

Abstract
Helicobacter pylori-mediated gastric carcinogenesis involves upregulation of the E3 ubiquitin ligase Siah2 and its phosphorylation-mediated stabilization. This study elucidates a novel mechanism of oxidative stress regulation by phosphorylated Siah2 in H. pylori-infected gastric epithelial cancer cells (GECs). We identify that H. pylori-mediated Siah2 phosphorylation at the 6th serine residue (P-S6-Siah2) enhances proteasomal degradation of the 78-kDa glucose-regulated protein (GRP78) possessing antioxidant functions. S6 phosphorylation stabilizes Siah2 and P-S6-Siah2 potentiates H. pylori-mediated reactive oxygen species (ROS) generation. However, infected S6A phospho-null Siah2-expressing cells have decreased cellular GRP78 level as surprisingly these cells release GRP78 to a higher extent and accumulate significantly higher ROS than the wild type (WT) Siah2 construct-expressing cells. Ectopic expression of GRP78 prevents the loss of mitochondrial membrane potential and cellular ROS accumulation caused by H. pylori. H. pylori-induced mitochondrial damage and mitochondrial membrane potential loss are potentiated in Siah2-overexpressing cells but these effects are further enhanced in S6A-expressing cells. This study also confirms that while phosphorylation-mediated Siah2 stabilization optimally upregulates aggresome accumulation, it suppresses autophagosome formation, thus decreasing the dependency on the latter mechanism in regulating cellular protein abundance. Disruption of the phospho-Siah2-mediated aggresome formation impairs proliferation of infected GECs. Thus, Siah2 phosphorylation has diagnostic and therapeutic significance in H. pylori-mediated gastric cancer (GC).
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-022-04437-5.
Keywords: Aggresome, Autophagosome, BiP/GRP78, E3 ubiquitin ligase, Siah, Oxidative stress
Introduction
One of the major factors responsible for human gastric cancer (GC) is colonization of the stomach by Helicobacter pylori, a microaerophilic, Gram-negative spiral bacterium which has been classified as a class I carcinogen [1]. H. pylori infection induces oxidative stress [2, 3]. Oxidative stress is associated with increased ROS production. Increased generation of ROS is the host cell’s primary attempt to eliminate the bacterium from the system. Although moderate level of ROS is essential for normal functioning of cells, excessive ROS may lead to many pathological conditions. Continuous generation of ROS increases DNA damage, impairs DNA repair processes and induces apoptosis. If a host cell with damaged DNA manages to evade apoptosis, that cell is more prone to become cancerous. Thus, ROS are key contributors in the progression of GC [4].
E3 ubiquitin ligases regulate ROS generation [5]. For example, HECT-type E3 ubiquitin ligase ITCH degrades thioredoxin-interacting protein, an inhibitor of the antioxidant thioredoxin and helps ameliorate ROS level in rat cardiomyocytes [6]. The E6AP E3 ubiquitin ligase controls ROS levels by regulating the expression of Prx1 in mouse embryo fibroblasts [5]. Knockdown of the E3 ubiquitin ligase c-CBL leads to ROS generation in cutaneous T-cell lymphoma [7]. The RING-type E3 ubiquitin ligase seven in absentia homolog 2 (Siah2) is also involved in ROS generation under hypoxia [8] and hypoglycaemia [9]. Siah2 promotes GC and several other cancers, such as breast, colon and stomach cancer [10]. Phosphorylated Siah2 enhances invasion and migration of cancer cells [11, 12]. However, Siah2 and its phosphorylation-mediated ROS regulation during H. pylori infection remain unexplored. ROS regulates multiple cellular processes [13, 14]. ROS-mediated protein modifications lead to the generation of protein aggregates in the cell that agglomerate and form perinuclear inclusion bodies—aggresomes [15, 16]. Aggresomes are induced in oxidative stress [17] and are substrates for macroautophagy [18, 19]. The connection of aggresome formation with ROS regulation makes it an interesting event to study in Helicobacter-mediated GC.
Suppression of GRP78 has been implicated in the upregulation of ROS in pancreatic cancer cells [20]. In addition, GRP78 protein promotes aggresome delivery to autophagosomes in multiple myeloma cells and proteasome blocking results in GRP78 upregulation [21]. This study unravels a novel mechanism wherein H. pylori-mediated Siah2 phosphorylation is found to downregulate GRP78, increase ROS generation, enhance aggresome formation as well as proliferation of H. pylori-infected GECs. Our results consistently present GRP78 decrease and P-S6-Siah2 increase as prominent features of Helicobacter-infected human and mouse GC tissues. These findings point to the importance of P-S6-Siah2 as a potential diagnostic and therapeutic target in H. pylori-mediated gastric carcinogenesis.
Materials and methods
Reagents, bacteria and cell culture
GECs (MKN45 and AGS), H. pylori (strains 26695 and 8-1) and H. felis (ATCC; VA, USA) were maintained as described previously [10, 22]. For infection, bacteria were inoculated in Brucella broth (BD Biosciences, NJ, USA; Cat. No. 211088) supplemented with 10% fetal bovine serum (HiMedia, Nashik, India; Cat. No. RM9970). Cells were infected with 200 multiplicity of infection (MOI) for 12 h, if not specified otherwise. Proteasomal degradation was inhibited using MG132 (Millipore-Sigma, MO, USA; Cat. No. M7449-200UL). Catalase (Sigma-Aldrich, MO, USA; Cat. No. C1345) and N-acetyl-l-cysteine (NAC; Sigma-Aldrich; Cat. No. A7250-50G) were used in assays to examine ROS. Autophagy inhibitor Bafilomycin A1 (Cell Signaling Technology, MA, USA; Cat. No. 54645) and microtubule polymerization inhibitor nocodazole (Sigma-Aldrich; Cat. No. M1404) were also used in this study.
Expression plasmids and site-directed mutagenesis
Eukaryotic expression vector pcDNA3.1+ (Thermo Fisher Scientific, IL, USA), the WT human siah2 (gene ID: 6478) construct (by Origene Technologies, MD, USA) and pDsRed2-Mito (Clontech, CA, USA; Cat. No. 632421) were purchased. siah2 phospho-null mutants S6A and T279A were generated by site-directed mutagenesis from the WT siah2 construct using specific primers as described earlier [12]. pcDNA3.1(+)-GRP78/BiP (grp78) was a gift from Dr. Richard C. Austin (Addgene plasmid #32701) [23].
Transfection and generation of stable cells
Lipofectamine 3000 and P3000 reagents (Invitrogen, CA, USA; Cat. No. L3000015) were used for transfection and generation of stable cells following standard protocols [10, 12, 22]. To select stably transfected clones, G418 solution (Millipore-Sigma; Cat. No. G8168-10ML) was used. Cells were transfected with human control siRNA (Santa Cruz Biotechnology, TX, USA; Cat. No. sc-37007), human grp78 siRNA (Santa Cruz Biotechnology; Cat. No. sc-29338), human control siRNA (Origene Technologies; Cat. No. SR30004) and human siah2 siRNA (Origene Technologies; Cat. No. SR304370) using Lipofectamine 3000.
Immunoblotting and co-immunoprecipitation assays
Whole cell lysates were subjected to SDS-PAGE followed by transfer onto the PVDF membrane. P-Ser (Sigma-Aldrich; Cat. No. P5747), P-Thr (Cell Signaling Technology; Cat. No. 9386S), CoxIV (Cell Signaling Technology; Cat. No. 4850S), custom-prepared human P-S6-Siah2 (Bioklone Biotech Pvt. Ltd., Chennai, India), GAPDH (Abgenex, Bhubaneswar, India; Cat. No. 10-10011), GRP78 (Abcam, MA, USA; Cat. No. ab21685), Siah2 (Santa Cruz Biotechnology; Cat. No. sc-5507), ubiquitin (Cell Signaling Technology; Cat. No. 3936P) and LC3B (Cell Signaling Technology; Cat. No. 3868) primary antibodies were used. The specificity of the custom-prepared P-S6-Siah2 antibody was assessed by performing western blotting of cell lysates of uninfected and infected pcDNA3.1+, Siah2 WT, S6A and T279A stably-expressing MKN45 cells. All blots were detected as described earlier [10, 12]. ImageLab software (Bio-Rad Laboratories; CA, USA; Cat. No. CCK003-1000) was used for densitometric analysis. Co-immunoprecipitation of whole cell lysates was performed using Siah2 antibody and goat IgG (Santa Cruz Biotechnology; Cat. No. sc-2755) was used to determine specificity.
Cytoplasmic and mitochondrial fractionation
Cytoplasmic and mitochondrial fractions of MKN45 cells were isolated following previously-described protocol [24].
ROS detection
0.166 × 106 AGS or MKN45 cells were either left uninfected or infected with H. pylori. Cells were then washed with 1X PBS and incubated with 1 µM 2, 7-dichlorodihydrofluorescein diacetate (DCFDA; Sigma-Aldrich; Cat. No. D6883) for 1 h or with 5 µM CellROX Deep Red reagent (Invitrogen, USA; Cat. no. C10422) for 30 min at 37 °C in the dark. Cells were fixed using 4% paraformaldehyde (PFA) for 10 min followed by nuclear counterstaining with 4′,6-diamidino-2-phenylindoledihydrochloride (DAPI; Invitrogen; Cat. No. D3571). ROS levels were detected by fluorescence microscopy (Nikon, Tokyo, Japan). Images were taken from three different fields for each biological repeat and the mean fluorescence intensity indicative of ROS levels were calculated.
Fluorescence microscopy and image analysis
Uninfected or infected 0.166 × 106 AGS or MKN45 cells were fixed using 4% PFA. Cells were permeabilized with 0.1% Triton X-100 in 1X PBS followed by blocking with 5% bovine serum albumin solution for 1 h at RT followed by incubation with the desired primary antibodies for overnight at 4 ºC. Incubation with specific Alexa Fluor-tagged secondary antibodies (Invitrogen) followed by counterstaining with DAPI was performed. Images were captured using either Nikon Eclipse TiU/Eeclipse Ni-E fluorescence microscopes (Nikon) or Leica DMi8 confocal microscope (Leica, Germany). Images were processed and analyzed using either the NIS Advanced Research software (Nikon) or Fiji [25].
Human gastric biopsy and murine tissue collection
Human and mouse gastric tissues were collected according to previously-established protocols [12, 26] and were ethically approved by NISER. Tissues were fixed with 4% PFA and cryosectioned at 5 µm thickness.
Enzyme-linked immunosorbent assay (ELISA)
Supernatants from uninfected or infected 0.166 × 106 empty vector, WT or S6A siah2-expressing MKN45 stable cells were collected and were used for assays. ELISA was performed using GRP78/BiP ELISA kit (Enzo Life Sciences, NY, USA; Cat. No. ADI-900-214) and results were analyzed using a four parameter logistic curve fitting program [27].
Aggresome formation
Empty vector, WT or S6A siah2-expressing MKN45 stable cells were plated and processed as described under the microscopy section. Cells were stained using aggresome detection kit (Abcam; Cat. No. ab139486) following the manufacturer-recommended protocol and were imaged using Leica DMi8 confocal microscope (Leica, Germany).
MTT assay
Uninfected or infected 1 × 104 empty vector, WT or S6A siah2-expressing MKN45 stable cells were quantified for cellular proliferation using EZcount MTT cell assay kit (HiMedia; Cat. No. CCK003-1000).
Mitochondrial membrane potential (Δψm) analysis
1 × 106 pcDNA3.1+, siah2 WT and siah2 S6A stably-expressing MKN45 cells were either left uninfected or were infected with H. pylori. Post infection, cells were loaded with 100 μM of tetramethylrhodamine, methyl ester (TMRM; Invitrogen, Cat. No. T668) and incubated for 30 min in CO2 incubator. Fluorescence from live cells was observed under Olympus FluoView FV300 confocal microscope (Olympus, Japan) and images were processed using Fiji [25].
Statistical analysis
Statistical significance was determined using unpaired two-tailed t test, one-way, two-way or three-way ANOVA followed by Tukey’s post hoc analysis. P < 0.05 was considered statistically significant. Statistical analysis was performed using GraphPad Prism software (Version 7; GraphPad Software Inc., CA, USA).
Results
Siah2 and its phosphorylation regulate ROS generation
S6 and T279 phosphorylation of Siah2 increases its stability and promotes GC [12]. To elucidate the role of Siah2 and its phosphorylation in H. pylori-mediated ROS generation, MKN45 cells stably-expressing the empty vector (pcDNA3.1+), siah2 WT, siah2 phospho-null mutants S6A and T279A were used. Cells were left uninfected or were infected with 200 MOI of H. pylori for 12 h followed by ROS detection. This MOI and time point were used for all following experiments, if not stated otherwise. Widefield fluorescence microscopy followed by quantification showed that siah2 significantly enhanced H. pylori-mediated ROS generation as compared to the empty vector-expressing stable cells. Interestingly, ROS generation was further increased in siah2 phospho-null mutant S6A-expressing cells than siah2 WT and siah2 T279A stable cells (Fig. 1A). These data established the importance of Siah2 and its phosphorylation at S6 in regulating ROS generation. Paired western blot data depicted P-Ser, P-Thr and total Siah2 levels in pcDNA3.1+, siah2 WT, siah2 S6A and siah2 T279A MKN45 stable cells from the same batch of the cells that were used to generate the micrographs (Fig. 1B). The western blot result also reaffirmed one of our previous findings that identified phosphorylation at Ser residue to be important in stabilizing Siah2 [12]. Due to the observed importance of Siah2 Ser phosphorylation in ROS regulation, only this phospho-null mutant was considered for further study.
Fig. 1.

Siah2 modulates ROS and interacts with GRP78 in H. pylori-infected GECs. A Micrographs depicting ROS generation in uninfected or H. pylori-infected MKN45 pcDNA3.1+, siah2 WT, siah2 phospho-null mutant S6A and T279A stably-expressed cells. Scale bars = 10 μm. The graph represents fold changes in ROS and depicts mean ± sem values. n = 3. Statistical significance is determined by two-way ANOVA followed by Tukey’s post hoc analysis. **P < 0.01, ***P < 0.001. B Above, representative western blots showing the level of P-Ser and P-Thr in uninfected or H. pylori-infected MKN45 pcDNA3.1+, siah2 WT, siah2 phospho-null mutant S6A and T279A stably-expressed cells. Below, Bar graphs represent level of P-Ser, P-Thr and Siah2 in these cells. Graphs = mean ± sem. Statistical significance is determined by two-way ANOVA followed by Tukey’s post hoc analysis (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Western blot showing the status of P-Ser, P-Thr and total Siah2 in uninfected and H. pylori-infected MKN45 pcDNA3.1+, siah2 WT, siah2 S6A and siah2 T279A stable cells. GAPDH is used as a loading control. C Western blot of the immuno-complexes representing the interaction of Siah2-GRP78 in uninfected and infected MKN45 cell whole cell lysates co-immuno-precipitated using Siah2 antibody. Non-specific band is used as the loading control. The input lanes are shown alongside to ascertain different protein expression level. D Western blot of the whole cell lysates from uninfected and infected (200 MOI) MKN45 cells showing GRP78 protein level at the indicated time points. GAPDH is used as a loading control (left). Bar graphs represent the change in GRP78 protein levels after various time intervals of infection (right). Graphs = mean ± sem. Statistical significance is determined by two-way ANOVA followed by Tukey’s post hoc analysis (n = 3). *P < 0.05. E Immunoblot representing GRP78 protein in whole cell lysates of MKN45 infected with 100, 200 and 300 MOIs for 12 h. GAPDH = loading control (left). Bar graphs represent GRP78 protein status at various MOIs (right). Graphs = mean ± sem. Statistical significance is determined by one-way ANOVA (n = 3). *P < 0.05; ***P < 0.001. F Western blots depicting GRP78 status in MKN45 cells infected with H. pylori cag PAI ( +) 26,695 and cag PAI (-) 8–1 strains (left). Bar graphs showing the cag PAI-independent decrease of GRP78 (right). Graphs = mean ± sem. Statistical significance is determined by one-way ANOVA (n = 3). ***P < 0.001
To identify the factor(s) responsible for the altered ROS production under the influence of Siah2, MKN45 cells were transfected with the empty vector or siah2 overexpression plasmid for 36 h and infected with H. pylori. Whole cell lysates were immuno-precipitated using Siah2 antibody. Lysates were subjected to SDS-PAGE followed by staining with Coomassie brilliant blue R-250. Bands were excised and analyzed by LC–MS/MS. GRP78, a cellular redox regulator [20, 28], was identified as the most prominent Siah2-interacting protein (Fig. S1A). GRP78 protein contains multiple “Siah degron motifs” which is also suggestive of possible GRP78-Siah2 interaction (Fig. S1B). Co-immunoprecipitation assay of whole cell lysates using Siah2 antibody from uninfected and infected MKN45 cells followed by western blotting confirmed of GRP78-Siah2 interaction and increased GRP78 downregulation in H. pylori-infected MKN45 cells (Fig. 1C).
H. pylori infection decreases GRP78
To investigate the status of GRP78 in H. pylori-infected GECs, MKN45 cells were challenged with H. pylori 26695 (MOI 200) for various durations. Representative western blot result (n = 3) showed that GRP78 decrease was optimal at 12 h post-infection (Fig. 1D). Further, to determine the optimal MOI to decrease GRP78, MKN45 cells were infected with various MOIs of H. pylori for 12 h. 200 MOI was optimal for GRP78 protein decrease as shown by a representative (n = 3) western blot (Fig. 1E). The presence or the absence of H. pylori cytotoxin-associated gene pathogenicity island (cag PAI) is associated with the degree of GC pathogenesis [10]. To unravel the effect of cag PAI on GRP78 protein, MKN45 cells were infected with a reference cag PAI (+) strain 26695 and cag PAI (−) strain 8–1 at 200 MOI for 12 h. Representative western blot (n = 3) confirmed that GRP78 decrease was independent of the cag PAI status of H. pylori (Fig. 1F). To elucidate the status of GRP78 in adenocarcinoma and metastatic GC, human gastric antral biopsy tissues were immuno-stained using GRP78 antibody. In comparison with their paired control tissues, both adenocarcinoma and metastatic tissues exhibited decreased GRP78 protein which were indicative of sustained GRP78 decrease in H. pylori-mediated GC (Fig. S1C).
GRP78 is marked for proteasomal degradation by Siah2
Siah2 is known to modulate the abundance of its interacting partners [29, 30]. Therefore, we were keen to investigate whether GRP78 decrease was in effect Siah2-mediated or not. For this, pcDNA3.1+ and siah2 WT MKN45 stable cells were infected. Confocal microscopy showed that GRP78 significantly decreased after H. pylori infection and was further decreased in siah2 WT-expressing infected cells (Fig. 2A). To examine proteasomal involvement in GRP78 downregulation, empty vector and siah2 WT MKN45 stable cells were treated with 50 µM MG132 in the presence or the absence of H. pylori infection. Confocal microscopy showed that GRP78 was significantly downregulated in WT Siah2-expressing H. pylori-infected cells and MG132-treatment could significantly rescue GRP78 from degradation (Fig. 2B). These results confirmed that GRP78 decrease in H. pylori-infected GECs was Siah2-dependent and proteasome-mediated. As polyubiquitination leads to proteasomal degradation, we further assessed the polyubiquitination status of GRP78. For this, MKN45 cells were infected with H. pylori in the presence or absence of 50 µM MG132. Western blots probed using anti-ubiquitin antibody found ubiquitin accumulation in H. pylori-infected and MG132-treated cells. This result indicated that the protein abundance of GRP78 was not mediated by ubiquitination-mediated proteasomal degradation. Membranes were then re-probed for GRP78 and GAPDH, revealing GRP78 rescue in MG132-treated H. pylori-infected cells (n = 3). This reversal of downregulation was statistically significant (Fig. 2C). As cellular protein abundance is regulated majorly by two pathways: autophagosome formation and proteasomal degradation [31–33], we wanted to examine the involvement of autophagy in GRP78 decrease. MKN45 cells were treated with 10 nM bafilomycin A1, a well-known inhibitor of lysosomal acidification, along with H. pylori infection for 12 h. A representative western blot (n = 3) showed that H. pylori-mediated GRP78 decrease was macroautophagy-independent (Fig. 2D). Interestingly, bafilomycin A1 treatment potentiated GRP78 degradation in both H. pylori-infected and uninfected cells. This result could be explained by enhanced Siah2 level in the presence of bafilomycin A1.
Fig. 2.

Proteasomal degradation of GRP78 is Siah2-mediated in H. pylori-infected GECs. A Confocal micrographs illustrating the levels of GRP78 protein in uninfected and infected pcDNA3.1+ and siah2 WT MKN45 stable cells. Scale bars represent 5 μm (left). Bar graphs represent fold change in the immunofluorescence of GRP78 (right). Graphs = mean ± sem. n = 3. Statistical significance is determined by two-way ANOVA followed by Tukey’s post hoc analysis. ****P < 0.0001. B Micrographs representing GRP78 protein levels in untreated, DMSO-treated and MG132-treated uninfected or infected pcDNA3.1+ and siah2 WT stably expressing MKN45 cells. Scale bars represent 5 μm. Graphs represent fold change in GRP78 levels. Graphs = mean ± sem. n = 3. Statistical significance is determined by three-way ANOVA followed by Tukey’s post hoc analysis. ****P < 0.0001. C Western blot representing ubiquitin aggregates in H. pylori-infected and MG132-treated MKN45 cells. Arrow indicates the position of ubiquitinated-GRP78 (left). Re-probed blots showing GRP78 status in H. pylori-infected and MG132-treated MKN45 cells (right). GAPDH = loading control. Bar graph represents GRP78 protein rescue in MG132-treated and H. pylori-infected MKN45 cells. Statistical significance is determined by one-way ANOVA (right). Graphs = mean ± sem. n = 3. **P < 0.01. D Western blot depicting autophagy-independent GRP78 decrease and Siah2 increase as assessed by infecting MKN45 cells in the presence or the absence of 10 nM autophagy inhibitor bafilomycin A1. GAPDH is used as the loading control. E Western blot of the whole cell lysates from uninfected and infected control or siah2 siRNA-transfected MKN45 cells depicting the levels of GRP78 and Siah2. GAPDH = loading control. Bar graphs represent GRP78 protein in the similar setup. Statistical significance is determined by two-way ANOVA followed by Tukey’s post hoc analysis. Graphs = mean ± sem. n = 3. **P < 0.01, ***P < 0.001
To identify the role of siah2 suppression on GRP78 protein level, MKN45 cells were transfected with siah2 siRNA for 36 h followed by H. pylori infection. A representative western blot (n = 3) and an accompanying graph showed that H. pylori-mediated proteasomal degradation of GRP78 was significantly hampered in siah2 siRNA-transfected and H. pylori-infected GEC (Fig. 2E). Taken together, these results confirmed that GRP78 proteasomal degradation is Siah2-mediated.
GRP78 decrease is associated with human and mouse Helicobacter-induced GC
Phosphorylation of Siah2 at S6 is a prominent feature of Helicobacter-infected human and mouse gastric epithelia [12]. In this context, we were interested to find out the correlation of GRP78 with Siah2 and its phosphorylation. Human GC biopsy samples from the gastric antrum (urease test-positive) were obtained from consenting patients (n = 9). For animal samples, uninfected (n = 16) or H. felis-infected (n = 16) C57BL/6 mouse were euthanized after 18 months and their gastric tissues were collected. Tissues were immuno-stained for Siah2, P-S6-Siah2 and GRP78. Fluorescence microscopy revealed prominent decrease in GRP78 protein level paired with enhanced Siah2 and P-S6-Siah2 in human metastatic GC samples (Fig. 3A). Similar results were also observed in uninfected and H. felis-infected murine gastric tissues (Fig. 3B). These findings further asserted the association of GRP78 decrease with P-S6-Siah2 increase in Helicobacter-infected GC.
Fig. 3.
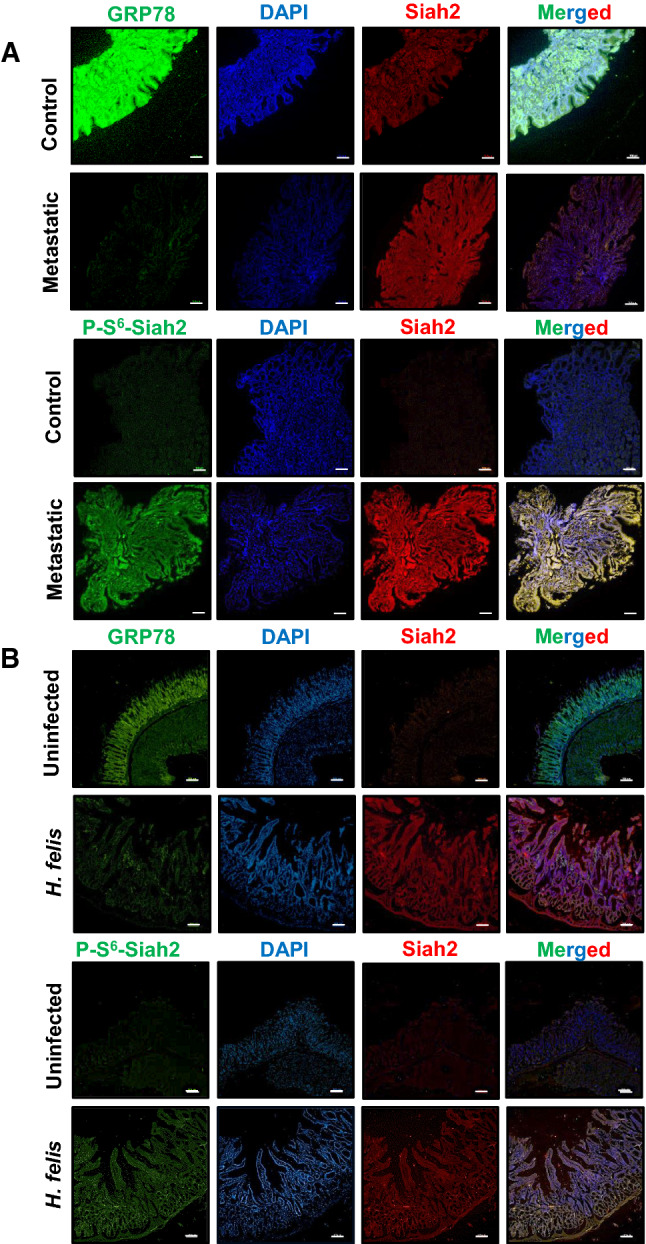
Decreased GRP78 and increased Siah2 are features of Helicobacter-infected gastric epithelia. A Human antral metastatic GC biopsy tissues depicting the status of GRP78, P-Ser6-Siah2 and Siah2. B Uninfected and H. felis-infected mouse gastric tissue sections representing GRP78 decrease and P-Ser6-Siah2 or Siah2 increase. Scale bars = 100 μm
GRP78 downregulates H. pylori-induced ROS generation in GECs
GRP78 regulates cellular ROS level [20]. However, the role of GRP78 in the regulation of ROS in H. pylori-infected GECs remains unknown. Since one of the major ROS generated by H. pylori is hydrogen peroxide (H2O2) [2], we aimed to identify the contribution of H2O2 in the H. pylori-mediated pool of ROS. For this, empty vector or grp78-expressing stable AGS cells were pre-treated with 350 units/ml of catalase, a H2O2 scavenger, 1 h prior to H. pylori infection and cells were incubated with DCFDA, a ROS-detecting fluorogenic dye. H. pylori-induced ROS was significantly lowered by GRP78 which was further potentiated by catalase confirming the formation of H2O2 in H. pylori-infected GECs (Fig. 4A). The accompanying western blot showed the level of GRP78 in transfected cells with or without infection. The role of grp78 in H. pylori-induced ROS regulation was further assessed by suppressing grp78. For this, one set of AGS cells were transfected with either control siRNA or grp78 siRNA followed by infection and another set was treated with catalase followed by ROS detection. ROS generation was significantly upregulated in grp78-suppressed cells as compared to the control siRNA-transfected cells. Catalase treatment could suppress the ROS upregulated by H. pylori in grp78-suppressed cells (Fig. 4B). Paired western blot confirmed the status of GRP78 suppression after siRNA transfection and infection. As we already found that suppression of siah2 could rescue GRP78 (Fig. 2E), next we wanted to understand the effect of siah2 suppression on ROS generation in H. pylori-infected GECs. For this, AGS cells were transfected with the control and human siah2 siRNA for 36 h followed by H. pylori infection. A similar setup of cells was treated with catalase. Micrographs showed that ROS level decreased in siah2 siRNA-transfected cells as compared with the control siRNA-transfected cells and catalase-treated cells exhibited significantly decreased ROS level (Fig. 4C). The accompanying western blot showed the status of Siah2 after siRNA transfection followed by infection. Taken together, these findings confirmed that Siah2-mediated GRP78 downregulation was crucial in the regulation of ROS generation in H. pylori-infected GECs. Significant suppression of ROS after catalase treatment also confirmed that H2O2 generation was predominant in H. pylori-infected cells.
Fig. 4.

GRP78 is crucial in the regulation of ROS in H. pylori-infected GECs. A Micrographs depicting ROS generation in uninfected or H. pylori-infected (200 MOI, 12 h) empty vector and grp78 stably-expressing AGS cells. Cat+/Cat− refer to catalase-treated/catalase-untreated cells, respectively. Western blots represent the status of GRP78 in uninfected or infected empty vector and grp78 stably-expressing AGS cells. GAPDH = loading control. Graphs represent fold change in ROS generation. B Fluorescence microscopy images representing ROS generation in AGS cells transfected with control and grp78 siRNA for 36 h followed by 12 h of H. pylori infection. Cat+/Cat− refer to catalase-treated/catalase-untreated cells, respectively. Western blots represent the status of GRP78 protein expression after grp78 suppression and H. pylori infection. GAPDH = loading control. Fold change in ROS generation is represented by bar graphs. C Micrographs depicting ROS generation in uninfected or infected control and siah2 suppressed AGS cells. Cat+/Cat− refer to catalase-treated/catalase-untreated cells, respectively. Western blots represent the status of Siah2 protein expression in the same experimental setup. GAPDH = loading control. Graphs represent fold change in ROS generation. For all images, scale bars = 10 μm. Graphs = mean ± sem. n = 3. Statistical significance is determined by three-way ANOVA followed by Tukey’s post hoc analysis. **P < 0.01, ***P < 0.001, ****P < 0.0001
GRP78 and Siah2 localize to mitochondria in H. pylori-infected GECs
ROS generation takes place during oxidative metabolism in mitochondria [34]. GRP78 gets localized in mitochondria and regulates ROS under various conditions [35, 36]. Mitochondrial function and homeostasis are also regulated by Siah2 [37–39], but its mitochondrial localization in H. pylori-infected GECs remains unknown. To explore this, uninfected or infected pDsRed2-Mito-expressing AGS stable cells were used to enable consistent visualization of mitochondria. Fluorescence micrographs showed that P-S6-Siah2 and Siah2 increased in mitochondria of infected cells but mitochondrial GRP78 was downregulated in infected cells as compared to uninfected cells (Fig. 5A). Further, MKN45 cells were infected with H. pylori followed by the mitochondrial and cytoplasmic fractionation. Representative western blots (n = 3) of mitochondrial and cytoplasmic fractions exhibited that P-S6-Siah2 or Siah2 significantly increased and GRP78 significantly decreased in both the subcellular fractions after H. pylori infection (Fig. 5B). Collectively, these results confirmed mitochondrial localization of P-S6-Siah2, Siah2 and GRP78 in H. pylori-infected GECs. Specificity of the custom-made P-S6-Siah2 antibody was assessed (n = 3) by performing western blot using whole cell lysates prepared from the uninfected and infected MKN45 stably-expressing pcDNA3.1+, siah2 WT, siah2 S6A and siah2 T279A cells. Results indicated decreased level of P-S6-Siah2 in case of siah2 S6A cells as compared to siah2 WT lanes (Fig. 5C).
Fig. 5.

Mitochondrial localization of GRP78 and Siah2. A Confocal microscopy images from uninfected and infected (200 MOI, 12 h) pDsRed2-Mito-expressing AGS stable cells depicting the mitochondrial expression of GRP78, P-S6-Siah2 and Siah2. Scale bars represent 50 μm. B Western blots from the cytoplasmic and the mitochondrial fractions of uninfected and infected MKN45 cells depicting the expression of GRP78, P-S6-Siah2 and Siah2. CoxIV is used as a loading control for the mitochondrial fraction and GAPDH for the cytoplasmic fraction, respectively. Bar graphs represent changes in the expression of GRP78, P-S6-Siah2 and Siah2 in the cytoplasmic and mitochondrial cellular fractions. Graphs = mean ± sem. n = 3. Statistical significance is determined by two-tailed unpaired t-test. *P < 0.05, **P < 0.01, ***P < 0.001. C Western blots form the whole cell lysates of uninfected and infected pcDNA3.1+, siah2 WT, siah2 S6A and siah2 T279A MKN45 stable cells representing specificity of the P-S6-Siah2 antibody indicated by the lowest level in S6A cells
P-Siah2 regulates mitochondrial morphology and GRP78 secretion
ROS imbalance alters mitochondrial morphology [40, 41]. We were eager to investigate whether Siah2 phosphorylation, which regulates ROS generation, affects the tubulo-reticular morphology of mitochondria. To examine this further, pDsRed2-Mito-expressing AGS stable cells were transfected with empty vector, siah2 WT and siah2 S6A overexpression plasmids followed by infection with H. pylori. Confocal micrographs (n = 3) showed that the tubulo-reticular morphology of mitochondria was disturbed and became smaller after H. pylori infection. Cells transfected with siah2 S6A exhibited the most damaged mitochondria after H. pylori infection. Mitochondrial stress, assessed by increased mitochondrial roundness and circularity, was also maximum in siah2 S6A-transfected cells after infection (Fig. 6A). These results confirmed that abrogation of Siah2 S6 phosphorylation significantly disrupted mitochondrial morphology. ROS is strongly correlated with changes in the Δψm [42]. To assess changes in the Δψm of uninfected and infected pcDNA3.1+, siah2 WT and siah2 phospho-null mutant S6A cells, these cells were treated with TMRM and image acquisition was done. Confocal microscopy followed by quantitation revealed that siah2 stable cells maximally retained the Δψm which was indicative of active/healthy mitochondria. (Fig. S2). The level of ROS in these cells were consistent with these findings (Fig. S3).
Fig. 6.

Siah2 phospho-null mutant S6A disrupts mitochondrial morphology and GRP78 abundance in H. pylori-infected GECs. A Confocal micrographs showing alteration of tubulo-reticular mitochondrial morphology in uninfected and infected pcDNA3.1+, siah2 WT and siah2 S6A-transfected pDsRed2-Mito AGS stable cells. Graphs represent mitochondrial length, circularity and roundness calculated using Fiji software from the above mentioned cells. Statistical significance is determined by two-way ANOVA followed by Tukey’s post hoc analysis. Graphs represent mean ± sem. n = 3. ****P < 0.0001. B Western blot of the whole cell lysates from uninfected or infected pcDNA3.1+, siah2 WT and siah2 S6A MKN45 stably expressed cells depicting the status of GRP78, P-S6-Siah2 and Siah2. GAPDH = loading control. Bar graphs represent densitometric analysis of GRP78 status in these cells. Statistical significance is determined by using two-way ANOVA followed by Tukey’s post hoc analysis. Graphs represent mean ± sem. n = 3. **P < 0.01. C Immunoblot of the Siah2 immuno-complexes obtained from untreated and MG132-treated H. pylori-infected pcDNA3.1+, siah2 WT and siah2 S6A MKN45 stable cells showing Siah2-GRP78 interaction. Non-specific band = loading control
The effect of Siah2 S6A mutant on mitochondrial stress noted in Fig. 6A necessitated the evaluation of GRP78 in S6A-expressing cells. For this, empty vector, siah2 WT and siah2 phospho-null mutant-expressing stable MKN45 cells were infected with H. pylori. Western blot analysis of the whole cell lysates showed that GRP78 protein decreased with siah2 WT overexpression. Surprisingly, despite the low level of Siah2 and P-S6-Siah2 in S6A cells, GRP78 remained significantly low in Siah2 S6A cells (Fig. 6B). This result explained the reason behind the upregulation of ROS in Siah2 S6A stable cells but opened a new question- whether Siah2 WT and Siah2 S6A differently interact with GRP78. To assess GRP78-Siah2 interaction, MG132-treated or untreated H. pylori-infected pcDNA3.1+, siah2 WT and siah2 S6A-expressing MKN45 stable cells were immunoprecipitated with Siah2 antibody. Western blot depicted that both Siah2 and GRP78 were less in S6A cells as compared to the WT cells (Fig. 6C). As reports suggestive of GRP78 secretion from cells exist [43, 44], we next evaluated the status of GRP78 secretion from H. pylori-infected pcDNA3.1+, siah2 WT and siah2 S6A-expressing MKN45 stable cells’ supernatant by ELISA. GRP78 secretion decreased after infection in empty vector-expressing cells (Fig. S4). GRP78 secretion further decreased after siah2 WT overexpression and infection. However, significantly increased GRP78 protein release was detected from both infected and uninfected siah2 S6A-expressing cells as compared to the other two transfection groups. These results reiterated the importance of P-S6-Siah2 in regulating the cellular abundance of GRP78 and explained why, in spite of low Siah2 level, the cellular level of GRP78 decreased in S6A cells. In summary, Siah2 S6 phosphorylation was found to be crucial for maintaining the cellular abundance of GRP78.
ROS leads to aggresome formation in H. pylori-infected GECs
ROS production leads to aggresome formation [17, 45]. As Siah2 and its phosphorylation altered cellular ROS level, we were interested to find out its role in aggresome formation in H. pylori-infected GECs. For this, pcDNA3.1+, siah2 WT and siah2 S6A-expressing MKN45 stable cells were infected with H. pylori. Cells were stained to detect Siah2 and aggresome. Confocal microscopy confirmed that aggresome formation was induced by H. pylori in all transfected groups. However, among the three transfected groups, siah2 WT stable cells showed the most aggresome formation which were further increased after infection. Aggresome formation was disrupted in siah2 S6A stable cells (Fig. 7A). To identify whether ROS were responsible for aggresome formation, pcDNA3.1+, siah2 WT and siah2 S6A MKN45 stable cells were treated with 10 mM of a ROS scavenger N-acteyl-l-cysteine (NAC) for 1 h followed by H. pylori infection. Confocal microscopy showed that NAC decreased aggresome formation in H. pylori-infected GECs (Fig. S5). These results confirmed the importance of ROS and Siah2 S6 phosphorylation in regulating aggresome formation. Aggresome formation is often coupled with macroautophagy [18, 19]. To elucidate the effect of aggresome formation on macroautophagy, pcDNA3.1+, siah2 WT and siah2 S6A stably-expressing MKN45 cells were infected with H. pylori. Western blotting (n = 3) confirmed that siah2 suppressed LC3B II accumulation which was indicative of disrupted autophagosome formation in both infected and uninfected cells when compared with the other transfection groups (Fig. 7B). To ascertain the correlation of autophagy with aggresome formation, pcDNA3.1+, siah2 WT and siah2 S6A MKN45 stable cells were infected with H. pylori in the presence or the absence of 10 nM bafilomycin A1. Confocal micrograph confirmed that aggresome formation was enhanced by bafilomycin A1 treatment (Fig. S6).
Fig. 7.

Siah2 S6 phosphorylation reciprocally regulates aggresome formation and autophagosome formation in H. pylori-infected GECs. A Confocal micrographs depicting the status of aggresome formation in uninfected and infected pcDNA3.1+, siah2 WT and siah2 S6A stably expressed MKN45 cells. 3D representation of the images, created using NIS software (Nikon), is shown to depict the disrupted aggresome formation in siah2 S6A MKN45 stable cells. Scale bars = 10 μm. B Immunoblots from the whole cell lysates of uninfected or infected pcDNA3.1+, siah2 WT and siah2 S6A stably expressing MKN45 cells showing the status of Siah2 and LC3B II. GAPDH is used as the loading control. C Graphical representation of the fold change in cellular proliferation after nocodazole treatment in uninfected and infected **** pcDNA3.1+, siah2 WT and siah2 S6A MKN45 stable cells. Statistical significance is determined by three-way ANOVA followed by Tukey’s post hoc analysis. n = 3. Graphs represent mean ± sem. ****P < 0.0001
Aggresome formation is cytoprotective and is dependent on microtubule retrograde transport [46–49]. Since Siah2 increased proliferative potential of GECs [10, 12, 26] and also enhanced aggresome formation as observed in this study, we next assessed the effect of aggresome disruption by nocodazole on the proliferative potential of H. pylori-infected cells. For this, MKN45 pcDNA3.1+, siah2 WT and siah2 S6A stable cells were either left uninfected or were infected and treated with 2 μM nocodazole for 12 h. MTT assay showed that disruption of aggresome formation decreased cellular proliferation. Empty vector and siah2 WT stable cells were more sensitive to the disruption of microtubule than siah2 S6A cells (Fig. 7C). These results confirmed that P-S6-Siah2-mediated aggresome formation imparted proliferative advantage to H. pylori-infected GECs.
Discussion
H. pylori-mediated Siah2 S6 phosphorylation increases its stability and thereby potentiates its function as an E3 ubiquitin ligase [12]. In the present study we identify that the increased level of Siah2 and P-Siah2 are accompanied by GRP78 decrease and increased ROS generation in H. pylori-infected GECs. Further, we establish that P-S6-Siah2 promotes aggresome formation in the infected cells. Thus, in spite of losing the intracellular antioxidant protein GRP78 and increased ROS generation, H. pylori-infected cells employ ROS in forming aggresomes which gives GECs survival advantage.
H. pylori colonization in the human stomach leads to severe inflammation, causing gastritis and initiating the Correa’s cascade of tumorigenic events leading to GC [1]. Chronic inflammation induces ROS generation. ROS promote mutations and initiate several carcinogenic events in the infected host cells [2]. Studies have shown that H. pylori infection promotes H2O2 generation [2] and induces DNA damage [50, 51]. We discover that H. pylori-mediated ROS generation is induced by Siah2. However, the phosphorylation null mutant-expressing Siah2 cells show significantly higher ROS generation than the WT Siah2-overexpressing cells. These observations strongly suggest that without the phosphorylation-mediated increased Siah2 stabilization in H. pylori-infected GECs, excessive ROS might affect the viability of GECs. In fact, we observe that H. pylori-induced cell proliferation is promoted with siah2 WT construct overexpression but the phosphorylation null S6A mutant of siah2 significantly hampers cell proliferation. Although it is known that ROS is regulated by Siah2 under various physiological conditions [52], this is the first report of Siah2-mediated ROS modulation in H. pylori-infected GECs. Therefore, controlling phosphorylation-dephosphorylation of Siah2 has a lot of potential in regulating oxidative stress.
GRP78 ubiquitination by Siah2 ensures the tight regulation of ROS in H. pylori-infected cells. Although enhanced GRP78 expression has been correlated with GC [53], H. pylori infection is associated with the downregulation of GRP78 [54]. Baird et al. have shown that GRP78 level decreases in H. pylori-induced gastritis but increases in mucous metaplasia [55]. The same study also reports that H. pylori suppresses GRP78 in AGS cells. A recent study by Wang et al. has identified that Nod-like receptor pyrin domain-containing protein 6 (NLRP6)-mediated ubiquitination of GRP78 suppresses GC but the study has not investigated H. pylori-induced GC [56]. Multiplexed mass spectrometry analysis also reveals GRP78 ubiquitination in bortezomib-treated colon cancer cells [57]. GRP78 is known for its role as an antioxidant. Silencing of grp78 in prostate cancer cells leads to ROS accumulation [58]. In line with these findings, we observe that grp78 expression decreases H. pylori-induced ROS. These results confirm that P-S6-Siah2-mediated GRP78 downregulation impacts the ROS-scavenging ability of H. pylori-infected GECs. Mitochondrial localization of GRP78 and Siah2 are reported in various pathological conditions [38, 39, 59]. Siah2 [60] and H. pylori [61, 62] are known for their ability to induce mitochondrial stress. We show for the first time that mitochondrial localization of P-S6-Siah2 decreases mitochondrial GRP78 in H. pylori-infected GECs. In the absence of Siah2 phosphorylation, GRP78 is not detected in the whole cell lysate but its release from the cell is promoted. This result explains why siah2 S6A-expressing cells have more ROS than siah2 WT-expressing cells. Secretion of GRP78 is induced by cellular stresses [44, 63, 64]. It would be interesting to find out the exact mechanism behind this heightened GRP78 decrease in Siah2 S6A-expressing cells.
We show for the first time that changes in the Δψm of H. pylori-infected GECs are strongly influenced by Siah2 and its phosphorylation at S6. Δψm has strong correlations with ROS, mitochondrial ATP generation and metabolic status. Generally, increased ROS generation is associated with a fall in Δψm. However, expression of oncogenes hyperpolarizes the Δψm [65]. Cancer cells with more aggressive nature have higher Δψm than their benign counterparts [66]. The enhancement in Δψm in Siah2 WT cells as compared to the other transfected cells indicates toward the enhanced invasive nature of the Siah2 WT cells which is already reported by our group [12]. It needs to be verified in future how exactly the mitochondrial membrane permeability transition, Δψm, mitochondrial metabolism and mitophagy are impacted by S6A Siah2 in H. pylori-infected GECs.
H. pylori infection [67] as well as ROS [16, 18] induce aggresome formation. GRP78 is involved in ROS regulation and is a known endoplasmic reticulum chaperone. Due to these attributes, it is not surprising that siah2 WT cells, which have less intracellularly available GRP78, show the highest aggresome formation. However, aggresomes are abrogated in the Siah2 phospho-null mutant-expressing cells which have the least intracellularly available GRP78. These results point to the importance of P-S6-Siah2 in aggresome formation in H. pylori-infected GECs. This is the first report showing that the availability of Siah2 suppresses macroautophagy. This is in agreement with studies confirming the reciprocal regulation of the ubiquitin–proteasome system and the macroautophagy [31]. Enhanced ROS level in S6A mutant-expressing cells can be associated with the formation of diffuse protein aggregates which are the major elicitors of ROS [68]. Since aggresome formation is cytoprotective and promotes cell proliferation [49, 69–72], the disruption of aggresome formation in S6A cells decreases the proliferative potential of infected GECs. This finding is also in agreement with the studies which suggest that disruption of aggresome formation might have a therapeutic potential in treating cancer [71, 73]. Further studies should be carried out to explore the efficacy of aggresome disruption in the prevention of H. pylori-mediated disease pathogenesis.
In summary, this study shows that Siah2 regulates the cellular abundance of antioxidant protein GRP78 and thus, the cellular redox status during H. pylori infection. Our study identifies that Helicobacter-mediated GRP78 downregulation is consistently tied with enhanced phospho-Siah2 in vitro as well as in vivo and finds their potential as diagnostic and therapeutic targets in H. pylori infection-induced GC.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
P.D., S.B.K., I.P. and D.C. obtained fellowships from DAE, India. S.S.S. obtained fellowship from KVPY, DST, India. S.S. obtained fellowship from CSIR, India. Central Instrumentation Facilities of NISER and Centre for Interdisciplinary Sciences, NISER are acknowledged for providing facilities.
Abbreviations
- cag PAI
Cytotoxin-associated gene pathogenicity island
- DCFDA
2′,7′-Dichlorofluorescin diacetate
- GC
Gastric cancer
- GECs
Gastric epithelial cancer cells
- GRP78
78-kDa glucose-regulated protein
- H2O2
Hydrogen peroxide
- H. pylori
Helicobacter pylori
- Δψm
Mitochondrial membrane potential
- MOI
Multiplicity of infection
- P-S6-Siah2
Phosphorylated Serine6 Siah2
- ROS
Reactive oxygen species
- Siah2
Seven in absentia homolog 2
- TMRM
Tetramethylrhodamine, methyl ester
- WT
Wild type
Author contributions
P.D. performed the experiments, analyzed the results and wrote the paper; S.S.S. performed some experiments and reviewed the manuscript; S.B.K. performed site-directed mutagenesis; I.P., D.C., S.S. assisted in manuscript preparation; A.S. gave crucial suggestions and helped in reviewing, N.R. and S.P.S. helped with GC biopsy collection and analysis; A.B. conceived the work, designed experiments, supervised the work, analyzed results and wrote the paper. All authors read and approved the final manuscript.
Funding
This work was supported by the Indian Science and Engineering Research Board grant (SB/SO/BB-0015/2014 to A.B.) and institutional funding by NISER (DAE, India).
Data availability
Data are available on reasonable request from the corresponding author.
Declarations
Conflict of interest
The authors declare no competing interests.
Ethics approval and consent to participate
Human GC tissue collection protocol was approved by Institutional Ethics Committee for Human Research, NISER. Institutional Animal Ethics Committee, NISER, approved animal experiments.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Wang F, Meng W, Wang B, Qiao L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014;345(2):196–202. doi: 10.1016/j.canlet.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 2.Butcher LD, Den Hartog G, Ernst PB, Crowe SE. Oxidative stress resulting from Helicobacter pylori infection contributes to gastric carcinogenesis. Cell Mol Gastroenterol Hepatol. 2017;3(3):316–322. doi: 10.1016/j.jcmgh.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wen J, Wang Y, Gao C, Zhang G, You Q, Zhang W, Zhang Z, Wang S, et al. Helicobacter pylori infection promotes aquaporin 3 expression via the ros-hif-1alpha-aqp3-ros loop in stomach mucosa: a potential novel mechanism for cancer pathogenesis. Oncogene. 2018;37(26):3549–3561. doi: 10.1038/s41388-018-0208-1. [DOI] [PubMed] [Google Scholar]
- 4.Gobert AP, Wilson KT. Polyamine- and nadph-dependent generation of ros during Helicobacter pylori infection: a blessing in disguise. Free Radic Biol Med. 2017;105:16–27. doi: 10.1016/j.freeradbiomed.2016.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolyniec K, Levav-Cohen Y, Jiang YH, Haupt S, Haupt Y. The e6ap e3 ubiquitin ligase regulates the cellular response to oxidative stress. Oncogene. 2013;32(30):3510–3519. doi: 10.1038/onc.2012.365. [DOI] [PubMed] [Google Scholar]
- 6.Otaki Y, Takahashi H, Watanabe T, Funayama A, Netsu S, Honda Y, Narumi T, Kadowaki S, et al. Hect-type ubiquitin e3 ligase itch interacts with thioredoxin-interacting protein and ameliorates reactive oxygen species-induced cardiotoxicity. J Am Heart Assoc. 2016 doi: 10.1161/JAHA.115.002485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu J, Salva KA, Wood GS. C-cbl e3 ubiquitin ligase is overexpressed in cutaneous t-cell lymphoma: Its inhibition promotes activation-induced cell death. J Investig Dermatol. 2015;135(3):861–868. doi: 10.1038/jid.2014.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baba K, Morimoto H, Imaoka S. Seven in absentia homolog 2 (siah2) protein is a regulator of nf-e2-related factor 2 (nrf2) J Biol Chem. 2013;288(25):18393–18405. doi: 10.1074/jbc.M112.438762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sajja RK, Green KN, Cucullo L. Altered nrf2 signaling mediates hypoglycemia-induced blood-brain barrier endothelial dysfunction in vitro. PLoS ONE. 2015;10(3):e0122358. doi: 10.1371/journal.pone.0122358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Das L, Kokate SB, Rath S, Rout N, Singh SP, Crowe SE, Mukhopadhyay AK, Bhattacharyya A. Ets2 and twist1 promote invasiveness of Helicobacter pylori -infected gastric cancer cells by inducing siah2. Biochem J. 2016;473(11):1629–1640. doi: 10.1042/BCJ20160187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarkar TR, Sharan S, Wang J, Pawar SA, Cantwell CA, Johnson PF, Morrison DK, Wang JM, et al. Identification of a src tyrosine kinase/siah2 e3 ubiquitin ligase pathway that regulates c/ebpdelta expression and contributes to transformation of breast tumor cells. Mol Cell Biol. 2012;32(2):320–332. doi: 10.1128/MCB.05790-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dixit P, Kokate SB, Poirah I, Chakraborty D, Smoot DT, Ashktorab H, Rout N, Singh SP, et al. Helicobacter pylori -induced gastric cancer is orchestrated by mrckbeta-mediated siah2 phosphorylation. J Biomed Sci. 2021;28(1):12. doi: 10.1186/s12929-021-00710-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Checa J, Aran JM. Reactive oxygen species: drivers of physiological and pathological processes. J Inflamm Res. 2020;13:1057. doi: 10.2147/JIR.S275595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sies H, Jones DP. Reactive oxygen species (ros) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21(7):363–383. doi: 10.1038/s41580-020-0230-3. [DOI] [PubMed] [Google Scholar]
- 15.Vasconcellos LR, Dutra FF, Siqueira MS, Paula-Neto HA, Dahan J, Kiarely E, Carneiro LA, Bozza MT, et al. Protein aggregation as a cellular response to oxidative stress induced by heme and iron. Proc Natl Acad Sci. 2016;113(47):E7474–E7482. doi: 10.1073/pnas.1608928113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Dam L, Dansen TB. Cross-talk between redox signalling and protein aggregation. Biochem Soc Trans. 2020;48(2):379–397. doi: 10.1042/BST20190054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bodas M, Vij N. Adapting proteostasis and autophagy for controlling the pathogenesis of cystic fibrosis lung disease. Front Pharmacol. 2019;10:20. doi: 10.3389/fphar.2019.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, Gavina M, Pulze L, et al. Defective cftr induces aggresome formation and lung inflammation in cystic fibrosis through ros-mediated autophagy inhibition. Nat Cell Biol. 2010;12(9):863–875. doi: 10.1038/ncb2090. [DOI] [PubMed] [Google Scholar]
- 19.Wang D-W, Peng Z-J, Ren G-F, Wang G-X. The different roles of selective autophagic protein degradation in mammalian cells. Oncotarget. 2015;6(35):37098. doi: 10.18632/oncotarget.5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dauer P, Sharma NS, Gupta VK, Durden B, Hadad R, Banerjee S, Dudeja V, Saluja A. Er stress sensor, glucose regulatory protein 78 (grp78) regulates redox status in pancreatic cancer thereby maintaining "stemness". Cell Death Dis. 2019;10(2):132. doi: 10.1038/s41419-019-1408-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abdel Malek MA, Jagannathan S, Malek E, Sayed DM, Elgammal SA, Abd El-Azeem HG, Thabet NM, Driscoll JJ (2015) Molecular chaperone grp78 enhances aggresome delivery to autophagosomes to promote drug resistance in multiple myeloma. Oncotarget 6(5):3098–3110. 10.18632/oncotarget.3075 [DOI] [PMC free article] [PubMed]
- 22.Das L, Kokate SB, Dixit P, Rath S, Rout N, Singh SP, Crowe SE, Bhattacharyya A. Membrane-bound beta-catenin degradation is enhanced by ets2-mediated siah1 induction in Helicobacter pylori-infected gastric cancer cells. Oncogenesis. 2017;6(5):e327. doi: 10.1038/oncsis.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Werstuck GH, Lentz SR, Dayal S, Hossain GS, Sood SK, Shi YY, Zhou J, Maeda N, et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Investig. 2001;107(10):1263–1273. doi: 10.1172/JCI11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rath S, Das L, Kokate SB, Pratheek BM, Chattopadhyay S, Goswami C, Chattopadhyay R, Crowe SE, et al. Regulation of noxa-mediated apoptosis in Helicobacter pylori-infected gastric epithelial cells. FASEB J. 2015;29(3):796–806. doi: 10.1096/fj.14-257501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kokate SB, Dixit P, Das L, Rath S, Roy AD, Poirah I, Chakraborty D, Rout N, et al. Acetylation-mediated siah2 stabilization enhances phd3 degradation in Helicobacter pylori -infected gastric epithelial cancer cells. FASEB J. 2018;32(10):5378–5389. doi: 10.1096/fj.201701344RRR. [DOI] [PubMed] [Google Scholar]
- 27.AAT Bioquest (2019) Quest graph™ four parameter logistic (4pl) curve calculator. AAT Bioquest. https://www.aatbio.com/tools/four-parameter-logistic-4pl-curve-regression-online-calculator
- 28.Teng J, Liu M, Su Y, Li K, Sui N, Wang S, Li L, Sun Y, et al. Down-regulation of grp78 alleviates lipopolysaccharide-induced acute kidney injury. Int Urol Nephrol. 2018;50(11):2099–2107. doi: 10.1007/s11255-018-1911-0. [DOI] [PubMed] [Google Scholar]
- 29.Chillappagari S, Belapurkar R, Möller A, Molenda N, Kracht M, Rohrbach S, Schmitz ML. Siah2-mediated and organ-specific restriction of ho-1 expression by a dual mechanism. Sci Rep. 2020;10(1):1–12. doi: 10.1038/s41598-020-59005-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kokate SB, Dixit P, Poirah I, Roy AD, Chakraborty D, Rout N, Singh SP, Ashktorab H, et al. Testin and filamin-c downregulation by acetylated siah2 increases invasiveness of Helicobacter pylori -infected gastric cancer cells. Int J Biochem Cell Biol. 2018;103:14–24. doi: 10.1016/j.biocel.2018.07.012. [DOI] [PubMed] [Google Scholar]
- 31.Kocaturk NM, Gozuacik D. Crosstalk between mammalian autophagy and the ubiquitin-proteasome system. Front Cell Dev Biol. 2018;6:128. doi: 10.3389/fcell.2018.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y, Le W-D. Autophagy and ubiquitin-proteasome system. Autophagy Biol Dis. 2019;1206:527–550. doi: 10.1007/978-981-15-0602-4_25. [DOI] [PubMed] [Google Scholar]
- 33.Pohl C, Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019;366(6467):818–822. doi: 10.1126/science.aax3769. [DOI] [PubMed] [Google Scholar]
- 34.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ni M, Zhang Y, Lee AS. Beyond the endoplasmic reticulum: atypical grp78 in cell viability, signalling and therapeutic targeting. Biochem J. 2011;434(2):181–188. doi: 10.1042/BJ20101569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun F-C, Wei S, Li C-W, Chang Y-S, Chao C-C, Lai Y-K. Localization of grp78 to mitochondria under the unfolded protein response. Biochem J. 2006;396(1):31–39. doi: 10.1042/BJ20051916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carlucci A, Adornetto A, Scorziello A, Viggiano D, Foca M, Cuomo O, Annunziato L, Gottesman M, et al. Proteolysis of akap121 regulates mitochondrial activity during cellular hypoxia and brain ischaemia. EMBO J. 2008;27(7):1073–1084. doi: 10.1038/emboj.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma B, Cheng H, Mu C, Geng G, Zhao T, Luo Q, Ma K, Chang R, et al. The siah2-nrf1 axis spatially regulates tumor microenvironment remodeling for tumor progression. Nat Commun. 2019;10(1):1–17. doi: 10.1038/s41467-019-08618-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, et al. Fine-tuning of drp1/fis1 availability by akap121/siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell. 2011;44(4):532–544. doi: 10.1016/j.molcel.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Picard M, Shirihai OS, Gentil BJ, Burelle Y. Mitochondrial morphology transitions and functions: implications for retrograde signaling? Am J Physiol Regul Integr Comp Physiol. 2013;304(6):R393–406. doi: 10.1152/ajpregu.00584.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willems PH, Rossignol R, Dieteren CE, Murphy MP, Koopman WJ. Redox homeostasis and mitochondrial dynamics. Cell Metab. 2015;22(2):207–218. doi: 10.1016/j.cmet.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 42.Suski JM, Lebiedzinska M, Bonora M, Pinton P, Duszynski J, Wieckowski MR. Relation between mitochondrial membrane potential and ros formation. Methods Mol Biol. 2012;810:183–205. doi: 10.1007/978-1-61779-382-0_12. [DOI] [PubMed] [Google Scholar]
- 43.Kern J, Untergasser G, Zenzmaier C, Sarg B, Gastl G, Gunsilius E, Steurer M. Grp-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood. 2009;114(18):3960–3967. doi: 10.1182/blood-2009-03-209668. [DOI] [PubMed] [Google Scholar]
- 44.Vig S, Buitinga M, Rondas D, Crèvecoeur I, Van Zandvoort M, Waelkens E, Eizirik DL, Gysemans C, et al. Cytokine-induced translocation of grp78 to the plasma membrane triggers a pro-apoptotic feedback loop in pancreatic beta cells. Cell Death Dis. 2019;10(4):1–13. doi: 10.1038/s41419-019-1518-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marambio P, Toro B, Sanhueza C, Troncoso R, Parra V, Verdejo H, Garcia L, Quiroga C, et al. Glucose deprivation causes oxidative stress and stimulates aggresome formation and autophagy in cultured cardiac myocytes. Biochim Biophys Acta. 2010;1802(6):509–518. doi: 10.1016/j.bbadis.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 46.Matsumoto G, Inobe T, Amano T, Murai K, Nukina N, Mori N. N-acyldopamine induces aggresome formation without proteasome inhibition and enhances protein aggregation via p62/sqstm1 expression. Sci Rep. 2018;8(1):9585. doi: 10.1038/s41598-018-27872-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hao R, Nanduri P, Rao Y, Panichelli RS, Ito A, Yoshida M, Yao TP. Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol Cell. 2013;51(6):819–828. doi: 10.1016/j.molcel.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garcia-Mata R, Bebok Z, Sorscher EJ, Sztul ES. Characterization and dynamics of aggresome formation by a cytosolic gfp-chimera. J Cell Biol. 1999;146(6):1239–1254. doi: 10.1083/jcb.146.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, Fischbeck KH. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet. 2003;12(7):749–757. doi: 10.1093/hmg/ddg074. [DOI] [PubMed] [Google Scholar]
- 50.Savoldi A, Carrara E, Graham DY, Conti M, Tacconelli E. Prevalence of antibiotic resistance in Helicobacter pylori: a systematic review and meta-analysis in world health organization regions. Gastroenterology. 2018;155(5):1372–1382 e17. doi: 10.1053/j.gastro.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xie C, Yi J, Lu J, Nie M, Huang M, Rong J, Zhu Z, Chen J, et al. N-acetylcysteine reduces ros-mediated oxidative DNA damage and pi3k/akt pathway activation induced by Helicobacter pylori infection. Oxid Med Cell Longev. 2018;2018:1874985. doi: 10.1155/2018/1874985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baba K, Miyazaki T. Novel function of e3 ubiquitin ligase siah2 to regulate ros metabolism. J Biochem Mol Biol Res. 2016;2(2):152–156. doi: 10.17554/j.issn.2313-7177.2016.02.20. [DOI] [Google Scholar]
- 53.Wang Y, Wang JH, Zhang XL, Wang XL, Yang L. Endoplasmic reticulum chaperone glucose-regulated protein 78 in gastric cancer: an emerging biomarker. Oncol Lett. 2018;15(5):6087–6093. doi: 10.3892/ol.2018.8114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Namba T, Hoshino T, Suemasu S, Takarada-Iemata M, Hori O, Nakagata N, Yanaka A, Mizushima T. Suppression of expression of endoplasmic reticulum chaperones by Helicobacter pylori and its role in exacerbation of non-steroidal anti-inflammatory drug-induced gastric lesions. J Biol Chem. 2010;285(48):37302–37313. doi: 10.1074/jbc.M110.148882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baird M, Woon Ang P, Clark I, Bishop D, Oshima M, Cook MC, Hemmings C, Takeishi S, et al. The unfolded protein response is activated in helicobacter-induced gastric carcinogenesis in a non-cell autonomous manner. Lab Investig. 2013;93(1):112–122. doi: 10.1038/labinvest.2012.131. [DOI] [PubMed] [Google Scholar]
- 56.Wang X, Wu X, Wang Q, Zhang Y, Wang C, Chen J. Nlrp6 suppresses gastric cancer growth via grp78 ubiquitination. Exp Cell Res. 2020;395(1):112177. doi: 10.1016/j.yexcr.2020.112177. [DOI] [PubMed] [Google Scholar]
- 57.Rose CM, Isasa M, Ordureau A, Prado MA, Beausoleil SA, Jedrychowski MP, Finley DJ, Harper JW, et al. Highly multiplexed quantitative mass spectrometry analysis of ubiquitylomes. Cell Syst. 2016;3(4):395–403 e4. doi: 10.1016/j.cels.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dauer P, Sharma NS, Gupta VK, Nomura A, Dudeja V, Saluja A, Banerjee S. Grp78-mediated antioxidant response and abc transporter activity confers chemoresistance to pancreatic cancer cells. Mol Oncol. 2018;12(9):1498–1512. doi: 10.1002/1878-0261.12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Casas C. Grp78 at the centre of the stage in cancer and neuroprotection. Front Neurosci. 2017;11:177. doi: 10.3389/fnins.2017.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sisalli MJ, Ianniello G, Savoia C, Cuomo O, Annunziato L, Scorziello A. Knocking-out the siah2 e3 ubiquitin ligase prevents mitochondrial ncx3 degradation, regulates mitochondrial fission and fusion, and restores mitochondrial function in hypoxic neurons. Cell Commun Signal. 2020;18(1):1–10. doi: 10.1186/s12964-020-0529-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Calvino-Fernández M, Parra-Cid T. H. pylori and mitochondrial changes in epithelial cells. The role of oxidative stress. Rev Esp Enferm Dig. 2010;102(1):41–50. doi: 10.4321/S1130-01082010000100006. [DOI] [PubMed] [Google Scholar]
- 62.Kim I-J, Lee J, Oh SJ, Yoon M-S, Jang S-S, Holland RL, Reno ML, Hamad MN, et al. Helicobacter pylori infection modulates host cell metabolism through vaca-dependent inhibition of mtorc1. Cell Host Microbe. 2018;23(5):583–593.e8. doi: 10.1016/j.chom.2018.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qin K, Ma S, Li H, Wu M, Sun Y, Fu M, Guo Z, Zhu H, et al. Grp78 impairs production of lipopolysaccharide-induced cytokines by interaction with cd14. Front Immunol. 2017;8:579. doi: 10.3389/fimmu.2017.00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rondas D, Crevecoeur I, D’hertog W, Ferreira GB, Staes A, Garg AD, Eizirik DL, Agostinis P, et al. Citrullinated glucose-regulated protein 78 is an autoantigen in type 1 diabetes. Diabetes. 2015;64(2):573–586. doi: 10.2337/db14-0621. [DOI] [PubMed] [Google Scholar]
- 65.Fantin VR, Berardi MJ, Scorrano L, Korsmeyer SJ, Leder P. A novel mitochondriotoxic small molecule that selectively inhibits tumor cell growth. Cancer Cell. 2002;2(1):29–42. doi: 10.1016/s1535-6108(02)00082-x. [DOI] [PubMed] [Google Scholar]
- 66.Grieco JP, Allen ME, Perry JB, Wang Y, Song Y, Rohani A, Compton SLE, Smyth JW, et al. Progression-mediated changes in mitochondrial morphology promotes adaptation to hypoxic peritoneal conditions in serous ovarian cancer. Front Oncol. 2020;10:600113. doi: 10.3389/fonc.2020.600113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu C-C, Yang J-C, Chang Y-C, Chuang J-G, Lin C-W, Wu M-S, Chow L-P. Vcp phosphorylation-dependent interaction partners prevent apoptosis in Helicobacter pylori-infected gastric epithelial cells. PLoS ONE. 2013;8(1):e55724. doi: 10.1371/journal.pone.0055724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Carija A, Navarro S, De Groot NS, Ventura S. Protein aggregation into insoluble deposits protects from oxidative stress. Redox Biol. 2017;12:699–711. doi: 10.1016/j.redox.2017.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hillert E-K, Brnjic S, Zhang X, Mazurkiewicz M, Saei AA, Mofers A, Selvaraju K, Zubarev R, et al. Proteasome inhibitor b-ap15 induces enhanced proteotoxicity by inhibiting cytoprotective aggresome formation. Cancer Lett. 2019;448:70–83. doi: 10.1016/j.canlet.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 70.Chin L, Olzmann J, Li L. Aggresome formation and neurodegenerative diseases: Therapeutic implications. Curr Med Chem. 2008;15(1):47–60. doi: 10.2174/092986708783330692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yehia M, Taha H, Salama A, Amer N, Mosaab A, Hassanain O, Refaat A, Yassin D, et al. Association of aggresomes with survival outcomes in pediatric medulloblastoma. Sci Rep. 2019;9(1):1–10. doi: 10.1038/s41598-019-49027-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Park J, Park Y, Ryu I, Choi M-H, Lee HJ, Oh N, Kim K, Kim KM, et al. Misfolded polypeptides are selectively recognized and transported toward aggresomes by a ced complex. Nat Commun. 2017;8(1):1–15. doi: 10.1038/ncomms15730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.An H, Statsyuk AV. An inhibitor of ubiquitin conjugation and aggresome formation. Chem Sci. 2015;6(9):5235–5245. doi: 10.1039/C5SC01351H. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available on reasonable request from the corresponding author.