from Trem2+/+ and Trem2−/− mice were stimulated with oxLDL at 50 µg/mL for 24 h. Then the cells were incubated with BODIPY in DMEM for 1 h. A flow analyzer evaluated the mFI of the two groups of cells (n = 3/group, student’s t-test). c, Quantitative real-time PCR analysis of the mRNA levels of MSR1, OLR1, ABCA1 and ABCG1 in macrophages and mSMCs in the LV-GFP or LV-TREM2 group. Data are normalized to ACTB expression (n = 6/group, student’s t-test). d, Relative expression of CD36 (green) at the aortic sinus in Trem2+/+/ApoE−/− and Trem2−/−/ApoE−/− mice by immunofluorescence (n = 4/group, student’s t-test). Scale bar: 50 µm. e, Quantitative real-time PCR analysis of the mRNA levels of CD36 in primary BMDMs and mSMCs isolated from Trem2+/+ and Trem2−/− mice (n = 4/group, student’s t-test). *P < 0.05, **P < 0.01, ****P < 0.0001; ns, P > 0.05; mFI, mean fluorescence intensity (PDF 1510 KB)
Supplementary file3Fig. S3 Knockout of the TREM2 gene inhibits lipid uptake. a, Flow analysis showed the mFI of BMDMs isolated from Trem2+/+ and Trem2−/− mice stimulated with DiI-oxLDL at 10 µg/mL for 4 h (n = 3/group, student’s t-test). b, WB and PCR levels of CD36 expression in macrophages and mSMCs transfected with the control vector (NC) or the CD36 siRNA (siCD36, n = 4-6/group, student’s t-test). *P < 0.05, **P < 0.01, ****P < 0.0001; mFI, mean fluorescence intensity (PDF 123 KB)
Data Availability Statement
All the data supporting the findings of this study are available within the paper. Data will be made available on reasonable request, not applicable for material.