

Abstract
Background
Hematoma leads to progressive neurological deficits and poor outcomes after intracerebral hemorrhage (ICH). Early clearance of hematoma is widely recognized as an essential treatment to limit the damage and improve the clinical prognosis. CD163, alias hemoglobin (Hb) scavenger receptor on microglia, plays a pivotal role in hematoma absorption, but CD163 on neurons permits Hb uptake and results in neurotoxicity. In this study, we focus on how to specially promote microglial but not neuronal CD163 mediated-Hb uptake and hematoma absorption.
Methods
RNA sequencing was used to explore the potential molecules involved in ICH progression, and hematoma was detected by magnetic resonance imaging (MRI). Western blot and immunofluorescence were used to evaluate the expression and location of fractalkine (FKN) after ICH. Erythrophagocytosis assay was performed to study the specific mechanism of action of FKN in hematoma clearance. Small interfering RNA (siRNA) transfection was used to explore the effect of peroxisome proliferator-activated receptor-γ (PPAR-γ) on hematoma absorption. Enzyme-linked immunosorbent assay (ELISA) was used to determine the serum FKN concentration in ICH patients.
Results
FKN was found to be significantly increased around the hematoma in a mouse model after ICH. With its unique receptor CX3CR1 in microglia, FKN significantly decreased the hematoma size and Hb content, and improved neurological deficits in vivo. Further, FKN could enhance erythrophagocytosis of microglia in vitro via the CD163/ hemeoxygenase-1 (HO-1) axis, while AZD8797 (a specific CX3CR1 inhibitor) reversed this effect. Moreover, PPAR-γ was found to mediate the increase in the CD163/HO-1 axis expression and erythrophagocytosis induced by FKN in microglia. Of note, a higher serum FKN level was found to be associated with better hematoma resolution in ICH patients.
Conclusions
We systematically identified that FKN may be a potential therapeutic target to improve hematoma absorption and we shed light on ICH treatment.
Keywords: Intracerebral hemorrhage, Microglia, Hematoma absorption, Fractalkine, CD163/HO-1, PPAR-γ
Introduction
Intracerebral hemorrhage (ICH) is one of the most devastating diseases throughout the world with 40–54% mortality and a 61–88% disability rate [1], which poses a heavy global burden on the patient’s family and society due to the lack of effective therapy [2]. Blood from the ruptured vessels rapidly accumulates in the brain parenchyma to form a hematoma, and its compression damage and neurotoxicity of hemolytic products are the main pathological mechanisms of injury closely related to the outcome and prognosis [3–5]. Early clearance of hematoma is widely recognized as an essential treatment to limit the direct compression damage and secondary neurotoxicity injury and to improve the outcomes after ICH [6–8]. However, previous randomized control trials (RCTs) of hematoma clearance by surgical interventions, such as STICH and STICH II, shed a glimmer of light in the darkness. STICH was a randomized trial comparing early surgical treatment and conservative treatment in patients with ICH. Unfortunately, patients who accepted early hematoma evacuation had no overall benefit compared with the conservative treatment. In the subsequent STICH II, it was confirmed that early surgery would not increase the mortality or disability rate within 6 months, especially for patients with superficial lobar intracerebral hemorrhage without intraventricular hemorrhage may have a small survival advantage [9, 10]. Therefore, medical treatment to promote hematoma absorption has recently been explored further.
Microglia are the intrinsic phagocytes in the brain, and after ICH, they can be activated within few minutes to promote brain repair and remove hematoma via some cytokines and chemokines [3, 8]. CD163, alias hemoglobin (Hb) scavenger receptor, is found to be mainly expressed on the microglial cytomembrane in the brain, and it is a major scavenger receptor that mediates endocytosis of Hb in a pathological condition [11, 12]. CD163 is also associated with neurological recovery and prognosis in animal models and patients, and it can be a biomarker of the outcome after ICH [13, 14]. After CD163-mediated endocytosis, Hb is catabolized by hemeoxygenase-1 (HO-1) [12]. HO-1 also mainly induced by microglia after ICH in the brain. As a key rate-limiting enzyme, HO-1 can control heme release from Hb and degrade to Fe (II) [12, 15]. However, the deficiency of CD163 was found to have distinct temporal influences on ICH that are initially beneficial and harmful later, which suggest the complexity of the CD163/HO-1 signal pathway in hematoma absorption [14]. Current further studies also showed that CD163 is expressed on neurons, and haptoglobin increases the CD163 expression of neurons to permit extracellular Hb uptake. Of note, Chen-Roetling et al. found that Hb was toxic to the neurons after endocytosis [16]. In neurons, heme is broken down by the constitutive HO-2, rather than the inducible HO-1, but the deficiency of neuronal iron sequestering systems leads to iron overload and results in an increase in neuronal death [12, 17]. Thus, here, we focus on how to specifically promote microglial but not neuronal CD163 mediated-Hb endocytosis and hematoma absorption.
To our knowledge, neurons can produce various "help me" molecular signals when exposed to harmful stimuli [18]. Recent studies showed that fractalkine (FKN) secreted from damaged neurons can activate the microglia to rescue neurons. FKN is a unique chemokine and the only representative of the CX3C group. FKN is mainly secreted by neurons, and occasionally, it is expressed on vascular endothelial cells, monocytes, and vascular smooth muscle cells [19, 20]. CX3CR1 is a unique receptor, which is exclusively expressed on resident microglia [19]. The FKN/CX3CR1 axis mediates neuron-microglia crosstalk in many CNS diseases, including traumatic brain injury (TBI), ischemic stroke, multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), and Alzheimer’s disease (AD) [20–25]. However, the role of the FKN/CX3CR1 axis in ICH remains unearthed.
In this study, we hypothesized that neurons responding to ICH would release a “help me” signal, FKN, which could promote hematoma absorption via PPAR-γ/CD163/HO-1-mediated phagocytosis in microglia. Thus, we aim to identify a potential therapeutic target to improve hematoma absorption in ICH.
Materials and methods
ICH model
All animal experiments followed the guidelines of the National Institutes of Health (NIH) and were confirmed by the Medical Ethics Committee of Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, China. Male C57BL/6 mice (weight 25–30 g) were obtained from the specialized institution of the Animal Care and Use Committee of Tongji Medical College, Huazhong University of Science and Technology, and mice died due to deep anesthesia and failed modeling were excluded. The ICH model was induced by intrastriatal injection of collagenase VII. Firstly, mice were immobilized on a stereotaxic frame after 1% pentobarbital sodium anesthesia, and then we drilled a 1-mm diameter hole in the skull and injected collagenase (0.08 U/5 min for mice) into the right corpus striatum with a 26-gauge needle (0.8 mm anterior to the bregma and 2.5 mm lateral to the midline, 3 mm deep to the skull).
Lateral ventricular injection: mice were divided into four groups using a randomization approach (odd/even numbers) before modeling ICH. Mice were injected with normal saline, FKN cytokine (R&D Systems, USA) (FKN cytokine was injected 30 min before ICH and mice received 100 nmol/μl FKN cytokine at 0.1 μl/min), and AZD8797 (MedChemExpress, USA) (AZD8797 was injected 30 min before ICH and mice received 100 μmol/μl of AZD8797 at 0.1 μl/min) in the lateral ventricular region respectively (0.0 mm anterior to the bregma and 1.0 mm lateral to the midline, 3 mm deep to the skull).
Cell culture
Mouse BV2 microglia cells (BV2 cells) were purchased from LiScien Biotechnology (Wuhan, China) and cultured in DMEM (Hyclone, Logan, USA) containing 10% FBS (Sciencell, San Diego, USA), 100 μg/ml Streptomycin, and 100 IU/ml Penicillin. Mouse N2a neuron cells (N2a cells) were purchased from LiScien Biotechnology (Wuhan, China) and cultured in MEM (Hyclone, Logan, USA) containing 10% FBS (Sciencell, San Diego, USA), 100 μg/ml Streptomycin, and 100 IU/ml Penicillin. Contamination, morphology, and motility were routinely assessed by light microscopy in normal culture.
ICH brain tissue harvesting
Animals were transcardially perfused with ice-cold phosphate-buffered saline (PBS) after anesthesia. The whole brain or tissue around the hematoma was separated and stored in a freezer at -80℃ for protein analysis and RNA isolation later.
Enzyme-linked immunosorbent assay (ELISA)
The levels of FKN in ICH patient serum, ICH mouse brains, and cell supernatant were measured with the human FKN ELISA kit (Boster, Wuhan, China) and the mouse FKN ELISA kit (Boster, Wuhan, China), and all operations were performed under the protocols of the manufacturer.
Hemoglobin content measurement
Hematoma volume was quantified by measuring the content of Hb in the ipsilateral striatum at day 3 after ICH and the Hb level in the ipsilateral striatum was measured using hemoglobin kit (Bestbio, Shanghai, China) under the guidance of the instructions of the manufacturer. First, mice were killed and perfused with ice-cold PBS to clear Hb in vessels. Then the hematoma-affected brain tissue was smashed and homogenated by sonication, centrifuged at 12,000 rpm and 4 ℃ for 30 min, placed in a 15 μl supernatant in a 96-well plate, and mixed reagents were added into the sample. The optical density (OD) value was measured by an enzyme-linked immune detector at 510 nm.
Western blot
Fresh perihematomal brain tissues were smashed and homogenated by sonication after adding protease inhibitor cocktail (Beyotime Biotechnology, Wuhan, China) and RIPA (Beyotime Biotechnology, Wuhan, China). Protein samples were added to 6–12% SDS-PAGE gel (Beyotime Biotechnology, Wuhan, China), transferred to 0.4 mm PVDF membrane (Millipore, Billerica, USA), blocked with 5% skim milk at RT for 2 h, and then incubated overnight at 4 ℃ with the following primary antibodies: rabbit antibody to FKN (1:1000, Abcam, USA), rabbit antibody to CX3CR1 (1:1000, Abcam, USA), mouse antibody to CD163 (1:500, Nourvous, USA), rabbit antibody to HO-1 (1:1000, Sanying Company, Wuhan, China), and rabbit antibody to PPAR-γ (1:500, ABclonal Technology, China). Then all membranes were visualized on a BioSpectrum Imaging System (UVP, Upland, USA) after incubation with the corresponding HRP-conjugated secondary antibodies (1:2000, ABclonal Technology, Wuhan, China) for 2 h at RT. Finally, the protein expression was quantified with ImageJ software.
Quantitative real-time PCR
The ipsilateral tissue surrounding the hematoma was processed for extracting total RNA with Trizol reagent (Takara, Kyoto, Japan). Then the cDNA of each group was synthesized from total RNA with Primescript RT Master kit (Takara, Kyoto, Japan) according to the instructions of the manufacturer. SYBR Premix Ex Taq™ kit (Takara, Kyoto, Japan) and Applied Biosystem StepOne Real-Time PCR System (Invitrogen, USA) were used to complete qRT-PCR. β-actin was used as an internal standard. The following CX3CR1: forward 5′-ACCTCACCATGTCCACCTCCTTC-3′, reverse 5′-TGAGAGCGAGGACCACCAACAG-3′; CD163: forward 5′-AATCACATCATGGCACAGGTCACC-3′, reverse 5′-TCGTCGCTTCAGAGTCCACAGG-3′; HO-1: forward 5′-ACCGCCTTCCTGCTCAACATTG-3′, reverse 5′-CTCTGACGAAGTGACGCCATCTG-3′; PPAR-γ: forward 5′-CGCCAAGGTGCTCCAGAAGATG-3′, reverse 5′-GGTGAAGGCTCATGTCTGTCTCTG-3′ primers were used.
Erythrophagocytosis assay
Erythrocytes were extracted form the whole blood of donor mice and centrifuged at 4 ℃ and 1200 rpm for 11 min. Then, isolated erythrocytes were labeled with the red fluorescent probe Dil (Beyotime Biotechnology, Wuhan, China) at 37 ℃ for 30 min according to the instructions of the manufacturer and remaining reagent was removed by PBS wash three times. Microglia (1 × 106) were incubated with serum-free medium for 12 h, and then fed with fluorescently labeled erythrocytes (1 × 107) for 6 h at 37 ℃ under the condition of avoiding light. After that, unengulfed erythrocytes were washed by PBS and the remaining microglia were stained using the FITC-labeled anti-mouse CD11b antibody (BD, USA) at 4 ℃ overnight. The erythrophagocytosis assay was measured with microscopy and calculated via the phagocytosis index (an index using for estimating the mean fluorescence intensity of engulfed erythrocytes per microglia).
MRI
All animal models were anesthetized with 3% isoflurane and maintained at 1.5% isoflurane during the whole MRI process. Mice received MRI scanning at day 3 after ICH on a 7.0 T/20 MRI scanner (Ettlingen, Germany) at the Wuhan Institute of Physics and Mathematics, Chinese Academy of Science. Two sequences were acquired in our study, including T1-weighted imaging (T1WI) and T2-weighted imaging (T2WI) to assess the hematoma size. The parameters are listed below and they remained consistent during the whole process. T1-weighted imaging was acquired as follows: field of view was 20 × 20 mm, slice thickness was 0.6 mm, matrix size was 128 × 128, echo time/repetition time was 13/500 ms, and number of averages was 8. T2-weighted imaging was acquired as follows: field of view was 20 × 20 mm, slice thickness was 0.6 mm, matrix size was 256 × 256, echo time/repetition time was 40/3000 ms, and number of averages was 4. The absolute volume of the hematoma was calculated by integrating the injured area in each hemorrhagic slice with the MRIcro software. The vital signs of mice were kept stable throughout the whole experiment.
Immunofluorescence staining
Sliced ICH mouse brain was placed at RT and blocked with 30% donkey serum for 1 h. Then sections were incubated with the following corresponding primary antibodies: rabbit antibody to FKN (1:50, Abcam, USA), mouse antibody to NeuN (1:100, Nourvous, USA), goat antibody to iba1 (1:100, Abcam, USA), mouse antibody to CD31 (1:100, R&D Systems, USA), and mouse antibody GFAP (1:100, R&D Systems, USA) at 4 ℃ overnight. Then, the samples were washed using TBST and incubated with the secondary fluorescein-conjugated antibodies (Life Technologies, USA) for 2 h at RT. The nucleus was stained with DAPI (Beyotime Biotechnology, Wuhan, China) for 8 min. All images were acquired with confocal microscope (Nikon, Tokyo, Japan).
Transwell migration assay
BV2 cells treated with and without AZD8797 were cultured with 200 μL serum-free DMEM in the upper chamber of a transwell insert and 600 μL DMEM containing 10% FBS was placed in the lower chamber with and without FKN. After incubation at 37 ℃ in 5% CO2 for 18 h, BV2 cells that had migrated through the membrane were stained with crystal violet (Beyotime Biotechnology, Wuhan, China) after being fixed with 4% paraformaldehyde, and they were quantified with an invert microscope.
Neurological deficit score (NDS)
Neurological deficits were measured at 3 days after ICH, and a 28-point neurological deficit score, including gait, body symmetry, climbing test, circling test, front limb symmetry, and forced circle and tentacle tests were used in this behavioral test. Scoring was performed in a quiet environment by two experimenters who were blind to the groups, and the final results were shown in the mean score of each mouse.
Participants, clinical data collection and hematoma volume detection
All patients were from the cohort of Chinese cerebral hemorrhage: mechanism and intervention study, and met the following inclusion criteria: (1) Over 18 years of age; (2) Patients were diagnosed as ICH according to the guidelines; (3) Within 7 days of onset, and will rule out when meet one of the exclusion criteria: (1) Subarachnoid hemorrhage; (2) Hemorrhagic transformation after cerebral infarction; (3) Bleeding after thrombolysis; (4) Traumatic intracerebral hemorrhage; (5) Epidural hematoma and subdural hematoma.
The clinical data of all patients include age, sex, hematoma volume on admission, systolic blood pressure (SBP)/ diastolic blood pressure (DBP) on admission, Glasgow coma score on admission, and modified ranking stroke scale (mRS) were copied from the medical record. The data of age were presented as mean (SD) and gender was presented as percentages. Of note, we expanded the sample size of patients to 30 in this revised version, and there is no statistical difference between hematoma expansion group and hematoma reduction group in age or gender. Volume of hematoma was based from MRI/CT image. The hematoma volume was detected by CT scanning and calculated with the following formula: hematoma volume (ml) = 1/2 × length (cm) × width (cm) × high (cm).
The clinical study protocol was conducted following the Declaration of Helsinki and were approved by the Research Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China (ethical approval number:2018-S485).
Statistical analysis
All data were analyzed by GraphPad Prism 7.0 software. Group size of the animal was calculated depending on the preliminary data of prior work to achieve a power of 0.8 and Type 1 error rate α = 0.05. All data were presented as mean ± SEM. All data were tested by Shapiro–Wilk normality test to confirm the normality of distribution. Statistical tests included two-tailed unpaired students’ t tests between two groups and one-way ANOVA test for multiple-group comparison. Brown–Forsythe test was used to test the homoscedasticity of data and Bonferroni correction was used to correct for multiple comparisons. For non-normally distributed data, Kruskal–Wallis test was applied instead of one-way ANOVA test. P value < 0.05 was considered as statistically significant.
Results
FKN/CX3CR1 expression was increased around the hematoma after ICH
To investigate the potentially crucial participants in ICH progression, we first performed RNA sequencing (RNA-seq) in an established mouse model of ICH. Significant difference was determined by fold change > 1.5 and FDR < 0.1. In total, 674 genes were differentially expressed in the perihematomal tissue compared with that in the sham exposed tissue. Kyoto Encyclopedia of Genes Genomes (KEGG) pathway enrichment analysis was performed to analyze the top 600 differently expressed genes, and the result showed cytokine–cytokine receptor interaction and chemokine signaling pathway as the key enriched gene ontology categories and CX3CR1 was identified in the two categories (Fig. 1A, B). CX3CR1, a special chemokine receptor and adhesion molecule, which is mainly expressed in microglia and its unique ligand FKN, is mainly expressed in neurons in the brain, thus, indicating that the FKN-CX3CR1 signaling pathway-mediated neuron–microglia interaction can respond to ICH and may play a key role in the pathophysiological process. To confirm the finding of RNA sequencing about FKN and CX3CR1, we detected the spatial–temporal expression of FKN and CX3CR1. Western blot analysis showed that FKN and CX3CR1 levels in the perihematomal region were increased as early as 6 h after the onset of ICH, peaked around 3 days, and then they decreased gradually over the next few days compared with those in the control and sham groups (Fig. 1C–E). Besides western blot analysis, PCR, ELISA, and immunofluorescence staining also displayed a similar change (Fig. 1F–I). Moreover, confocal immunofluorescence staining with NeuN (neuron marker), iba1 (microglia marker), CD31 (endothelial cell marker), and GFAP (astrocyte marker) were used to identify the colocalization of FKN and CX3CR1. Fluorescent staining revealed that FKN was mainly colocalized to the neurons and CX3CR1 was mainly colocalized to microglia in the perihematomal region at 3 days after ICH (Fig. 1J–M). In addition, we performed neuronal culture, stimulated with Hb (from 0 to 10 μmol/L), and then, we detected the expression of FKN secreted from neurons by ELISA assay. The result showed that FKN from neurons increased gradually over the Hb concentration (Fig. 1N).
Fig. 1.

FKN/CX3CR1 expression was increased around the hematoma after ICH. A Heatmap showing differentially expressed genes between ICH mouse models and sham mouse models in part, and data are colored according to the row minimum and maximum. B KEGG analysis of the top 600 differentially expressed genes in ICH mouse models and sham mouse models. Top 20 KEGG clusters are shown with their key words. C Representative immunoblots of FKN and CX3CR1 in the ipsilateral area around the hematoma at 0 h (sham group) to 7 days compared with the control group after ICH in C57BL/6 mice. D, E Quantification analysis of FKN and CX3CR1 protein expression levels normalized to β-actin, ****P < 0.0001, (n = 6/group). F Quantification real-time PCR of CX3CR1 mRNA expression in the ipsilateral area around the hematoma at 0 h (sham group) to 7 days compared with the control group after ICH in C57BL/6 mice. β-actin was used as an internal control, ****P < 0.0001, (n = 5/group). G ELISA results showed the level of FKN in the ipsilateral area around the hematoma over the time course, ***P < 0.001, (n = 5/group). H, I Representative confocal immunofluorescence microscopy images of the ipsilateral area around the hematoma at 6 h to 7 days in C57BL/6 mice after ICH by staining with FKN (red), CX3CR1 (red), and DAPI (blue), scale bar 100 μm, (n = 3/group). J–M Representative immunofluorescent images and quantification for FKN (green), CX3CR1 (green) with either NeuN (red, neuron marker), iba1 (red, microglia marker), CD31 (red, endothelial cell marker), or GFAP (red, astrocyte marker) at 3 days after ICH, scale bar 200 μm, **P < 0.01, ****P < 0.0001 (n = 4/group). N ELISA results showed the level of FKN secreted from N2a cells stimulated with hemoglobin (from 0 to 10 μmol/L), ****P < 0.0001, (n = 6/group)
FKN augmented erythrophagocytosis, accelerated hematoma absorption, and improved neurological function recovery
To test the role of the FKN/CX3CR1 signaling pathway in erythrophagocytosis and hematoma absorption, normal saline, FKN, and AZD8797 (a high-affinity inhibitor of CX3CR1) were used before the ICH model in vitro or in vivo, respectively. In vivo MRI images, Hb content, and brain tissue section showed that hematoma volume in the FKN group was significantly smaller than that in the vehicle group, and the hematoma volume in the AZD8797 group was larger at day 3 after ICH (Fig. 2A–D). With respect to the neurological function recovery, the FKN group showed a better neurological deficit score (NDS) at day 3 after ICH and the AZD8797 group showed a worse score (Fig. 2E). In vitro erythrophagocytosis assay showed that FKN significantly increased the phagocytosis (mean fluorescence intensity of engulfed erythrocytes per microglia) and AZD8797 played an opposite role (Fig. 2F, G). Meanwhile, transwell assay revealed that FKN could promote cell migration in vitro, which may confirm that it could increase the chemotactic ability of microglia towards the hematoma after ICH (Fig. 2H–I). These data jointly demonstrated that FKN could promote hematoma clearance and neurological recovery.
Fig. 2.

FKN augmented erythrophagocytosis, accelerated hematoma absorption, and improved neurological function recovery. A, B Representative MRI images of T1W1 and T2W1 for hematoma volume at 3 days after ICH in mice injected with saline, FKN cytokine, and inhibitor AZD8797 via the lateral ventricle and sham group, quantification analysis of hematoma volume, *P < 0.05, **P < 0.01, ***P < 0.001, (n = 3/group). C Quantification analysis of hemoglobin content at 3 days after ICH in mice injected with saline, FKN cytokine, and inhibitor AZD8797 via the lateral ventricle and sham group, *P < 0.05, ****P < 0.0001, (n = 5/group). D Representative photograph of brain slice at 3 days after ICH in mice injected with saline, FKN cytokine, and inhibitor AZD8797 via the lateral ventricle and sham group, scale bar 5 mm, (n = 3/group). E The NDS score in ICH mice injected with saline, FKN cytokine, and inhibitor AZD8797 via the lateral ventricle and sham group, **P < 0.01, ***P < 0.001, ****P < 0.0001, (n = 5/group). F, G Transwell migration assay of BV2 cells incubated with saline, FKN cytokine and inhibitor AZD8797 and quantitative analysis, scale bar 50 μm, *P < 0.05, **P < 0.01, (n = 4/group). H, I Representative confocal immunofluorescence microscopy images showed phagocytosis of Dil-labeled erythrocytes (red) in CD11b-positive (green) BV2 cells with FKN cytokine and inhibitor AZD8797 incubation, and the phagocytosis index showed the quantification analysis of phagocytosis by microglia, scale bar 10 μm, ***P < 0.001, ****P < 0.0001, (n = 6/group)
FKN increased CD163 and HO-1 expression in mice and microglia after ICH
As CD163 (the hemoglobin scavenging receptor) and HO-1 (a heme decomposing enzyme) are the key molecules for hematoma absorption/hemoglobin scavenging after ICH, to explore whether FKN could promote hematoma absorption by upregulating CD163 and HO-1 expression in microglia, we used western blot and PCR to detect the protein expressions of CD163 and HO-1 in microglia treated with FKN and AZD8797 in vitro and in vivo. The results revealed that FKN could increase CD163 and HO-1 expressions surrounding the hematoma, whereas AZD8797 had an opposite effect (Fig. 3A–C). Moreover, the in vitro experiment revealed a similar result in microglia (Fig. 3D–H).
Fig. 3.

FKN increased the expressions of CD163 and HO-1 in mice and microglia after ICH. A–C Representative immunoblots of CD163 and HO-1 at 3 days after ICH in mice injected with saline, FKN cytokine, and inhibitor AZD8797 via the lateral ventricle and sham group, and quantitative analysis of CD163 and HO-1 protein expression level, normalized to β-actin *P < 0.05, **P < 0.01, (n = 4/group). D–F Representative immunoblots of CD163 and HO-1 in BV2 cells treated with saline, FKN cytokine, and inhibitor AZD8797, and quantitative analysis of CD163 and HO-1 protein levels normalized to β-actin, *P < 0.05, **P < 0.01, ***P < 0.001, (n = 6/group). G, H Quantification real-time PCR was used to detect the expressions of CD163 and HO-1 in BV2 cells incubated with saline, FKN cytokine, and inhibitor AZD8797, *P < 0.05, **P < 0.01, (n = 4/group)
FKN improved erythrophagocytosis and migration of microglia after ICH via PPAR-γ
To explore the mediators between FKN and CD163/HO-1 and to better understand the changes in microglia after ICH, a temporal transcriptional analysis of genes from microglia treated with and without FKN was performed. RNA sequence analysis highlighted the transcription factor PPAR-γ as an enriched gene in microglia treated with FKN compared with the control group (Fig. 4A). Then, western blot and PCR confirmed the effect of FKN on PPAR-γ expression (Fig. 4B–D). Furthermore, the phagocytosis assay showed that FKN facilitated the engulfment of microglia and this ability was reversed by PPAR-γ siRNA (Fig. 4G, H), which showed that the phagocytosis of microglia modulated by FKN was largely dependent on the regulation of PPAR-γ. Furthermore, the transwell assay result also displayed that PPAR-γ siRNA could reverse the increase in cell migration by FKN (Fig. 4E, F).
Fig. 4.

FKN improved erythrophagocytosis and migration of microglia after ICH via PPAR-γ. A Heatmap of differentially expressed genes in BV2 cells incubated with FKN cytokine and control group in part, and data are colored according to row minimum and maximum. B, C Representative immunoblots of PPAR-γ in BV2 cells incubated with FKN cytokine and the control group, and quantitative analysis of PPAR-γ protein level normalized to β-actin, *P < 0.05, (n = 4/group). D Quantification real-time PCR of PPAR-γ in BV2 cells incubated with FKN cytokine and the control group, and quantitative analysis of PPAR-γ mRNA expression, **P < 0.01, (n = 4/group). E, F Transwell migration assay in normal and FKN-stimulated BV2 cells with and without siRNA transfection, and quantitative analysis, scale bar 50 μm, *P < 0.05, **P < 0.01, (n = 4/group). G, H Representative immunofluorescence images showed phagocytosis of Dil-labeled erythrocytes (red) in normal and FKN-stimulated CD11b-positive (green) BV2 cells with and without siRNA transfection, and the phagocytosis index showed the quantification analysis, scale bar 10 μm, **P < 0.01, ***P < 0.001, (n = 4/group)
FKN increased CD163/HO-1 expression after ICH via PPAR-γ
To verify whether PPAR-γ could mediate the FKN/CD163/HO-1 signaling pathway to influence erythrophagocytosis of microglia, we used western blot and PCR to measure the expressions of CD163 and HO-1 in microglia treated with PPAR-γ siRNA. The results displayed that PPAR-γ siRNA reversed FKN-induced CD163 and HO-1 protein and mRNA expressions (Fig. 5A–G). These results indicated that PPAR-γ increased hematoma clearance via augmenting the expressions of CD163 and HO-1 in microglia after ICH.
Fig. 5.

FKN increased the CD163/HO-1 expression after ICH via PPAR-γ. A–D Representative immunoblots of CD163, HO-1, and PPAR-γ in normal and FKN-stimulated BV2 cells with and without siRNA transfection and quantitative analysis, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, (n = 4/group). E–G Quantification real-time PCR of CD163, HO-1, and PPAR-γ in normal and FKN-stimulated BV2 cells with and without siRNA transfection, and quantitative analysis of CD163, HO-1 and PPAR-γ mRNA expression, *P < 0.05, **P < 0.01, ***P < 0.001, (n = 4/group)
Serum FKN concentration was relative to smaller hematoma volume and better outcome
To confirm the role of FKN in hematoma absorption in clinical practice, 30 patients with ICH were divided into the hematoma expansion group and hematoma reduction group according to the progress of hematoma, and then the serum FKN concentration was detected by ELISA. The results showed that the hematoma expansion group had a lower concentration of serum FKN and a higher modified Rankin Score (mRS score) than the hematoma reduction group (Fig. 6A, Table. 1), which suggested that FKN might be involved in hematoma absorption and functional recovery after ICH. The clinical characteristics are shown in Table 1.
Fig. 6.
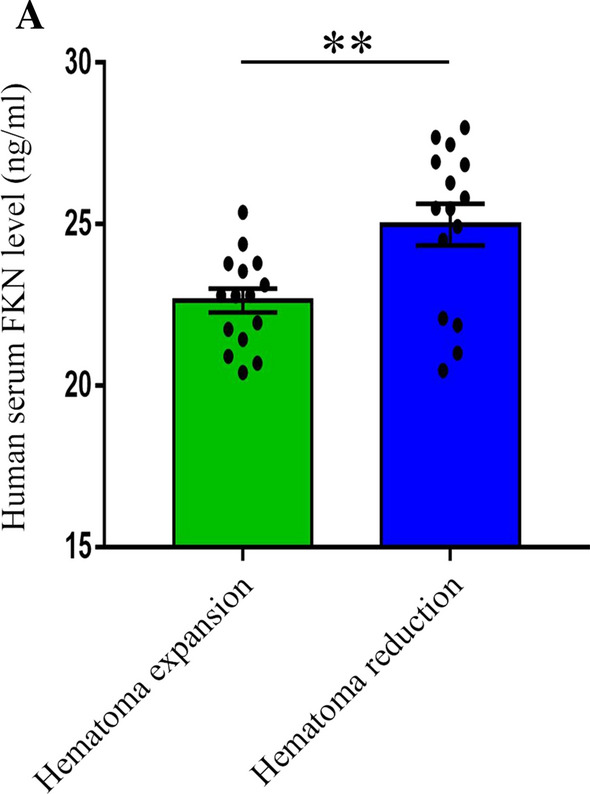
Serum FKN concentration was relative to smaller hematoma volume and better outcome. A ELISA results showed the level of serum FKN in ICH patients, **P < 0.01, (n = 15/group)
Table 1.
The clinical characteristics of ICH patients
| Hematoma expansion (n = 15) | Hematoma reduction (n = 15) | P value | |
|---|---|---|---|
| Age, years | 55.47 (12.21) | 54.80 (11.24) | NS |
| Male, % | 80 | 80 | NS |
| Admission hematoma, volume (ml) | 17.77 (9.14) | 20.13 (9.01) | NS |
| Admission systolic blood pressure, mmHg | 159.00 (27.89) | 157.6 (18.00) | NS |
| Admission diastolic blood pressure, mmHg | 93.07 (15.09) | 94.87 (11.13) | NS |
| Admission Glasgow Coma Score | 11 (10–14) | 12 (8–15) | NS |
| 1 month mRS score | 4 (4–5) | 3 (1–4) | P < 0.05 |
NS: P > 0.05. The definition of hematoma expansion is more than 33% relative increase of hematoma volume; the definition of hematoma reduction is decreased area and intensity of hematoma than admission
GCS Glasgow coma scale, mRS modified Rankin scale
Discussion
In this study, we found that (a) the FKN/CX3CR1 pair was expressed in the brain and was increased after ICH, (b) FKN promoted microglia hematoma absorption and improved the neurological recovery in mice, (c) FKN increased CD163 and HO-1 expressions of microglia via PPAR-γ to promote hematoma absorption after ICH, and (d) serum FKN was associated with good hematoma absorption in ICH patients. The mechanisms of our study are shown in Fig. 7 (Fig. 7).
Fig. 7.

FKN promoted hematoma absorption via PPAR-γ/CD163/HO-1-mediated phagocytosis in microglia after ICH. Neurons produced FKN to accumulate PPAR-γ via binding to its receptor CX3CR1 in microglia, which in turn increased the expressions of CD163 and HO-1 to promote microglia-induced hematoma clearance and improve neurological recovery after ICH
Hematoma causes primary injury due to its mass effect and mechanical compression and subsequently, hematoma and its lysate also lead to secondary injury, including neurotoxicity, inflammation, and oxidative stress. Hematoma-induced primary and secondary injuries are the crucial pathological changes after ICH [26, 27]. Efficient clearance of hematoma could limit ICH injury and can be regarded as a potential treatment strategy. There are a series of studies that attempted to verify the effectiveness of evacuation of hematoma; unfortunately, the results are not positive [7, 9, 10]. Thus, recent studies have focused on how to promote endogenous hematoma clearance, and some of them have received preclinical success, such as the exogenous application of VK-28 (a brain-permeable iron chelator) or LTF and the Fc domain of human IgG (FcLTF) can promote the hematoma absorption of microglia after ICH [28, 29]. In fact, microglia in the brain can be activated and they can clear the hematoma and its debris [8, 30]. However, the natural phagocytosis of microglia is not adequate to deal with hematoma. Researches have confirmed that the microglia are activated within hours after ICH and they continued to function up to several weeks. But it may be a double-edged sword, the production of “pro-inflammatory” and “anti-inflammatory” chemokines and cytokines making the microglial role become complicated; it even exacerbated brain injury via crosstalk with other cells such as astrocytes in ICH [3, 31, 32]; thus, regulation of microglia may be a potential and effective target for brain repair [33].
In our study, we found that CX3CR1 and its unique ligand FKN were significantly increased around the hematoma after ICH, but the peak of the increase occurred at about the third day after ICH, which is somewhat delayed, and exogenous administration may be needed.
FKN was reported to be an intriguing chemokine, which played a central role in the nervous system [34] and further research found that FKN also had the function of neuroprotection and anti-inflammation. FKN mediated neuron-microglia crosstalk in physiological conditions or pathological conditions [19]. The FKN/CX3CR1 axis played an anti-inflammatory role in a rat model of Parkinson’s disease (PD) treated with deep brain stimulation in the subthalamic nucleus [35]. FKN also exerted neuroprotection by promoting microglia polarization in a radiation-induced brain injury model [36]. The current study demonstrated that FKN from neurons is a pivotal chemokine in regulating the phagocytosis of microglia and appears to be important for hematoma cleanup after ICH [22]. However, some studies showed that FKN could sustain the neuroinflammatory status and lead to neurotoxicity. For example, FKN could induce dopaminergic cell exhaustion and motor dysfunction after treatment with 1-methyl-4-phenylpyridinium (MPP+), a rat model of PD [37]. The controversy may be explained as follows: the dual role of the FKN/CX3CR1 axis dependent on the immune status of different diseases induces different signaling pathways. Moreover, when and where to intervene in the FKN/CX3CR1 axis is the key point as the complicated signaling pathway undermined. Of note, it was found that AZD8797 treatment even doubled the hematoma expansion, which suggested that the role of FKN/CX3CR1 signaling in ICH is beyond phagocytosis, and some other mechanisms, such as inflammation, cell protection, and vascular permeability, may also be involved, and further research will be performed in the next step.
CD163 and HO-1 are well-recognized main molecules, which control hematoma absorption after ICH and the CD163/HO-1 axis also regulates inflammation around the hematoma [12, 38]. Thus, CD163 could be a good target for ICH therapy and increased CD163 expression may be of great benefit to ICH treatment. However, it was reported that CD163 is also expressed in neurons, it helps neuronal uptake of Hb, and then it leads to cell death of neurons due to iron overload [39]. Our results showed that FKN treatment increased CD163 and HO-1 expressions in microglia after ICH in vivo and in vitro, while FKN special antagonist reversed this effect. It is likely that FKN treatment promotes hematoma absorption after ICH through CD163 and HO-1 in microglia, which could avoid the adverse effect of a general increase in the expression of CD163 in the neurons. To further explore the specific molecular mechanism, RNA sequence analysis was used to detect the changes in microglia with or without FKN treatment, and the results showed that PPAR-γ was significantly increased after FKN treatment. As already known, PPAR-γ is a member belonging to the nuclear hormone receptor superfamily of transcription factors, and it is not only protective for neurons, astrocytes, oligodendrocytes, endothelial cells, and microglia, but is also beneficial to reduce inflammation, decrease oxidative damage, and attenuate cell death [40, 41].
PPAR-γ has been previously reported to increase phagocytosis of RBCs in microglia, accelerate hematoma absorption, and attenuate brain injury [42–44], which is in accordance with our study showing that PPAR-γ siRNA could significantly decrease phagocytosis of RBCs and expressions of CD163 and HO-1 in microglia. Thus, we propose the view that FKN could promote hematoma clearance after ICH via the PPAR-γ/CD163/HO-1 axis in microglia. Of note, our study about the role of FKN and its receptor CX3CR1 in hematoma absorption after ICH provides a fine example of the fact that communication between neurons and microglia could attenuate potential injury in the CNS.
Conclusions
In summary, our study found that FKN is more than a chemotactic factor and could mediate the communication between neurons and microglia to act against ICH. FKN could promote hematoma absorption via the PPAR-γ/CD163/HO-1 signaling pathway in microglia. Thus, FKN may be a potential therapeutic target to improve hematoma absorption and it may shed light on ICH treatment.
Abbreviations
- ICH
Intracerebral hemorrhage
- FKN
Fractalkine
- Hb
Hemoglobin
- CD163
Hemoglobin scavenger receptor
- HO-1
Hemeoxygenase-1
- PPAR-γ
Peroxisome proliferator-activated receptor-γ
- RCTs
Randomized control trials
- TBI
Traumatic brain injury
- MS
Multiple sclerosis
- ALS
Amyotrophic lateral sclerosis
- AD
Alzheimer’s disease
- PD
Parkinson’s disease
- NIH
National Institutes of Health
- BV2 cells
Mouse BV2 microglia cells
- N2a cells
Mouse N2a neuron cells
- PBS
Phosphate-buffered saline
- ELISA
Enzyme-linked immunosorbent assay
- T1WI
T1-weighed imaging
- T2WI
T2-weighed imaging
- NDS
Neurological deficit score
- RNA-seq
RNA sequencing
- KEGG
Kyoto encyclopedia of genes genomes
- SN
Substantia nigra
- MPP+
1-Methyl-4-phenylpyridinium
- RBC
Red blood cell
- GFAP
Glial fibrillary acidic protein
- Iba-1
Ionized calcium-binding adaptormolecule-1
- NeuN
Neuronal nuclei
- CD31
Platelet endothelial cell adhesion molecule-1
- MRI
Magnetic resonance imaging
- siRNA
Small interfering RNA
Author contributions
MFY, CNL and YW performed the ICH model, western blots, MRI, qRT-PCR, ELISA, erythrophagocytosis assay and immunofluorescence staining. HXG performed behavioral tests. JS performed hemoglobin content measurement. YW performed transwell migration assay. ML performed cell culture. MFY analyzed the data. CNL collected clinical data. MFY, QWH and BH conceived and designed the experiments, wrote and edited the manuscript. All authors reviewed and approved the final manuscript version.
Funding
This work was supported by National Key Research & Development Program of China (No.2018YFC1312200 to Bo Hu), National Natural Science Foundation of China (No. 82071335 to Quanwei He, No. 81571119 to Bo Hu, No. 81901214 to Yan Wan).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
All animal experiments followed the guidelines of the National Institutes of Health (NIH) and were confirmed by the Medical Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology, China. Acquisition of human blood samples and subsequent experiments were approved by the Medical Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology, China.
Consent for publication
This manuscript has been approved for publication by all authors.
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Mingfeng You, Chunnan Long, and Yan Wan contributed equally to this work.
Contributor Information
Quanwei He, Email: 2013XH0954@hust.edu.cn.
Bo Hu, Email: hubo@mail.hust.edu.cn.
References
- 1.An SJ, Kim TJ, Yoon BW. Epidemiology, risk factors, and clinical features of intracerebral hemorrhage: an update. J Stroke. 2017;19(1):3–10. doi: 10.5853/jos.2016.00864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gross BA, Jankowitz BT, Friedlander RM. Cerebral intraparenchymal hemorrhage: a review. JAMA. 2019;321(13):1295–1303. doi: 10.1001/jama.2019.2413. [DOI] [PubMed] [Google Scholar]
- 3.Lan X, Han X, Li Q, Yang QW, Wang J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat Rev Neurol. 2017;13(7):420–433. doi: 10.1038/nrneurol.2017.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 2012;11(8):720–731. doi: 10.1016/S1474-4422(12)70104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhu H, Wang Z, Yu J, Yang X, He F, Liu Z, et al. Role and mechanisms of cytokines in the secondary brain injury after intracerebral hemorrhage. Prog Neurobiol. 2019;178:101610. doi: 10.1016/j.pneurobio.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 6.Bulters D, Gaastra B, Zolnourian A, Alexander S, Ren D, Blackburn SL, et al. Haemoglobin scavenging in intracranial bleeding: biology and clinical implications. Nat Rev Neurol. 2018;14(7):416–432. doi: 10.1038/s41582-018-0020-0. [DOI] [PubMed] [Google Scholar]
- 7.Wilkinson DA, Keep RF, Hua Y, Xi G. Hematoma clearance as a therapeutic target in intracerebral hemorrhage: From macro to micro. J Cereb Blood Flow Metab. 2018;38(4):741–745. doi: 10.1177/0271678X17753590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang CF, Wan J, Li Q, Renfroe SC, Heller NM, Wang J. Alternative activation-skewed microglia/macrophages promote hematoma resolution in experimental intracerebral hemorrhage. Neurobiol Dis. 2017;103:54–69. doi: 10.1016/j.nbd.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mendelow AD, Gregson BA, Fernandes HM, Murray GD, Teasdale GM, Hope DT, et al. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet (London, England) 2005;365(9457):387–397. doi: 10.1016/S0140-6736(05)70233-6. [DOI] [PubMed] [Google Scholar]
- 10.Mendelow AD, Gregson BA, Rowan EN, Murray GD, Gholkar A, Mitchell PM. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial lobar intracerebral haematomas (STICH II): a randomised trial. Lancet (London, England) 2013;382(9890):397–408. doi: 10.1016/S0140-6736(13)60986-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abraham NG, Drummond G. CD163-Mediated hemoglobin-heme uptake activates macrophage HO-1, providing an antiinflammatory function. Circ Res. 2006;99(9):911–914. doi: 10.1161/01.RES.0000249616.10603.d6. [DOI] [PubMed] [Google Scholar]
- 12.Garton T, Keep RF, Hua Y, Xi G. CD163, a hemoglobin/haptoglobin scavenger receptor, after intracerebral hemorrhage: functions in microglia/macrophages versus neurons. Transl Stroke Res. 2017;8(6):612–616. doi: 10.1007/s12975-017-0535-5. [DOI] [PubMed] [Google Scholar]
- 13.Xie WJ, Yu HQ, Zhang Y, Liu Q, Meng HM. CD163 promotes hematoma absorption and improves neurological functions in patients with intracerebral hemorrhage. Neural Regen Res. 2016;11(7):1122–1127. doi: 10.4103/1673-5374.187047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leclerc JL, Lampert AS, Loyola Amador C, Schlakman B, Vasilopoulos T, Svendsen P, et al. The absence of the CD163 receptor has distinct temporal influences on intracerebral hemorrhage outcomes. J Cereb Blood Flow Metab. 2018;38(2):262–273. doi: 10.1177/0271678X17701459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schallner N, Pandit R, LeBlanc R, 3rd, Thomas AJ, Ogilvy CS, Zuckerbraun BS, et al. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J Clin Investig. 2015;125(7):2609–2625. doi: 10.1172/JCI78443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen-Roetling J, Regan RF. Haptoglobin increases the vulnerability of CD163-expressing neurons to hemoglobin. J Neurochem. 2016;139(4):586–595. doi: 10.1111/jnc.13720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garton T, Keep RF, Hua Y, Xi G. Brain iron overload following intracranial haemorrhage. Stroke Vasc Neurol. 2016;1(4):172–184. doi: 10.1136/svn-2016-000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xing C, Lo EH. Help-me signaling: non-cell autonomous mechanisms of neuroprotection and neurorecovery. Prog Neurobiol. 2017;152:181–199. doi: 10.1016/j.pneurobio.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Réaux-Le Goazigo A, Van Steenwinckel J, Rostène W, Mélik PS. Current status of chemokines in the adult CNS. Prog Neurobiol. 2013;104:67–92. doi: 10.1016/j.pneurobio.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Grosse GM, Tryc AB, Dirks M, Schuppner R, Pflugrad H, Lichtinghagen R, et al. The temporal dynamics of plasma fractalkine levels in ischemic stroke: association with clinical severity and outcome. J Neuroinflamm. 2014;11:74. doi: 10.1186/1742-2094-11-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finneran DJ, Nash KR. Neuroinflammation and fractalkine signaling in Alzheimer's disease. J Neuroinflamm. 2019;16(1):30. doi: 10.1186/s12974-019-1412-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cipriani R, Villa P, Chece G, Lauro C, Paladini A, Micotti E, et al. CX3CL1 is neuroprotective in permanent focal cerebral ischemia in rodents. J Neurosci. 2011;31(45):16327–16335. doi: 10.1523/JNEUROSCI.3611-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gyoneva S, Ransohoff RM. Inflammatory reaction after traumatic brain injury: therapeutic potential of targeting cell-cell communication by chemokines. Trends Pharmacol Sci. 2015;36(7):471–480. doi: 10.1016/j.tips.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Liu Y, Liu X, Li S, Cheng C, Chen S, et al. Dynamic changes of CX3CL1/CX3CR1 axis during microglial activation and motor neuron loss in the spinal cord of ALS mouse model. Transl Neurodegener. 2018;7:35. doi: 10.1186/s40035-018-0138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ridderstad Wollberg A, Ericsson-Dahlstrand A, Juréus A, Ekerot P, Simon S, Nilsson M, et al. Pharmacological inhibition of the chemokine receptor CX3CR1 attenuates disease in a chronic-relapsing rat model for multiple sclerosis. Proc Natl Acad Sci USA. 2014;111(14):5409–5414. doi: 10.1073/pnas.1316510111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shao A, Zhu Z, Li L, Zhang S, Zhang J. Emerging therapeutic targets associated with the immune system in patients with intracerebral haemorrhage (ICH): from mechanisms to translation. EBioMedicine. 2019;45:615–623. doi: 10.1016/j.ebiom.2019.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bai Q, Xue M, Yong VW. Microglia and macrophage phenotypes in intracerebral haemorrhage injury: therapeutic opportunities. Brain. 2020;143(5):1297–1314. doi: 10.1093/brain/awz393. [DOI] [PubMed] [Google Scholar]
- 28.Li Q, Wan J, Lan X, Han X, Wang Z, Wang J. Neuroprotection of brain-permeable iron chelator VK-28 against intracerebral hemorrhage in mice. J Cereb Blood Flow Metab. 2017;37(9):3110–3123. doi: 10.1177/0271678X17709186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao X, Kruzel M, Ting SM, Sun G, Savitz SI, Aronowski J. Optimized lactoferrin as a highly promising treatment for intracerebral hemorrhage: Pre-clinical experience. J Cereb Blood Flow Metab. 2021;41(1):53–66. doi: 10.1177/0271678X20925667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang QW. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol. 2014;115:25–44. doi: 10.1016/j.pneurobio.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 31.Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol. 2010;92(4):463–477. doi: 10.1016/j.pneurobio.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi SX, Li YJ, Shi K, Wood K, Ducruet AF, Liu Q. IL (Interleukin)-15 bridges astrocyte-microglia crosstalk and exacerbates brain injury following intracerebral hemorrhage. Stroke. 2020;51(3):967–974. doi: 10.1161/STROKEAHA.119.028638. [DOI] [PubMed] [Google Scholar]
- 33.Chen ZQ, Yu H, Li HY, Shen HT, Li X, Zhang JY, et al. Negative regulation of glial Tim-3 inhibits the secretion of inflammatory factors and modulates microglia to antiinflammatory phenotype after experimental intracerebral hemorrhage in rats. CNS Neurosci Ther. 2019;25(6):674–684. doi: 10.1111/cns.13100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cardoso AL, Fernandes A, Aguilar-Pimentel JA, de Angelis MH, Guedes JR, Brito MA, et al. Towards frailty biomarkers: candidates from genes and pathways regulated in aging and age-related diseases. Ageing Res Rev. 2018;47:214–277. doi: 10.1016/j.arr.2018.07.004. [DOI] [PubMed] [Google Scholar]
- 35.Chen Y, Zhu G, Liu D, Zhang X, Liu Y, Yuan T, et al. Subthalamic nucleus deep brain stimulation suppresses neuroinflammation by Fractalkine pathway in Parkinson's disease rat model. Brain Behav Immun. 2020;90:16–25. doi: 10.1016/j.bbi.2020.07.035. [DOI] [PubMed] [Google Scholar]
- 36.Wang J, Pan H, Lin Z, Xiong C, Wei C, Li H, et al. Neuroprotective effect of fractalkine on radiation-induced brain injury through promoting the M2 polarization of microglia. Mol Neurobiol. 2021;58(3):1074–1087. doi: 10.1007/s12035-020-02138-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shan S, Hong-Min T, Yi F, Jun-Peng G, Yue F, Yan-Hong T, et al. New evidences for fractalkine/CX3CL1 involved in substantia nigral microglial activation and behavioral changes in a rat model of Parkinson's disease. Neurobiol Aging. 2011;32(3):443–458. doi: 10.1016/j.neurobiolaging.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 38.Ren H, Han R, Chen X, Liu X, Wan J, Wang L, et al. Potential therapeutic targets for intracerebral hemorrhage-associated inflammation: an update. J Cereb Blood Flow Metab. 2020;40(9):1752–1768. doi: 10.1177/0271678X20923551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu R, Cao S, Hua Y, Keep RF, Huang Y, Xi G. CD163 expression in neurons after experimental intracerebral hemorrhage. Stroke. 2017;48(5):1369–1375. doi: 10.1161/STROKEAHA.117.016850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zolezzi JM, Santos MJ, Bastías-Candia S, Pinto C, Godoy JA, Inestrosa NC. PPARs in the central nervous system: roles in neurodegeneration and neuroinflammation. Biol Rev Camb Philos Soc. 2017;92(4):2046–2069. doi: 10.1111/brv.12320. [DOI] [PubMed] [Google Scholar]
- 41.Cai W, Yang T, Liu H, Han L, Zhang K, Hu X, et al. Peroxisome proliferator-activated receptor γ (PPARγ): a master gatekeeper in CNS injury and repair. Prog Neurobiol. 2018;163–164:27–58. doi: 10.1016/j.pneurobio.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhao X, Grotta J, Gonzales N, Aronowski J. Hematoma resolution as a therapeutic target: the role of microglia/macrophages. Stroke. 2009;40(3 Suppl):S92–S94. doi: 10.1161/STROKEAHA.108.533158. [DOI] [PubMed] [Google Scholar]
- 43.Zhao X, Sun G, Zhang J, Strong R, Song W, Gonzales N, et al. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol. 2007;61(4):352–362. doi: 10.1002/ana.21097. [DOI] [PubMed] [Google Scholar]
- 44.Flores JJ, Klebe D, Rolland WB, Lekic T, Krafft PR, Zhang JH. PPARγ-induced upregulation of CD36 enhances hematoma resolution and attenuates long-term neurological deficits after germinal matrix hemorrhage in neonatal rats. Neurobiol Dis. 2016;87:124–133. doi: 10.1016/j.nbd.2015.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.