


Abstract
The cDNA encoding the human RNA lariat debranching enzyme (hDBR1) was identified and cloned by searching the Expressed Sequence Tag (EST) database and screening a HeLa cDNA library, based on predicted amino acid sequence homologies with the Saccharomyces cerevisiae, Schizosaccharomyces pombe and Caenorhabditis elegans debranching enzymes. The hDBR1 cDNA expressed in Escherichia coli showed debranching activity in vitro and was also shown to be functional in an interspecies specific complementation experiment. hDBR1 cDNA in a S.cerevisiae expression vector complemented the intron accumulation phenotype of a S.cerevisiae dbr1 null mutant. Integration of the cDNA for hDBR1 into the ura4 locus of S.pombe also complemented both the intron accumulation and slow growth phenotypes of a S.pombe dbr1 null mutant strain. Comparison of the amino acid sequence of hDBR1 with the other DBR protein sequences showed several conserved regions, with 40, 44 and 43% identity to the S.cerevisiae, S.pombe and C.elegans debranching enzymes, respectively.
INTRODUCTION
An enzymatic activity that specifically hydrolyzes 2′-5′ branched phosphodiester bonds was first identified in HeLa cell extracts and named RNA lariat debranching enzyme (1). This RNA lariat debranching enzyme (DBR) cleaves the 2′-5′ phosphodiester linkage at the branch point of excised lariat intron RNA and converts them into linear molecules. These linear molecules are subsequently degraded in vivo, with a half-life of only a few seconds, by as yet undefined exonucleases (2). In mammalian cells enormous numbers of lariat intron RNAs are generated by a two-step pre-mRNA splicing reaction and discarded during the process of normal gene expression. Thus lariat intron RNA degradation is likely to be an important pathway for normal cellular function. Only one component of the intron turnover pathway, the RNA lariat debranching enzyme, has been identified in mammalian cells (1) and in both budding and fission yeasts (3–5). We will refer to the human debranching enzyme as hDBR1, fission yeast debranching enzyme as SpDBR1 and budding yeast debranching enzyme as ScDBR1.
The gene encoding RNA lariat debranching enzyme, ScDBR1, was identified and first cloned in the budding yeast Saccharomyces cerevisiae using a genetic screen aimed at identifying cellular factors involved in Ty1 retrotransposition (4). The ScDBR1 protein was purified to near homogeneity from a bacterial overexpression strain (6). The purified ScDBR1 enzyme was shown to digest a variety of branched nucleic acid substrates, including lariat RNAs derived from pre-mRNA splicing, self-splicing group II introns, multi-copy satellite DNAs (msDNA) and a variety of synthetic branched RNAs (6). The cDNAs encoding ScDBR1 homologs were subsequently cloned from Schizosaccharomyces pombe (SpDBR1) and Caenorhabditis elegans (CeDBR1) and shown to represent authentic debranching enzymes by interspecies specific complementation experiments (5).
The dbr1 mutation in S.cerevisiae reduces Ty1 transposition frequency ∼10-fold and also produces a severe defect in intron degradation, i.e. excised introns accumulate to very high levels in the form of lariat structures lacking their 3′-tail (4). Thus, debranching apparently represents a rate limiting step in the intron degradation pathway. Furthermore, several recent studies have shown that efficient processing of certain intronically encoded small nucleolar RNAs (snoRNAs U24, snR38, U18 and snR39) also requires debranching enzyme activity in the yeast S.cerevisiae (7,8). In contrast to the budding yeast S.cerevisiae dbr1 null mutant strain, which has a relatively normal growth rate and cell morphology, the dbr1 null mutant in the fission yeast S.pombe has a severe growth defect and shows an aberrant elongated cell shape in addition to an intron accumulation phenotype. In S.cerevisiae and S.pombe 2.5 and 40% of genes contain introns, respectively (9–11). Furthermore, S.pombe genes often have multiple introns, while S.cerevisiae genes usually have a single intron. Thus it is not surprising that DBR1 is a non-essential gene in S.cerevisiae and that the accumulation of excised introns has no deleterious effect on cell growth, i.e. the rapid turnover of excised introns is not critical in S.cerevisiae (4). However, efficient turnover of excised introns may be important or essential in more complex eukaryotes. In the S.pombe dbr1 null mutant strain blockage of intron degradation showed a severe cell growth defect and an unusual elongated cell shape (5). Thus the growth phenotype of the S.pombe dbr1 mutant suggests that the debranching activity may be much more critical in higher eukaryotes, in which nearly all genes contain more intron than exon sequences.
As a first step to evaluating the role of debranching enzyme in multicellular eukaryotes we sought to identify the cDNA encoding it. In this paper we describe a human cDNA and show that it indeed encodes a debranching activity through an interspecies specific complementation test in budding and fission yeasts.
MATERIALS AND METHODS
The nucleotide sequence of the hDBR1 cDNA reported in this paper has been submitted to the GenBank database with accession no. AF180919.
Strains and growth media
The Escherichia coli, S.cerevisiae and S.pombe strains used in this study are described in Table 1. The yeast strains were grown at 30°C in YPD (10 g/l yeast extract, 20 g/l BactoTryptone, 20 g/l glucose and 0.32 g/l tryptophan) for S.cerevisiae, YEC (5 g/l yeast extract, 2 g/l Casamino acids and 30 g/l glucose) for S.pombe or synthetic complete (SC) medium complemented with 250 µg/ml uracil, adenine or amino acid additives. All yeast media contained 2% (w/v) glucose for S.cerevisiae or 3% for S.pombe as the carbon source unless otherwise specified. SC medium plates were prepared as previously described by Rose et al. (12). The yeast strains were transformed by the lithium acetate method (13). All E.coli strains were grown at 37°C in LB (10 g/l BactoTryptone, 5 g/l yeast extract, 10 g/l NaCl) with 100 µg/ml ampicillin or 50 µg/ml tetracycline.
Table 1. Strains used in this study.
| Strain | Genotype |
|---|---|
| Saccharomyces cerevisiae | |
| JB224 | MATa his3Δ200 ura3-167GAL+ |
| KC106 | MATa trp1Δ1 his3Δ200 leu2Δ1 ura3-167 dbr1-1 |
| Schizosaccharomyces pombe | |
| YKN163 (BP427) | h– ura4D-18 leu1-32 ade6-M210 |
| YKN164 | h+ ade6-M210 ura4-294 leu1-32 dbr1::leu1+ |
| YKN165 | h+ ade6-M210 ura4-294::pJK210 leu1-32 dbr1::leu1+ |
| YKN204 | h+ ade6-M210 ura4-294::pKN207/StuI leu1-32 dbr1::leu1+ |
| Escherichia coli | |
| XL1-Blue | Δ(mcrA)183, Δ(mcrCB-hsdSMR-mrr)173, endA1, supE44, thi-1, recA1, gyrA96, relA1, lac[F′proAB, lacIqZΔM15, Tn(tetr)] |
| SOLR™ | e14-(mrcA), Δ(mcrCB-hsdSMR-mrr)171, sbcC, recB, recJ, umuC::Tn5(kanr), uvrC, lac, gyrA96, relA1, thi-1, endA1, λR[F′proAB, lacIq,ZΔM15]Su– (non-suppressing) |
| BL21(DE3) | F– ompT hsdSB (rB–mB–) gal dcm (DE3) |
Plasmid preparation
The plasmids used in this study are listed in Table 2 and were prepared with a Qiagen kit. The oligonucleotides used in this study are listed in Table 3. Plasmid pKN200 containing full-length hDBR1 cDNA was obtained by in vivo excision from a positive clone obtained from a λ ZAP II HeLa cDNA library screen. In vivo excision was performed according to the manufacturer’s instructions (Stratagene). Plasmid pKN201 was constructed by inserting PCR amplified full-length hDBR1 product into the pCRRII-TOPO TA cloning vector (Invitrogen). To amplify hDBR1 by PCR, KBN62 (sense primer containing a NdeI site) and KBN55 (antisense primer containing a BamHI site) were used with pKN200 as template. For construction of E.coli expression plasmid pKN203 a NdeI–BamHI full-length hDBR1 cDNA fragment from pKN201 was subcloned into NdeI and BamHI digested pET28a (Novagen). The yeast expression plasmids pKN205 and pKN206 were constructed by inserting an EcoRI–BamHI full-length hDBR1 cDNA fragment from pKN201 into S.cerevisiae expression vectors pRS314GU (14) and pRS316GU (14) digested with EcoRI and BamHI, respectively. The plasmids pRS314GU (Trp+) and pRS316GU (Ura+) are basically the same except for the different selection marker. To integrate full-length hDBR1 cDNA into the S.pombe genome at the ura4 locus, the EcoRI and BamHI digested hDBR1 fragment from pKN201 was subcloned into the S.pombe integration vector pJK210 (15) digested with EcoRI and BamHI (pKN207). Then linearized pKN207 (pKN207 cut with StuI) was used to transform S.pombe dbr1 mutant strain YKN164 (5). The hDBR1 cDNA was without its own promoter and was placed under control of the S.pombe ura4+ promoter.
Table 2. Plasmids used in this study.
| Plasmid | Description |
|---|---|
| pKN200 | hDBR1 in pBlueScript (from λ ZAP II library by in vivo excision) |
| pKN201 | hDBR1 PCR product in pCRRII-TOPO TA cloning vector |
| pKN203 | NdeI–BamHI fragment of hDBR1 in pET28a |
| pKN205 | EcoRI–BamHI fragment of hDBR1 in pRS314GU |
| pKN206 | EcoRI–BamHI fragment of hDBR1 in pRS316GU |
| pKN207 | EcoRI–BamHI fragment of hDBR1 in pJK210 |
| pCRRII-TOPO | TA cloning vector (Invitrogen) |
| pET28a | Escherichia coli expression vector (Novagen) |
| pJK210 | Schizosaccharomyces pombe integration vector (15) |
| pRS314GU | Saccharomyces cerevisiae expression vector (14) |
| pRS316GU | Saccharomyces cerevisiae expression vector (14) |
Table 3. Primers used in this study.
| Primer | Sequence |
|---|---|
| KBN19 | 5′-GGA AAA TAA CAC ATT AGG AAG T-3′ |
| KBN20 | 5′-CCA AGT AAG TCA GGA GCA CTG G-3′ |
| KBN55 | 5′-CCT AGT CTA GAG GAT CCA GTC TAG GTG CAA GAG TGA AAC-3′ |
| KBN62 | 5′-ATA TGG AAT TCC ATA TGC GGG TGG CTG TGG CTG G-3′ |
Human cDNA library screen
A HeLa cDNA library was purchased from Stratagene. Escherichia coli strain XL-1 Blue was infected with the λ ZAP II HeLa cDNA library and screened by plaque hybridization as suggested by the manufacturer (Stratagene).
Complementation of S.cerevisiae and S.pombe dbr1 mutant strains by hDBR1 cDNA
To test whether hDBR1 could complement a S.cerevisiae dbr1 mutant, pKN205 (full-length hDBR1 cDNA in pRS314GU) or pKN206 (full-length hDBR1 cDNA in pRS316GU) were used to transform S.cerevisiae dbr1 mutant strain KC106 by selecting for Trp+ for pKN205 or for Ura+ for pKN206 colonies. SC Trp– or SC Ura– medium containing 1% raffinose as the carbon source was inoculated with positive transformants and they were grown at 30°C overnight. Cells were harvested and washed with distilled water. The washed cells were grown in SC Trp– or SC Ura– medium in the presence of 2% glucose for repression or 2% galactose for induction of the hDBR1 gene for 1–2 days at 30°C. RNAs were then prepared by a glass bead method (4) from glucose-repressed or galactose-induced cells and intron accumulation was monitored by RNA blotting. For complementation of the S.pombe dbr1 mutant, full-length hDBR1 cDNA was integrated into the ura4 locus on the S.pombe chromosome. Plasmid pKN207 was constructed by inserting full-length hDBR1 cDNA into S.pombe integration vector pJK210. For integration, plasmid pKN207 were linearized with StuI, which cuts only ura4, and used to transform Sp-dbr1::leu1+ knockout strain YKN164. For the positive integrants, Ura+ transformants were selected and analyzed by PCR and Southern blot analysis. RNAs were prepared by a glass bead method (4) and intron accumulation was monitored by northern blot analysis.
Expression and purification of His-tagged hDBR1 enzyme
To express hDBR1 protein, full-length hDBR1 cDNA was coupled to a His tag and placed under control of the T7 promoter in E.coli expression vector pET28a (pKN203). A single colony harboring pKN203 was inoculated and grown in LB with tetracycline at 37°C. When the culture reached an OD600 of 0.6, 1 mM IPTG was added to induce hDBR1. Cells were harvested 3–4 h after IPTG induction, disrupted and purified on a His-Bind Resin (Novagen) column. All steps for purifying hDBR1 enzyme were performed at 0–4°C and purification was carried out according to the manufacturer’s instructions (Novagen). The purified protein was further concentrated with Centriprep PM10 (Amicon). Crude and purified hDBR1 were separated by 10–20% gradient SDS–PAGE, transferred to Immobilon-P membrane (Millipore) and subjected to western blot analysis using anti-His tag antibody (Qiagen). Western blotting was performed with an ECL system according to the manufacturer’s instructions (Amersham Pharmacia Biotech).
In vitro RNA lariat debranching enzyme assay
The in vitro debranching enzyme assay was performed as previously described by Nam et al. (6). The substrate for hDBR1 enzyme was purified from an E.coli strain harboring plasmid pDB808 which contained msDNA Ec86 (6). The msDNA was gel purified after labeling with [γ-32P]ATP. The induced (+IPTG) and uninduced (–IPTG) cell extracts (S30) were prepared from cells harboring pKN203 (full-length hDBR1 cDNA coupled to a His tag and placed under control of the T7 promoter in E.coli expression vector pET28a). Briefly, cells were harvested, resuspended with 0.1 M Tris–HCl, pH 7.5, 2 mM EDTA, 1% Triton X-100 and 100 µg/ml lysozyme and lysed by incubating for 30 min at 37°C. The membrane fraction was removed by centrifugation at 30 000 g for 1 h (S30 extract). For in vitro debranching enzyme assay the gel purified msDNA was incubated with S30 extract in a 20 µl reaction mixture containing 20 mM HEPES–KOH, pH 7.6, 40 mM KCl, 3 mM MgCl2, 1 mM DTT and 10% glycerol (v/v) for 30 min at 30°C. The reaction was stopped by adding an equal volume of formamide dye and the product of the debranching reaction was analyzed on a 20% polyacrylamide gel with 8 M urea.
RESULTS
Cloning and expression of hDBR1 cDNA and in vitro debranching enzyme assay
A 388 bp partial cDNA sequence of hDBR1, homologous to the S.cerevisiae DBR1 gene, was identified by searching the Expressed Sequence Tag (EST) database (human pancreatic islet cell clone zc90d2.r1, dbEST accession no. W53026). Based on the DNA sequence of this partial cDNA clone, we designed two primers (KBN19 and KBN20) and amplified a 240 bp hDBR1 cDNA fragment from HeLa cell mRNA by RT–PCR. Using this RT–PCR product as probe, a HeLa cDNA library was screened. Two positive plaques were isolated out of ∼1 000 000 individual plaques screened. The 2.4 kb λ ZAP II cloned hDBR1 cDNA fragment was rescued by in vivo excision (pKN200) and sequenced by the dideoxy chain termination method and the sequence reconfirmed using an ABI automatic sequencer (GenBank accession no. AF180919). The hDBR1 cDNA sequence contains a single 545 amino acid open reading frame (ORF) encoding a protein of 62 kDa. Comparison of the predicted amino acid sequence of hDBR1 to the other DBR sequences showed several regions of conserved sequence, especially within the N-terminal portion of the first 200 amino acids (Fig. 1). Since the alignment of hDBR1 with the other DBR proteins suggested that the hDBR1 clone represents a homolog of ScDBR1, we checked whether this protein could be expressed and had debranching activity. To express the hDBR1 gene, the full-length cDNA of hDBR1 was coupled to a His tag and placed under control of the T7 promoter in an E.coli expression vector. The hDBR1 protein expressed in E.coli was purified on a His-Bind® Resin column and showed >80% homogeneity after one step of purification (Fig. 2A). Since the expressed hDBR1 protein was coupled to a His tag and a thrombin cleavage site, purified hDBR1 protein ran a little slower than the expected size of the native protein, 62 kDa on a 10–20% gradient SDS–PAGE (Fig. 2A). The purified hDBR1 protein was confirmed by western blot analysis using an anti-His tag antibody (Fig. 2B). To test whether the expressed hDBR1 had debranching activity, we prepared cell extracts from induced (+IPTG) and uninduced (–IPTG) cultures and performed a debranching enzyme assay in vitro with E.coli msDNA Ec86 as substrate (6). Figure 3 shows that the msDNA Ec86 incubated with induced extract was cleaved and generated a pAGCp trinucleotide debranching product (lanes 2 and 3). However, the msDNA Ec86 incubated with uninduced extract showed intact msDNA without the debranched product (lanes 4 and 5). The western blot analysis and in vitro debranching enzyme assay showed that the cloned hDBR1 cDNA sequence encodes an authentic human RNA lariat debranching enzyme that is a homolog of ScDBR1, SpDBR1 and CeDBR1.
Figure 1.

Amino acid sequence alignment of S.cerevisiae (ScDBR1), S.pombe (SpDBR1), C.elegans (CeDBR1), A.thaliana (AtDBR1), mouse (mDBR1) and human (hDBR1) RNA lariat debranching enzymes. Amino acid sequences were aligned using the Genetics Computer group PILEUP program and identities boxed using BOXSHADE.
Figure 2.

Expression and western blot analysis of hDBR1. (A) Expression of hDBR1. To overexpress hDBR1, hDBR1 cDNA was subcloned into E.coli overexpression vector pET28a and expressed by adding IPTG. Total E.coli extract (S30) was directly loaded onto a His-Bind® Resin column and hDBR1 protein eluted with a high concentration of imidazole. Then the purified protein was concentrated, loaded on a 10–20% gradient SDS–PAGE gel and stained with Coomassie brilliant R staining solution. Lane M, size standard marker; lane 1, uninduced cell lysate (–IPTG); lane 2, induced cell lysate (+IPTG); lane 3, purified His-tagged hDBR1 protein. (B) Western blot analysis. Proteins separated by SDS–PAGE were transferred to Immobilon-P membrane and subjected to western blot analysis using Penta- and Tetra-His antibodies. Lane 1, uninduced cell lysate (–IPTG); lane 2, induced cell lysate (+IPTG); lane 3, purified His-tagged hDBR1 protein.
Figure 3.
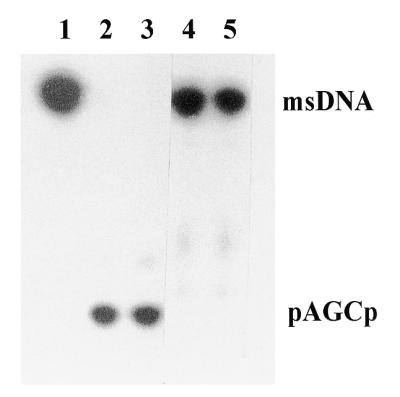
In vitro debranching enzyme assay. 32P-labeled msDNA Ec86 was incubated with the cell extract (S30) for 30 min at 30°C in a 20 µl reaction mixture containing 20 mM HEPES, pH 7.6, 40 mM KCl, 3.0 mM MgCl2, 1.0 mM DTT and 10% glycerol. The debranched trinucleotide products were analyzed on a 20% polyacrylamide gel with 8 M urea. Lane 1, 32P-labeled msDNA Ec86; lanes 2 and 3, 32P-labeled msDNA Ec86 incubated with cell extract prepared from induced (+IPTG) E.coli harboring pKN203 (hDBR1 cDNA under control of the T7 promoter in pET28a); lanes 4 and 5, 32P-labeled msDNA Ec86 incubated with cell extract prepared from uninduced (–IPTG) E.coli harboring pKN203.
The intron accumulation phenotype of both S.cerevisiae and S.pombe dbr1 null mutant strains implies that there is only one copy of a functional DBR1 gene in both budding and fission yeasts. A low stringency Southern blot analysis also supported this result, i.e. in a genomic Southern blot no additional dbr1+-related sequences were detected in S.pombe genomic DNA (K.B.Nam and J.D.Boeke, unpublished data). The complete genomic sequences of S.cerevisiae and C.elegans contain only a single copy of the DBR1 gene. A Southern blot analysis of human genomic DNA was performed using the entire hDBR1 cDNA as probe and the data suggest that the human genome contains one or, at most, two copies of the hDBR1 gene (data not shown).
The complete cDNA sequence was used to query the NCBI EST database. There were numerous low scoring hits against the 3′-UTR sequence, in which we found a reverse complement of an Alu repeat sequence at nt 1777–2067. However, there were only a few perfect matches to the hDBR cDNA: AA765401 and AA765829 from tonsil, W53026 and AA159840 from pancreas, N67797 from melanocytes, T80130 from brain, H85388 from eye, R66803 from placenta and AA069408 and AA069435 from pineal gland. These results suggest that hDBR1 mRNA is of extremely low abundance, may be enriched in certain tissues and, given the many introns it must debranch, is likely to encode an enzyme of high specific activity. However, we cannot rule out the possibility that the protein is very stable.
Complementation of the lariat intron RNA accumulation phenotype of a S.cerevisiae dbr1 mutant by hDBR1 cDNA
To demonstrate the function of the cloned hDBR1, a complementation test was carried out. First, the full-length hDBR1 cDNA was placed under control of the S.cerevisiae GAL1 promoter in a single copy CEN plasmid (pRS314GU or pRS316GU) and these constructs (pKN205 and pKN206, respectively) were introduced into an S.cerevisiae dbr1 mutant strain to determine whether the lariat intron RNA accumulation defect in this mutant strain could be complemented. The hDBR1 cDNA in a S.cerevisiae dbr1 mutant strain in a single copy CEN plasmid was induced by adding galactose and assayed for restoration of lariat intron degradation. Total RNAs were prepared from cells harboring a single copy CEN plasmid grown with glucose for repression or with galactose for induction of hDBR1. The intron accumulation phenotype was analyzed by RNA blot using a S.cerevisiae actin intron-specific probe (4). Accumulation of the actin lariat intron RNA was markedly decreased and was similar to the wild-type DBR1 strain (JB224) when hDBR1 cDNA expression was induced by galactose, relative to lariat intron RNA accumulation observed in cells grown in glucose or in a dbr1 mutant strain (KC106) (Fig. 4). This result demonstrates that the human cDNA clone encodes an RNA lariat debranching enzyme.
Figure 4.

Complementation of the S.cerevisiae intron accumulation defect by expression of hDBR1 cDNA. Total RNAs were prepared from S.cerevisiae strain KC106 (dbr1-1) carrying the hDBR1 cDNA subcloned in pRS314GU (pKN205) or pRS316GU (pKN206) and fractionated on a 5% polyacrylamide–8 M urea gel. Expression of hDBR1 was induced by adding 2% galactose and incubation for 48 h. RNAs were transferred to GeneScreen Plus nylon membrane and probed with a S.cerevisiae actin intron-specific probe. Lane 1, JB224 (DBR1); lane 2, KC106 (dbr1-1, no plasmid); lanes 3 and 4, hDBR1 in pRS314GU (pKN205) and grown with glucose (repression); lanes 5 and 6, hDBR1 in pRS316GU (pKN206) and grown with glucose (repression); lanes 7 and 8, hDBR1 in RS314GU (pKN205) and grown with galactose (induction); lanes 9 and 10, hDBR1 in RS316GU (pKN206) and grown with galactose (induction). The minor bands below the lariat intron RNAs are linearized actin introns. Plasmids pRS314GU and pRS316GU are basically the same except for different selection markers.
A single copy of hDBR1 complements the growth defect and intron accumulation phenotypes of a S.pombe dbr1 knockout mutation
ScDBR1 is a non-essential gene and accumulation of excised introns has no deleterious effect on cell growth. However, knockout of dbr1 in S.pombe produces a severe growth defect and an aberrant elongated cell shape in addition to an intron accumulation phenotype (5). To test whether the full-length hDBR1 cDNA can complement the growth and cell shape defects of a dbr1 knockout S.pombe mutant strain, the hDBR1 cDNA was integrated into the ura4 locus of the S.pombe chromosome. To integrate hDBR1 cDNA into the ura4 locus of the S.pombe chromosome, an integrating plasmid (pKN207) was constructed by inserting full-length hDBR1 cDNA into integration vector pJK210 (15). The integrating plasmid was then digested with StuI and used to transform a dbr1 null mutant strain (Spdbr1::leu1+ knockout strain YKN164). Positive transformants in which the full-length hDBR1 cDNA successfully integrated into the ura4 locus of the S.pombe genome by homologous recombination were selected on SC Ura– medium. Integration of full-length hDBR1 cDNA into the ura4 locus of the Spdbr1::leu1+ knockout strain was confirmed by PCR and Southern blot analysis (data not shown). As shown in Figure 5, the parental dbr1::leu1+ knockout strain (YKN164; Fig. 5a) and the strain containing only the parental plasmid pJK210 at the ura4 locus (YKN165; Fig. 5c) showed growth defects, a slow growing phenotype and an aberrant elongated cell shape. However, in the integrant strain the full-length hDBR1 cDNA integrated into the ura4 locus (YKN204) complemented the slow growth phenotype and elongated cell shape (Fig. 5a and d), i.e. the growth rate and the cell shape of the integrant strain (YKN204) was almost the same as the wild-type dbr1+ strain (YKN163; Fig. 5a and b). Also, to test whether the hDBR1 full-length cDNA integrant (YKN204) can also complement the intron accumulation phenotype of the S.pombe dbr1 knockout strain, northern blot analysis was performed. For the RNA blot, total RNAs were prepared from a wild-type dbr1+ strain (YKN163), a dbr1 knockout strain (YKN164), a dbr1 knockout strain with integration vector pJK210 at the ura4 locus (YKN165) and a full-length hDBR1 cDNA integrant strain (YKN204) grown on SC or SC Ura– and probed with a S.pombe rpl7 intron-specific probe. As shown in Figure 6, the accumulation of rpl7 lariat intron RNA was reduced in the strain with the integrated copy of hDBR1 (YKN204), while S.pombe dbr1 mutant strains (YKN164 and YKN165) showed the intron accumulation phenotype. However, the reduction in intron accumulation was not fully complemented to wild-type levels. This could be due to low level expression or low debranching activity of the integrated hDBR1 in S.pombe. The same result was obtained when S.cerevisiae DBR1 on a CEN plasmid was introduced into a S.cerevisiae dbr1 mutant strain (K.B.Chapman and J.D.Boeke, unpublished data). These data demonstrate that the full-length cDNA clone of hDBR1 can complement the mutant phenotypes of intron accumulation and growth defects due to knockout of dbr1 in S.pombe.
Figure 5.

Complementation of the S.pombe dbr1::leu1+ mutant growth defect by integrating hDBR1 cDNA into the S.pombe chromosome. To integrate full-length hDBR1 cDNA into the S.pombe chromosome, hDBR1 cDNA was subcloned into S.pombe integration vector pJK210 (pKN207). Linearized pKN207 (digested with StuI) was integrated into the ura4 locus. The hDBR1 cDNA integrated into the ura4 locus is expressed under control of the ura4 promoter. (a) Restoration of the growth defect of a S.pombe dbr1 mutant was observed by serial dilution. Schizosaccharomyces pombe strains YKN163 (dbr1+), YKN164 (dbr1::leu1+) and YKN204-1, 204-2 and 204-3 (ura4-294::pKN207/StuI dbr1::leu1+) were grown on YEC medium at 30°C; (b) YKN163 (dbr1+); (c) YKN165 (ura4-294::pJK210 dbr1::leu1+); (d) YKN204 (ura4-294::pKN207/StuI dbr1::leu1+). Cells grown on a YEC plate at 30°C were resuspended in distilled water and observed under a phase contrast microscope at 1000× magnification.
Figure 6.

Complementation of the S.pombe intron accumulation defect by integration of hDBR1 cDNA into the S.pombe chromosome. Total RNAs were prepared from S.pombe strains YKN163 (dbr1+), YKN164 (dbr1::leu1+), YKN165 (ura4-294::pJK210 dbr1::leu1+) and YKN204 (ura4-294::pKN207/StuI dbr1::leu1+) and fractionated on a 5% polyacrylamide–8 M urea gel. The RNAs were transferred to a GeneScreen Plus nylon membrane and probed with the S.pombe rpl7 intron-specific probe. Lanes 1–3, YKN204-3, YKN204-2 and YKN204-1 (ura4-294::pKN207/StuI dbr1::leu1+); lane 4, YKN165 (dbr1::leu1+ ura4-294::pJK210); lane 5, YKN164 (dbr1::leu1+); lane 6, YKN163 (dbr1+).
DISCUSSION
By an EST database search and cDNA library screening we identified a human cDNA sequence that was likely to be a homolog of ScDBR1. The full-length hDBR1 cDNA sequence revealed a single ORF encoding a 545 amino acid protein with 40, 44 and 43% identity to S.cerevisiae, S.pombe and C.elegans debranching enzymes, respectively. This suggests that hDBR1 is a functional homolog of the RNA lariat debranching enzymes. The amino acid sequence of human debranching enzyme also showed 79 and 58% identity to the mouse (J.-W.Kim, H.-C.Kim, J.-M.Yang and K.B.Nam, unpublished data) and Arabidopsis thaliana (EMBL locus ATF28M20, accession no. AL031004.1) DBR1 proteins (Fig. 1). Furthermore, hDBR1 cDNA expressed in E.coli showed debranching enzyme activity in vitro and could complement the DBR1 null mutant phenotype in S.cerevisiae and S.pombe in vivo, i.e. could restore debranching activity and the normal intron degradation phenotype of S.cerevisiae and the severe growth defect of S.pombe dbr1 null mutants.
The absence of debranching activity in vivo results in accumulation of the excised lariat intron molecule but cell viability is apparently not affected in S.cerevisiae. However, lariat intron accumulation in higher eukaryotes, which have more intron than exon sequences, might be expected to be deleterious to the cell. Indeed, in S.pombe, where 40% of genes contain introns, the dbr1 knockout strain showed a very different growth phenotype from that of the S.cerevisiae dbr1 mutant, i.e. the S.pombe dbr1 null mutant grew much more slowly and had an elongated cell shape.
Eukaryotic cells contain a large number of snoRNAs involved in the maturation of pre-rRNA and modification of rRNA nucleotides (reviewed in 16,17). Although some snoRNAs are derived from independent transcription units, many mammalian snoRNAs are encoded in intronic sequences of protein genes. In S.cerevisiae the U18 (18) and U24 (19) snoRNAs are synthesized from the intronic sequences of the host genes EFB1 (elongation factor EF-1β) and BEL1 (a G-β-like protein of unknown function), respectively. U24 is synthesized exclusively from the debranched lariat intron RNA; in the dbr1 mutant strain of S.cerevisiae, U24 snoRNA was found almost entirely in lariat form (7). In the case of U18, the level of mature snoRNA was reduced to ∼30% of the wild-type level in the S.cerevisiae dbr1 mutant strain (7). Recently, Ooi et al. (8) have also shown that the synthesis of certain snoRNAs (U24, snR38, U18 and snR39) requires debranching enzyme activity in the yeast S.cerevisiae, i.e. in a dbr1 mutant strain the levels of mature intronic snoRNAs are reduced and varied among different intronic snoRNAs depending on the size of the introns in which they are encoded. These data suggest that lariat introns are the normal source of certain snoRNAs such as U24, snR38, U18 and snR39. In the dbr1 mutant strain these intronic snoRNAs are trapped as lariat introns and may not produce proper U RNAs efficiently. A similar defect could potentially lead to the observed growth defect in the S.pombe dbr1 mutant. The growth phenotype of the dbr1 mutant in vertebrates could be more severe or even lethal.
Tycowski et al. (20) discovered an unusual RNA, the human UHG (U22 host gene) transcript, which contains nine introns, eight of which encode snoRNAs. Furthermore, the spliced UHG transcript has little protein encoding capacity. Thus the introns rather than exons encode the critical functions of UHG. This unique RNA sequence emphasizes the possible important role of so-called junk intron sequences and raises the possibility that the lariat debranching enzyme might be required for efficient processing of these snoRNAs in mammalian cells.
The cloned full-length cDNA of the human debranching enzyme can be tested for possible involvement in human snoRNA processing and may also provide a unique opportunity to study the relationship between hDBR1 and retroviral replication.
Acknowledgments
ACKNOWLEDGEMENTS
We thank K.-H. Han and J.-H. Sung for their technical assistance. K.B.N. and J.W.K. wish to acknowledge the financial support of the Korea Research Foundation in the program year 1997. This research was also partially supported by Samsung Biomedical Research Institute grant C98003 (J.-M.Y.).
DDBJ/EMBL/GenBank accession no. AF180919
REFERENCES
- 1.Ruskin B. and Green,M. (1985) Science, 229, 135–140. [DOI] [PubMed] [Google Scholar]
- 2.Sharp P.A., Konarska,M.M., Grabowski,P.J., Lamond,A.I., Marciniak,R. and Seiler,S.R. (1987) Cold Spring Harbor Symp. Quant. Biol., LII, 277–285. [DOI] [PubMed] [Google Scholar]
- 3.Jacquier A. and Rosbash,M. (1986) Proc. Natl Acad. Sci. USA, 83, 5835–5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chapman K.B. and Boeke,J.D. (1991) Cell, 65, 483–491. [DOI] [PubMed] [Google Scholar]
- 5.Nam K.B., Lee,G., Trambley,J., Devine,S.E. and Boeke,J.D. (1997) Mol. Cell. Biol., 17, 809–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nam K.B., Hudson,R.H.E., Chapman,K.B., Ganeshan,K., Damha,M.J. and Boeke,J.D. (1994) J. Biol. Chem., 269, 20613–20621. [PubMed] [Google Scholar]
- 7.Petfalski E., Dandekar,T., Henry,Y. and Tollervey,D. (1998) Mol. Cell. Biol., 18, 1181–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ooi S.L., Samarsky,D.A., Fournier,M.J. and Boeke,J.D. (1998) RNA, 4, 1096–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prabhala G., Rosenberg,G.H. and Kaeufer,N.F. (1992) Yeast, 8, 171–182. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez-Mendina J.R. and Rymond,B.C. (1994) Mol. Gen. Genet., 243, 532–539. [DOI] [PubMed] [Google Scholar]
- 11.Kalogeropoulos A. (1995) Yeast, 11, 555–565. [DOI] [PubMed] [Google Scholar]
- 12.Rose M.D., Winston,F. and Hieter,P. (1990) Methods in Yeast Genetics: A Laboratory Course Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 13.Gietz R.D. and Schiestle,R.H. (1991) Yeast, 7, 253–263. [DOI] [PubMed] [Google Scholar]
- 14.Kroll E.S., Hyland,K.M., Hieter,P. and Li,J.J. (1996) Genetics, 143, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keeny J. and Boeke,J.D. (1994) Genetics, 136, 849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maxwell E.S. and Fournier,M.J. (1995) Annu. Rev. Biochem., 35, 897–934. [DOI] [PubMed] [Google Scholar]
- 17.Tollervey D. and Kiss,T. (1997) Curr. Opin. Cell Biol., 9, 337–342. [DOI] [PubMed] [Google Scholar]
- 18.Balakin A.G., Smith,L. and Fournier,M.J. (1996) Cell, 85, 823–834. [Google Scholar]
- 19.Qu L.H., Henry,Y., Nicoloso,M., Michot,B., Azum,M.C., Renalier,M.H., Caizergues-Ferrer,M. and Bachellerie,J.P. (1995) Nucleic Acids Res., 23, 2669–2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tycowski K.T., Shu,M.M. and Steitz,J.A. (1996) Nature, 379, 464–466. [DOI] [PubMed] [Google Scholar]