

Abstract
Bacteria that occupy an intracellular niche can evade extracellular host immune responses and antimicrobial molecules. In addition to classic intracellular pathogens, other bacteria including uropathogenic Escherichia coli (UPEC) can adopt both extracellular and intracellular lifestyles. UPEC intracellular survival and replication complicates treatment, as many therapeutic molecules do not effectively reach all components of the infection cycle. In this study, we explored cell-penetrating antimicrobial peptides from distinct structural classes as alternative molecules for targeting bacteria. We identified two β-hairpin peptides from the horseshoe crab, tachyplesin I and polyphemusin I, with broad antimicrobial activity toward a panel of pathogenic and non-pathogenic bacteria in planktonic form. Peptide analogs [I11A]tachyplesin I and [I11S]tachyplesin I maintained activity toward bacteria, but were less toxic to mammalian cells than native tachyplesin I. This important increase in therapeutic window allowed treatment with higher concentrations of [I11A]tachyplesin I and [I11S]tachyplesin I, to significantly reduce intramacrophage survival of UPEC in an in vitro infection model. Mechanistic studies using bacterial cells, model membranes and cell membrane extracts, suggest that tachyplesin I and polyphemusin I peptides kill UPEC by selectively binding and disrupting bacterial cell membranes. Moreover, treatment of UPEC with sublethal peptide concentrations increased zinc toxicity and enhanced innate macrophage antimicrobial pathways. In summary, our combined data show that cell-penetrating peptides are attractive alternatives to traditional small molecule antibiotics for treating UPEC infection, and that optimization of native peptide sequences can deliver effective antimicrobials for targeting bacteria in extracellular and intracellular environments.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-021-04041-z.
Keywords: Antimicrobial peptide, Selective membrane-active mechanism, Host defence, Macrophages, Intracellular niche, Uropathogenic Escherichia coli
Introduction
Antimicrobial resistance and tolerance pose a serious threat to global health, with the Centre for Disease Control and Prevention reporting more than 2.8 million cases of antibiotic-resistant infection and more than 35,000 associated deaths in the United States annually [1]. Due to intrinsic or acquired mechanisms of resistance, common and important antibiotics are becoming less effective [2, 3], leading to a growing need for new classes of antimicrobial compounds with different mechanisms of action to treat bacterial infections [4–7].
Antibiotic tolerance is a phenomenon where bacteria are genetically susceptible but phenotypically tolerant to antibiotic treatments [8, 9]. One mechanism of antibiotic tolerance is adoption of an intracellular lifestyle [10–12], where bacteria are protected from extracellular host defence mechanisms and antimicrobial therapy [11]. Pathogens with the ability to occupy intracellular niches promote their uptake by host cells, can escape or resist cellular antimicrobial mechanisms, adapt to the new host cell environment, and modulate host cell biology [10–12]. Depending on the bacterial pathogen, this intracellular niche can either be in the cytosol or inside vacuoles where they are shielded from antibiotic treatment and can facilitate recurrent infection (Fig. 1) [13, 14].
Fig. 1.
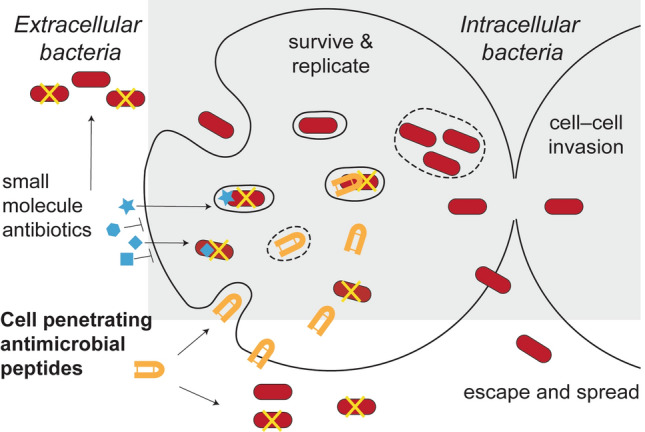
Challenges of targeting bacteria with an intracellular lifestyle. Pathogenic bacteria that survive and replicate inside cells can hide from cell-impermeable antimicrobial agents. Peptides able to cross host cell membranes to reach bacteria in the cytosol and/or vacuoles can overcome these challenges, representing candidates for developing new therapies for this challenging subset of bacteria
The first barrier for a drug targeting intracellular bacteria is crossing the host cell membrane at non-toxic concentrations [10, 11, 15]. As host cell membranes are impermeable to most polar or charged molecules [11], this barrier precludes a large number of conventional therapeutics [10, 11]. The second barrier is the challenge of accessing bacteria located in the host cell cytosol, or sequestered inside membrane-bound vesicles, and maintaining antimicrobial activity in these environments (Fig. 1) [11, 15, 16]. Also, therapeutic molecules which can successfully reach the bacteria may not be at concentrations high enough to eradicate infection [11].
Classic examples of intracellular bacterial pathogens include Mycobacterium tuberculosis, Salmonella enterica serovar Typhimurium and Burkholderia pseudomallei. However, there is growing evidence that other bacteria, including Pseudomonas aeruginosa, Staphylococcus aureus and Escherichia coli, can occupy intracellular niches [17–20]. A pertinent example is uropathogenic E. coli (UPEC), which is the leading cause of urinary tract infections (UTIs) [21] that affect 40–50% of women during their lifetime [22, 23]. UPEC is primarily an extracellular pathogen that infects the bladder epithelium, but can also survive in intracellular niches such as within bladder epithelial cells where it proliferates to form intracellular bacterial communities [24, 25] and within neutrophils and macrophages, which are recruited to the bladder in response to UPEC infection [26–29]. The establishment of infection within immune cells is problematic as these cells can act as ‘Trojan horses’, leading to dissemination of the pathogen in the whole organism [10, 30]. Intracellular UPEC can also become dormant and form quiescent intracellular bacterial reservoirs which persist within the host cell, possibly contributing to recurrent UTIs [31]. Despite their previous use in the treatment of UTIs, widespread emergence of resistance to antibiotics such as trimethoprim–sulfamethoxazole, fluoroquinolones, and β-lactams has led to a decrease in their therapeutic efficacy [32–34].
Peptide-based therapeutics are an attractive alternative to conventional small molecule drugs. Current glycopeptide [35] and lipopeptide drugs [36] have potent antimicrobial activity, but are restricted to use as last-line therapies due to their narrow therapeutic index and undesirable off-target toxicity [37, 38]. For new molecule discovery, naturally occurring antimicrobial peptides are gaining traction due to their broad spectrum of antimicrobial activity, relative inactivity toward eukaryotic cells at concentrations that inhibit bacterial growth, and efficacy at low and sub-micromolar concentrations [39, 40]. A subset of antimicrobial peptides with cell-penetrating properties, herein referred to as antimicrobial cell-penetrating peptides (CPPs), are especially attractive due to their ability to cross mammalian cell membranes and access intracellular targets [40–42].
Antimicrobial CPPs are typically composed of 5–30 amino acids and have an amphipathic arrangement of hydrophobic and positively charged residues that promotes interaction with cell membranes [40, 43]. Furthermore, the arrangement of positively charged residues on one side of the peptide promotes electrostatic interactions with negatively charged phospholipids that are more commonly found in bacterial cell membranes, compared to neutral mammalian host cell membranes [40, 44–46]. Peptides are also thought to be less prone to common mechanisms of resistance observed for small molecule antibiotics because of their direct actions on microbial membranes, in contrast to affecting single, smaller molecular targets [41, 47].
Another strategy for avoiding antibiotic resistance is to enhance the effect of innate antimicrobial mechanisms. For example, innate immune cells such as macrophages and neutrophils use zinc poisoning as an antimicrobial weapon [48], and the zinc ionophore PBT2 reversed antibiotic resistance for several bacterial pathogens [49]. Interestingly, macrophage-mediated zinc toxicity is enhanced against E. coli mutants that are defective in zinc export (ΔzntA, ΔzntR), and also mutants with reduced membrane integrity (ΔcpxR, ΔpstB) [50]. Some pathogens, including Salmonella [51] and UPEC [50], can subvert intracellular zinc toxicity. We aimed to determine if amphipathic antimicrobial CPPs could enhance antimicrobial zinc toxicity due to their ability to reduce the membrane integrity of bacterial pathogens.
In this study, we examined the antimicrobial efficacy of known antimicrobial CPPs, tachyplesin I and polyphemusin I, toward pathogenic and non-pathogenic Gram-negative bacteria and demonstrate enhanced zinc toxicity at sublethal peptide concentrations in vitro. Modification of native tachyplesin I improved selective toxicity toward UPEC, resulting in significantly reduced intracellular bacterial loads in an in vitro macrophage infection model. Our findings highlight the potential of CPPs as candidates for developing new therapeutics that target bacteria in both extracellular and intracellular environments.
Materials and methods
Bacterial strains
Pathogenic bacterial strains included in this study were: B. pseudomallei MSHR10517, MSHR2154 and MSHR1364 [52], S. Typhimurium SL1344, and UPEC ST131 strains EC958 (wild type, WT), EC958ΔzntA [50], and CTF073 [53]. Non-pathogenic strains were: Burkholderia humptydooensis (MSMB043) [52], and E. coli strains K-12 MG1655, ATCC 25922 and CSGC 7139 [54].
Peptide synthesis and purification
Synthesis and purification of peptides were achieved using previously described methods [55]. Briefly, the peptides were synthesized using 9-fluroenylmethoxycarbonyl (Fmoc) solid-phase peptide synthesis on an automated peptide synthesizer (Symphony, Protein Technologies Inc, Tucson, USA). Rink amide resin was used for tachyplesin I, polyphemusin I, protegrin I, Sub3, while 2-chlorotrityl (2-CTC) resin was used for cyclic gomesin, BP100 and analogs. The peptides were oxidized in 20% (v/v) solvent B with dropwise addition of a concentrated solution of iodine in acetic acid (persistent yellow color) for 1 h. The reaction was quenched with ascorbic acid. Peptides were purified (> 95%) using reverse-phase high-performance liquid chromatography (Solvent A: H2O, 0.05% (v/v) trifluoracetic acid, solvent B: 90% (v/v) acetonitrile, 0. 05% (v/v) trifluoracetic acid). The correct peptide mass was confirmed using electrospray ionization mass spectrometry. The peptide concentration in aqueous solution was determined from the absorbance at 280 nm using the extinction coefficient based on the contribution of tyrosine and tryptophan resides, as well as disulfide bonds [56]. Hydrophobic moment was calculated from the amino acid sequence using the hmoment function of the R package [57].
Peptide structure analysis
Correct fold and overall structure of peptides was determined by comparing one-dimensional 1H spectra with previously reported peptide structures. Additional two-dimensional structural information was obtained for [I11A]tachyplesin I and [I11S]tachyplesin I. Peptides (1 mg/mL) were dissolved in H2O/D2O (10:1, v/v) and the pH was adjusted to pH 4–5. One dimensional 1H spectra, and two-dimensional total correlated spectroscopy (TOCSY) and nuclear Overhauser effect spectroscopy (NOESY) were acquired at 298 K with a Bruker Avance III HD 600 MHz NMR spectrometer. Spectra were referenced externally to 2,2-dimethyl-2-silapentone-5-sulfonate (DSS) at 0 ppm. After processing with TOPSPIN 3.6 (Bruker), spectra were assigned using CcpNmr software [58]. Secondary αH shifts were calculated as the difference between observed αH chemical shifts and those of the corresponding residues in random coil peptides [59].
Antimicrobial activity screen
The minimum inhibitory concentrations (MICs) of the peptides against Burkholderia, E. coli, UPEC and Salmonella strains was determined using a plate-based broth microdilution method [60]. Overnight bacterial cultures were subcultured until mid-log phase then diluted to OD600 = 0.01 in broth (Mueller Hinton Broth for Burkholderia strains, or lysogeny broth (LB) for E. coli, UPEC and S. Typhimurium strains). Peptides were serially diluted starting at 64 µM. Plates were incubated at 37 °C for 24 h and the MIC was determined by the lowest concentration of compound that inhibited visible bacterial growth. Data represent the MIC determined from three independent experiments for each bacterial strain.
Bone marrow-derived macrophage cytotoxicity
Bone marrow cells were harvested from the femurs and tibias of C57Bl/6 mice aged between 7 and 12 weeks old, as previously described [61]. All studies involving animals were approved by The University of Queensland animal ethics committee (AEC IMB/123/18). Bone marrow-derived macrophages (BMMs) were obtained by in vitro differentiation of the bone marrow cells in complete RPMI 1640 (media supplemented with 10% heat-inactivated fetal bovine serum, 2 mM l-glutamine, 50 U/ mL penicillin and 50 μg/mL streptomycin with recombinant human CSF-1 (10,000 U/mL, UQ Protein Expression Facility) [61]. BMMs were differentiated at 37 °C in a humidified incubator at 5% CO2 for 6 days prior to experimentation.
Day 6 BMMs were seeded at a density of 80,000 cells/well in a 96-well flat bottom cell culture plate. Cells were left overnight in complete RPMI to rest. On day 7, the media were removed, and cells were washed twice with phosphate buffered saline (PBS). Cell toxicity was determined following peptide treatment using a resazurin assay as before [62]. Serum-free and antibiotic-free media (RPMI supplemented with 2 mM l-glutamine and recombinant human CSF-1 (10,000 U/mL)) was added to the well with serial dilutions of compounds starting at 64 µM. Untreated cells and cell lysed by 0.1% (v/v) Triton X-100 were used as controls to establish 0% and 100% of cell death, respectively. The plate was returned to the incubator for 2 h, before addition of 0.01% (w/v) resazurin and return to the incubator for a further 22 h. Fluorescence emission intensity (excitation at 560 nm, emission at 585 nm) was then measured on a Tecan Infinite M1000 Pro. The percentage of cell death was calculated compared to 100% death induced by the Triton X-100 control. Data were collected from three independent experiments.
Red blood cell hemolysis assay
Red blood cells (RBCs) were isolated from healthy adult donors and washed three times in PBS (centrifuged at 500g for 1 min). Peptides were diluted in PBS and incubated with 0.25% (v/v) RBCs at 37 °C for 1 h. Samples with 0.1% (v/v) Triton X-100 were included as a 100% hemolysis control, and PBS was included as a 0% hemolysis control. Plates were centrifuged (500g for 5 min) to pellet the RBCs. The supernatant was transferred to a flat-bottomed 96-well plate, and the absorbance was read at 415 nm on a Tecan Infinite M1000 Pro to detect released hemoglobin. The percentage hemolysis was calculated based on the hemolysis of the 0.1% Triton X-100 sample [63]. Data were collected from a single experiment using RBCs from two individual donors.
Synthetic lipid vesicle preparation
The synthetic lipids 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), POPC/ 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) (4:1 molar ratio mixture) and POPC/ 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) (4:1 molar ratio mixture) were prepared as a lipid film by mixing in chloroform and then drying under both nitrogen gas and a vacuum desiccator overnight. The lipid film was resuspended at 1 mM in surface plasmon resonance (SPR) running buffer (10 mM HEPES, 150 mM NaCl, pH 7.4) and subjected to repeated freeze/ thaw cycles then extruded through membranes with 50 nm pores to form small unilamellar lipid vesicles as previously described [64].
Membrane extract vesicle preparation
To extract bacterial cell membranes, mid-log phase bacterial cultures were centrifuged at 2000g for 5 min. The supernatant was removed, and cells were washed in PBS. Bacterial cells were then centrifuged at 2000g for 5 min and the supernatant was removed. The membranes of RBCs and BMMs were extracted by similar methods. After washing with PBS and removing the supernatant, 110 µL of methanol and 390 µL of methyl tert-butyl ether (MTBE) with 0.01% butylated hydroxytoluene (BHT) was added to each sample [65]. Samples were vortexed to mix, followed by shaking incubation for 1 h. Next, 100 µL of 150 mM of ammonium acetate was added, and the samples were vortexed for 20 s before 5 min of centrifugation at 2000g. The top (MTBE) phase was removed and stored in glass vial, taking care not to disturb the aqueous phases. Cell membrane extracts were dried under nitrogen gas and a vacuum desiccator overnight. Vesicles were prepared using the same method of freeze/thaw and extrusion as described for the synthetic lipid vesicles.
Surface plasmon resonance
SPR (Biacore T200) was used to measure the affinity of peptides for lipid membranes composed of synthetic lipids or of cell membrane extracts. SUVs prepared as described above were deposited onto a Biacore L1 chip at a flow rate of 2 µL/min for 40 min, to reach a steady-state plateau and surface coverage with lipid bilayer. Serial dilutions of peptides (in SPR running buffer) were injected over the lipid bilayer at 5 µL/min for 180 s. The peptide–lipid disassociation was monitored for 600 s.
The response units (RUs) of the sensorgrams are proportional to the mass and to the amount of bound peptide, and dependent on the total amount of lipid deposited to cover the chip surface. Therefore, for a direct comparison of the sensorgrams obtained with the various peptides and lipid systems tested, the RUpeptide was divided with the molecular weight of the respective peptide, and the RUlipid obtained for the corresponding lipid bilayer deposition was divided by the average molecular weight of the lipid mixture, to obtain a peptide-to-lipid ratio (P/L = (RUpeptide/mwpeptide)/(RUlipid/avg mwlipid), as before [62, 66–68] (see Supplementary Table 1 for average lipid mass of cell membrane extracts).
Dose–response curves were plotted using the RUpeptide obtained when peptide–lipid binding is in equilibrium (i.e., at the end of the association phase) versus the concentration of peptide solution injected. Peptide-to-lipid ratio when the binding reaches saturation (P/Lmax) was determined by fitting P/L dose–response curves with saturation binding and variable Hill slope. The membrane partition coefficient (Kp), i.e., the ratio of molar fractions of peptide molecules in the lipid and aqueous phases when peptide–lipid association/dissociation reaches equilibrium, and σ (i.e., lipid-to-peptide ratio when the binding reaches saturation) were calculated for each peptide–lipid system using a steady-state model adapted to SPR and by fitting the membrane partition equation (see equation 1 in Supplementary Fig. 1) [69, 70]. To validate the use of these two approaches to fit the data sets, the fitted P/Lmax obtained with P/L dose–response curves was directly compared to the inverse of σ, fitted using the membrane partition equation (see Supplementary Table 2 and Supplementary Table 3).
The peptide–lipid dissociation constants (koff) were calculated by fitting the dissociation phase of sensorgrams obtained with peptide at 16 μM. Association constants (kon) were not calculated because kinetic models (designed for one-to-one interaction between analyte and ligand) provide a poor fit for peptide–lipid associations.
Bacterial cell permeability
SYTOX™ Green nucleic acid stain in combination with flow cytometry was used to investigate the integrity of bacterial outer and inner membranes [71]. Samples were prepared in a 96-well plate, performing serial dilutions from the determined MIC. Peptide vehicle treated (10% water) bacteria were used as the 0% permeability control and bacteria treated with 50% isopropanol were used as the 100% permeabilized control.
After 1 h of treatment, 200 µL of ice-cold PBS was added to each well. Samples were transferred to FACS tubes and kept at 4 °C while previous samples were run. 1.3 µM final concentration of SYTOX™ Green was added to each sample prior to loading into a CytoFLEX S Flow Cytometer (Beckman Coulter). 100,000 gated events were recorded for each condition. The percentage of permeabilized cells was calculated relative to the isopropanol control. Data were collected from three independent experiments.
Flow cytometry to measure peptide internalization
Peptides were labeled with Alexa Fluor® 488 (A488) 5-sulfodichlorophenol ester (SDP) (Thermo Fisher Scientific) using previously described methods [72]. Peptide (0.5–1 mg) was dissolved in dimethylformamide, A488 5-SDP (dissolved to 10 mM in dimethylsulfoxide (DMSO)) and N,N-diisopropylethylamine at a ratio of 78:20:2 (v/v/v). The peptide labeling reaction was performed at room temperature, protected from light for 2 h. A488-labeled peptide was purified using analytical scale HPLC. The correct peptide mass was confirmed using mass spectrometry as above. Stocks of labeled peptide were prepared, and their concentration quantified by measuring the absorbance at 495 nm (ε495 = 73,000 M–1 cm–1).
Fluorescently labeled peptide was used to determine the percentage of BMM cells with peptide internalized. BMMs were seeded and incubated as per the cytotoxicity assay. Media was removed and replaced with 90 µL serum free media and 10 µL of labeled peptide stock. Cells were incubated at 37 °C and 5% CO2 for 1 h. Cells were lifted from the plate using ice-cold PBS, transferred to tubes, and analyzed using a CytoFLEX S Flow Cytometer. Trypan blue (0.02% w/v final) was added to each sample to quench the fluorescence of labeled peptide accessible to aqueous environment (e.g., bound onto the surface of cells, but not internalized) before the recording was repeated [67]. The percentage of fluorescent cells with fluorescence above a background threshold for cells without peptide was determined from 10,000 gated events. Data were collected from three independent experiments.
Live cell imaging to examine internalization of tachyplesin I into BMMs
Day 7 BMMs were seeded at 80,000 cells per well in a Nunc™ Lab-Tek™ Chambered coverglass and incubated overnight at 37 °C and 5% CO2. Before the assay, cells were washed with PBS, then incubated with serum free RPMI media containing 2 µM tachyplesin I labeled with A488 and 100 µg/mL dextran-tetramethylrhodamine (TMRE) (Thermo Fisher Scientific) for 15 min. The media were removed and replaced with FluoroBrite DMEM media (Thermo Fisher Scientific). Cells were incubated at 37 °C and 5% CO2 during imaging. Localization of A488-labeled tachyplesin I inside BMMs was examined using a Zeiss LSM 880 inverted confocal microscope using a 63 × 1.4 NA C-Plan Apochromat oil immersion objective and acquired with Zen Black imaging software with a pinhole of 1 AU set for all channels and a pixel dwell time of 0.25 µs. A488-labeled peptide was excited with the 488 nm line of an Argon Ion laser and the fluorescence emission detected with the internal GaAsP detector gated to 492–556 nm, whereas dextran-TMRE was excited with the 561 nm line of a solid-state laser and fluorescence emission detected with the internal GaAsP detector gated to 566–685 nm with a gain voltage of 500. Images were post processed using Fiji software [73].
Microscopy to determine location of peptides inside infected BMMs
Samples for microscopy were prepared using day 7 BMMs seeded at 80,000 cells per well in a Nunc™ Lab-Tek™ Chambered coverglass. Bacterial infection assays were performed as previously described [26]. Briefly, EC958 was cultured overnight at 37 °C in LB under static growth conditions to enrich for type 1 fimbriae expression, which was confirmed by yeast-agglutination as previously described [74]. Cells were regrown for 4 h under the same conditions and then used to infect BMMs (MOI 100:1) for 1 h. Media was removed and replaced with fresh media containing 200 µg/mL gentamicin for an additional 1 h. The media were then removed and replaced with media containing 20 µg/mL gentamicin and 8 µM of fluorescently A488-labeled [I11A]tachyplesin I or [I11S]tachyplesin I. BMMs were treated with peptide for 1 h before being washed three times with PBS. Cells were fixed used 4% paraformaldehyde (15 min at room temperature). The fixative was removed and replaced with PBS. Cells were stained with Wheat Germ Agglutinin (WGA)-TMRE (Thermo Fisher Scientific) and 4′,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific).
Multichannel confocal images were acquired using a Zeiss LSM 880 inverted confocal microscope using a 63 × 1.4NA C-Plan Apochromat oil immersion objective and acquired with Zen Black imaging software with a pinhole of 1 AU set for all channels and a pixel dwell time of 0.5 µs. Peptide (A488-labeled) was excited with the 488 nm line of an Argon Ion laser and its fluorescence emission detected with the internal GaAsP detector gated to 492–623 nm, while WGA-TMRE and DAPI were co-excited with the 405 nm and 561 nm lines of individual lasers and detected with the internal GaAsP detectors gated to 410–483 nm, and 566–685 nm with voltage gains of 650 and 713, respectively. Images were acquired as Z-stacks covering the full height of the cell at intervals of 250 nm and are displayed as maximum intensity projections.
Co-treatment of planktonic EC958 with peptides and metal ions
A checkerboard strategy was employed to examine the effect of peptide co-treatment with metal ions on the growth of planktonic bacteria. Peptides were serially diluted with zinc sulfate, copper (II) sulfate or iron (II) sulfate on a 96-well plate. Starting concentrations of peptides against each bacterial strain was determined from the antimicrobial peptide screen. The broth microdilution assay was performed as described above. After the addition of bacteria, the plate was placed in a POLARstar Omega (BMG LABTECH) at 37 °C. Absorbance was read at 600 nm every 30 min for 12 h, with orbital shaking performed before the readings. Data points were collected from three independent experiments for each bacterial strain.
Peptide treatment of bone marrow-derived macrophages infected with EC958
Day 7 BMMs were infected with static cultures of type I fimbriae enriched EC958 WT or EC958ΔzntA bacteria (enriched as described for EC958 WT, with MOI 100:1) for 1 h. Media (RPMI supplemented with 10% FCS, 2 mM Glutamax and colony stimulating factor 1 (CSF-1) was replaced with fresh media containing 200 µg/mL gentamicin for an additional 1 h. The media were then removed and replaced with media containing 20 µg/mL gentamicin and tachyplesin I, [I11A]tachyplesin I or [I11S]tachyplesin I. At 6 h post-infection (4 h post peptide treatment) samples of each condition were lysed using 0.1% (v/v) Triton X-100 in PBS. Dilutions of the lysates were plated on agar (LB agar for EC958 WT cells and LB agar with 30 mg/chloramphenicol for EC958ΔzntA). This step was repeated 12 h post-infection. Agar plates were placed in a 37 °C incubator overnight (~ 18 h). CFU were determined by enumerating the number of colonies per condition. Data were collected from three independent experiments.
For each experiment, a lactate dehydrogenase activity assay (LDH) (using the CytoTox 96® Non-Radioactive Cytotoxicity Assay LDH Kit (Promega)) was performed in parallel to determine host cell toxicity during the infection. BMMs were infected and treated in the same manner as the infection plates. Controls for 100% lysis were included by adding 0.1% (v/v) Triton X-100 into control wells and incubating for 10 min before removing the supernatant sample. At 6 and 12 h post-infection, 25 µL of the supernatant from each condition was removed and transferred to a new plate. The LDH assay was then performed as per the manufacturer’s instructions. Absorbance at 480 nm was read on a Tecan infinite M1000Pro and the percentage cell death was calculated using the ratio of media subtracted conditions divided by subtracted absorbance of 100% lysis controls.
Results and discussion
Antimicrobial peptides with cell-penetrating properties
The panel of peptides in this study (see Fig. 2), herein referred to as antimicrobial CPPs, predominately comprises antimicrobial peptides with cell-penetrating properties and reported membrane-active mechanisms of action [62, 71, 75–77]. It includes peptides representing the two major secondary structures (i.e., α-helix and β-hairpin), with differences in amino acid sequence, amount of positive charge, and distinct amphiphilicity (hydrophobic moment). These properties are likely to impact antimicrobial activity [78]. Two analogs were prepared for tachyplesin I by modifying an Ile residue (position 11) to either Ala or Ser to produce [I11A]tachyplesin I and [I11S]tachyplesin I, respectively. Tachyplesin I analogs with substitutions at this position ([I11A]TI [76], [Y8S,I11S]cTI [72]) have been previously shown to improve selectivity for bacterial membranes compared to mammalian cells. However, the impact of single substitution at position 11 on cell-penetration has not yet been investigated.
Fig. 2.

Characteristics of the peptide panel used for antimicrobial susceptibility screening. a Cys residues and disulfide bonds are shown in red. Backbone cyclization is illustrated by a dashed black line. * Indicates C-terminal amide. Sequence reference: tachyplesin I and [I11A]tachyplesin I [76], polyphemusin I and protegrin I [75], cyclic gomesin [71], BP100 and analogs [62], Sub3 [77]. b Peptide mass (Da) is the average theoretical mass calculated from the amino acid sequence. c Peptide overall charge was calculated from contributions of charged amino acid side chains. d Hydrophobic moment was calculated from the amino acid sequence using the hmoment function of the R package [57]
Comparison of the overall secondary structure of tachyplesin I and analogs
Differences in peptide structure can affect peptide–membrane interactions and activity; therefore, structural studies were undertaken to investigate whether the Ile to Ser substitution affected the overall structure of [I11S]tachyplesin I compared to the native tachyplesin I. The secondary αH chemical shift for tachyplesin I [42, 76] and [I11A]tachyplesin I [76] were previously described, and are compared here to the novel secondary αH chemical shift for [I11S]tachyplesin I (Fig. 3A). All three peptides show a defined β-hairpin structure, with a high conservation of αH chemical shifts. [I11A]tachyplesin I and [I11S]tachyplesin I show similar deviation at position 11. In the native tachyplesin I peptide, the side chain of I11 is positioned at the opposite end of the peptide to the hydrophobic residue W2 that inserts into lipid bilayers [42] (Fig. 3B) and is solvent exposed in the lowest energy structures (Fig. 3C). Ala has a shorter hydrophobic side chain than Ile, and Ser has a polar side chain with a hydroxyl group, which decreases local hydrophobicity and can establish H-bonds with phospholipid headgroups. Thus, while the overall structure of the peptide is conserved, the different characteristics of the amino acid side chain at position 11 (Ala or Ser) may affect peptide binding and insertion into biological membranes, possibly enhancing antibacterial activity.
Fig. 3.
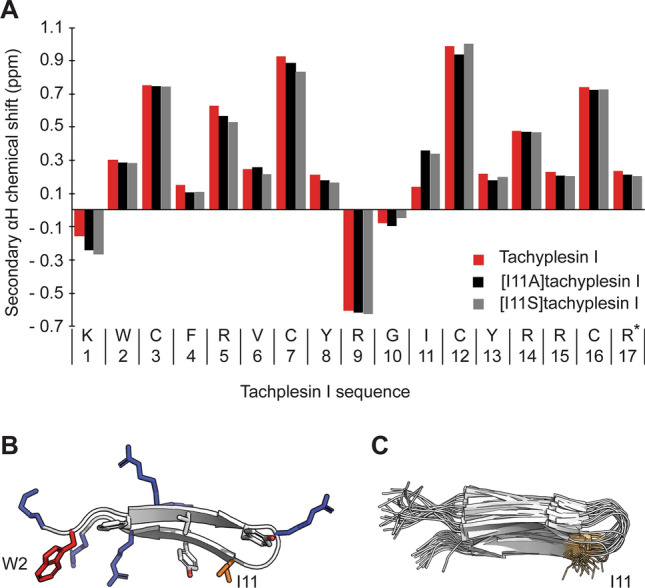
Structural characteristics of tachyplesin I and analogs. A Secondary αH chemical shift at 298 K determined from 1H NMR spectra. The secondary αH shifts of tachyplesin I (red), [I11A]tachyplesin I (black) and [I11S]tachyplesin I (gray) were calculated as the difference between observed αH chemical shifts and those of the corresponding residues in random coil peptides [72]. B Cartoon representation of tachyplesin I (PDB ID: 2RTV) showing amphipathic arrangement of positively charged (blue) and hydrophobic residues (orange). W2 and I11 are situated at opposite ends of the peptide. C Overlay of the 20 lowest energy forms of tachyplesin I (PDB ID: 2RTV) showing the consistent solvent exposed positioning of the side chain of I11
Peptide susceptibility screen against planktonic bacteria
The peptides were tested for activity against representative pathogenic and non-pathogenic bacteria. The minimum inhibitory concentration (MIC) was determined for planktonic bacteria (Table 1) and used to identify the most potent candidates for progression in this study. The pathogenic bacteria—B. pseudomallei, UPEC strain EC958, and S. Typhimurium (Table 1)—share the ability to survive and replicate within host cells, making them the most suitable bacteria for this study. CFT073 was included to account for UPEC strain differences in virulence and innate immune responses [79], particularly with respect to interactions with macrophages [80, 81]. The non-pathogenic bacteria—Burkholderia humptydooensis and E. coli strains—(Table 1) were included as control strains to illustrate the broader context of antimicrobial susceptibility and provide information on the specificity and mechanism of action. Notably, we have previously demonstrated that the non-pathogenic species B. humptydooensis recapitulates the antimicrobial susceptibility of B. pseudomallei, and can be used as a lower-risk group model organism in planktonic studies [52].
Table 1.
MIC of peptides against pathogenic and non-pathogenic bacteria strains, compared to toxicity in host mammalian cells
| Peptide | Antimicrobial activity: MIC (µM)a | Mammalian cell toxicity CC50 (µM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Burkholderia pseudomalleib | Burkholderia humptydooensis | Uropathogenic E. coli | Non-pathogenic E. coli | Salmonella enterica Typhimurium | ||||||
| MSMB 043 |
EC958 | CFT073 | K-12 MG1655 |
ATCC 25,922 |
CSGC 7139 |
SL1344 | BMMc | RBCd | ||
| β-hairpin | ||||||||||
| Tachyplesin I | > 64 | 32 | 0.25–0.5 | 0.25–0.5 | 0.125–0.25 | 0.125–0.5 | 0.5 | 0.25–0.5 | 11.1 ± 1.3 | 42.1 ± 5.9 |
| [I11A]tachyplesin Ie | nd | nd | 0.5–2 | nd | 0.5–2 | nd | nd | nd | 26.7 ± 1.5 | > > 128 |
| [I11S]tachyplesin Ie | nd | nd | 0.5–2 | nd | 0.5–2 | nd | nd | nd | 29.3 ± 1.4 | > > 128 |
| Polyphemusin I | > 64 | 32 – 64 | 1–2 | 0.5–2 | 0.5–1 | 4 | 0.5–1 | 1–4 | 12.6 ± 2.3 | 52.3 ± 7.0 |
| Protegrin-I | > 64 | 64 | 4–16 | 16–64 | 2–4 | 4–8 | 2–4 | 8–64 | nd | nd |
| Cyclic gomesin | nd | > 64 | 2–16 | 4–8 | 8 | 2 | 1–2 | 32 | ||
| α-helix | ||||||||||
| BP100 | nd | > 64 | 16–32 | 32–64 | 16–64 | 1–2 | 0.5–1 | 8–16 | ||
| R-BP100 | nd | > 64 | 16–32 | 16–32 | 16–64 | 2–4 | 1–2 | 8–16 | ||
| RW-BP100 | nd | > 64 | 32–64 | 32–64 | 32 to > 64 | 2–16 | 1–4 | 8–32 | ||
| Sub3 | nd | > 64 | 16–32 | 32–64 | 8–16 | 2–4 | 0.5–1 | 32–64 | ||
aMICs were determined using broth microdilution of planktonic bacteria in growth phase. Data represent the MIC range determined from a minimum of three independent experiments. 64 µM was the highest concentration tested. nd indicates where an MIC was not determined
bMIC was determined for B. pseudomallei strains MSHR1364, MSHR2154 and MSHR10517
cBMMs were treated with serial dilutions of tachyplesin I, polyphemusin I, [I11A]tachyplesin I or [I11S]tachyplesin I at 37 °C for 24 h and viability was determined using resazurin (0.01% w/v). Triton X-100 (0.1% v/v) was included to establish 100% cell death. The cytotoxic concentration required to kill 50% of the cells (CC50) was determined from dose–response curves, where the nonlinear regression was fit using saturation binding with Hill slope (GraphPad Prism 8). Data represent a minimum of three independent experiments
dRBCs were treated with serial dilutions of tachyplesin I, polyphemusin I, [I11A]tachyplesin I or [I11S]tachyplesin I at 37 °C for 1 h. Triton X-100 (0.1% v/v) was included to establish 100% hemolysis. Hemolysis was determined by monitoring hemoglobin in the supernatant. The concentration required to lyse 50% of the cells (CC50) was determined from the dose–response curves, where the nonlinear regression was fit using [inhibitor] vs response with a variable slope (GraphPad Prism 8). Data represent a single experiment with RBCs isolated from two healthy donors. 128 µM was the highest concentration tested
e[I11A] and [I11S]tachyplesin I were designed as non-toxic analogs for targeting intracellular bacteria. Therefore, antimicrobial activity was determined for EC958 and control non-pathogenic MG1655 strains
Tachyplesin I and polyphemusin I were the two most active peptides across the entire bacteria panel (Table 1). Antimicrobial activity of [I11A]tachyplesin I [76], and [I11S]tachyplesin I (new analog) was tested against EC958 and non-pathogenic MG1655. Both analogs retained low micromolar potency toward these bacterial strains.
Overall, the β-hairpin peptides (tachyplesin I and analogs, polyphemusin I, protegrin I, and cyclic gomesin) were more active against the tested bacterial strains than the α-helical peptides (BP100 and analogs, and Sub3), suggesting that the β-hairpin structure contributes to more potent antimicrobial activity; especially toward pathogenic UPEC and S. Typhimurium strains. Comparing the β-hairpin peptides, there appeared to be a relationship between a higher hydrophobic moment value (a measure of amphiphilicity, where a large hydrophobic moment indicates a structure that is predominantly hydrophobic on one side and predominantly hydrophilic on the other) [82] and a lower MIC. For example, polyphemusin I, and tachyplesin I and analogs (hydrophobic moment range 0.73–0.76) were consistently more active than protegrin I and cyclic gomesin (hydrophobic moments of 0.60 and 0.44). All tested peptides had an overall positive charge (+ 5 to + 7) and no distinction in antimicrobial activity was correlated with difference in charge within this range.
Host cell toxicity
When developing new therapies to target bacteria within intracellular niches, it is important to consider toxicity of the active antimicrobials against host cells. To this end, we determined the toxicity of the most potent antimicrobial peptides from the planktonic activity assays (Table 1); tachyplesin I, polyphemusin I, [I11A]tachyplesin I and [I11S]tachyplesin I, toward human RBCs and murine BMMs.
The two tachyplesin I analogs, [I11A] and [I11S], were the least toxic peptides, with CC50 (peptide concentration required to kill 50% of BMMs) values approximately twofold higher than tachyplesin I and polyphemusin I. Notably, these peptide concentrations are 10- to 100-fold higher than the concentrations required to inhibit growth of 100% of E. coli and S. Typhimurium strains (see Table 1). For RBC toxicity, we confirmed previously reported concentrations of tachyplesin I [72, 76, 83] and polyphemusin I [83, 84] required to induce hemolysis, and reduced toxicity of [I11A]tachyplesin I [76] (see Table 1). Incubation with 128 µM [I11S]tachyplesin I and [I11A]tachyplesin I resulted in < 50% lysis of RBCs; therefore, the tachyplesin I analogs were at least threefold less hemolytic than the native tachyplesin I (42.1 ± 5.9 µM) and polyphemusin I (52.2 ± 7.0 µM). These differences in effective concentration underline the greater therapeutic possibilities of [I11A]tachyplesin I and [I11S]tachyplesin I during bacterial infection as they are better tolerated by mammalian cells.
Peptide–lipid binding saturation and membrane partition coefficient
Peptide–lipid membrane binding studies were performed using SPR to determine whether differences in selective antimicrobial activity of tachyplesin I, polyphemusin I and the two tachyplesin I analogs relate to differences in their ability to interact with the phospholipid bilayer in cell membranes. Lipid model membranes composed of synthetic zwitterionic phospholipid POPC were used to represent the neutral outer leaflet in the cell membrane of healthy eukaryotic cells. Phospholipids containing phosphatidylethanolamine (PE)- or phosphatidylglycerol (PG)-headgroups are the major lipid components of bacterial membranes [44]; therefore, model membranes composed of POPC/POPE (4:1 molar ratio) and POPC/POPG (4:1 molar ratio) were also investigated.
All four peptides reached the highest binding saturation with POPC/POPG (4:1) bilayers, followed by POPC, then POPC/POPE (4:1) (Fig. 4), as suggested by the peptide/lipid (P/L, mol/mol) dose–response curves (Fig. 4A) and the fitted peptide-to-lipid ratio for binding at saturation (P/Lmax) (Fig. 4B, fitted from the P/L curves;). A similar trend was observed for membrane partition coefficients (Kp) (Fig. 4B, from fitted curves and equation in Supplementary Fig. 1) [69], suggesting that binding and partition into lipid bilayers containing PG-phospholipids may drive interaction of these peptides with bacterial membranes.
Fig. 4.

Peptide–lipid binding of tachyplesin I, polyphemusin I, [I11A]tachyplesin I and [I11S]tachyplesin I toward synthetic lipid model membranes. Comparison of the binding affinity of each peptide for model membranes composed of POPC, POPC/POPE (4:1) and POPC/POPG (4:1). SPR sensorgrams were obtained for peptides injected over lipid bilayers deposited on an L1 chip for 180 s, with dissociation monitored for 600 s. The response units at the end of the association phase were converted to peptide-to-lipid molar ratios (P/L (mol/mol)). A Dose–response curves allow comparison of the amount of peptide bound to different lipid mixtures. B The peptide-to-lipid ratio when the binding reaches saturation (P/Lmax) was determined by fitting P/L dose–response curves (saturation binding with Hill slope, GraphPad Prism 8). Membrane partition coefficient, Kp, determined by fitting the dose–response data using a partition formalism (see equation and fitted curves in Supplementary Fig. 1), is the ratio of molar fractions of peptide in the lipid and aqueous phases [69]. C Representative sensorgrams for 16 µM peptide show peptide–lipid association (50–230 s) and dissociation (230–830 s)
Kp values (Fig. 4B) show that tachyplesin I and polyphemusin I had similar membrane partition to each other, for the three model membranes, and had approximately twofold higher partition than [I11A]tachyplesin I and [I11S]tachyplesin I for POPC/POPG and POPC. This is further demonstrated with the peptide–membrane binding association/dissociation, as shown by the representative sensorgrams for 16 µM peptides in Fig. 4C. All peptides showed a fast initial association to tested lipid bilayers, but with differences in the amount bound at equilibrium (see P/L signal obtained in the sensorgram at the end of the association phase (t = 230 s in Fig. 4C). The lower amount of bound [I11A]tachyplesin I and [I11S]tachyplesin I to POPC/POPG (4:1) bilayers at equilibrium is consistent with the lower Kp, compared to tachyplesin I and polyphemusin I. Dissociation of the peptides from the lipid bilayers occurred at a similar rate with near identical dissociation constants determined from the 16 μM sensorgrams (0.005–0.009 s−1 range).
The peptide–lipid binding affinity of tachyplesin I, polyphemusin I, [I11A]tachyplesin I and [I11S]tachyplesin I was further examined with lipid bilayers prepared using lipid extracts from pathogenic and non-pathogenic bacteria, and from mammalian cells. The average lipid molar mass was determined from reported lipid profiles for each of the cell types (see Supplementary Table 1) and used to determine P/L at the end of peptide association, as above. Dose–response curves (P/L), fitted P/Lmax and Kp values (from curves and equation in Supplementary Fig. 2), and representative SPR sensorgrams for 16 μM peptides are shown for EC958 (pathogenic bacteria), MG1655 (non-pathogenic bacteria) and BMMs (mammalian cells) in Fig. 5. Additional peptide–lipid binding curves with fitted P/Lmax and Kp values are shown for S. Typhimurium, B. humptydooensis and RBC membranes in Supplementary Fig. 3.
Fig. 5.

Peptide–lipid binding of tachyplesin I, polyphemusin I, [I11A]tachyplesin I and [I11S]tachyplesin I toward membrane extracts from bacterial and mammalian cells. Membranes extracts were deposited on an L1 chip. SPR sensorgrams were obtained for peptides injected over lipid bilayers for 180 s, with dissociation monitored for 600 s. The response units at the end of the association phase were converted to peptide-to-lipid ratios (P/L (mol/mol)). A Dose–response curves allow comparison of peptide binding to EC958, MG1655 and BMMs membrane extracts. Peptide binding to S. Typhimurium (S. Ty), B. humptydooensis (B. hu) and RBC membranes are shown in Supplementary Fig. 3. B Peptide-to-lipid ratio when the binding reaches saturation (P/Lmax) was determined by fitting P/L dose–response curves (saturation binding with Hill slope, GraphPad Prism 8). Membrane partition coefficient, Kp, determined by fitting the dose–response curves using a partition formalism (see fitted curves in Supplementary Fig. 2). C Representative sensorgrams for 16 µM peptide show peptide–lipid association and dissociation
Each of the tested peptides has higher partition into bilayers (Kp) prepared using membrane lipids from bacterial cells and reach binding saturation at higher peptide-to-lipid ratio (P/Lmax), than those prepared with lipids from mammalian cell lipids (BMMs or RBCs). Within the bacterial strains tested, all four peptides had greater membrane partition and reach binding saturation at higher peptide-to-lipid ratio for bilayers composed of EC958 WT and MG1655 lipids, compared to bilayers with S. Typhimuirum and B. humptydooensis lipids.
In general, the peptides showed higher binding affinity toward lipid bilayers with higher amounts of anionic phospholipids (including those with PG headgroups), as shown with bilayers composed with bacterial versus mammalian cell lipids, and with relative differences in the cytotoxic activity of peptides to corresponding cell types. Greater variability was observed between different peptides across the tested lipid bilayers composed with cell membrane extracts, than was observed for the model membranes composed with synthetic lipids, which is consistent with the higher degree of complexity present for the cell membrane extracts. However, the trend confirms that binding to anionic PG headgroups is likely to play an important role in antimicrobial activity of the peptides.
Membrane partition coefficient (Kp), but not the peptide-to-lipid ratio when binding reaches saturation (P/Lmax), were consistently higher for tachyplesin I and polyphemusin I, compared to [I11A]tachyplesin I and [I11S]tachyplesin, for all tested model membranes (see Fig. 4B). Differences in membrane partition coefficients between parent and analog peptides were less pronounced for the membrane extracts (Fig. 5B). Therefore, differences in antimicrobial activity observed between peptides for some bacterial strains, and the two-fold difference in host cell toxicity between tachyplesin I and polyphemusin I, and the two tachyplesin I analogs (see Table 1) were not directly explained by membrane partition coefficient, or by the peptide-to-lipid ratio when binding reaches saturation. However, the reduction in toxicity of [I11A]tachyplesin I and [I11S]tachyplesin I toward BMMs and RBCs might be explained by the smaller size of Ala, and increased polarity of Ser side chains compared to Ile at position 11, (opposite end of the peptide compared to membrane-inserting Trp at position 2) that reduce hydrophobic interactions and insertion into mammalian cell membranes.
Membrane permeabilization
To determine whether the antimicrobial activity of the peptides involves bacterial membrane permeabilization, bacterial cells were treated with tachyplesin I, polyphemusin I, [I11A]tachyplesin I and [I11S]tachyplesin I, then exposed to SYTOX™ Green nucleic acid stain. SYTOX™ Green is impermeant to intact membranes; however, it readily penetrates cells with compromised membranes and binds to nucleic acids, causing > 500-fold fluorescence enhancement. The percentage of bacteria cells that were permeabilized after treatment with peptide was determined by monitoring the fluorescence of SYTOX™ Green using flow cytometry. Dose–response curves prepared using serial dilutions, starting from the MIC of peptide for each strain (Table 1), are shown in Fig. 6. Isopropanol was used as a control and shown to permeabilize 100% of bacterial cells.
Fig. 6.

Comparison of peptide concentrations required to permeabilize bacterial cells. Bacterial cells were treated with serial dilutions of tachyplesin I, polyphemusin I, [I11A]tachyplesin I or [I11S]tachyplesin I and incubated in a shaking incubator for 1 h at 37 °C. SYTOX® Green added to each sample only enters cells with damaged membranes to bind DNA. Fluorescent cells were detected using flow cytometry, excitation = 488 nm and emission 530/30 nm. 100,000 gated events were recorded for each condition. Dose–response curves show the percentage of permeabilized cells, normalized using a control sample with cells treated with isopropanol (100% of permeabilized cells) and plotted using GraphPad Prism 8. Data represent the mean ± SEM from a minimum of three independent experiments. The red arrows on S. Typhimurium indicate the MIC
EC958 and MG1655 cells were completely permeabilized by treatment with tachyplesin I, polyphemusin I, [I11A]tachyplesin I and [I11S]tachyplesin I at concentrations equal to the MICs (the highest concentrations tested for each peptide in this assay, Fig. 6). Concentrations below the MICs were found to cause permeabilization of a significant percentage of bacterial cells. For example, [I11A]tachyplesin I, which inhibits 100% of EC958 growth at 0.5–2 µM, permeabilizes approximately 100% of cells at 0.25 µM. [I11A]tachyplesin I permeabilized E. coli with the highest efficiency, while the lowest efficiency was observed for polyphemusin I. However, it is important to note that recorded cell populations only included intact cells.
In contrast to the high degree of permeabilization observed for E. coli, only ~ 80% of S. Typhimurium cells were permeabilized following treatment with 1–2 µM polyphemusin I (equivalent to the MIC), and concentrations greater than the MIC for tachyplesin I. B. humptydooensis membranes appeared to remain intact, despite treatment with up to 64 µM tachyplesin I or polyphemusin I (Fig. 6).
Peptide internalization of host cells
For peptides to be effective at targeting intracellular bacteria, they must be able to penetrate host cell membranes at concentrations that are toxic to the bacteria (e.g., EC958 0.5–2 μM, see Table 1), but are not toxic to host cells. The ability of tachyplesin I, polyphemusin I, [I11A]tachyplesin I and [I11S]tachyplesin I to internalize BMMs was examined using flow cytometry with peptides labeled with Alexa Fluor® 488, from a starting concentration of 1 µM. The percentage of fluorescent BMMs was determined in the absence and presence of trypan blue (TB, Fig. 7A), a membrane impermeable quencher of fluorescence. The identical fluorescence percentage in the absence and presence of trypan blue indicates that all the cells have their cell membrane intact, and all cells have internalized fluorescent peptide (and, therefore, not accessible to the quencher). All four peptides internalized into ~ 100% BMMs at 1 μM (Fig. 7A). The [I11A] and [I11S]tachyplesin I analogs internalized more efficiently into BMMs, with > tenfold lower concentrations required to internalize 50% of cells compared to tachyplesin I (Fig. 7A).
Fig. 7.
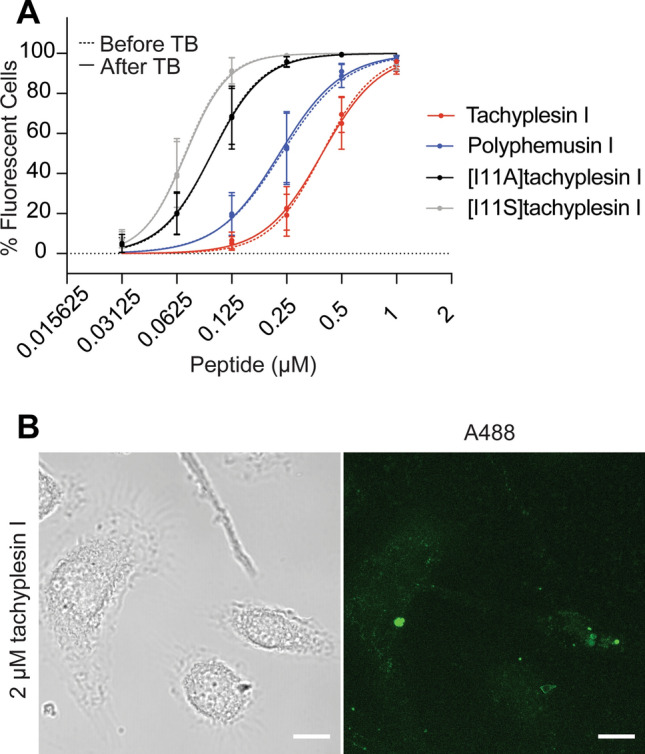
Internalization of fluorescently labeled peptides into BMMs. A Tachyplesin I, polyphemusin I, [I11A]tachyplesin I and [I11S]tachyplesin I were labeled with Alexa Fluor® 488 and incubated with BMMs for 1 h. Mean fluorescence emission intensity and the population of fluorescent cells were detected using flow cytometry with excitation at 488 nm and emission at 530/30 nm. Trypan blue (TB) was added to quench the fluorescence of solvent accessible peptide (e.g., bound to the cell surface). The fluorescence emission intensity of 10,000 gated events were screened per peptide and the percentage of fluorescent cells were plotted, compared to unstained cells. Data represent the mean ± SEM from a minimum of three independent experiments. B BMM cells were plated onto micro Nunc™ Lab-Tek™ Chambered coverglass and treated with 2 µM tachyplesin I labeled with Alexa Fluor® 488 (green) and 100 µg/mL dextran-TMRE for 15 min. The medium was removed and replaced with FluoroBrite DMEM media. Cells were incubated at 37 °C and 5% CO2 during imaging. Confocal images were acquired using a Zeiss LSM 880 and are displayed here as brightfield and a maximum intensity projection. Images were processed and cropped in Fiji. Scale bar represents 10 µm
Internalization of tachyplesin I was verified using confocal fluorescence microscopy of live BMMs (Fig. 7B). After treatment with 2 µM A488-tachyplesin I for 15 min, simultaneous uptake of peptide and dextran-TMRE was observed (see Supplementary Movie 1). The presence of fluorescent peptide and dextran-TMRE in discrete vesicles suggests that the predominant mechanism of cell entry under these conditions was via endocytosis. The more disperse fluorescence observed in some cells suggests that tachyplesin I can access the cytosol of BMMs within minutes. It is still not clear whether the cytosolic localization is a result of direct entry of peptide through the plasma membrane of BMMs, or whether the peptide within the vesicles is able to escape into the cytosol. It is possible that the observed cytosolic tachyplesin I is a result of both mechanisms of cell entry, similar to observations for other CPPs including the β-hairpin peptide cyclic gomesin [70] and cyclotide kalata B1 [67].
Localization of tachyplesin I analogs inside BMMs infected with EC958
[I11A]tachyplesin I and [I11S]tachyplesin I, the two peptides that showed higher internalization efficiency (Fig. 7A), were used to investigate if they could target intracellular bacteria. BMMs were infected with EC958 and treated for 1 h with peptides at the highest concentration that was non-toxic to BMMs (8 μM). EC958 were observed within intracellular vesicle compartments (Fig. 8, DAPI and TMRE panels) in agreement with our previous findings [26]. Treatment of infected BMMs with 8 µM labeled [I11A]tachyplesin I for 1 h resulted in a punctate appearance of fluorescence inside cells, suggesting that like tachyplesin I (Fig. 7B), [I11A]tachyplesin I locates mainly within vesicles (Fig. 8, A488 panel and merge inset). In contrast, treatment of infected BMMs with 8 µM labeled [I11S]tachyplesin I for 1 h produced diffuse fluorescence throughout the BMM cytosol and strong co-localization with intracellular EC958 bacteria (Fig. 8).
Fig. 8.

Intracellular localization of tachyplesin analogs in BMM cells infected with EC958. [I11A]tachyplesin and [I11S]tachyplesin were labeled with Alexa Fluor®488 (A488) (green). BMM cells were plated onto micro Nunc™ Lab-Tek™ Chambered coverglass and infected with EC958 (multiplicity of infection 100:1) for 2 h. Infected cells (media aspirated after treatment with 200 µg/mL gentamicin) were treated with 8 µM peptides for 1 h. The media were removed, and cells were fixed with 4% paraformaldehyde for 15 min. The fixative was removed and replaced with PBS and cells were stained with WGA-TMRE (staining the cell membranes, red) and DAPI (staining the DNA, blue). Untreated (no peptide) infected cells were included as a control. Confocal images were acquired using a Zeiss LSM 880 and are displayed here as maximum intensity projections. For clarity, the DAPI signal has been inverted. Images were processed and cropped in Fiji. Scale bar represents 10 µm
Investigating peptide ability to enhance zinc toxicity
Macrophages employ a variety of antimicrobial responses against pathogens. One mechanism relies on the modulation of metal ion concentrations, inducing starvation or direct toxicity, to eradicate bacteria during infection [50, 85–87]. In particular, zinc is essential for maintaining structure and catalytic activity of bacterial enzymes [88] and plays a role in signal transduction [89]. However, excessive zinc is cytotoxic, and immune cells can mobilize and deliver toxic concentrations of zinc to intracellular bacterial pathogens [48, 90].
Genome-wide analysis of EC958, employing a transposon directed insertion site (TraDIS) approach, identified several genes critical for survival in the presence of high zinc concentrations. Notably, this list included the bacterial zinc exporter ZntA, that counteracts intracellular zinc accumulation, along with other proteins required for maintaining cell membrane integrity [50]. Thus, due to the ability of tachyplesin I and polyphemusin I to bind and perturb bacterial membrane integrity, we hypothesized that co-treatment with zinc and peptides might have an additive antimicrobial effect, and that an EC958 strain lacking the zinc exporter ZntA (EC958ΔzntA) may be more sensitive than the wild type strain (EC958 WT) to zinc stress in presence of the peptides.
To test this hypothesis, a co-treatment analysis was performed with sublethal concentrations (below the MIC, see Table 1) of tachyplesin I, polyphemusin I, [I11A]tachyplesin I and [I11S]tachyplesin I and 0.1 mM zinc, which does not significantly inhibit EC958 WT or EC958ΔzntA growth (Fig. 9 and Supplementary Fig. 4). Except for a slight delay in onset of bacterial growth with polyphemusin I, treatment with a sublethal dose of peptide alone did not alter bacterial growth. Conversely, growth inhibition was observed after 6 h of co-treatment with 0.1 mM zinc and polyphemusin I, [I11A]tachyplesin I, or [I11S]tachyplesin I, which was more pronounced for EC958ΔzntA compared to EC958 WT (Fig. 9, 300 min). Despite the observed growth inhibition, co-treated EC958 WT returned to within 80–90% of the bacterial density that was observed for untreated bacteria after 12 h.
Fig. 9.

Treatment of EC958 WT and EC958ΔzntA with sublethal concentrations of peptides enhances zinc toxicity. Sublethal concentrations of tachyplesin I, polyphemusin I, [I11A]tachyplesin I or [I11S]tachyplesin I were determined from MIC data (Table 1). EC958 WT (A) and EC958ΔzntA (B) were treated with peptides in the presence and absence of 0.1 mM zinc. Samples were incubated at 37 °C inside a PolarStar Omega plate reader and optical density was monitored by measuring absorbance at 600 nm every 30 min for 12 h. Controls included bacteria treated with 0.1 mM zinc, 1 mg/mL gentamicin (for 100% growth inhibition) or untreated. Data represent the mean ± SEM from a minimum of three independent experiments
To investigate whether the observed additive effects were specific to zinc, EC958 WT was treated with 0.25 µM polyphemusin I in the presence of copper or iron. Copper accumulation within the phagolysosome is thought to contribute to bacterial killing by mechanisms such as inhibition of bacterial metabolic processes, and by causing damage to proteins, lipids and DNA [85]. Iron is required by bacteria for survival as it participates in major biological functions, and it is important that it is maintained at non-toxic levels and in non-toxic forms [91, 92]. Indeed, accumulation of Fe(III) is reported to alter membrane integrity and lead to DNA damage via an excess production of hydroxyl radicals [93]. However, under similar experimental conditions to zinc, no additive effects were observed for polyphemusin I with either copper or iron at concentrations up to 0.5 mM (Supplementary Fig. 5). Together the peptide–metal ion co-treatment data suggest that polyphemusin I, [I11A]tachyplesin I and [I11S]tachyplesin I exert an additive effect on EC958 that is specific to zinc. Further mechanistic studies, including investigation into the effect of sublethal concentrations of peptide on expression levels of zinc exporter ZntA, and the effect on zinc stress reporter strains of EC958 [50] are warranted.
The effect of peptides in an in vitro intracellular infection model
We established that tachyplesin I, polyphemusin I, [I11A]tachyplesin I, and [I11S]tachyplesin I exhibit antimicrobial activity toward EC958 WT and EC958ΔzntA at concentrations that are non-toxic to BMMs (Table 1); that the peptides can enter BMMs without damaging their membrane, at concentrations that permeabilize bacteria; and that sublethal concentrations of the latter three peptides enhance susceptibility of planktonic bacteria to zinc toxicity. EC958 can evade macrophage-mediated zinc toxicity, through mechanisms that may involve a higher resistance to zinc toxicity but also the capacity to protect themselves in LAMP1+ vacuoles [50]. Notably, EC958ΔzntA, which lacks its zinc exporter, can still subvert macrophage antibacterial responses and survive intracellularly. Therefore, EC958 WT and EC958ΔzntA were included in in vitro infection assays to determine whether peptide treatment enhances the zinc toxicity to affect intracellular bacterial survival.
To measure the effect on intramacrophage bacterial load, BMMs infected with EC958 WT or EC958ΔzntA were treated with peptide at concentrations that were lethal to bacteria (> MIC) but non-toxic to BMMs (< CC50, see Table 1). After 6 h and 12 h post-infection, BMMs were lysed and CFU were counted to determine the number of viable intracellular bacteria in all conditions. Treatment of BMMs with 0.5 µM tachyplesin I or 2 µM polyphemusin I did not reduce the number of intracellular EC958 WT at 6 h or 12 h post-infection (Fig. 10A). However, when using the same concentration of polyphemusin I (2 µM) or slightly increased tachyplesin I (4 µM) on BMM infected with EC958ΔzntA, there was a trend for reduced bacterial loads after 12 h of infection, suggesting an enhancement of zinc toxicity (Fig. 10B). More dramatically, treatment with a higher concentration (8 µM) of the less toxic analogs, [I11A]tachyplesin I and [I11S]tachyplesin I, significantly reduced the bacterial load of both EC958 WT and EC958ΔzntA at both timepoints. Notably, the bacterial load of BMMs treated with [I11S]tachyplesin I was reduced to < 5% of the control treatment after 10 h of peptide treatment (12 h post-infection). We confirmed that this significant reduction in the number of bacteria (CFU) was not associated with toxicity to BMMs, as there was no increase in cell death measured by lactate dehydrogenase (LDH) released into the culture supernatant (Fig. 10C). The absence of toxicity to infected BMMs at these peptide concentrations is also consistent with uninfected BMM toxicity data (Table 1).
Fig. 10.

Reduction of intracellular bacterial load following treatment of BMMs with peptides. BMMs were infected with either EC958 WT or EC958ΔzntA for 1 h (multiplicity of infection (MOI) 100:1). Extracellular bacteria were removed by removing the supernatant and treating the replaced media with gentamicin (200 µg/mL final concentration) for 1 h. Media was removed and replaced with media containing 20 µg/mL gentamicin and peptides at MICs (tachyplesin I and polyphemusin I) or the highest possible non-toxic dose of 8 µM ([I11A]tachyplesin I and [I11S]tachyplesin I). After 4 h or 10 h of peptide treatment (equivalent to 6 h or 12 h post-infection), BMMs were lysed with 0.01% (v/v) Triton X-100. Supernatant was diluted and plated on agar. Agar plates were incubated at 37ºC for 12 h. Colonies were counted and converted to CFUs. The percentage of CFU compared to untreated controls were plotted ± SEM from three independent experiments for A EC958 WT or B EC958ΔzntA. C A lactate dehydrogenase activity assay (LDH) was performed in parallel to the infection assay. BMMs were infected with MOI 100:1 EC958 WT or EC958ΔzntA and treated in the same manner as the infection assay. The LDH assay was performed as per manufacturer’s instructions, taking samples at 6 h and 12 h post-infection. Absorbance at 480 nm was read on a Tecan infinite M1000Pro and the percentage cell death was calculated using the ratio of media subtracted conditions divided by the subtracted absorbance of 100% lysis controls. The LDH assay for EC958ΔzntA is presented here. Significance for all panels was determined by one-way ANOVA. P values are denoted by: *P ≤ 0.05, **P ≤ 0.01, ****P ≤ 0.0001
Taken together with the planktonic co-treatment study, it is proposed that the enhanced intracellular antimicrobial activity of these peptides against EC958ΔzntA compared to EC958 WT may be due to the additive effect of intramacrophage zinc toxicity, and the diminished ability of EC958ΔzntA to overcome this effect. This finding is important as it was previously shown that UPEC evades the zinc toxicity response within human macrophages [50]. Species differences could be one explanation, as human and murine macrophages trigger evolutionarily divergent inflammatory responses [94], but a more exciting possibility is that the low level zinc exposure of UPEC in macrophages, usually non-toxic, is amplified when CPPs exert their effect on bacterial membranes.
Conclusions
The growing problem of antibiotic resistance is made more challenging by ability of some pathogenic bacteria to occupy intracellular niches. To effectively control these bacteria, new therapies must be able to reach and kill bacteria that sequester within vesicles to survive and replicate, at concentrations that do not damage host cells. Here we demonstrate the potential for antimicrobial CPPs, especially β-hairpin peptides from the horseshoe crab, tachyplesin I and polyphemusin I, to inhibit the growth of UPEC bacteria in both extracellular and intracellular environments. Moreover, a single amino acid substitution to produce [I11S]tachyplesin I reduced toxicity towards host BMM cells and greatly increased the ability to access intracellular bacteria and clear infection with multidrug-resistant UPEC strain EC958.
Polyphemusin I, tachyplesin I and analogs [I11A]tachyplesin I and [I11S]tachyplesin I inhibit bacterial growth at concentrations at least ten-fold lower than concentrations that are toxic to mammalian cells. This selectivity is related to their preferential binding to biological membranes that are rich in negatively charged phospholipid headgroups. Furthermore, these peptides permeabilize membranes of both pathogenic and non-pathogenic E. coli at sublethal concentrations. This membrane-active mechanism is particularly important when targeting drug-resistant pathogens such as EC958 that are known to avoid antimicrobial pathways and to remain quiescent in macrophages for long periods of time. Indeed, compared to the non-pathogenic E. coli, EC958 manifests a high tolerance to zinc toxicity, through both evasion and resistance. Our data suggest that the CPPs polyphemusin I, [I11A]tachyplesin I and [I11S]tachyplesin I, target and fragilize bacterial membranes, enabling sensitivity to zinc at concentrations that are usually non-toxic for EC958.
[I11A]tachyplesin I and [I11S]tachyplesin I analogs provided at least a two-fold increase in therapeutic window compared to native tachyplesin I, thereby affording treatment of BMMs infected with UPEC at higher concentrations, and significant reduction of intramacrophage bacterial load. The basis of this improved efficacy remains unclear, as overall structure and affinity of the peptide analogs for model and extracted bacterial membranes was similar to native tachyplesin I and polyphemusin I. Nevertheless, these data highlight the potential for cell-penetrating peptides as new modalities for targeting bacteria with intracellular niches. Overall, this study contributes to the growing body of evidence that antimicrobial peptides are an attractive alternative to conventional small molecule drugs, especially for pathogenic bacteria that can adopt an intracellular lifestyle.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by funding from the Australian Government scholarships (ASA. Research Training Program Scholarship), the Australian Research Council (Centre of Excellence for Innovations in Peptide and Protein Science CE200100012; DJC. Laureate Fellowship FL150100146; STH. Future Fellowship FT150100398), the National Health and Medical Research Council grants (NL. 1183927; JRW. and BJC. 1098337). NDC is supported as a CZI Imaging Scientist by grant number 2020-225648 from the Chan Zuckerberg Initiative DAF, an advised fund of Silicon Valley Community Foundation.
Author contributions
AA, NL, STH, and RK contributed to the study conception and design. Material preparation, data collection and analysis were performed by AA, JP, JW, NL, STH, NC, and PH. Funding and resources were provided by NL, STH, DC, BC, MS, and MS. The manuscript was written by AA, NL, STH, and RK. All authors commented on the previous versions of the manuscript and read and approved the final manuscript. The authors would like to thank Dr Kaustav Gupta at The Institute for Molecular Bioscience, Centre for Inflammation and Disease Research and Australian Infectious Diseases Research Centre, The University of Queensland, for harvesting and culturing bone marrow macrophages.
Availability of data and material
Peptide sequences are provided herein. Average data are provided, with processing and curve fits as reported for specific experiments.
Code availability
Not applicable.
Declarations
Conflict of interest
The authors declare that they do not have any conflicts or competing interests.
Ethical approval
The University of Queensland Institutional animal ethics committee approved the use of primary mouse cells, herein referred to as BMM cells (IMB/123/18). RBCs were collected from healthy adult donors following protocols approved by the Human Research Ethics Committees (University of Queensland approval number 2013000582) Identity of human blood donors was not recorded or reported.
Consent to participate
Human donors consented to use of their blood for research purposes.
Consent for publication
Human donors consented to publication of research using their blood.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Ronan Kapetanovic, Email: ronan.kapetanovic@fmi.ch.
Sónia Troeira Henriques, Email: sonia.henriques@qut.edu.au.
Nicole Lawrence, Email: n.lawrence@imb.uq.edu.au.
References
- 1.Centers for Disease Control and Prevention . Antibiotic resistance threats in the United States. Atlanta: U.S. Department of Health and Human Services; 2019. [Google Scholar]
- 2.McDermott PF, Walker RD, White DG. Antimicrobials: modes of action and mechanisms of resistance. Int J Toxicol. 2003;22:135–143. doi: 10.1080/10915810305089. [DOI] [PubMed] [Google Scholar]
- 3.Munita JM, Arias CA. Mechanisms of antibiotic resistance. Microbiol Spectr. 2016;4:VMBF-0016-2015. doi: 10.1128/microbiolspec.VMBF-0016-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laxminarayan R, Duse A, Wattal C, Zaidi AK, Wertheim HF, Sumpradit N, Vlieghe E, Hara GL, Gould IM, Goossens H, Greko C, So AD, Bigdeli M, Tomson G, Woodhouse W, Ombaka E, Peralta AQ, Qamar FN, Mir F, Kariuki S, Bhutta ZA, Coates A, Bergstrom R, Wright GD, Brown ED, Cars O. Antibiotic resistance-the need for global solutions. Lancet Infect Dis. 2013;13:1057–1098. doi: 10.1016/S1473-3099(13)70318-9. [DOI] [PubMed] [Google Scholar]
- 5.Hancock RE, Patrzykat A. Clinical development of cationic antimicrobial peptides: from natural to novel antibiotics. Curr Drug Targets Infect Disord. 2002;2:79–83. doi: 10.2174/1568005024605855. [DOI] [PubMed] [Google Scholar]
- 6.Pasupuleti M, Schmidtchen A, Malmsten M. Antimicrobial peptides: key components of the innate immune system. Crit Rev Biotechnol. 2012;32:143–171. doi: 10.3109/07388551.2011.594423. [DOI] [PubMed] [Google Scholar]
- 7.Luepke KH, Mohr JF., 3rd The antibiotic pipeline: reviving research and development and speeding drugs to market. Expert Rev Anti Infect Ther. 2017;15:425–433. doi: 10.1080/14787210.2017.1308251. [DOI] [PubMed] [Google Scholar]
- 8.Brauner A, Fridman O, Gefen O, Balaban NQ. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat Rev Microbiol. 2016;14:320–330. doi: 10.1038/nrmicro.2016.34. [DOI] [PubMed] [Google Scholar]
- 9.Grant SS, Hung DT. Persistent bacterial infections, antibiotic tolerance, and the oxidative stress response. Virulence. 2013;4:273–283. doi: 10.4161/viru.23987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Jia Y, Yang K, Wang Z. Heterogeneous strategies to eliminate intracellular bacterial pathogens. Front Microbiol. 2020;11:563. doi: 10.3389/fmicb.2020.00563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamaruzzaman NF, Kendall S, Good L. Targeting the hard to reach: challenges and novel strategies in the treatment of intracellular bacterial infections. Br J Pharmacol. 2017;174:2225–2236. doi: 10.1111/bph.13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silva MT, Pestana NT. The in vivo extracellular life of facultative intracellular bacterial parasites: role in pathogenesis. Immunobiology. 2013;218:325–337. doi: 10.1016/j.imbio.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 13.Mitchell G, Chen C, Portnoy DA. Strategies used by bacteria to grow in macrophages. Microbiol Spectr. 2016;4:MCHD-0012-2015. doi: 10.1128/microbiolspec.MCHD-0012-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abed N, Couvreur P. Nanocarriers for antibiotics: a promising solution to treat intracellular bacterial infections. Int J Antimicrob Agents. 2014;43:485–496. doi: 10.1016/j.ijantimicag.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 15.Tulkens PM. Intracellular distribution and activity of antibiotics. Eur J Clin Microbiol Infect Dis. 1991;10:100–106. doi: 10.1007/BF01964420. [DOI] [PubMed] [Google Scholar]
- 16.Nemeth A, Orgovan N, Sodar BW, Osteikoetxea X, Paloczi K, Szabo-Taylor KE, Vukman KV, Kittel A, Turiak L, Wiener Z, Toth S, Drahos L, Vekey K, Horvath R, Buzas EI. Antibiotic-induced release of small extracellular vesicles (exosomes) with surface-associated DNA. Sci Rep. 2017;7:8202. doi: 10.1038/s41598-017-08392-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garzoni C, Kelley WL. Staphylococcus aureus: new evidence for intracellular persistence. Trends Microbiol. 2009;17:59–65. doi: 10.1016/j.tim.2008.11.005. [DOI] [PubMed] [Google Scholar]
- 18.Angus AA, Lee AA, Augustin DK, Lee EJ, Evans DJ, Fleiszig SMJ. Pseudomonas aeruginosa induces membrane blebs in epithelial cells, which are utilized as a niche for intracellular replication and motility. Infect Immun. 2008;76:1992–2001. doi: 10.1128/Iai.01221-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garzoni C, Kelley WL. Return of the Trojan horse: intracellular phenotype switching and immune evasion by Staphylococcus aureus. Embo Mol Med. 2011;3:115–117. doi: 10.1002/emmm.201100123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dikshit N, Bist P, Fenlon SN, Pulloor NK, Chua CEL, Scidmore MA, Carlyon JA, Tang BL, Chen SL, Sukumaran B. Intracellular uropathogenic E. coli exploits host Rab35 for iron acquisition and survival within urinary bladder cells. Plos Pathog. 2015;11:1005083. doi: 10.1371/journal.ppat.1005083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ronald A. The etiology of urinary tract infection: traditional and emerging pathogens. Am J Med. 2002;113:14s–19s. doi: 10.1067/mda.2003.8. [DOI] [PubMed] [Google Scholar]
- 22.Flores-Mireles AL, Walker JN, Caparon M, Hultgren SJ. Urinary tract infections: epidemiology, mechanisms of infection and treatment options. Nat Rev Microbiol. 2015;13:269–284. doi: 10.1038/nrmicro3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foxman B. Epidemiology of urinary tract infections: Incidence, morbidity, and economic costs. Am J Med. 2002;113:5s–13s. doi: 10.1016/s0002-9343(02)01054-9. [DOI] [PubMed] [Google Scholar]
- 24.Anderson GG, Palermo JJ, Schilling JD, Roth R, Heuser J, Hultgren SJ. Intracellular bacterial biofilm-like pods in urinary tract infections. Science. 2003;301:105–107. doi: 10.1126/science.1084550. [DOI] [PubMed] [Google Scholar]
- 25.Hannan TJ, Totsika M, Mansfield KJ, Moore KH, Schembri MA, Hultgren SJ. Host–pathogen checkpoints and population bottlenecks in persistent and intracellular uropathogenic Escherichia coli bladder infection. FEMS Microbiol Rev. 2012;36:616–648. doi: 10.1111/j.1574-6976.2012.00339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bokil NJ, Totsika M, Carey AJ, Stacey KJ, Hancock V, Saunders BM, Ravasi T, Ulett GC, Schembri MA, Sweet MJ. Intramacrophage survival of uropathogenic Escherichia coli: differences between diverse clinical isolates and between mouse and human macrophages. Immunobiology. 2011;216:1164–1171. doi: 10.1016/j.imbio.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 27.Ingersoll MA, Kline KA, Nielsen HV, Hultgren SJ. G-CSF induction early in uropathogenic Escherichia coli infection of the urinary tract modulates host immunity. Cell Microbiol. 2008;10:2568–2578. doi: 10.1111/j.1462-5822.2008.01230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Engel DR, Maurer J, Tittel AP, Weisheit C, Cavlar T, Schumak B, Limmer A, van Rooijen N, Trautwein C, Tacke F, Kurts C. CCR2 mediates homeostatic and inflammatory release of Gr1(high) monocytes from the bone marrow, but is dispensable for bladder infiltration in bacterial urinary tract infection. J Immunol. 2008;181:5579–5586. doi: 10.4049/jimmunol.181.8.5579. [DOI] [PubMed] [Google Scholar]
- 29.Song J, Bishop BL, Li G, Grady R, Stapleton A, Abraham SN. TLR4-mediated expulsion of bacteria from infected bladder epithelial cells. Proc Natl Acad Sci USA. 2009;106:14966–14971. doi: 10.1073/pnas.0900527106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan NC, Foreman A, Jardeleza C, Douglas R, Vreugde S, Wormald PJ. Intracellular Staphylococcus aureus: the Trojan horse of recalcitrant chronic rhinosinusitis? Int Forum Allergy Rhinol. 2013;3:261–266. doi: 10.1002/alr.21154. [DOI] [PubMed] [Google Scholar]
- 31.Ting K, Aitken KJ, Penna F, Samiei AN, Sidler M, Jiang JX, Ibrahim F, Tolg C, Delgado-Olguin P, Rosenblum N, Bagli DJ. Uropathogenic E. coli (UPEC) infection induces proliferation through enhancer of zeste homologue 2 (EZH2) PLoS ONE. 2016;11:0149118. doi: 10.1371/journal.pone.0149118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Al-Badr A, Al-Shaikh G. Recurrent urinary tract infections management in women: a review. Sultan Qaboos Univ Med J. 2013;13:359–367. doi: 10.12816/0003256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zowawi HM, Harris PN, Roberts MJ, Tambyah PA, Schembri MA, Pezzani MD, Williamson DA, Paterson DL. The emerging threat of multidrug-resistant Gram-negative bacteria in urology. Nat Rev Urol. 2015;12:570–584. doi: 10.1038/nrurol.2015.199. [DOI] [PubMed] [Google Scholar]
- 34.Wagenlehner FME, Bjerklund Johansen TE, Cai T, Koves B, Kranz J, Pilatz A, Tandogdu Z. Epidemiology, definition and treatment of complicated urinary tract infections. Nat Rev Urol. 2020;17:586–600. doi: 10.1038/s41585-020-0362-4. [DOI] [PubMed] [Google Scholar]
- 35.Van Bambeke F, Van Laethem Y, Courvalin P, Tulkens PM. Glycopeptide antibiotics: from conventional molecules to new derivatives. Drugs. 2004;64:913–936. doi: 10.2165/00003495-200464090-00001. [DOI] [PubMed] [Google Scholar]
- 36.Cui AL, Hu X-X, Chen Y, Jin J, Yi H, Wang X-K, He Q-Y, You X-F, Li Z-R. Design, synthesis, and bioactivity of cyclic lipopeptide antibiotics with varied polarity, hydrophobicity, and positive charge distribution. ACS Infect Dis. 2020;6:1796–1806. doi: 10.1021/acsinfecdis.0c00056. [DOI] [PubMed] [Google Scholar]
- 37.Nation RL, Rigatto MHP, Falci DR, Zavascki AP. Polymyxin acute kidney injury: dosing and other strategies to reduce toxicity. Antibiotics. 2019;8:8010024. doi: 10.3390/antibiotics8010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cisneros JM, Rosso-Fernandez CM, Roca-Oporto C, De Pascale G, Jimenez-Jorge S, Fernandez-Hinojosa E, Matthaiou DK, Ramirez P, Diaz-Miguel RO, Estella A, Antonelli M, Dimopoulos G, Garnacho-Montero J, Magic Bullet Working Group WP Colistin versus meropenem in the empirical treatment of ventilator-associated pneumonia (Magic Bullet study): an investigator-driven, open-label, randomized, noninferiority controlled trial. Crit Care. 2019;23:383. doi: 10.1186/s13054-019-2627-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neundorf I. Antimicrobial and cell-penetrating peptides: how to understand two distinct functions despite similar physicochemical properties. Adv Exp Med Biol. 2019;1117:93–109. doi: 10.1007/978-981-13-3588-4_7. [DOI] [PubMed] [Google Scholar]
- 40.Henriques ST, Melo MN, Castanho MA. Cell-penetrating peptides and antimicrobial peptides: how different are they? Biochem J. 2006;399:1–7. doi: 10.1042/BJ20061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Steinberg DA, Hurst MA, Fujii CA, Kung AH, Ho JF, Cheng FC, Loury DJ, Fiddes JC. Protegrin-1: a broad-spectrum, rapidly microbicidal peptide with in vivo activity. Antimicrob Agents Chemother. 1997;41:1738–1742. doi: 10.1128/AAC.41.8.1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vernen F, Harvey PJ, Dias SA, Veiga AS, Huang YH, Craik DJ, Lawrence N, Henriques ST. Characterization of tachyplesin peptides and their cyclized analogues to improve antimicrobial and anticancer properties. Int J Mol Sci. 2019;20:4184. doi: 10.3390/ijms20174184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guidotti G, Brambilla L, Rossi D. Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol Sci. 2017;38:406–424. doi: 10.1016/j.tips.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 44.Murzyn K, Rog T, Pasenkiewicz-Gierula M. Phosphatidylethanolamine-phosphatidylglycerol bilayer as a model of the inner bacterial membrane. Biophys J. 2005;88:1091–1103. doi: 10.1529/biophysj.104.048835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Teixeira V, Feio MJ, Bastos M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog Lipid Res. 2012;51:149–177. doi: 10.1016/j.plipres.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 46.Hoskin DW, Ramamoorthy A. Studies on anticancer activities of antimicrobial peptides. Biochim Biophys Acta. 2008;1778:357–375. doi: 10.1016/j.bbamem.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seo MD, Won HS, Kim JH, Mishig-Ochir T, Lee BJ. Antimicrobial peptides for therapeutic applications: a review. Molecules. 2012;17:12276–12286. doi: 10.3390/molecules171012276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.von Pein JB, Stocks CJ, Schembri MA, Kapetanovic R, Sweet MJ. An alloy of zinc and innate immunity: galvanising host defence against infection. Cell Microbiol. 2021;23:e13268. doi: 10.1111/cmi.13268. [DOI] [PubMed] [Google Scholar]
- 49.Bohlmann L, De Oliveira DMP, El-Deeb IM, Brazel EB, Harbison-Price N, Ong CY, Rivera-Hernandez T, Ferguson SA, Cork AJ, Phan MD, Soderholm AT, Davies MR, Nimmo GR, Dougan G, Schembri MA, Cook GM, McEwan AG, von Itzstein M, McDevitt CA, Walker MJ. Chemical synergy between ionophore PBT2 and zinc reverses antibiotic resistance. MBio. 2018;9:e02391. doi: 10.1128/mBio.02391-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stocks CJ, Phan MD, Achard MES, Nhu NTK, Condon ND, Gawthorne JA, Lo AW, Peters KM, McEwan AG, Kapetanovic R, Schembri MA, Sweet MJ. Uropathogenic Escherichia coli employs both evasion and resistance to subvert innate immune-mediated zinc toxicity for dissemination. Proc Natl Acad Sci USA. 2019;116:6341–6350. doi: 10.1073/pnas.1820870116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kapetanovic R, Bokil NJ, Achard ME, Ong CL, Peters KM, Stocks CJ, Phan MD, Monteleone M, Schroder K, Irvine KM, Saunders BM, Walker MJ, Stacey KJ, McEwan AG, Schembri MA, Sweet MJ. Salmonella employs multiple mechanisms to subvert the TLR-inducible zinc-mediated antimicrobial response of human macrophages. FASEB J. 2016;30:1901–1912. doi: 10.1096/fj.201500061. [DOI] [PubMed] [Google Scholar]
- 52.Amiss AS, Webb JR, Mayo M, Currie BJ, Craik DJ, Henriques ST, Lawrence N. Safer in vitro drug screening models for melioidosis therapy development. Am J Trop Med Hyg. 2020;103:1846–1851. doi: 10.4269/ajtmh.20-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mobley HL, Green DM, Trifillis AL, Johnson DE, Chippendale GR, Lockatell CV, Jones BD, Warren JW. Pyelonephritogenic Escherichia coli and killing of cultured human renal proximal tubular epithelial cells: role of hemolysin in some strains. Infect Immun. 1990;58:1281–1289. doi: 10.1128/iai.58.5.1281-1289.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bachmann B. Derivations and genotypes of some mutant derivatives of Escherichiacoli K12. In: Neidhardt FC, editor. Escherichiacoli and Salmonella: cellular and molecular biology. 2. Washington, DC: ASM Press; 1996. pp. 2460–2488. [Google Scholar]
- 55.Cheneval O, Schroeder CI, Durek T, Walsh P, Huang YH, Liras S, Price DA, Craik DJ. Fmoc-based synthesis of disulfide-rich cyclic peptides. J Org Chem. 2014;79:5538–5544. doi: 10.1021/jo500699m. [DOI] [PubMed] [Google Scholar]
- 56.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 57.R_package (2021) hmoment: compute the hydrophobic moment of a protein sequence. https://rdrr.io/cran/Peptides/man/hmoment.html. Accessed 18 Mar 2021
- 58.Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas P, Ulrich EL, Markley JL, Ionides J, Laue ED. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- 59.Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. J Biomol NMR. 1995;5:67–81. doi: 10.1007/BF00227471. [DOI] [PubMed] [Google Scholar]
- 60.Cockerill F, Wikler M, Alder J, Dudley M, Eliopoulos G, Ferraro M, Hardy D, Hecht D, Hindler J, Patel J. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically: approved standard. Clin Lab Stand Inst. 2012;32:M07-A09. [Google Scholar]
- 61.Ravasi T, Mavromatis CH, Bokil NJ, Schembri MA, Sweet MJ. Co-transcriptomic analysis by RNA sequencing to simultaneously measure regulated gene expression in host and bacterial pathogen. Methods Mol Biol. 2016;1390:145–158. doi: 10.1007/978-1-4939-3335-8_10. [DOI] [PubMed] [Google Scholar]
- 62.Torcato IM, Huang YH, Franquelim HG, Gaspar D, Craik DJ, Castanho MA, Henriques ST. Design and characterization of novel antimicrobial peptides, R-BP100 and RW-BP100, with activity against Gram-negative and Gram-positive bacteria. Biochim Biophys Acta. 2013;1828:944–955. doi: 10.1016/j.bbamem.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 63.Lawrence N, Philippe GJB, Harvey PJ, Condon ND, Benfield AH, Cheneval O, Craik DJ, Troeira HS. Cyclic peptide scaffold with ability to stabilize and deliver a helical cell-impermeable cargo across membranes of cultured cancer cells. RSC Chem Biol. 2020;1:405–420. doi: 10.1039/D0CB00099J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Henriques ST, Pattenden LK, Aguilar M-I, Castanho MARB. PrP(106–126) does not interact with membranes under physiological conditions. Biophys J. 2008;95:1877–1889. doi: 10.1529/biophysj.108.131458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Young RSE, Bowman AP, Williams ED, Tousignant KD, Bidgood CL, Narreddula VR, Gupta R, Marshall DL, Poad BLJ, Nelson CC, Ellis SR, Heeren RMA, Sadowski MC, Blanksby SJ. Apocryphal FADS2 activity promotes fatty acid diversification in cancer. Cell Rep. 2021;34:108738. doi: 10.1016/j.celrep.2021.108738. [DOI] [PubMed] [Google Scholar]
- 66.Henriques ST, Huang Y-H, Rosengren KJ, Franquelim HG, Carvalho FA, Johnson A, Sonza S, Tachedjian G, Castanho MARB, Daly NL, Craik DJ. Decoding the membrane activity of the cyclotide kalata B1: the importance of phosphatidylethanolamine phospholipids and lipid organization on hemolytic and anti-HIV activities. J Biol Chem. 2011;286:24231–24241. doi: 10.1074/jbc.M111.253393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Henriques ST, Huang Y-H, Chaousis S, Sani M-A, Poth AG, Separovic F, Craik DJ. The prototypic cyclotide kalata B1 has a unique mechanism of entering cells. Chem Biol. 2015;22:1087–1097. doi: 10.1016/j.chembiol.2015.07.012. [DOI] [PubMed] [Google Scholar]
- 68.Henriques ST, Peacock H, Benfield AH, Wang CK, Craik DJ. Is the mirror image a true reflection? Intrinsic membrane chirality modulates peptide binding. JACS. 2019;141:20460–20469. doi: 10.1021/jacs.9b11194. [DOI] [PubMed] [Google Scholar]
- 69.Figueira TN, Freire JM, Cunha-Santos C, Heras M, Goncalves J, Moscona A, Porotto M, Veiga AS, Castanho MARB. Quantitative analysis of molecular partition towards lipid membranes using surface plasmon resonance. Sci Rep. 2017 doi: 10.1038/srep45647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benfield AH, Defaus S, Lawrence N, Chaousis S, Condon N, Cheneval O, Huang Y-H, Chan LY, Andreu D, Craik DJ, Henriques ST. Cyclic gomesin, a stable redesigned spider peptide able to enter cancer cells. BBA Biomembranes. 2021;1863:183480. doi: 10.1016/j.bbamem.2020.183480. [DOI] [PubMed] [Google Scholar]
- 71.Henriques ST, Lawrence N, Chaousis S, Ravipati AS, Cheneval O, Benfield AH, Elliott AG, Kavanagh AM, Cooper MA, Chan LY, Huang YH, Craik DJ. Redesigned spider peptide with improved antimicrobial and anticancer properties. ACS Chem Biol. 2017;12:2324–2334. doi: 10.1021/acschembio.7b00459. [DOI] [PubMed] [Google Scholar]
- 72.Vernen F, Craik DJ, Lawrence N, Henriques ST. Cyclic analogues of horseshoe crab peptide tachyplesin I with anticancer and cell penetrating properties. ACS Chem Biol. 2019;14:2895–2908. doi: 10.1021/acschembio.9b00782. [DOI] [PubMed] [Google Scholar]
- 73.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schembri MA, Sokurenko EV, Klemm P. Functional flexibility of the FimH adhesin: insights from a random mutant library. Infect Immun. 2000;68:2638–2646. doi: 10.1128/IAI.68.5.2638-2646.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Edwards IA, Elliott AG, Kavanagh AM, Zuegg J, Blaskovich MA, Cooper MA. Contribution of amphipathicity and hydrophobicity to the antimicrobial activity and cytotoxicity of beta-hairpin peptides. ACS Infect Dis. 2016;2:442–450. doi: 10.1021/acsinfecdis.6b00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Edwards IA, Elliott AG, Kavanagh AM, Blaskovich MAT, Cooper MA. Structure-activity and -toxicity relationships of the antimicrobial peptide tachyplesin-1. ACS Infect Dis. 2017;3:917–926. doi: 10.1021/acsinfecdis.7b00123. [DOI] [PubMed] [Google Scholar]
- 77.Torcato IM, Huang YH, Franquelim HG, Gaspar DD, Craik DJ, Castanho MA, Henriques ST. The antimicrobial activity of Sub3 is dependent on membrane binding and cell-penetrating ability. ChemBioChem. 2013;14:2013–2022. doi: 10.1002/cbic.201300274. [DOI] [PubMed] [Google Scholar]
- 78.Kumar P, Kizhakkedathu JN, Straus SK. Antimicrobial peptides: diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules. 2018;8:8010004. doi: 10.3390/biom8010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ulett GC, Totsika M, Schaale K, Carey AJ, Sweet MJ, Schembri MA. Uropathogenic Escherichia coli virulence and innate immune responses during urinary tract infection. Curr Opin Microbiol. 2013;16:100–107. doi: 10.1016/j.mib.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 80.Schaale K, Peters KM, Murthy AM, Fritzsche AK, Phan MD, Totsika M, Robertson AA, Nichols KB, Cooper MA, Stacey KJ, Ulett GC, Schroder K, Schembri MA, Sweet MJ. Strain- and host species-specific inflammasome activation, IL-1β release, and cell death in macrophages infected with uropathogenic Escherichia coli. Mucosal Immunol. 2016;9:124–136. doi: 10.1038/mi.2015.44. [DOI] [PubMed] [Google Scholar]
- 81.Murthy AMV, Sullivan MJ, Nhu NTK, Lo AW, Phan MD, Peters KM, Boucher D, Schroder K, Beatson SA, Ulett GC, Schembri MA, Sweet MJ. Variation in hemolysin A expression between uropathogenic Escherichia coli isolates determines NLRP3-dependent vs. -independent macrophage cell death and host colonization. Faseb J. 2019;33:7437–7450. doi: 10.1096/fj.201802100R. [DOI] [PubMed] [Google Scholar]
- 82.Silverman BD. Hydrophobic moments of protein structures: spatially profiling the distribution. Proc Natl Acad Sci USA. 2001;98:4996–5001. doi: 10.1073/pnas.081086198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Marggraf MB, Panteleev PV, Emelianova AA, Sorokin MI, Bolosov IA, Buzdin AA, Kuzmin DV, Ovchinnikova TV. Cytotoxic potential of the novel horseshoe crab peptide polyphemusin III. Mar Drugs. 2018 doi: 10.3390/md16120466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang L, Scott MG, Yan H, Mayer LD, Hancock RE. Interaction of polyphemusin I and structural analogs with bacterial membranes, lipopolysaccharide, and lipid monolayers. Biochemistry. 2000;39:14504–14514. doi: 10.1021/bi0011173. [DOI] [PubMed] [Google Scholar]
- 85.Hood MI, Skaar EP. Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol. 2012;10:525–537. doi: 10.1038/nrmicro2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stafford SL, Bokil NJ, Achard MES, Kapetanovic R, Schembri MA, McEwan AG, Sweet MJ. Metal ions in macrophage antimicrobial pathways: emerging roles for zinc and copper. Bioscience Rep. 2013;33:541–554. doi: 10.1042/BSR20130014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schaible ME, Kaufmann SHE. Iron and microbial infection. Nat Rev Microbiol. 2004;2:946–953. doi: 10.1038/nrmicro1046. [DOI] [PubMed] [Google Scholar]
- 88.Andreini C, Bertini I. A bioinformatics view of zinc enzymes. J Inorg Biochem. 2012;111:150–156. doi: 10.1016/j.jinorgbio.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 89.Haase H, Rink L. Signal transduction in monocytes: the role of zinc ions. Biometals. 2007;20:579. doi: 10.1007/s10534-006-9029-8. [DOI] [PubMed] [Google Scholar]
- 90.Lonergan ZR, Skaar EP. Nutrient zinc at the host–pathogen interface. Trends Biochem Sci. 2019;44:1041–1056. doi: 10.1016/j.tibs.2019.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Andrews SC, Robinson AK, Rodriguez-Quinones F. Bacterial iron homeostasis. FEMS Microbiol Rev. 2003;27:215–237. doi: 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- 92.Park S, Imlay JA. High levels of intracellular cysteine promote oxidative DNA damage by driving the Fenton reaction. J Bacteriol. 2003;185:1942–1950. doi: 10.1128/Jb.185.6.1942-1950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chamnongpol S, Dodson W, Cromie MJ, Harris ZL, Groisman EA. Fe(III)-mediated cellular toxicity. Mol Microbiol. 2002;45:711–719. doi: 10.1046/j.1365-2958.2002.03041.x. [DOI] [PubMed] [Google Scholar]
- 94.Schroder K, Irvine KM, Taylor MS, Bokil NJ, Le Cao K-A, Masterman K-A, Labzin LI, Semple CA, Kapetanovic R, Fairbairn L, Akalin A, Faulkner GJ, Baillie JK, Gongora M, Daub CO, Kawaji H, McLachlan GJ, Goldman N, Grimmond SM, Carninci P, Suzuki H, Hayashizaki Y, Lenhard B, Hume DA, Sweet MJ. Conservation and divergence in toll-like receptor 4-regulated gene expression in primary human versus mouse macrophages. Proc Natl Acad Sci USA. 2012;109:E944. doi: 10.1073/pnas.1110156109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Peptide sequences are provided herein. Average data are provided, with processing and curve fits as reported for specific experiments.
Not applicable.