

Abstract
The fusion oncoprotein RUNX1/ETO which results from the chromosomal translocation t (8;21) in acute myeloid leukemia (AML) is an essential driver of leukemic maintenance. We have previously shown that RUNX1/ETO knockdown impairs expression of the protein component of telomerase, TERT. However, the underlying molecular mechanism of how RUNX1/ETO controls TERT expression has not been fully elucidated. Here we show that RUNX1/ETO binds to an intergenic region 18 kb upstream of the TERT transcriptional start site and to a site located in intron 6 of TERT. Loss of RUNX1/ETO binding precedes inhibition of TERT expression. Repression of TERT expression is also dependent on the destabilization of the E3 ubiquitin ligase SKP2 and the resultant accumulation of the cell cycle inhibitor CDKN1B, that are both associated with RUNX1/ETO knockdown. Increased CDKN1B protein levels ultimately diminished TERT transcription with E2F1/Rb involvement. Collectively, our results show that RUNX1/ETO controls TERT expression directly by binding to its locus and indirectly via a SKP2—CDKN1B—E2F1/Rb axis.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-023-04713-y.
Keywords: RUNX1/ETO, Acute Myeloid Leukaemia (AML), Transcription factor, Telomerase Reverse Transcriptase (TERT), Self renewal, Cell cycle
Introduction
Telomerase maintains chromosomal integrity by preventing telomere shortening and attrition caused by chromosome replication and DNA damage, respectively. It, thus, is required for normal and malignant self-renewal by preventing replicative senescence in proliferating cells [1].
The telomerase complex comprises TERT, TERC, dyskerin and other associated proteins including NHP2, NOP10 and GAR1 [2]. However, the core components of the telomerase complex are the RNA and reverse transcriptase subunits encoded by the TERC and TERT genes, respectively. The catalytic subunit TERT constitutes the rate-limiting factor in the telomerase complex thus acting as a major regulator in maintaining active telomerase levels [2, 3] TERT expression is regulated by a GC-rich promoter without TATA or CCAAT consensus sequences. Instead, it contains SP1 binding sites in addition to CpG islands that are mostly methylated in normal cells. Typically, TERT is repressed in somatic cells and activated in most cancer cells [4, 5]. Transcription factors such as MYC, STAT3 and members of the E2F family have been described to activate TERT transcription [6–8]. Point mutations within the TERT promoter have been described for several tumors including glioblastomas and melanomas that generate de novo binding sites for ETS factors. A recent study demonstrated that GABPA binding to such de novo ETS sites facilitates a long-range interaction with an enhancer 300 kb upstream of TERT [1, 3]. In contrast to its mutated variants, the regulation of the wildtype TERT promoter by enhancer elements is only incompletely understood.
Oncogenic fusion proteins are also known to promote malignant self-renewal by maintaining telomerase activity. We and others have previously shown that TERT is a downstream target for oncogenic fusion proteins including EWS/FLI1, MLL/AF4 and RUNX1/ETO [9, 10]. RUNX1/ETO is the most common fusion protein in AML; it both represses genes essential for myeloid differentiation and promotes leukemic self-renewal by maintaining cell cycle regulators such as D-cyclins and CDK6 [11]. Notably, loss of RUNX1/ETO impairs TERT expression and induces telomere shortening and ultimately replicative senescence in AML cells [8, 9]. However, the mechanism of RUNX1/ETO-exerted regulation of TERT has remained unknown. Here we report that RUNX1/ETO affects TERT expression directly by binding to an upstream enhancer sequence and indirectly by controlling SKP2 and CDKN1B (p27) protein levels.
Materials and methods
Cell culture
Kasumi-1 and SKNO-1 cells were cultivated in Roswell Park Memorial Institute Medium (RPMI 1640) containing 10% (v/v) or 20% fetal bovine serum, respectively. SKNO-1 medium was further supplemented with 10 ng/µl of granulocyte macrophage stimulating growth factor (GM-CSF). Cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2. For telomerase assay in SKNO-1 after knockdown of RUNX1/ETO, cells were cultured in RPMI + 20% FBS without GM-CSF.
Gene knockdown
Electroporation of Kasumi-1 and SKNO-1 cells was essentially performed as described previously [12]. Cells (107 cells/ml) were electroporated at room temperature in complete culture medium with 50 – 200 nM siRNA in 4 mm cuvettes using a 10 ms square wave of 330 V (Kasumi-1) or 350 V (SKNO-1) using a Bio-Rad Gene Pulser Xcell. In case of prolonged knockdown, electroporations were repeated every 3–4 days. The siRNAs used in this study are listed in Table 1 in supplementary material.
RNA and protein extraction
Total RNA was extracted from Kasumi-1 and SKNO-1 cells using Qiagen RNeasy according to the manufacturer’s protocol and stored at – 80 °C. The first RNeasy flow-through was used for protein extraction by precipitation with 2 volumes of acetone. After careful removal of the supernatants, the protein pellets were dissolved in urea lysis buffer (9 M urea, 4% CHAPS, 1%DTT) and stored at – 80 °C.
Real time qPCR
Quantitative reverse transcription polymerase chain reactions (RT-qPCR) were carried out in a StepOne Plus instrument (ABI) using the Quantifast SYBR Green one step RT-PCR master mix (QIAGEN). Primers used in this study is shown in Table 2 in supplementary material and synthesized by BIONEER, Korea. Reactions were set up in triplicates with 20 ng of RNA for each sample. The composition of each reaction is shown in Table 3 in supplementary material. Data were analyzed using the ΔCT approach using HPRT1 for normalization.
Western blots
A rapid semidry transfer method was carried out using Trans-Blot Turbo blotter (BIORAD). Transfer was carried out using a mixed molecular weight protocol which used a constant current of 1.5A up to 25 V. Western blot was done as described [13, 14]. Antibodies used are listed in Table 4 in supplementary material. To image the targeted protein on the membrane, Clarity Western ECL Substrate (BioRad) was used as per manufacturer’s protocol. Membranes were imaged using VersaDoc system (BioRad). Band intensity was analyzed using Quantity One/ Image One software.
Cell cycle analysis
Cell cycle status of Kasumi-1 and SKNO-1 cells was analyzed on days 0, 4 and 7 after siRNA electroporation. Cells were centrifuged at 400xg for seven minutes followed by cell staining according to the manufacturer’s protocol in the BD Cycle TESTTM PLUS DNA Reagent Kit. Samples were kept cold and analyzed within three hours. Results were analyzed using Mod Fit LT for Windows version 3.3 (VSH, USA).
Colony formation assay
Colony formation assay was carried out as described [14].
Senescence associated β-galactosidase assay
Cellular senescence was measured using β-galactosidase staining. Staining was carried out as described [14] or using cellular senescence assay kit from Cell Biolabs, Inc.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was carried out as described. Immunoprecipitation was carried out using the Dynabeads Protein G kit (ThermoFisher). Briefly, following cross-linking and sonication, chromatin was subjected to immunoprecipitation as previously described [15]. Subsequently, DNA purification using the Qiaquick PCR purification kit was carried out. Real Time-qPCR was carried out using the StepOne Plus instrument (ABI) and data were analyzed using the ΔCT approach with ChIP input being used for normalization. ChIP-qPCR primers used for E2F1 occupancy on the TERT promoter and a 3 kb upstream sequence are listed in Table 2 in supplementary materail.
Co-immunoprecipitation
Co-immunoprecipitation was carried out as previously described with slight modifications [15]. Briefly, following cross-linking, purified nuclei were subjected to sonication and immunoprecipitation using Dynabeads Protein G kit (ThermoFisher) linked to underphosphorylated Rb antibody. The isolated purified complex was blotted using antibodies against E2F1 and Rb. IgG served as the isotype control.
Telomerase activity
Telomerase activity was determined with RQ-TRAP assays as previously described [16]. HEK 293 T was used to generate a standard for telomerase activity quantitation. Primers used for RQ TRAP are listed in Table 2 in supplementary material.
Statistical analysis
All statistical analysis was carried out using SPSS version 22. Comparison between two groups was carried out using paired t-test while comparison involving more than two groups was analyzed using one-way ANOVA. Difference between single groups in one-way ANOVA was compared using a post hoc test with Dunnett’s corrections. Levine’s test for equality of variance was performed as well.
Results
TERT is a direct target gene of RUNX1/ETO
Knockdown of RUNX1/ETO caused G1 cell cycle arrest, induction of senescence and telomere shortening in the t(8;21) AML cell lines Kasumi-1 and SKNO-1 [9]. Connected to these changes were reduced levels of TERT transcript and protein (Figs. 1A–D, S1A, B). This loss of TERT resulted in a five-fold reduction in telomerase activity as demonstrated by RQ-TRAP assays (Figs. 1E, S1C). Interestingly, genome-wide ChIP-Seq experiments identified two locations bound by RUNX1/ETO with one weaker binding located at the 3’-end of intron 6 and a second, substantial peak 18 kb upstream of TERT in proximity to the transcriptional start site of CLPTM1L (Fig. 1F) [17, 18]. RUNX1/ETO knockdown led to a complete loss of its binding to both locations within 48 h. Moreover, and in agreement with the recruitment of class I histone deacetylases by RUNX1/ETO, its knockdown increased levels of H3K9 acetylation at the 18 kb element. Nevertheless, this is linked with a decreased H3K27 acetylation at the TERT promoter. Interestingly, this -18 kb binding site is located in enhancer GH05J001312, [17] which has been found to interact with the TERT promoter (Figs. 1F, S1E).
Fig. 1.

TERT is a direct target gene of RUNX1/ETO. A, B Knockdown of RUNX1/ETO transcript and protein levels in Kasumi-1 cells. Left panel, qPCR analysis; right panel, western blot. C, D TERT RNA (left panel) and protein (right panel) levels after RUNX1/ETO knockdown. E Telomerase Activity after RUNX1/ETO knockdown after 10 days in Kasumi-1. F UCSC screenshot displaying changes in RUNX1/ETO binding on the TERT locus two days after RUNX1/ETO knockdown. G, UCSC screenshot displaying changes in transcription factor binding at the TERT locus upon RUNX1/ETO knockdown. H RUNX1/ETO and CEBPA transcript levels 48 h post siRE mediated suppression. I Lack of RUNX1/ETO binding on the TERT CLPTM1L locus (− 18 kb region from TERT TSS) in primary AML samples. J Publicly available RNASeq datasets of CLPTM1L levels post RUNX1/ETO suppression via several methods. (shAE; small hairpin RNA mediated RUNX1/ETO suppression, siRE; small interfering RNA mediated RUNX1/ETO suppression; dTAG-47: direct protein mediated RUNX1/ETO degradation). siRE RUNX1/ETO siRNA; siMM mismatch control. Error bars indicate standard deviations from three independent experiments. Asterisk indicates p-value < 0.05
Further inspection revealed that the -18 kb site was co-occupied by RUNX1 and PU.1 (also known as SPI1), two crucial transcriptional regulators of myeloid differentiation. However, RUNX1/ETO knockdown did not substantially affect their binding to the -18 kb element (Fig. 1F, G). Similarly, recruitment of LMO2 to this site was not affected by the RUNX1/ETO status (Fig. 1G) Since LMO2 has been shown to form a complex with RUNX1/ETO, this finding suggests either a RUNX1/ETO-independent recruitment of LMO2 at this site or that other factors such as RUNX1 keep LMO2 on site. However, loss of RUNX1/ETO binding was paralleled by an increased binding of C/EBPα, a well-established driver of myeloid differentiation and inhibitor of TERT expression at the -18 kb element (Figs. 1G, S1D) [15]. It is also interesting as suppression of RUNX1/ETO resulted in the changes in C/EBPα transcript levels (Fig. 1H). These combined data strongly suggest that TERT is a direct target gene of RUNX1/ETO and that its loss causes a reorganization of the chromatin ultimately impairing TERT expression (Figs. 1G, S1E).
Surprisingly, analyses of publicly available datasets of AML patients with t(8;21) chromosomal translocation(GSE23730) [20] hardly showed binding peaks of RUNX1/ETO on the TERT/CLPTM1L locus − 18 kb site with mostly background signals observed (Fig. 1I). This pattern was different from the one obtained from cell lines (Figs. 1F, S1E). Furthermore, to ascertain that RUNX1/ETO knockdown only affects TERT expression and does not affect CLPTM1L, publicly available RNA-seq datasets (GSE100446, GSE55478 and GSE153281) generated from experiments in which RUNX1/ETO was suppressed was via different methods; shRNA. siRNA and direct RUNX1/ETO targeted degradation were analyzed [19, 21, 22]. Data showed no significant changes in CLPTM1L levels post RUNX1/ETO suppression (Fig. 1J) which strongly points to RUNX1/ETO mediated TERT expressional control.
RUNX1/ETO controls TERT via posttranslational regulation of CDKN1B
However, loss of TERT expression trailed loss of RUNX1/ETO both at RNA and protein level reaching a minimum after 96–120 h, which was accompanied by a concomitant loss of telomerase activity in the t(8;21)-positive AML cell lines Kasumi-1 and SKNO-1 [18, 19] (Figs. 1A–E, S1A-C). This delay suggests that RUNX1/ETO controls TERT expression also in an indirect fashion.
Since TERT expression has been linked to the G1/S progression in the cell cycle [23], we first examined whether a G1 block would affect TERT transcript levels. RUNX1/ETO promotes G1 progression by directly binding to and activating expression of CCND2 [24, 25]. However, neither CCND2 knockdown nor inhibition of the G1 associated CDKs, CDK4 and CDK6 by palbociclib for four days significantly affected TERT expression in RUNX1/ETO-expressing AML cells (Fig. 2A). These data argue against substantial roles of CDK4, CDK6 or CCND2 on RUNX1/ETO-mediated control of TERT.
Fig. 2.

RUNX1/ETO controls TERT via posttranslational regulation of CDKN1B. A TERT transcript levels after palbociclib treatment, CCND2 or RUNX1/ETO knockdown. Graph shows normalized RNA-seq data from n = 3. B CDKN1B protein expression level after gene knockdown with siRE in Kasumi-1 cells at various time points. Graph shows normalized protein data from n = 2. C Cell cycle distribution of Kasumi-1 cells after RUNX1/ETO and CDKN1B knockdown. FACS analysis of PI-stained cells was performed 10 days after first siRNA treatment. D Induction of senescence by RUNX1/ETO knockdown with and without concomitant CDKN1B knockdown. Left panel, graph showing quantitation of SA-βGAL -positive cells; right panel, Kasumi-1 cells stained for senescence-associated β-galactosidase (SA-βGAL). E RUNX1/ETO RNA and protein levels, respectively, after RUNX1/ETO knockdown with and without concomitant CDKN1B knockdown (RNA; day 4, 7 and 10) (protein; day 10). Upper panel, qPCR analyses; lower panel, western blot. F CDKN1B transcript and protein levels in Kasumi-1 cells after RUNX1/ETO knockdown with and without concomitant CDKN1B knockdown (RNA;day 4, 7 and 10) (protein; day 10). Upper panel, qPCR analyses; lower panel, western blot. G, TERT RNA and protein levels, respectively, after RUNX1/ETO knockdown with and without concomitant CDKN1B knockdown (RNA; day 4, 7 and 10) (protein; day 10). Upper panel, qPCR analyses; lower panel, western blot. siRE, RUNX1/ETO siRNA; siCDKN1B, CDKN1B siRNA; siCCND2-2 and siCCND2-4, two independent CCND2 siRNAs; siMM, mismatch control. Error bars indicate standard deviations from three independent experiments. Asterisk indicates p value < 0.05
Next, we asked if inhibition of passing the G1/S restriction point would affect TERT levels. Here we focused on CDKN1B, which was previously found to repress TERT expression in cervical cancer cell lines by downregulation of HPV E7 protein [26]. Indeed, prolonged loss of RUNX1/ETO caused an accumulation of CDKN1B protein; after 7–10 days of RUNX1/ETO knockdown, CDKN1B protein levels raised three- to fourfold without affecting its transcript levels (Figs. 2B, S2A). These data suggest a posttranslational regulation of CDKN1B by RUNX1/ETO. Since the kinetics of CDKN1B accumulation paralleled that of TERT inhibition, we directly examined its involvement in the control of TERT by RUNX1/ETO. While RUNX1/ETO knockdown caused an accumulation of Kasumi-1 cells in the G1 phase of the cell cycle and ultimately senescence as indicated by staining for senescence-associated β-galactosidase (SA-βGAL), concomitant knockdown of RUNX1/ETO and CDKN1B rescued cell cycle progression and prevented senescence (Figs. 2C – F). Inhibiting CDKN1B accumulation also restored both TERT RNA and protein expression (Fig. 2G). These combined findings demonstrate that RUNX1/ETO controls TERT through modulation of CDKN1B protein levels thus expanding the role of RUNX1/ETO in controlling G1/S progression as CDKN1B is an important CDK inhibitor for G1/S progression [27, 28].
RUNX1/ETO controls TERT via SKP2 protein stabilization and CDKN1B down modulation
CDKN1B stability is controlled by the ubiquitin ligase SKP2, which ubiquitinates CDKN1B upon Cyclin E/CDK2-mediated phosphorylation at T187 leading to its proteasomal degradation [29, 30]. We therefore postulated that RUNX1/ETO affects the SKP2 status in leukemic cells and examined the impact of RUNX1/ETO knockdown on RNA and protein levels of SKP2. siRNA-mediated RUNX1/ETO depletion for 10 days was associated with an up to sevenfold decrease in SKP2 protein levels in Kasumi-1 cells (Fig. 3A). As in the case of CDKN1B, SKP2 transcript levels did not change pointing at stabilization of SKP2 by RUNX1/ETO (Figure S2B). Single knockdown of CDKN1B increased SKP2 levels twofold (Fig. 3B) suggesting a feedback loop where accumulating CDKN1B facilitates SKP2 degradation, possibly by arresting cells in G1 and thereby promoting APC-dependent SKP2 ubiquitination and degradation [30]. Inverse correlations between SKP2 and p27 levels have been reported in other subtypes of leukemia [32, 33] and cancers in general as well [34]. Interestingly, combined knockdown of RUNX1/ETO and CDKN1B hardly restored SKP2 showing that RUNX1/ETO-mediated control of SKP2 protein levels does not involve a CDKN1B-driven feed-back loop (Fig. 3B).
Fig. 3.

RUNX1/ETO controls TERT via SKP2 protein stabilization and CDKN1B down modulation. A Western blot showing SKP2 protein levels in Kasumi-1 cells upon RUNX1/ETO knockdown. B SKP2 protein levels after RUNX1/ETO knockdown with and without concomitant CDKN1B knockdown. C SKP2 protein levels after SKP2 knockdown D CDKN1B protein levels after SKP2 knockdown. E, Induction of senescence by RUNX1/ETO, CDKN1B and SKP2 knockdown, respectively. Senescent cells were stained for SA-βGAL. F Clonogenic growth of Kasumi-1 cells upon RUNX1/ETO, CDKN1B and SKP2 knockdown, respectively. G, H, TERT transcript (G) and protein (H) levels after SKP2 knockdown. siRE RUNX1/ETO siRNA; siSKP2, SKP2 siRNA: siCDKN1B, CDKN1B siRNA; siMM mismatch control. Error bars indicate standard deviations from three independent experiments. Asterisk indicates p value < 0.05
Next, we asked if suppression of SKP2 affected CDKN1B and TERT in RUNX1/ETO-expressing cells. SKP2 depletion caused a threefold accumulation of CDKN1B without changing its transcript levels (Figs. 3C, D, S2C). Similar to RUNX1/ETO, knockdown of SKP2 induced senescence and inhibited colony formation in Kasumi-1 cells (Figs. 3E, F, S1F). This was paralleled by twofold lower transcript and threefold lower protein amounts of TERT (Figs. 3G, H). SKP2 loss did not affect expression levels of the telomerase subunit TERC (Figures S2D, E). These combined findings establish a hierarchically organized RUNX1/ETO–SKP2–CDKN1B axis that controls TERT transcription.
Loss of RUNX1/ETO causes dissociation of E2F1 from the TERT promoter via CDKN1B
Several transcription factor complexes including MYC and E2F–RB1 complexes have been described to bind to the TERT promoter thereby linking TERT transcription to the cell cycle [35]. Specifically, E2F1 forms a repressive complex with hypo-phosphorylated RB at position -174/-170 of the TERT transcriptional start site [6, 7, 20]. Since CDKN1B is interfering with the CDK2-dependent dissociation of RB1 from E2F1 and has also been found to induce proteasomal degradation of RB1 [36], we investigated the significance of RUNX1/ETO and CDKN1B on E2F1 and RB1 expression and function. Gene set enrichment analysis of the RUNX1/ETO knockdown signature revealed a loss of genes harboring E2F binding sites flanking transcriptional start sites. Furthermore, RUNX1/ETO knockdown decreased total E2F1 protein levels fivefold, which was also paralleled by a loss of RB1 protein (Figs. 4A–C). This effect was dependent on CDKN1B accumulation: depletion of CDKN1B alone even increased RB1 and E2F1 levels 1.7 and 2.5-fold, respectively (Fig. 4D, E). Nevertheless, the increase in CDKN1B does not translate to its increase in TERT promoter occupation (Fig. 4I).
Fig. 4.

RUNX1/ETO causes dissociation of E2F1 from the TERT promoter via CDKN1B thus affecting the expression of TERT. A Gene set enrichment analyses (GSEA) of E2F1 flanked genes B, C E2F1 and RB1 protein levels upon 10 days of RUNX1/ETO knockdown. D, E E2F1 and RB1 protein levels upon CDKN1B knockdown, respectively. F, G graph showing ChIP assay data for E2F1 occupancy on the TERT promoter (F) and a control site located 3 kb upstream (G) after 10 days of RUNX1/ETO and/or CDKN1B knockdown. H CDKN1B occupation on TERT promoter levels in Kasumi-1 cells after RUNX1/ETO knockdown with and without concomitant CDKN1B knockdown. I E2F1 protein levels in Kasumi-1 cells after RUNX1/ETO knockdown with and without concomitant CDKN1B knockdown at day 10. J Under phosphorylated RB levels in Kasumi-1 cells after RUNX1/ETO knockdown with and without concomitant CDKN1B knockdown. K Co-IP of E2F1/Rb complex, levels in Kasumi-1 cells after RUNX1/ETO knockdown with and without concomitant CDKN1B knockdown Ip underphosphorylated Rb blot E2F1. Input sample (blot for E2F1 and Rb), IgG was used as the isotype control. siRE RUNX1/ETO siRNA; siCDKN1B CDKN1B siRNA; siMM mismatch control. Error bars indicate standard deviations from three independent experiments. Asterisk indicates p value < 0.05
These changes in protein levels led to reduced E2F1 binding to the TERT promoter but not at a control site located 3 kb upstream of the promoter as shown by chromatin immunoprecipitation (ChIP) experiments (Figs. 4F, G). RUNX1/ETO knockdown caused a threefold diminished binding of E2F1 to the TERT promoter (p < 0.001). Co-knockdown of RUNX1/ETO and CDKN1B restored occupation of the TERT promoter by E2F1, while CDKN1B knockdown alone led to a slightly increased E2F1 binding (Figs. 4F, G). This is further supported by reduced E2F1 protein levels (Fig. 4B, I) and a slight increase in underphosphorylated Rb levels by 1.2-fold upon RUNX1/ETO suppression (Fig. 4J).
Co-immunoprecipitation showed an increase in the RB/E2F1 complex upon RUNX1-ETO knockdown (Fig. 4K). Complex formation could be observed as E2F1 and RB signal increased upon RUNX1-ETO suppression following immunoprecipitation using underphosphorylated RB antibody. IgG was used as the isotype control.
Discussion
Because of its pivotal role in maintaining chromosome integrity and self-renewal, maintaining telomerase activity by activating TERT expression is central to most malignant transcription programs. TERT transcription is maintained by multiple mechanisms including promoter mutations, epigenetic changes and differential transcription factor binding. Here we show that the leukemic RUNX1/ETO fusion protein drives TERT expression directly by modulating histone acetylation and indirectly by facilitating E2F1 binding to the TERT promoter by destabilizing CDKN1B (Fig. 5).
Fig. 5.
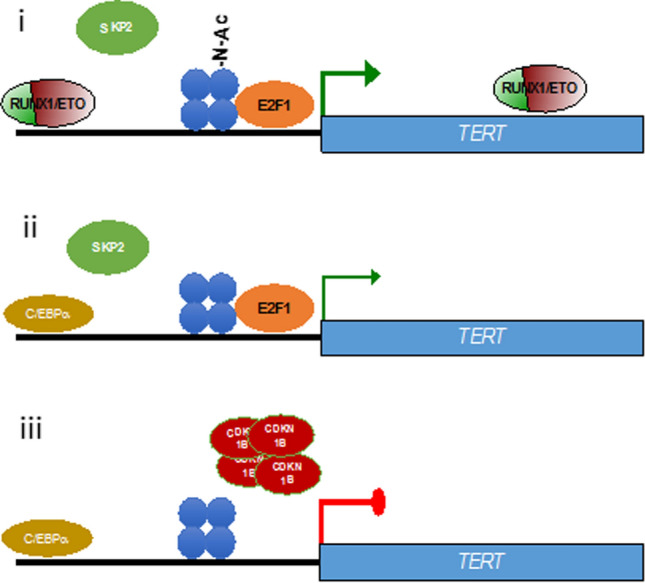
Model for TERT regulation. Model for regulation of TERT expression by RUNX1/ETO. (i) RUNX1/ETO binds to a -18 kb upstream element and to intron 6. Loss of RUNX1/ETO diminishes H3K27 acetylation at the TERT promoter (ii) and (iii). Subsequent depletion of SKP2 and accumulation of CDKN1B causes E2F1 destabilization; the associated loss of E2F1 binding on the TERT promoter represses TERT expression. Blue dots; nucleosome (H3K27), Blue dots with N-AC; H3K27 acetylation
Our ChIP-seq data revealed a RUNX1/ETO binding site located 18 kb upstream of the TERT TSS, which is also occupied by additional hematopoietic factors including RUNX1, PU.1 (SPI1) and LMO2. Intriguingly, loss of RUNX1/ETO binding results also in recruitment of C/EBPα to this location. C/EBPα has been previously identified as a transcriptional repressor of TERT and important factor for the differentiation of myeloid progenitors [37]. On the other hand, depletion of RUNX1/ETO is also associated with reduced binding of E2F1 to the TERT promoter region. E2F1 coordinates gene expression with the cell cycle. In complex with RB1 it represses gene expression. Phosphorylation of RB1 by CDK4, 6 and later 2 leads to its dissociation and the activation of gene expression by E2F1. Our data suggest that RUNX1/ETO keeps E2F1 in the active state by preventing the accumulation of the CDK2 inhibitor CDKN1B. In conclusion, RUNX1/ETO maintains TERT expression by establishing an active chromatin by direct occupation of the − 18 kb element and by facilitating E2F1-mediated transcription via an SKP2-CDKN1B axis. Nevertheless, the dominant mechanism by which RUNX1/ETO regulates TERT merits further investigation and require additional experimental validations. Furthermore, the more heterogeneous results regarding TERT expression in AML patients as reported by BloodSpot and Vizome databases [38, 39] as well as the binding of RUNX1/ETO on the − 18 kb loci upstream of the TERT TSS could be due to clonal heterogeneity and the lower quality of AML primary samples which require further scrutiny. This discrepancy could be resolved in part by examining single cell RNA seq data of AML patients’ which has recently been made available [40] and will constitute part of future validation works. Additionally, it would also be interesting to examine of regulation of TERT in other AML sub-types which lack RUNX1/ETO.
It is currently unknown how precisely RUNX1/ETO maintains SKP2 levels. SKP2 is being polyubiquitinated by the APC-CDH1 complex [31]. Vice versa, CDH1 levels are controlled by several cullin E3 ligases including SKP2 [41]. It is tempting to speculate that RUNX1/ETO might shift the balance of this reciprocal feedback loop to the favor of SKP2.
Higher expression of TERT is generally linked to higher proliferative capacity and enhanced oncogenic potential. Impairing TERT expression by targeting RUNX1/ETO or the SKP2-CDKN1B-E2F1 axis may offer new therapeutic possibility for the treatment of AML in future.
In summary, we show here that the leukemic RUNX1/ETO controls telomerase activity by directly and indirectly modulating the expression of its reverse transcriptase subunit TERT. These findings provide a paradigm for how an initiator and driver of leukemia corrupts a central component of genome integrity and self-renewal and further highlight the significance of continuous RUNX1/ETO expression for leukemic propagation.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file2 Figure S1: TERT is a direct target gene of RUNX1/ETOA,B RUNX1/ETO and TERT levels after 10 days of RUNX1/ETO knockdown in SKNO-1 cells. C, Telomerase Activity after RUNX1/ETO knockdown for 10 days in SKNO-1. D, UCSC screenshot displaying changes in RUNX1/ETO binding and gene expression at the TERT locus upon 2 days of RUNX1/ETO knockdown in Kasumi-1 cells. E, UCSC screenshot displaying changes in RUNX1/ETO binding, histone modifications and DNase hypersensitive site on TERT Locus upon RUNX1/ETO knockdown. siRE, RUNX1/ETO siRNA; siMM, mismatch control. Asterisk indicates p-value<0.05. F, Clonogenic growth of Kasumi-1 cells upon RUNX1/ETO, CDKN1B and SKP2 knockdown, respectively (Absolute Count).Figure S2: CDKN1B, SKP2 and TERC Transcript LevelsA, CDKN1B transcript levels in Kasumi-1 cells with siRE B, SKP2 transcript levels in Kasumi-1 cells with siRE. C, CDKN1B transcript levels with siSKP2. D, E, TERC transcript levels upon RUNX1/ETO and SKP2 knockdown, respectively. siRE, RUNX1/ETO siRNA; siSKP2, SKP2 siRNA siCDKN1B, CDKN1B siRNA; siMM, mismatch control. Error bars indicate standard deviations from three independent experiments. (PPTX 2467 kb)
Author contributions
Concept and supervision were contributed by OH and NMY. Experiments were contributed by EJM, AA, SM, KKZ, AS and FB. Data analysis was contributed by EJM, AA, MYY, FB, NMY and OH. Manuscript writing was contributed by EJM, AA, MYY, FB and OH.
Funding
This study was supported by Research University grants 1001/CIPPT/8012265 and 1001/CIPPT/813064 from Universiti Sains Malaysia to EJM and NMY, and Children with Cancer UK project grant 17–245 and Kika programme grant 329 to OH.
Availability of data and materials
Will be made available upon request.
Declarations
Conflict of interest
There are no disclosures or conflict of interest statement to be made.
Ethical approval and consent to participate
Not applicable.
Consent for publication
All authors have read the manuscript and consented for publication.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Emmanuel J. Moses, Email: emmanuel_jm@usm.my
Olaf Heidenreich, Email: o.t.heidenreich@prinsesmaximacentrum.nl, Email: olaf.heidenreich@ncl.ac.uk.
References
- 1.Trybek T, Kowalik A, Góźdź S, Kowalska A. Telomeres and telomerase in oncogenesis (review) Oncol Lett. 2020;20(2):1015–1027. doi: 10.3892/ol.2020.11659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yik MY, Azlan A, Rajasegaran Y, Rosli A, Yusoff NM, Moses EJ. Mechanism of human telomerase reverse transcriptase (hTERT) regulation and clinical impacts in leukemia. Genes (Basel) 2021;12(8):1188. doi: 10.3390/genes12081188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yuan X, Larsson C, Xu D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: old actors and new players. Oncogene. 2019;38(34):6172–6183. doi: 10.1038/s41388-019-0872-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu T, Yuan X, Xu D. Cancer-specific telomerase reverse transcriptase (TERT) promoter mutations: biological and clinical implications. Genes. 2016;7(7):38. doi: 10.3390/genes7070038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shay JW, Wright WE. Telomeres and telomerase: three decades of progress. Nat Rev Genet. 2019;20(5):299–309. doi: 10.1038/s41576-019-0099-1. [DOI] [PubMed] [Google Scholar]
- 6.Crowe DL, Nguyen DC. Rb and E2F–1 regulate telomerase activity in human cancer cells. Biochim et Biophys Acta (BBA) Gene Struct Expr. 2001;1518(1–2):1–6. doi: 10.1016/s0167-4781(00)00296-7. [DOI] [PubMed] [Google Scholar]
- 7.Won J, Yim J, Kim TK. Opposing regulatory roles of E2F in human telomerase reverse transcriptase (hTERT) gene expression in human tumor and normal somatic cells. FASEB J. 2002;16(14):1943–1945. doi: 10.1096/fj.02-0311fje. [DOI] [PubMed] [Google Scholar]
- 8.Endoh T, Tsuji N, Asanuma K, Yagihashi A, Watanabe N. Survivin enhances telomerase activity via up-regulation of specificity protein 1-and c-Myc-mediated human telomerase reverse transcriptase gene transcription. Exp Cell Res. 2005;305(2):300–311. doi: 10.1016/j.yexcr.2004.12.014. [DOI] [PubMed] [Google Scholar]
- 9.Gessner A, Thomas M, Castro PG, Büchler L, Scholz A, Brümmendorf TH, et al. Leukemic fusion genes MLL/AF4 and AML1/MTG8 support leukemic self-renewal by controlling expression of the telomerase subunit TERT. Leukemia. 2010;24(10):1751–1759. doi: 10.1038/leu.2010.155. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi A, Higashino F, Aoyagi M, Yoshida K, Itoh M, Kyo S, et al. EWS/ETS fusions activate telomerase in Ewing’s tumors. Can Res. 2003;63(23):8338–8344. [PubMed] [Google Scholar]
- 11.Martinez-Soria N, McKenzie L, Draper J, Ptasinska A, Issa H, Potluri S, et al. The oncogenic transcription factor RUNX1/ETO corrupts cell cycle regulation to drive leukemic transformation. Cancer Cell. 2018;34(4):626–642. doi: 10.1016/j.ccell.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heidenreich O, Krauter J, Riehle H, Hadwiger P, John M, Heil G, et al. AML1/MTG8 oncogene suppression by small interfering RNAs supports myeloid differentiation of t(8;21)-positive leukemic cells. Blood. 2003;101(8):3157–3163. doi: 10.1182/blood-2002-05-1589. [DOI] [PubMed] [Google Scholar]
- 13.Bradford NA. A rapid and sensitive method for the quantitation microgram quantities of a protein isolated from red cell membranes. Anal Biochem. 1976;72(248):e254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 14.Martinez N, Drescher B, Riehle H, Cullmann C, Vornlocher HP, Ganser A, et al. The oncogenic fusion protein RUNX1-CBFA2T1 supports proliferation and inhibits senescence in t(8;21)-positive leukaemic cells. BMC Cancer. 2004;6(4):44. doi: 10.1186/1471-2407-4-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ptasinska A, Pickin A, Assi SA, Chin PS, Ames L, Avellino R, et al. RUNX1-ETO depletion in t(8;21) AML leads to C/EBPα- and AP-1-mediated alterations in enhancer-promoter interaction. Cell Rep. 2019;28(12):3022–3031.e7. doi: 10.1016/j.celrep.2019.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wege H, Chui MS, Le HT, Tran JM, Zern MA. SYBR Green real-time telomeric repeat amplification protocol for the rapid quantification of telomerase activity. Nucleic Acids Res. 2003;31(2):E3–3. doi: 10.1093/nar/gng003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fishilevich S, Nudel R, Rappaport N, Hadar R, Plaschkes I, Iny Stein T, Rosen N, Kohn A, Twik M, Safran M, Lancet D, Cohen D. GeneHancer: genome-wide integration of enhancers and target genes in genecards. Database (Oxford) 2017 doi: 10.1093/database/bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ptasinska A, Assi SA, Mannari D, James SR, Williamson D, Dunne J, et al. Depletion of RUNX1/ETO in t(8;21) AML cells leads to genome-wide changes in chromatin structure and transcription factor binding. Leukemia. 2012;26(8):1829–1841. doi: 10.1038/leu.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ptasinska A, Assi SA, Martinez-Soria N, Imperato MR, Piper J, Cauchy P, et al. Identification of a dynamic core transcriptional network in t(8;21) AML that regulates differentiation block and self-renewal. Cell Rep. 2014;8(6):1974–1988. doi: 10.1016/j.celrep.2014.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martens JH, Mandoli A, Simmer F, Wierenga BJ, Saeed S, Singh AA, Altucci L, Vellenga E, Stunnenberg HG. ERG and FLI1 binding sites demarcate targets for aberrant epigenetic regulation by AML1-ETO in acute myeloid leukemia. Blood. 2012;120(19):4038–4048. doi: 10.1182/blood-2012-05-429050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Y, Man N, Karl D, Martinez C, Liu F, Sun J, Martinez CJ, Martin GM, Beckedorff F, Lai F, Yue J, Roisman A, Greenblatt S, Duffort S, Wang L, Sun X, Figueroa M, Shiekhattar R, Nimer S. TAF1 plays a critical role in AML1-ETO driven leukemogenesis. Nat Commun. 2019;10(1):4925. doi: 10.1038/s41467-019-12735-z.PMID:31664040;PMCID:PMC6820555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stengel KR, Ellis JD, Spielman CL, Bomber ML, Hiebert SW. Definition of a small core transcriptional circuit regulated by AML1-ETO. Mol Cell. 2021;81(3):530–545. doi: 10.1016/j.molcel.2020.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gizard F, Nomiyama T, Zhao Y, Findeisen HM, Heywood EB, Jones KL, et al. The PPARα/p16INK4a pathway inhibits vascular smooth muscle cell proliferation by repressing cell cycle–dependent telomerase activation. Circ Res. 2008;103(10):1155–1163. doi: 10.1161/CIRCRESAHA.108.186205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez-Soria N, McKenzie L, Nakjang S, Forster V, Isa A, Blair HJ, et al. CCND2 is a RUNX1/ETO target required for leukaemic propagation. Klin Padiatr. 2017;229(03):A10. [Google Scholar]
- 25.McKenzie L, Martinez-Soria N, Draper J, Nakjang S, Blair HJ, Wichmann C, et al. Identification of CCND2 as a RUNX1/ETO target required for leukaemic propagation. Blood. 2016;128(22):835. doi: 10.1182/blood.V128.22.835.835. [DOI] [Google Scholar]
- 26.Lee SH, Kim JW, Oh SH, Kim YJ, Rho SB, Park K, et al. IFN-γ/IRF-1-induced p27kip1 down-regulates telomerase activity and human telomerase reverse transcriptase expression in human cervical cancer. FEBS Lett. 2005;579(5):1027–1033. doi: 10.1016/j.febslet.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Satoh T, Kaida D. Upregulation of p27 cyclin-dependent kinase inhibitor and a C-terminus truncated form of p27 contributes to G1 phase arrest. Sci Rep. 2016;6(1):1–8. doi: 10.1038/srep27829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hume S, Dianov GL, Ramadan K. A unified model for the G1/S cell cycle transition. Nucleic Acids Res. 2020;48(22):12483–12501. doi: 10.1093/nar/gkaa1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999;1(4):193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- 30.Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27Kip1 ubiquitination and degradation is regulated by the SCFSkp2 complex through phosphorylated Thr187 in p27. Curr Biol. 1999;9(12):661–S2. doi: 10.1016/S0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- 31.Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 2004;428(6979):194–8. doi: 10.1038/nature02381. [DOI] [PubMed] [Google Scholar]
- 32.Bretones G, Acosta JC, Caraballo JM, Ferrándiz N, Gómez-Casares MT, Albajar M, et al. SKP2 oncogene is a direct MYC target gene and MYC down-regulates p27KIP1 through SKP2 in human leukemia cells. J Biol Chem. 2011;286(11):9815–25. doi: 10.1074/jbc.M110.165977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez S, Abundis C, Boccalatte F, Mehrotra P, Chiang MY, Yui MA, et al. Therapeutic targeting of the E3 ubiquitin ligase SKP2 in T-ALL. Leukemia. 2020;34(5):1241–52. doi: 10.1038/s41375-019-0653-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu T, Gu X, Cui H. Emerging roles of SKP2 in cancer drug resistance. Cells. 2021;10(5):1147. doi: 10.3390/cells10051147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alonso MM, Fueyo J, Shay JW, Aldape KD, Jiang H, Lee OH, et al. Expression of transcription factor E2F1 and telomerase in glioblastomas: mechanistic linkage and prognostic significance. J Natl Cancer Inst. 2005;97(21):1589–600. doi: 10.1093/jnci/dji340. [DOI] [PubMed] [Google Scholar]
- 36.Broude EV, Swift ME, Vivo C, Chang BD, Davis BM, Kalurupalle S, et al. p21 Waf1/Cip1/Sdi1 mediates retinoblastoma protein degradation. Oncogene. 2007;26(48):6954–8. doi: 10.1038/sj.onc.1210516. [DOI] [PubMed] [Google Scholar]
- 37.Kumar M, Witt B, Knippschild U, Koch S, Meena JK, Heinlein C, et al. CEBP factors regulate telomerase reverse transcriptase promoter activity in whey acidic protein-T mice during mammary carcinogenesis. Int J Cancer. 2013;132(9):2032–43. doi: 10.1002/ijc.27880. [DOI] [PubMed] [Google Scholar]
- 38.Bagger FO, Kinalis S. Rapin N (2019) BloodSpot: a database of healthy and malignant haematopoiesis updated with purified and single cell mRNA sequencing profiles. Nucleic Acids Res. 2019;47(D1):D881–5. doi: 10.1093/nar/gky1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Expression stratification: Vizome [Internet] (2022). http://vizome.org/aml/expression_strat/. Accessed 07 May 2022
- 40.Zhai Y, Singh P, Dolnik A, Brazda P, Atlasy N, Del Gaudio N, Döhner K, Döhner H, Minucci S, Martens J, Altucci L, Megchelenbrink W, Bullinger L, Stunnenberg HG. Longitudinal single-cell transcriptomics reveals distinct patterns of recurrence in acute myeloid leukemia. Mol Cancer. 2022;21(1):166. doi: 10.1186/s12943-022-01635-4.PMID:35986270;PMCID:PMC9389773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choudhury R, Bonacci T, Arceci A, Lahiri D, Mills CA, Kernan JL, et al. APC/C and SCFcyclin F constitute a reciprocal feedback circuit controlling S-phase entry. Cell Rep. 2016;16(12):3359–72. doi: 10.1016/j.celrep.2016.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file2 Figure S1: TERT is a direct target gene of RUNX1/ETOA,B RUNX1/ETO and TERT levels after 10 days of RUNX1/ETO knockdown in SKNO-1 cells. C, Telomerase Activity after RUNX1/ETO knockdown for 10 days in SKNO-1. D, UCSC screenshot displaying changes in RUNX1/ETO binding and gene expression at the TERT locus upon 2 days of RUNX1/ETO knockdown in Kasumi-1 cells. E, UCSC screenshot displaying changes in RUNX1/ETO binding, histone modifications and DNase hypersensitive site on TERT Locus upon RUNX1/ETO knockdown. siRE, RUNX1/ETO siRNA; siMM, mismatch control. Asterisk indicates p-value<0.05. F, Clonogenic growth of Kasumi-1 cells upon RUNX1/ETO, CDKN1B and SKP2 knockdown, respectively (Absolute Count).Figure S2: CDKN1B, SKP2 and TERC Transcript LevelsA, CDKN1B transcript levels in Kasumi-1 cells with siRE B, SKP2 transcript levels in Kasumi-1 cells with siRE. C, CDKN1B transcript levels with siSKP2. D, E, TERC transcript levels upon RUNX1/ETO and SKP2 knockdown, respectively. siRE, RUNX1/ETO siRNA; siSKP2, SKP2 siRNA siCDKN1B, CDKN1B siRNA; siMM, mismatch control. Error bars indicate standard deviations from three independent experiments. (PPTX 2467 kb)
Data Availability Statement
Will be made available upon request.