

Abstract
The successful treatment of human cancers by immunotherapy has been made possible by breakthroughs in the discovery of immune checkpoint regulators, including CTLA-4 and PD-1/PD-L1. However, the immunosuppressive effect of the tumor microenvironment still represents an important bottleneck that limits the success of immunotherapeutic approaches. The tumor microenvironment influences the metabolic crosstalk between tumor cells and tumor-infiltrating immune cells, creating competition for the utilization of nutrients and promoting immunosuppression. In addition, tumor-derived metabolites regulate the activation and effector function of immune cells through a variety of mechanisms; in turn, the metabolites and other factors secreted by immune cells can also become accomplices to cancer development. Immune-metabolic checkpoint regulation is an emerging concept that is being studied with the aim of restoring the immune response in the tumor microenvironment. In this review, we summarize the metabolic reprogramming of various cell types present in the tumor microenvironment, with a focus on the interaction between the metabolic pathways of these cells and antitumor immunosuppression. We also discuss the main metabolic checkpoints that could provide new means of enhancing antitumor immunotherapy.
Keywords: Antitumor immunotherapy, Immune checkpoint, Tumor metabolism, Metabolic reprogramming, Metabolic checkpoint
Background
Extensive interactions occur between tumor cells and other cell types in the tumor microenvironment (TME), in which immune cells have a particularly important role in the malignant progression of tumors [1]. As a defense system against tumors, the immune system can recognize and activate immune clearance mechanisms to specifically kill malignant cells, thereby preventing them from further developing into malignant tumors; this mechanism is called tumor immune surveillance. However, immune escape is one of the basic biological characteristics of malignant tumors [2]. Tumor cells can survive and proliferate by evading or resisting the recognition and attack of the immune system through a variety of mechanisms [3, 4]. The TME comprises diverse immune cell types, including T cells, B cells, tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), neutrophils and mast cells [5, 6]. They establish a complex network of intercellular interactions that enhances and maintains the immunosuppressive microenvironment and promotes immune escape, which ultimately enhances tumor progression [7, 8].
Increasing the immunogenicity of tumor cells and the cytotoxic susceptibility of targeted effector cells can effectively enhance the antitumor immune response [9]. Therefore, tumor immunotherapy has become a powerful and promising strategy for the treatment of advanced, recurrent and refractory tumors. Several immunotherapy approaches have been developed, such as tumor vaccines, adoptive T cell transfer (ACT) therapy, and immune checkpoint therapy. ACT involves the isolation, ex vivo expansion and reinfusion of immune cells to mediate tumor regression and remission [10]. Presently, several types of ACT have been developed for tumor immunotherapy, such as tumor-infiltrating lymphocytes (TILs), T cell receptor (TCR) T cells, and chimeric antigen receptor (CAR) T cells; CART cells were the first gene therapy approved by the US Food and Drug Administration (FDA) [11].
With the ongoing advances in antitumor immune research, the discovery of immune checkpoints, including cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed death-1 (PD-1) and programmed death ligand-1 (PD-L1), has opened a new chapter in tumor immunotherapy [12–15]. CTLA-4 is a CD28 homolog mainly expressed on T cells, and it binds with high affinity to B7 on antigen-presenting cells (APCs) to play a negative regulatory role in T cell activation [16]. PD-1 is an immune checkpoint receptor present on T cells, and it induces T cell exhaustion and immunosuppression by binding to its ligand PD-L1, which is highly expressed on tumor cells [17]. Thus, CTLA-4 and PD-1 have similar negative effects on T cell immune function, and inhibition of these checkpoints can reactivate the immune system and result in immune-mediated antitumor activity [18]. The inhibitors of immune checkpoints (such as antibodies that block the CTLA-4 and PD-1/PD-L1 proteins) have led to durable responses in some patients with various tumor types [19–21]. For example, the breakthrough results of a phase III clinical trial (KEYNOTE-042) of patients with PD-L1-expressing, previously untreated, locally advanced or metastatic non-small-cell lung cancer showed that, compared with chemotherapy, pembrolizumab (anti-PD-1) monotherapy led to better response rates and fewer side effects as a first-line treatment and could greatly improve overall survival (OS) [22]. However, some patients have no response to antitumor immunotherapy, or have tolerance and recurrence, leading to immunotherapy resistance and relapse [23–25]. Metabolic reprogramming is one of the important hallmarks of tumors and has been found to be associated with immunotherapy resistance [26–29]. Tumor cells continuously adjust their metabolism and nutrient acquisition to maintain sustained proliferation and thus alter the metabolic landscape of the TME [30]. The production of immunosuppressive metabolites in the TME can inhibit immune cell infiltration and other antitumor immune functions. Therefore, based on the relationship between tumor metabolism and the immune response, combined therapies that target metabolic and immune checkpoints may enhance the effectiveness of antitumor therapy and overcome resistance to immunotherapy [31].
This review covers the most recent literatures on the metabolic characteristics of tumor cells and immune cells in the TME. We also discuss the complex relationships between antitumor immunosuppression and metabolic crosstalk in the TME. The potential metabolic checkpoint inhibitors that can be used in combination with immune checkpoint inhibitors will help us find the next key breakthroughs for improving antitumor immunotherapy.
Cancer is a metabolic disease
A specific physical microenvironment is formed during tumor development, including hypoxia, low potential of hydrogen (pH), nutrient pressure and oxidative stress [32]. The altered metabolic landscape of the TME can induce the metabolic reprogramming of immune cells, which further interferes with immune surveillance and promotes cancer progression [33, 34]. Considering that immune cells have similar metabolic needs as tumor cells, here we first summarize the characteristics of cancer metabolism. Cancer is a heterogeneous and metabolic disease, and the heterogeneity of tumor cells gives rise to their complex metabolic patterns [33, 35]. Tumor cells predominantly use glycolysis for adenosine triphosphate (ATP) production. Glutamine and lipids are also essential for tumor cell proliferation [36].
Hypoxia
Balanced oxygen sensing and oxygen uptake are indispensable for cells to maintain normal function [33]. In 1955, Thomlinson first proposed the concept of tumor hypoxia [37]. The following decades of clinical and experimental confirmation established that hypoxia is a widespread feature in many solid tumors [38]. The unlimited expansion and abnormal vascular structure of tumor cells cause increased oxygen consumption and inadequate oxygen supply [39]. In response to hypoxia, hypoxia-inducible factors (HIFs) are triggered to allow cells to adapt to low-oxygen conditions [40]. As a transcriptional regulator, HIF can regulate the transcription of downstream target genes that mediate diverse cancer hallmarks, such as proliferation, differentiation, metabolism, angiogenesis, invasion and metastasis [41–45]. Considering that HIFs are an effective target in tumor therapy, many HIF inhibitors have been developed, and they can inhibit the expression and/or function of HIF-1α and HIF-2α through direct or indirect mechanisms [46, 47]. Among these inhibitors, everolimus and temsirolimus have been used to treat metastatic renal cell carcinoma with significant effects. Another important HIF-1α inhibitor, digoxin, has mainly been used to treat heart failure in the past. Recently, some studies showed that digoxin can effectively inhibit the growth and metastasis of breast cancer and is a potential therapy for medulloblastoma [48, 49]. In addition, early clinical trials of other HIF inhibitors, such as 2-methoxyestradiol (NCT00030095), 17-AAG (NCT00118248), PT2385 (NCT02293980), and EZN-2208 (NCT01295697), are ongoing or have been completed [50, 51].
Hypoxia is also a major cause of resistance to antitumor treatments, such as radiotherapy, chemotherapy and immunotherapy [52–56]. In terms of immunotherapy, the hypoxic microenvironment can contribute to the immunosuppression phenotype and thereby promote anti-PD-L1 therapy resistance [57]. Previous studies have shown that patients that co-expressed HIF-1α and PD-L1 had a high risk of tumor recurrence, metastasis and mortality [58, 59]. Mechanically, PD-L1 is a direct target of HIF-1α, and blocking PD-L1 under hypoxic conditions can enhance T cell activation [60]. Thus, a combinational therapy blocking PD-L1 along with HIF-1α inhibitors may represent a novel approach to tumor immunotherapy. In addition, as the root cause of tumor recurrence, tumor-initiating stem cells can also enhance resistance to immunotherapy, such as ACT treatment [61]. The hypoxic microenvironment is conducive to the proliferation of these stem cells, which involve the same mechanisms as in embryonic stem cells, such as the HIF-ALKBH5-NANOG signaling pathway [62]. Moreover, hypoxia can inhibit oxygen-dependent ten–eleven translocation (TET) enzyme activity, which then increases epigenetic abnormality, such as hypermethylation, leading to the inhibition of tumor suppressor gene expression [63]. In conclusion, targeting tumor hypoxia or maintaining the oxygen supply in tumors may be an effective method for improving immunotherapy. A recent study first defined the molecular hallmarks of tumor hypoxia across cancer types, which influence tumor evolution and aggressivity; thus, the identification of these hallmarks may help to develop new therapies that target multiple hypoxia-related therapeutic tolerance and metastases [64].
Glycolysis
As the main source of cellular energy, glucose is oxidized in respiration through glycolysis and the tricarboxylic acid (TCA) cycle. Glucose is maintained in an equilibrium state in normal cells. When the oxygen content is normal, pyruvate enters the TCA cycle and undergoes oxidative phosphorylation (OXPHOS), which uses oxygen as the final receptor of the respiratory electron-transport chain to generate energy; however, in the absence of oxygen, glucose converts pyruvate into lactate [65]. In malignant tumor cells, even in the case of sufficient oxygen content, glucose uptake and lactate accumulation are abnormally high, showing active glycolysis and decreased OXPHOS. This metabolic state is called aerobic glycolysis or the “Warburg effect” [66, 67]. Glycolysis provides a large amount of energy and intermediates for tumor metabolism [68]. The intermediates of glycolysis, namely, glucose-6-phosphate (G-6-P), glyceraldehyde-3-phosphate (G-3-P) and fructose-6-phosphate (F-6-P), can synthesize nicotinamide adenine dinucleotide phosphate (NADPH) via the pentose phosphate pathway (PPP), thereby reducing intracellular reactive oxygen species (ROS) levels and increasing tumor dependence on glycolysis [66, 69, 70]. Tumor glycolysis greatly influences the T cell-mediated antitumor response and the activity of tumor-infiltrating myeloid cells [71]. As the end product of glycolysis, lactate is delivered to the external environment of cancer cells by a monocarboxylic acid transporter (MCT). The accumulation of lactic acid in the TME disrupts T cell metabolism via blockade of lactate transport in T cells, thereby suppressing their proliferation and activation [72]. In addition, tumor-derived lactate can also modulate and inhibit tumor necrosis factor (TNF) secretion by human monocytes by blocking glycolytic flux [73].
The Warburg effect represents the transformation of glucose utilization from OXPHOS to glycolysis. The regulatory network of such energy metabolism changes is extremely complex and includes genetic changes, such as alterations in mitochondrial DNA, some oncogenes and tumor suppressors [74]. Activation of some oncogenes (such as RAS and MYC) and inactivation of several tumor suppressors (such as PTEN and p53) can abnormally regulate key metabolic enzymes, thereby promoting glycolysis and reducing OXPHOS [75–78]. These key metabolic enzymes for glycolysis, such as glucose transporter 1 (GLUT1), hexokinase (HK), phosphofructokinase (PFK) isoforms, phosphoglycerate kinase 1 (PGK1) and pyruvate kinase (PK), are tumor markers and can significantly affect the development of tumors [79, 80]. The aggressive glycolytic rates lead to clear proliferative advantages in the process of carcinogenesis, which might contribute to other hallmarks of cancer, such as invasion and metastasis [81]. Thus, inhibition of glycolysis can effectively control the proliferation of tumor cells and even play a role in killing tumor cells, and targeting glycolysis-related metabolic enzymes has become a focus of antitumor therapy [82]. For example, lonidamine is an oral HK inhibitor with significant antitumor activity, especially when used in combination with chemotherapy, and it has been studied in preclinical and clinical trials. In addition, shikonin is a naphthoquinone derivative that inhibits the expression of PK, and it has entered a clinical phase II trial for treatment of bladder carcinoma [83]. Although interventions that target glycolysis are being studied in several clinical trials, it should be noted that since tumor cells and normal cells use several of the same glycolytic enzymes, targeting these enzymes may cause other clinical responses.
Glutaminolysis
Glutamine is a secondary essential nutrient for tumor cells besides glucose, and the uptake of glutamine by tumor cells is approximately 10 times than that of other amino acids [84]. As the most abundant free amino acid in the human body, glutamine can be catabolized to α-ketoglutarate (α-KG), which is an intermediate product of the TCA cycle. Specifically, glutamine enters cells through transporters, such as solute carrier family 1 member 5 (SLC1A5) and solute carrier family 7 member 5 (SLC7A5), and is then deaminated to produce glutamate and ammonia by glutaminase (GLS). Glutamate is then converted to α-KG by glutamate dehydrogenase (GDH) or transaminase [85]. Tumor cells rely on glutaminolysis to maintain biosynthesis, energy metabolism, and cellular homeostasis that are necessary for rapid growth and proliferation [86]. In addition to transporting glutamine from the extracellular space, tumor cells can also obtain glutamine through the breakdown of macromolecules under nutrient-eficient conditions. Several oncogenes, such as MYC and KRAS, have been shown to mediate glutamine reprogramming, leading to tumor growth and survival [87–90]. Moreover, a recent study identified a homeostatic role for the tumor suppressor p53 in adaptation to glutamine starvation [91].
Due to the dependence of tumor cells on glutamine metabolism, the levels of glutamine are correlated with the sensitivity of tumors to antitumor therapies [92]. For example, tumor cells can utilize glutamine to acquire a nitrogen source and synthesize nucleotides, thereby promoting efficient DNA damage repair and ultimately leading to radiotherapy resistance [93]. For immunotherapy, glutamine consumption by tumors can inhibit T cell proliferation, activation, and regulate the transition of CD4+ T cells towards to inflammatory subtypes [94]. Thus, interventions targeting glutamine metabolism in tumor cells may affect the immune states of TME. GLS controls the first step of glutaminolysis, and high GLS expression is closely related to various types of tumors, such as pancreatic cancer, breast cancer and lung cancer [95–97]. Therefore, GLS is considered to be a potential effective target for tumor treatment. CB-839 is a small-molecule allosteric inhibitor of GLS that is undergoing clinical trials in different stages for various cancers [98]. CB-839 is a very well-tolerated drug and shows promising results in combination therapies with other drugs, such as MLN128 (mTOR inhibitor) and everolimus (mTOR inhibitor) [99]. In addition to CB-839, a recent study reported that a new glutamine antagonist, JHU083, could overcome tumor immune evasion and enhance antitumor responses [100]. JHU083 treatment can enhance ACT immunotherapy, and combined treatment with JHU083 and anti-PD-1 antibody can significantly enhance the antitumor effects in different mouse models, which means that JHU083 has the potential to become a broad-spectrum antitumor inhibitor [101].
In addition to glutaminolysis, the high consumption of other amino acids, such as arginine and tryptophan, is also a metabolic feature of many tumor cells [102–104]. Thus, tumor cells can choose different metabolic reprogramming pathways to produce available ATP and biomacromolecules according to the concentration and content of external nutrients, such as glucose, glutamine, serine, arginine and fatty acids [105]. From the perspective of systems biology and evolutionary biology, studies have quantitatively analyzed the use of amino acids for protein synthesis in tumor cells and showed that tumor cells evolve to minimize the biosynthetic energy cost of amino acids by optimizing gene expression [106].
De novo fatty acid synthesis
De novo synthesis of fatty acids often takes place during embryogenesis; in contrast, most adult cells preferentially utilize fatty acids in the circulatory system to synthesize functional lipids. However, proliferating cancer cells have increased fatty acid synthesis regardless of whether the fatty acids in the circulation are sufficient [107]. Thus, uncontrolled de novo fatty acid synthesis is an important metabolic hallmark of cancer cells [108, 109]. The metabolite acetyl-coenzyme A (acetyl-CoA) is the required carbon source and synthetic precursor for de novo fatty acid synthesis [110]. Tumor cells synthesize a large number of lipids to promote the formation of cell membranes and accelerate cell division; the intermediates of lipid metabolism also positively regulate tumor proliferation and growth signaling pathways [111]. De novo fatty acid synthesis in tumor cells also contributes to the production of lipids involved in the regulation of proto-oncogene activity, such as phosphatidylinositol, phosphatidylserine and lecithin, which are crucial for the activation and mediation of the proliferation and growth signaling pathways [112]. Many lipogenesis-related enzymes, such as ATP-citrate lyase (ACL), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FAS), are highly expressed in many cancers [113, 114]. In tumor cells, most fatty acids are produced primarily by FAS catalysis; therefore, FAS is a novel antitumor drug target. Although blocking fatty acid metabolism can be used as a strategy for treating tumors, certain specific cancer types are resistant to fatty acid metabolism inhibition. The synthesis of unsaturated fatty acids requires stearoyl-CoA desaturase (SCD), and previous studies showed that SCD is overexpressed in prostate cancer, liver cancer, kidney cancer and breast cancer. However, liver cancer and lung cancer cells are not affected by SCD expression, and they can utilize alternative fatty acid desaturation pathways to promote membrane biosynthesis during proliferation [115], which can explain the tolerance of cancer to fatty acid metabolism inhibition.
Immune cell metabolism in the TME
The metabolic reprogramming of tumor cells is influenced by the complex and variable TME. Tumor cells and immune cells are exposed to a similar TME, and thus they share many metabolic features. The immune system contains a variety of immune cells, which are in a static state when the body is immunologically stable, and will be activated quickly when the body is stimulated by infection, inflammation or other external stimuli. Studies have shown that immune cells have significant differences in energy utilization between their quiescent and activated states [8]. T lymphocytes, especially CD8+ cytotoxic T cells (CTLs), are considered the major effector cells involved in the antitumor immunity process; this process is associated with the presentation of tumor antigens by dendritic cells (DCs) [116]. However, T cell dysfunction-associated immune escape occurs because of different immune-suppressive cell populations in the TME, such as TAMs, MDSCs and Tregs (Fig. 1). This review mainly focuses on these immune cell types to describe how metabolic alterations in these cells regulate tumor progression or dormancy.
Fig. 1.

Overview of the complex interactions between tumor cells and immune cells and the metabolic characteristics of these cells. During tumorigenesis and malignant progression, a complex intercellular interaction network is established between tumor-infiltrated immune cells and tumor cells that improves and maintains the immunosuppressive microenvironment and promotes immune escape. Black arrow recruit or promote, Pink T-line inhibit, Red up arrow upregulate, Red down arrow downregulate, TME tumor microenvironment, TAM tumor-associated macrophage, Treg regulatory T cell, DC dendritic cell, MDSC myeloid-derived suppressor cell
Tumor-infiltrating T cells
Although T cells are essential for immune defense, they frequently have low activity in TME and are generally unable to control tumor development [117]. T cell dysfunction in the TME is mainly caused by T cell anergy, exhaustion and senescence (Fig. 1). T cells exhibit dynamic metabolic patterns depending on their activation states. Naïve T cells maintain a quiescent state and only exhibit basic nutrient uptake and a minimal rate of glycolysis; they mainly generate energy through fatty acid oxidation (FAO) and OXPHOS [118]. Under normal circumstances, upon activation, these cells (differentiating into effector T cells) exhibit increased dependence on glycolysis and glutaminolysis to meet the energy and biosynthesis demands required for their rapid proliferation and effector function. Activated T cells also increase OXPHOS and lipid synthesis but suppress FAO [119]. Mechanistically, TCR-mediated signaling pathways (e.g., the PI3K-AKT-mTOR pathway) activate transcription factors, such as HIF-1α and c-Myc, which in turn promote the expression of glucose and amino acid transporters on the T cell surfaces and direct metabolic reprogramming to glycolysis and glutaminolysis [120]. However, in the TME, glucose and glutamine consumption by tumors can inhibit and reduce glycolysis and glutaminolysis in effector T cells. Memory T cells undergo a metabolic pattern similar to that of naïve T cells, but they exhibit a higher aerobic respiration that facilitates their rapid activation after re-infection [121].
CD8+ cytolytic T lymphocytes have been shown to be key regulators of antitumor immunity. CD8+ T cell expansion, activation and effector function strongly rely on glycolysis. Acylglycerol kinase (AGK) is a lipid metabolic enzyme that is indispensable for CD8+ T cell metabolic programming, which occurs via the activation of PTEN and PI3K-mTOR signaling [122]. Similar to CD8+ effector T cells, CD4+ effector T cells (Th1, Th2, and Th17) also predominantly use aerobic glycolysis to generate energy, whereas CD4 regulatory T cells are more reliant on FAO and OXPHOS [123]. In addition, as a nutritional sensor, mTOR signaling is extremely important for the differentiation of CD4+ T cells into effector cells and the formation of memory CD8+ T cells [124, 125]. Of course, there are distinct metabolic differences between CD8+ and CD4+ T cells [126]. The TCR-induced downstream signaling pathways in these effector T cell types are different [127]. CD8+ effector T cells are less dependent on GLUT1 and environmental oxygen levels compared to CD4+ effector T cells [128]. In addition, CD8+ T cells exhibit a higher metabolic flexibility than their CD4+ counterparts, which make CD8+ T cells proliferate more rapidly, even under conditions of nutrition starvation [129]. For example, CD8+ T cells have a higher glycolysis flux and do not rely solely on glucose, while in the glucose-deficient TME, they can oxidize glutamine to support survival and effector function [130]. Furthermore, OXPHOS is a key metabolic pathway that distinguishes T cell subtypes; CD4+ T cells have higher OXPHOS levels than their CD8+ counterparts in melanoma and squamous cell carcinoma of the head and neck [131].
Tumor-associated macrophages (TAMs)
TAMs are the most abundant component in the TME and are characterized by high heterogeneity and plasticity [132]. TAMs play key roles in accelerating the proliferation of cancer cells, promoting angiogenesis and the formation of an immunosuppressive TME (Fig. 1). Macrophages are a heterogeneous cell population with a wide range of activation states. Ideally, macrophages can be classified into two extreme types: classically activated (pro-inflammatory) M1 macrophages and alternatively activated (anti-inflammatory) M2 macrophages. Macrophages undergo polarization between M1 and M2 phenotypes depending on changes in the environmental stimuli. M1 macrophages arise from stimulation with LPS and Th1 cytokine interferon-γ (IFN-γ). In contrast, Th2 cytokines, such as interleukin 4 (IL-4), IL-13, glucocorticoids and macrophage colony-stimulating factor (M-CSF), induce the polarization to M2 macrophages [133]. M1 and M2 macrophages play opposite roles in tumor development by secreting diverse growth factors, chemokines and cytokines. The polarization of macrophages changes in different stages of the tumor: TAMs tend towards M1-like phenotypes during tumor initiation. M1 macrophages secrete high amounts of ROS, inflammatory cytokines (e.g., IL-1, IL-6 and IL-12) and have a high antigen presentation rate; thus, they can promote an antitumor immune response [134]. During tumor development, TAMs are hijacked by tumor cells and are gradually reprogrammed into M2-like macrophages that express high levels of anti-inflammatory cytokines (e.g., IL-10 and transforming growth factor-β (TGF-β)), growth factors, and extracellular matrix degrading enzymes, which support tumor growth and immunosuppression [135].
Metabolic reprogramming is considered to be the material foundation for the phenotypic transformation between M1-like macrophages and M2-like macrophages. The enhancement of nitric oxide (NO) synthesis is a characteristic of pro-inflammatory M1-like macrophages, while tumor-promoting M2-type macrophages show increased glutamine metabolism [136]. Compared to the M1 type, the M2 type has an enhanced ability to biosynthesize other molecules from glucose [137, 138]. The metabolic patterns of TAMs change according to the TME, and are often characterized by enhanced glycolysis and fatty acid synthesis [139]. The glucose metabolism process of TAMs undergoes a similar change to that of tumor cells, that is, acquiring highly active aerobic glycolysis, which is related to the maintenance of angiogenic and metastatic functions [140]. Although in the TME of ischemia, hypoxia, and nutritional competition, TAMs have a large amount of lipid accumulation. Studies have shown that saturated free fatty acids (FFAs) activate macrophages via toll-like receptor 4 (TLR4) and downstream nuclear factor kappa B (NF-κB) and JNK pathways [141, 142]. The inhibition of monoacylglycerol lipase (MGLL) is also a major cause of lipid accumulation in TAMs [143]. TAM-derived metabolites play a non-negligible role in tumor resistance to chemotherapy or other targeted therapies. Gemcitabine is a first-line chemotherapy drug for the treatment of pancreatic ductal adenocarcinoma (PDA), and TAM abundance is closely correlated with a worse response to therapy in PDA [144]. Recently, Halbrook et al. identified that TAMs release a spectrum of metabolites, including pyrimidine nucleosides, which are structurally similar to gemcitabine and may therefore reduce the efficacy of gemcitabine [145].
Regulatory T cells (Tregs)
Tregs are generally considered the brakes that restrict the function of effector T cells, thereby negatively regulating the pathological and physiological immune responses. Under normal circumstances, Tregs can inhibit excessive immune responses and prevent autoimmune diseases. However, Tregs can secrete a variety of inhibitory cytokines into the TME, which in turn suppresses antitumor immunity, allowing tumor cells to escape immune surveillance (Fig. 1) [146]. The migration of activated Tregs to inflammatory sites is an important prerequisite for their immune regulation. Glucokinase (GCK)-dependent glycolysis contributes to the migration of Tregs through the PI3K-mTORC2-mediated pathway. In addition, mTORC1-dependent cholesterol biosynthesis is important for Treg proliferation and immunosuppressive functions [147, 148]. Researchers have previously tried to relieve immunosuppression by eliminating Tregs, but surprisingly, the failed clinical trial showed that the immunosuppressive effect did not disappear, rather it was strengthened. A recent study addressed this question and found that Tregs are highly apoptotic in the TME, and these apoptotic Tregs mediate immunosuppression via an oxidative-stress-associated mechanism [149]. Tregs can migrate to the tumor from throughout the body, resulting in a large number of Tregs being recruited to the TME. However, when the Tregs proliferate, their apoptosis rate also increases. Thus, there are many live and apoptotic Tregs in the tumor at the same time [150, 151]. Such apoptotic tumor-infiltrating Tregs are powerful immune suppressors, which can eliminate spontaneous and PD-L1 blockade-mediated T cell antitumor immune responses. Mechanistically, apoptotic Tregs release and convert large amounts of ATP to adenosine via CD39 and CD73, thereby mediating immunosuppression [149]. The apoptosis of Tregs is usually associated with a weak antioxidant system and a high susceptibility to free reactive oxygen species in the TME. Therefore, the oxidative pathway can be used as a metabolic checkpoint to control the function of Tregs, improving the efficacy of PD-L1 checkpoint therapy. At present, the conventional therapeutic approaches that target Tregs still aim for Treg depletion and functional modulation; these approaches include small molecules and immune checkpoints inhibitors with Treg-depleting effects and agonistic antibodies that suppress Tregs [152]. However, considering the immunosuppressive effect of apoptotic Tregs, researchers will next develop an approach to prevent Tregs from traveling into the TME, thereby reducing or controlling immunosuppression and allowing more cancer patients to benefit from immunotherapy. In addition, a recent study proposed a method different from the Treg depletion treatments, that is, reprogramming tumor-infiltrating Tregs to create a local inflammatory autoimmune reaction, thereby increasing the sensitivity of tumors to immunotherapy [146].
Myeloid-derived suppressor cells (MDSCs)
MDSCs represent a heterogeneous population of immature myeloid cells that are activated and expanded in response to a variety of growth factors and pro-inflammatory cytokines secreted by tumor cells. MDSCs have the ability to promote the formation of an immunosuppressive TME, which contributes to tumor progression and immune escape (Fig. 1) [153]. MDSCs mainly consist of two subsets: polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs). PMN-MDSCs exert relatively modest immunosuppressive activity via an antigen-specific mechanism, whereas M-MDSCs exhibit strong suppressive activity in a non-specific manner [154]. There is increased lipid accumulation in tumors, and thus, the uptake of exogenous lipid promotes tumor-infiltrating MDSC metabolic reprogramming from glycolysis to FAO, thereby forming a metabolic symbiotic relationship with tumor cells in the TME [155, 156]. Mechanically, some tumor-derived cytokines, such as granulocyte-colony stimulating factor (G-CSF) and granulocyte–macrophage colony stimulating factor (GM-CSF), can lead to an increase in lipid transport receptors in tumor-infiltrating MDSCs, resulting in increased lipid uptake [157]. The accumulation of lipids in MDSCs increases oxidative metabolism, shifting MDSCs towards an immunosuppressive and anti-inflammatory phenotype [158]. MDSCs mediate immunosuppression through several primary mechanisms: (1) MDSCs increase the production of NO and ROS by expressing different enzymes, such as inducible nitric oxide synthase (iNOS) and NADPH oxidase, thereby inhibiting T cell proliferation and activation; (2) MDSCs induce the depletion of L-arginine and L-cysteine, which are two key nutrients required for T cell proliferation and activation; and (3) MDSCs induce the expansion of Tregs and macrophage M2 polarization through the secretion of inhibitory cytokines, ultimately promoting and maintaining an immunosuppressive microenvironment [159]. Furthermore, a recent study reported that the uptake of arachidonic acid (AA) and the synthesis of prostaglandin E2 play important roles in the acquisition of immunosuppressive activity by MDSCs [160]. Hypoxia in the TME and tumor glycolysis can also mediate the differentiation of functional MDSCs. Therefore, MDSCs are an important target in tumor immunotherapy, and the most promising approach is based on the promotion of myeloid differentiation.
Dendritic cells (DCs)
DCs are the most powerful professional APCs and play a key regulatory role in activating the immune response and maintaining autoimmune tolerance [161]. Cell metabolism has an important influence on the activation and function of DCs. DCs in a resting state acquire energy mainly by mitochondrial OXPHOS, while activated DCs rely on Warburg metabolism to survive, at which point lactate production increases and the TCA cycle decreases [162]. DCs are potent activators of the T cell immune response via the uptake, processing, and presentation antigens in the TME, which can effectively eliminate target tumor cells. However, many studies have found that the function of tumor-infiltrating DCs usually changes, which in turn affects the antitumor immunity of T cells, leading to tumor immune escape (Fig. 1) [163]. Tumor-associated DCs produce ROS, which in turn induces endoplasmic reticulum (ER) stress and further activates the transcription factor X-box binding protein 1 (XBP1) to induce lipid synthesis. The accumulation of lipids in DCs can reduce the antigen presentation ability, thereby inhibiting the antitumor response [164]. In addition to lipid accumulation, the increased decomposition of arginine and tryptophan in tumor-associated DCs can also alter the immune function of T cells [165]. Moreover, tumor cells also secrete several factors that act on DCs to induce the production of adenosine- and lactate-tolerant DCs.
Relationship between antitumor immunosuppression and metabolic crosstalk
The success of tumor immunotherapy reveals the critical role of the host immune system in the antitumor immune responses; however, the immunosuppressive TME is still the bottleneck restricting the success of tumor immunotherapy. Due to the common demand for nutrition, there is metabolic competition between tumor cells and immune cells in the TME, which has been proved to be closely related to the occurrence of antitumor immunosuppression. The activation and effector function of immune cells are regulated by metabolites or waste from tumor cells through various mechanisms. Meanwhile, immune cell-derived metabolites or secretions can be utilized by tumor cells to promote their survival and proliferation.
Metabolic competition dampens antitumor immunity
Immune cells and tumor cells have similar metabolic patterns. For example, they both preferentially utilize glycolysis to obtain the energy required for rapid proliferation and acquire the raw materials for cytoskeleton formation, such as carbon and oxaloacetic acid [119, 166]. Due to the urgent need for cell proliferation and activation during the antitumor process, immune cells need to compete with tumor cells for energy utilization [167], and the failure of immune cells in this competition is an important reason for antitumor immunosuppression (Fig. 2). Given the critical role of tumor-infiltrating T cells in immunotherapy, we will focus on the competition of tumor cells and T cells for nutrition in the TME.
Fig. 2.
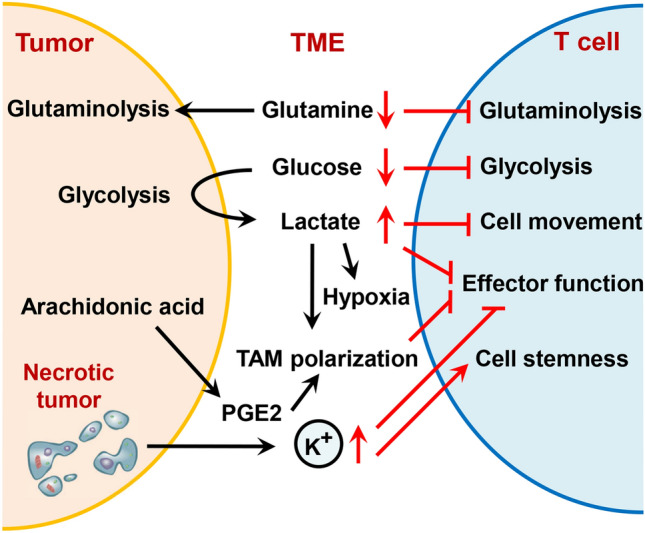
Metabolic competition and tumor-derived metabolite-mediated immunosuppression in the tumor microenvironment. Tumor cells compete with immune cells to preferentially utilize glucose and glutamine in the microenvironment. Tumor-derived metabolites, such as lactate, PGE2 and K+, can inhibit T cell movement, effector function and TAM polarization and promote stemness, ultimately promoting immune escape
Glucose consumption by tumors can affect the metabolic reprogramming of T cells to directly dampen their antitumor function and promote tumor progression; this process involves many mechanisms and pathways [128, 168]. Using a mouse sarcoma model, researchers revealed that the large uptake of glucose by tumors in the TME inhibits calcium signaling and the activity of nutrient sensor mTOR in T cells, thereby restricting glycolysis and IFN-γ production, inducing T cell exhaustion and immune escape [167]. Blockade antibodies against CTLA-4, PD-1 and PD-L1 can weaken the glycolysis of tumor cells to restore the glucose levels in the TME, thereby promoting glycolysis in T cells [167]. In addition, glucose consumption by tumor cells can metabolically restrict T cells via downregulating the expression of methyltransferase, which epigenetically inhibits the multifunctional cytokine production and T cell survival [169, 170]. Furthermore, the accelerating depletion of extracellular glucose by tumor cells suppresses glycolysis in T cells and the downstream metabolite phosphoenolpyruvate (PEP), which eventually inhibits the effector function of T cells. The overexpression of phosphoenolpyruvate carboxykinase 1 (PCK1) can promote gluconeogenesis and increase PEP production, thereby restoring TCR-induced Ca2+ flow and the antitumor function. Therefore, manipulating metabolism to improve immune function may be a new direction for future immunotherapy [171]. Although glucose is the preferred energy source for T cells, in the face of low oxygen and glucose conditions in the TME, T cells can also survive by generating energy through fatty acid oxidation [172].
In addition to glucose, glutamine, tryptophan and arginine can also provide nutrition for immune cells [85]. Glutamine is a key nutrient for cell proliferation; therefore, a lack of glutamine inhibits the activation and differentiation of T cells [94, 173]. By blocking the glutamine pathway in tumor cells, this amino acid content will increase in the TME, which can effectively enhance antitumor immune responses [94]. Tryptophan is another essential amino acid for the effector function of T cells. Tumor cells can inhibit T cell activity by depleting the tryptophan required for T cell survival or triggering Tregs to induce immunosuppression [174]. In addition to tumor cells, other immunosuppressive cells also compete with T cells for nutrients in the TME. For example, MDSCs increase sharply during tumorigenesis, leading to the upregulation of arginase 1 (ARG1), which reduces the extracellular levels of L-arginine, inhibiting T cell activation [175, 176]. The depletion of L-arginine also can cause G0-G1 cell cycle arrest in T cells [177]. Collectively, these studies suggest that amino acid deficiency can lead to many immunosuppressive factors in the TME through various mechanisms, which together suppress antitumor immune responses.
Tumor cell-generated metabolites induce immune cell incompetence and immunotherapy resisitance
Tumor cells produce a variety of immunosuppressive metabolites that accelerate the dysfunction of immune cells (Fig. 2). Lactate, a metabolite of glycolysis, has multiple roles in the TME, including intercellular signaling and epigenetic reprogramming [178]. Tumor cells and tumor-associated fibroblasts (CAFs) are the main sources of lactate production. Tumor-derived lactate inhibits the movement, cytotoxicity and effector function of T cells [179], and targeting L-lactate dehydrogenase A (LDHA)-associated lactic acid can restore T cell infiltration [180]. In addition, high lactate concentrations disrupt the immune surveillance of NK cells and promote the survival of Tregs and a tolerogenic phenotype in DCs, thus creating an environment in which tumor cells are not attacked by the immune system [181]. Moreover, the lactate-rich TME also mediates macrophage polarization to promote tumor growth. For example, tumor-derived lactate suppresses ATP6V0d2 expression in macrophages by activating the mTOR-TFEB axis, resulting in macrophage reprogramming into a pro-tumoral phenotype [182]. Collectively, the above studies indicate that tumor derived-lactate inhibits the function of immune cells and lead to immune escape. In addition to lactate, tumor-derived lipid metabolites and amino acids also have profound effects on immune cells in the TME. Prostaglandin E2 (PGE2) is a metabolite of arachidonic acid and is in a class of highly active inflammatory mediators [183]. Tumor-derived PGE2 can stimulate the secretion of CXCL1 and IL-6 and is necessary for the development of suppressive TAMs [184]. In addition, the extracellular shedding of gangliosides from tumor cell membranes can recruit immunosuppressive MDSCs into the TME [185]. Moreover, tumor cells and CAFs promote immunosuppression by degrading tryptophan to kynurenine [186]. Kynurenine can act as a ligand to activate the aryl hydrocarbon receptor (AHR), leading to AHR-dependent Treg generation [187].
Many other tumor cell-generated metabolites can also induce immune cell incompetence, directly or indirectly. Due to insufficient nutrient supply, tumors usually contain areas of cellular necrosis. Necrotic tumors release potassium ions into the TME, leading to the suppression of T cell effector function [188]. Surprisingly, a recent study revealed that elevated potassium also triggers stem cell-like properties of some antitumor T cells. Elevated potassium ions trigger functional caloric restriction and autophagy, which drive metabolic and epigenetic reprogramming of T cells, thereby limiting the T cell effector function and remaining stemness [189]. This mechanism may lead to novel therapeutic strategies that enhance cancer immunotherapies. T cell senescence can also induce a potent suppression function that aids in the immune escape of tumors. Tumor-derived cAMP has been shown to directly induce T cell senescence and is also a key regulator for Treg cells to induce T cell senescence [190, 191]. In summary, these studies have shown that tumor cells can not only deprive immune cells of nutrients in the TME, but they also produce metabolites that inhibit immune defense.
In general, tumor-derived metabolites can induce immune cell incompetence, which further affects the efficacy of immunotherapy and may cause resistance and recurrence [192, 193]. For example, high levels of lactate are closely related to ACT therapy resistance [71]. Tumor-derived lactate can induce the upregulation of PD-L1, thereby affecting the efficacy of immunotherapy [194]. Increased serum LDH is negatively related to the clinical efficacy of immunotherapy, so targeting LDH-associated L-lactate is a potential method for overcoming immunotherapy resistance [195]. High levels of fatty acids can prevent T cells from killing tumor cells so that the tumor can resist the attack of the immune system, thereby reducing the efficacy of antitumor drugs that rely on the functional immune system [196]. In addition, some tumor-derived amino acids (such as kynurenine) and nucleotides (such as adenosine) also have an inhibitory effect on the efficacy of immunotherapy, and many inhibitors targeting these molecules can be used in combination with PD-1 monoclonal antibodies to alleviate treatment resistance to a certain extent (which will be discussed in the following section).
Immune cell-derived metabolites and secretions are complicit in tumor progression and metabolic reprogramming
Immune cell functions can be regulated by metabolites derived from tumors. In turn, some types of immunosuppressive cells can also directly or indirectly assist tumor progression and metastasis by secreting cytokines, growth factors and proteases. In melanoma, TAMs can produce arachidonic acid metabolites, TGF-β and TNF-α, thereby inducing tumor cells to secrete IL-8 and vascular endothelial growth factor-A (VEGF-A), promoting tumor angiogenesis [197]. TAM-secreted IL-6 can induce PGK1 phosphorylation in tumor cells to enhance tumor cell glycolysis, ultimately promoting the malignant progression [198]. Moreover, TAM-secreted extracellular vesicle (EV)-packaged lncRNA can be taken up by cancer cancer cells to promote aerobic glycolysis [199].
A new method of cancer immunotherapy: immunometabolic checkpoints
Cancer immunotherapy refers to treatments that aim to kill or suppress tumors by activating and increasing the immune response (enhancement tumor immunotherapy) or restoring a lost antitumor immunity (normalization tumor immunotherapy) [200]. “Immune checkpoint” refers to the proteins on the surface of immune cells that are responsible for regulating the degree of immune cell activation, and the most widely studied immune checkpoints are CTLA-4 and PD-1/PD-L1 [17]. Although immune checkpoint therapy has led to a paradigm shift in cancer treatment, the majority of patients still have no response to such therapy, which is inseparably related to the metabolically suppressive TME [201]. Tumor cells always adjust their metabolism and nutrient acquisition to maintain sustained proliferation and, thus, alter the metabolic landscape of the TME, which can impose metabolic stress on infiltrating immune cells and affect the presentation and recognition of antigens, resulting in immunosuppression and immune escape [30]. The limited therapeutic efficacy of immune checkpoints is presumably due to the insufficient metabolic reprogramming of the immunosuppressive TME and thus impacts the reinvigoration of the antitumor immune response [27, 193]. “Metabolic checkpoints” refer to the important enzymes or receptors in metabolic pathways, and their activity levels can affect immune cell function [100]. Therefore, the modulation of various metabolic functions of immune cells is another important branch of immunotherapy. Effective interventions that target dysregulated metabolic checkpoints may reprogram the immune status of the TME, regulate the activation and function of T cells, increase the immunogenicity of tumor cells, and synergistically enhance effectiveness of immune checkpoint therapy.
IDO
Tumor cells metabolize tryptophan to kynurenine by indoleamine 2,3-dioxygenase (IDO). Tryptophan is important for T cells to exert an immune function, while kynurenine inhibits the antitumor immune response of T cells [202]. Therefore, IDO ultimately promotes immune escape as an immunomodulatory enzyme that can dampen the proliferation and function of effector T cells and form a positive feedback loop with Tregs [203, 204]. IDO is overexpressed in tumor cells and in a variety of inhibitory immune cells in the TME, including TAMs and MDSCs [193]. Highly expressed IDO can inhibit T cell activity through two mechanisms, direct inhibition and indirect inhibition (Fig. 3). IDO can directly deplete the tryptophan required for T cell survival and can also form a positive feedback regulation loop with Tregs to suppress the immune response [205]. Blocking IDO can reduce the number of Tregs and restore the function of T cells.
Fig. 3.
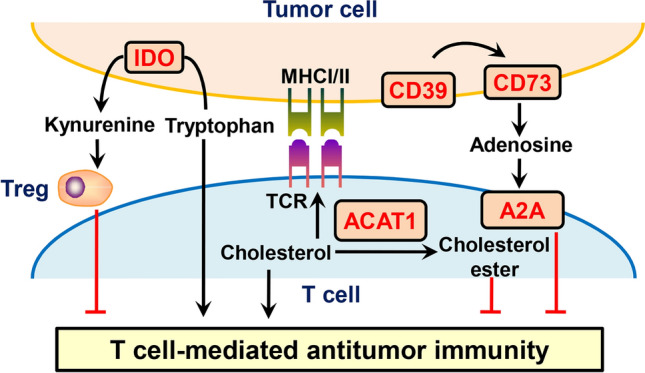
Modulation of T cell-mediated antitumor immunity by targeting metabolic checkpoints. Targeted inhibition of metabolic checkpoints, such as IDO, ACAT1, CD39, CD73 and A2A, can enhance the antitumor effects of T cells. IDO indoleamine 2,3-dioxygenase, ACAT1 acetyl-CoA acetyltransferase 1
IDO inhibitors are new type of immunotherapeutic drug that blocks tryptophan metabolism in cancer cells, activates T cells, and improves the lethality of immune cells [206]. Currently, several IDO inhibitors are being combined with immunotherapy or chemotherapy in clinical research, including epacadostat, indoximod, navoximod, EOS200271 and BMS-986205 [207]. As an oral drug, epacadostat has shown a very significant therapeutic effect in early trials, especially in melanoma [208]. Indoximod is a small-molecule compound that directly reverses the IDO inhibition of immune cells, and its strong activity has also been observed in early clinical trials [209]. IDO inhibition combined with anti-PD-1 antibodies was once considered to be the most promising treatment for cancers, and the early clinical data showed a high efficiency. Epacadostat was the first IDO inhibitor to be combined with anti-PD-1 antibody to treat solid tumors in phase III clinical trials. Regrettably, Incyte and Merck have recently announced that epacadostat in combination with keytruda (PD-1 antibody) for phase III clinical trials of malignant melanoma had failed [210]. Compared to the anti-PD-1 antibody alone, the combination therapy has more side effects and does not prolong the patient’s progression-free survival (PFS) or estimated OS. Although this clinical trial has failed, IDO inhibitors combined with anti-PD-1 antibodies still provide a new treatment model for reversing immunotherapy resistance.
ACAT1
Cholesterol is the main component of membrane lipids and is closely related to the TCR clustering and the formation of immunological synapses [211]. As a key cholesterol esterification enzyme, acetyl-CoA acetyltransferase 1 (ACAT1) converts free cholesterol into cholesterol ester. Knocking out or inhibiting ACAT1 can increase the level of cholesterol in CD8+ T cells membranes, thereby making them more sensitive to antigens and eventually improving the immune response (Fig. 3) [212, 213]. Treatment with the small-molecule ACAT1 inhibitor avasimibe in a mouse model of melanoma achieved a good antitumor effect [213]. Moreover, the combination of avasimibe and anti-PD-1 antibody effectively enhanced the antitumor immunotherapy. Most importantly, avasimibe underwent phase III clinical trials for the treatment of cardiovascular and neurodegenerative diseases and showed no obvious side effects [214]. Therefore, avasimibe may be a good candidate for cancer immunotherapy. In addition, a recent study found that the high expression level of a cholesterol transesterase called sterol O-acyltransferase 1 (SOAT1) is closely related to the poor prognosis of hepatocellular carcinoma (HCC) patients [215]. As a SOAT1 inhibitor, avasimibe could also significantly reduce the size of tumors in a patient-derived tumor xenograft (PDX) mouse model of HCC [215]. Therefore, avasimibe is expected to become a new drug for the precise treatment of HCC.
Adenosine signaling
Adenosine signaling is one of the major immunosuppressive mechanisms in the TME, and it is also a potential therapeutic target for tumor therapy (Fig. 3). Adenosine is an endogenous nucleoside distributed throughout human cells and can be used as an important intermediate for the synthesis of ATP and adenine [27]. Adenosine is produced in many different types of tumors and is maintained at a high level in the TME, which is beneficial for tumor development and immune escape [216]. The ectonucleotidases CD39 and CD73 are key mediators of adenosine accumulation in the TME. Preclinical animal model studies have shown that both tumor cells and immune cells can express CD39 and CD73 to promote tumor immune escape, development and metastasis [217]. Thus, targeting CD39 and CD73 activity to reduce adenosine production can effectively block tumor growth and metastasis [218]. Several CD73 monoclonal antibodies are currently undergoing clinical trials, including MEDI9447, TJD5 and BMS-986179. In addition to antibodies, small-molecule inhibitors have been developed, such as AB680 and hydrochlorothiazide derivatives. The clinical trials targeting CD73 in solid tumors primarily focus on the efficacy and safety of these inhibitors when used in combination with PD-(L)1 monoclonal antibody.
Tumor-derived adenosine can bind to any of four adenosine receptors (adenosine receptor A1 (A1R), A2AR, A2BR, and A3R) on the surface of infiltrating immune cells, and of these receptors, A2AR and A2BR are usually associated with immunosuppression [219]. A2A is a G protein-coupled receptor and is often expressed in many immune cells, including T cells, NK cells, TAMs, and DCs. Adenosine binds to A2AR and inhibits the ability of immune cells to attack tumors [220]; this occurs through two primary mechanisms. First, adenosine can block the activation and function of antitumor immune cells. Second, adenosine increases the number of Tregs and contributes to Treg-mediated immunosuppression. Studies have shown that apoptotic Tregs release large amounts of ATP via CD39 and CD73 and convert them to adenosine, thereby mediating immunosuppression via the A2AR pathway [149]. Small-molecule oral A2AR antagonists that have been clinically tested include preladenant, CPI-444, AB928, AZD4635 and NIR178. Among these antagonists, CPI-444 has shown good clinical efficacy and tolerance, both as a monotherapy and in combination with PD-L1 antibody [221]. In general, A2AR antagonists can enhance the activity of immune cells and promote antitumor immunity, and their prospects in immunotherapy are promising. In addition to the above purinergic receptors selective for adenosine (P1 receptors), another type of purinergic receptor selective for ATP (P2 receptor) also plays a pivotal role in the tumor immune response [222]. P2 receptors contain two subfamilies: P2X ion channels and G-protein coupled P2Y receptors, among which the P2X7 receptor (P2X7R) is highly regulated by ATP levels [223]. The expression and function of P2X7R can modulate the infiltration of immune cells and the concentration of ATP in the TME [224]. Some preclinical models indicated that P2X7R antagonists can inhibit tumor proliferation and metastasis. In addition, P2X7R blockade (A740003) increases the infiltration of CD4+ T effector cells and reduces the expression of CD39 and CD73, thereby inhibiting immunosuppression in the TME [225].
Microbial metabolites
The intestine is the largest immune organ in our body and is the home of 70% of immune lymphocytes [226]. Therefore, there is a direct relationship between the intestinal flora and the immune system. The intestinal flora of patients with malignant tumors is often dysregulated, which can affect intestinal homeostasis, intestinal metabolism, immune function and promote cancer development [227]. Many studies have found that the intestinal microbiota is closely associated with the effectiveness of antitumor immunosurveillance. In 2015, two studies first indicated that the composition of intestinal microbes in cancer patients influenced the effectiveness of immunotherapy targeting CTLA-4 and PD-L1 checkpoints. The antitumor immune responses following CTLA-4 blockade are associated with B. thetaiotaomicron and B. fragilis in mice and patients. Tumors in germ-free mice have little response to CTLA-4 blockade, which can be overcome by the transplantation of B. fragilis [228]. Another study found that Bifidobacterium is beneficial for the therapeutic effects of antibodies targeting PD-L1 [229]. Recently, three more studies have focused on the key role of intestinal microbes in immunotherapy. Researchers have discovered that Akkermansia muciniphila can promote the efficacy of PD-1-based immunotherapy in lung cancer and kidney cancer [230]. Another study in the same journal conducted oral and intestinal microbial analysis of melanoma patients treated with PD-1 inhibitors and found that Ruminococcaceae is involved in the immunotherapy responses [231]. In addition, Bifidobacterium longum, Collinsellaaerofaciens, and Enterococcus faecium are more abundant in patients who respond to anti-PD-1-based immunotherapy [232]. Therefore, the composition of intestinal microbiota affects the therapeutic activity of immune checkpoint inhibitors targeting CTLA-4 or PD-1/PD-L1, and the regulation of the microbiome is expected to improve the efficacy of tumor immunotherapy in the future.
Conclusions and perspectives
Antitumor immunotherapy provides possibilities for the clinical treatment of cancer, but a considerable number of patients do not respond to or become resistant to these treatments; thus, it is necessary to fully understand the mechanisms of immune escape in the TME. Cancer is a metabolic disease in which nutrients and metabolic signaling changes can affect the fate and function of tumor cells and immune cells. In addition to macromolecules such as glucose, amino acids and lipids, there are many vitamins and trace metals in the TME, including vitamin C, zinc and copper. These micronutrients may be cofactors of various metabolic enzymes and may also form complexes with intracellular proteins to participate in metabolic cycles and exert signal transduction functions. However, the function of these cofactors in tumors and immune cells remains unclear and requires further research. Regulating metabolic reprogramming to promote the immune response has gradually become the frontier of current antitumor immunotherapy. Although scientists have achieved encouraging results in the field of immunometabolism in recent years, there is still a great deal of work to be done to conquer tumors. First, the composition of the TME is extremely complex and dynamic, and the metabolic landscape and relationships (cooperativity and competition) within this microenvironment remain poorly understood. Few metabolic pathways are confined to tumor cells, which increases the potential of toxic effects on normal cells. Therefore, it is of great significance to develop metabolic inhibitors that can preserve T cell function and have high specificity for tumor cells. Moreover, targeting metabolism-based immunotherapy aims to reprogram the immunosuppressive TME and thus reinvigorate antitumor immunity. In light of the high metabolic adaptability of tumor cells, when any metabolic pathways encounter obstacles, the tumor cells will automatically switch or activate other pathways to escape stress damage. Thus, tumor metabolic regulation should jointly block or regulate multiple metabolic pathways to maximize synergy, which requires a comprehensive understanding of the metabolic mechanisms of immune evasion and the metabolic needs of immune cells in the future research.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81903138, 81972776, 81803025, 81772928, 81702907, 81772901, 81672993, 81672683), and the Natural Science Foundation of Hunan Province (2019JJ50778, 2018SK21210, 2018SK21211, 2018JJ3704, 2018JJ3815).
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Shanshan Zhang, Email: zhangshanshan@csu.edu.cn.
Zhaoyang Zeng, Email: zengzhaoyang@csu.edu.cn.
References
- 1.Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, Gong Z, Zhang S, Zhou J, Cao K, Li X, Xiong W, Li G, Zeng Z, Guo C. Role of tumor microenvironment in tumorigenesis. J Cancer. 2017;8(5):761–773. doi: 10.7150/jca.17648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duan S, Guo W, Xu Z, He Y, Liang C, Mo Y, Wang Y, Xiong F, Guo C, Li Y, Li X, Li G, Zeng Z, Xiong W, Wang F. Natural killer group 2D receptor and its ligands in cancer immune escape. Molecular Cancer. 2019;18(1):29. doi: 10.1186/s12943-019-0956-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 2018;32(19–20):1267–1284. doi: 10.1101/gad.314617.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peng M, Mo Y, Wang Y, Wu P, Zhang Y, Xiong F, Guo C, Wu X, Li Y, Li X, Li G, Xiong W, Zeng Z. Neoantigen vaccine: an emerging tumor immunotherapy. Molecular Cancer. 2019;18(1):128. doi: 10.1186/s12943-019-1055-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, Lichtor T, Decker WK, Whelan RL, Kumara H, Signori E, Honoki K, Georgakilas AG, Amin A, Helferich WG, Boosani CS, Guha G, Ciriolo MR, Chen S, Mohammed SI, Azmi AS, Keith WN, Bilsland A, Bhakta D, Halicka D, Fujii H, Aquilano K, Ashraf SS, Nowsheen S, Yang X, Choi BK, Kwon BS. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35(Suppl):S185–S198. doi: 10.1016/j.semcancer.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Saleh R, Elkord E. Acquired resistance to cancer immunotherapy: Role of tumor-mediated immunosuppression. Semin Cancer Biol. 2019 doi: 10.1016/j.semcancer.2019.07.017. [DOI] [PubMed] [Google Scholar]
- 7.Herbel C, Patsoukis N, Bardhan K, Seth P, Weaver JD, Boussiotis VA. Clinical significance of T cell metabolic reprogramming in cancer. Clin Transl Med. 2016;5(1):29. doi: 10.1186/s40169-016-0110-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Biswas SK. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity. 2015;43(3):435–449. doi: 10.1016/j.immuni.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 9.Chodon T, Koya RC, Odunsi K. Active Immunotherapy of Cancer. Immunol Invest. 2015;44(8):817–836. doi: 10.3109/08820139.2015.1096684. [DOI] [PubMed] [Google Scholar]
- 10.Tey SK. Adoptive T-cell therapy: adverse events and safety switches. Clin Transl Immunol. 2014;3(6):e17. doi: 10.1038/cti.2014.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545(7655):423–431. doi: 10.1038/nature22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong H, Zhu G, Tamada K, Chen L. B7–H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5(12):1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 13.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 14.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren D, Hua Y, Yu B, Ye X, He Z, Li C, Wang J, Mo Y, Wei X, Chen Y, Zhou Y, Liao Q, Wang H, Xiang B, Zhou M, Li X, Li G, Li Y, Zeng Z, Xiong W. Predictive biomarkers and mechanisms underlying resistance to PD1/PD-L1 blockade cancer immunotherapy. Molecular Cancer. 2020;19(1):19. doi: 10.1186/s12943-020-1144-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walker LS, Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol. 2015;36(2):63–70. doi: 10.1016/j.it.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buchbinder EI, Desai A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am J Clin Oncol. 2016;39(1):98–106. doi: 10.1097/COC.0000000000000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hahn AW, Gill DM, Pal SK, Agarwal N. The future of immune checkpoint cancer therapy after PD-1 and CTLA-4. Immunotherapy. 2017;9(8):681–692. doi: 10.2217/imt-2017-0024. [DOI] [PubMed] [Google Scholar]
- 19.Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC, David O, Burow M, Gordon A, Dhurandhar N, Myers L, Berggren R, Hemminki A, Alvarez RD, Emilie D, Curiel DT, Chen L, Zou W. Blockade of B7–H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9(5):562–567. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 20.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mok TSK, Wu YL, Kudaba I, Kowalski DM, Cho BC, Turna HZ, Castro G, Jr, Srimuninnimit V, Laktionov KK, Bondarenko I, Kubota K, Lubiniecki GM, Zhang J, Kush D, Lopes G, Investigators K. Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet. 2019;393(10183):1819–1830. doi: 10.1016/S0140-6736(18)32409-7. [DOI] [PubMed] [Google Scholar]
- 23.Jenkins RW, Barbie DA, Flaherty KT. Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer. 2018;118(1):9–16. doi: 10.1038/bjc.2017.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, Iyer AK. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front Pharmacol. 2017;8:561. doi: 10.3389/fphar.2017.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee A, Sun S, Sandler A, Hoang T. Recent progress in therapeutic antibodies for cancer immunotherapy. Curr Opin Chem Biol. 2018;44:56–65. doi: 10.1016/j.cbpa.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 26.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 27.Ramapriyan R, Caetano MS, Barsoumian HB, Mafra ACP, Zambalde EP, Menon H, Tsouko E, Welsh JW, Cortez MA. Altered cancer metabolism in mechanisms of immunotherapy resistance. Pharmacol Ther. 2019;195:162–171. doi: 10.1016/j.pharmthera.2018.11.004. [DOI] [PubMed] [Google Scholar]
- 28.Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17(4):351–359. doi: 10.1038/ncb3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, Wu X, Ma J, Zhou M, Li X, Li Y, Li G, Xiong W, Guo C, Zeng Z. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Molecular Cancer. 2019;18(1):10. doi: 10.1186/s12943-018-0928-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang XJ, Wang J, Deng XY, Li XL, Li XY, Zeng ZY, Xiong W, Li GY, Xiong F, Guo C. Immunotherapy targeted to immune checkpoint: a revolutionary breakthrough in cancer therapy. Prog Biochem Biophys. 2018;45(11):1178–1186. doi: 10.16476/j.pibb.2018.0264. [DOI] [Google Scholar]
- 32.Phan LM, Yeung SC, Lee MH. Cancer metabolic reprogramming: importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol Med. 2014;11(1):1–19. doi: 10.7497/j.issn.2095-3941.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23(1):27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rinaldi G, Rossi M, Fendt SM. Metabolic interactions in cancer: cellular metabolism at the interface between the microenvironment, the cancer cell phenotype and the epigenetic landscape. Wiley Interdiscip Rev Syst Biol Med. 2018 doi: 10.1002/wsbm.1397. [DOI] [PubMed] [Google Scholar]
- 35.Mo Y, Wang Y, Zhang L, Yang L, Zhou M, Li X, Li Y, Li G, Zeng Z, Xiong W, Xiong F, Guo C. The role of Wnt signaling pathway in tumor metabolic reprogramming. J Cancer. 2019;10(16):3789–3797. doi: 10.7150/jca.31166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, Kreutz M. Metabolic Hallmarks of Tumor and Immune Cells in the Tumor Microenvironment. Front Immunol. 2017;8:248. doi: 10.3389/fimmu.2017.00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer. 1955;9(4):539–549. doi: 10.1038/bjc.1955.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munoz-Pinedo C, El Mjiyad N, Ricci JE. Cancer metabolism: current perspectives and future directions. Cell Death Dis. 2012;3:e248. doi: 10.1038/cddis.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun RC, Denko NC. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014;19(2):285–292. doi: 10.1016/j.cmet.2013.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang CT, Guo WH, Tan L, He YB, Xiong F, Zhang SS, Zeng ZY, Xiong W, Li GY, Guo C. Hypoxia-inducible Factor-1: a Key Protein for Cells Adapting to Changes in Oxygen Supply. Prog Biochem Biophys. 2019;46(11):1041–1049. doi: 10.16476/j.pibb.2019.0250. [DOI] [Google Scholar]
- 41.Qiu GZ, Jin MZ, Dai JX, Sun W, Feng JH, Jin WL. Reprogramming of the Tumor in the Hypoxic Niche: The Emerging Concept and Associated Therapeutic Strategies. Trends Pharmacol Sci. 2017;38(8):669–686. doi: 10.1016/j.tips.2017.05.002. [DOI] [PubMed] [Google Scholar]
- 42.Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, Gimotty PA, Gilks CB, Lal P, Zhang L, Coukos G. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475(7355):226–230. doi: 10.1038/nature10169. [DOI] [PubMed] [Google Scholar]
- 43.Tang T, Yang L, Cao Y, Wang M, Zhang S, Gong Z, Xiong F, He Y, Zhou Y, Liao Q, Xiang B, Zhou M, Guo C, Li X, Li Y, Xiong W, Li G, Zeng Z. LncRNA AATBC regulates Pinin to promote metastasis in nasopharyngeal carcinoma. Mol Oncol. 2020 doi: 10.1002/1878-0261.12703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang D, Zeng Z, Zhang S, Xiong F, He B, Wu Y, Li W, Tang L, Wei F, Xiang B, Li Z, Zhou Y, Zhou M, Li X, Li Y, Li G, Xiong W, Guo C. Epstein-Barr virus-encoded miR-BART6-3p inhibits cancer cell proliferation through the LOC553103-STMN1 axis. FASEB J. 2020 doi: 10.1096/fj.202000039RR. [DOI] [PubMed] [Google Scholar]
- 45.Wang D, Tang L, Wu Y, Fan C, Zhang S, Xiang B, Zhou M, Li X, Li Y, Li G, Xiong W, Zeng Z, Guo C (2020) Abnormal X chromosome inactivation and tumor development. CMLS, Cellular and molecular life sciences [DOI] [PMC free article] [PubMed]
- 46.Wigerup C, Pahlman S, Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther. 2016;164:152–169. doi: 10.1016/j.pharmthera.2016.04.009. [DOI] [PubMed] [Google Scholar]
- 47.Meijer TW, Kaanders JH, Span PN, Bussink J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin Cancer Res. 2012;18(20):5585–5594. doi: 10.1158/1078-0432.CCR-12-0858. [DOI] [PubMed] [Google Scholar]
- 48.Huang L, Garrett Injac S, Cui K, Braun F, Lin Q, Du Y, Zhang H, Kogiso M, Lindsay H, Zhao S, Baxter P, Adekunle A, Man TK, Zhao H, Li XN, Lau CC, Wong STC. Systems biology-based drug repositioning identifies digoxin as a potential therapy for groups 3 and 4 medulloblastoma. Sci Transl Med. 2018 doi: 10.1126/scitranslmed.aat0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gkountela S, Castro-Giner F, Szczerba BM, Vetter M, Landin J, Scherrer R, Krol I, Scheidmann MC, Beisel C, Stirnimann CU, Kurzeder C, Heinzelmann-Schwarz V, Rochlitz C, Weber WP, Aceto N. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell. 2019;176(98–112):e114. doi: 10.1016/j.cell.2018.11.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 51.Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, Homayoun F, Ma Y, Patel N, Yell P, Hao G, Yousuf Q, Joyce A, Pedrosa I, Geiger H, Zhang H, Chang J, Gardner KH, Bruick RK, Reeves C, Hwang TH, Courtney K, Frenkel E, Sun X, Zojwalla N, Wong T, Rizzi JP, Wallace EM, Josey JA, Xie Y, Xie XJ, Kapur P, McKay RM, Brugarolas J. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature. 2016;539(7627):112–117. doi: 10.1038/nature19796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harada H. Hypoxia-inducible factor 1-mediated characteristic features of cancer cells for tumor radioresistance. J Radiat Res. 2016;57(Suppl 1):i99–i105. doi: 10.1093/jrr/rrw012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harada H, Kizaka-Kondoh S, Li G, Itasaka S, Shibuya K, Inoue M, Hiraoka M. Significance of HIF-1-active cells in angiogenesis and radioresistance. Oncogene. 2007;26(54):7508–7516. doi: 10.1038/sj.onc.1210556. [DOI] [PubMed] [Google Scholar]
- 54.McNamee EN, Korns Johnson D, Homann D, Clambey ET. Hypoxia and hypoxia-inducible factors as regulators of T cell development, differentiation, and function. Immunol Res. 2013;55(1–3):58–70. doi: 10.1007/s12026-012-8349-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Westendorf AM, Skibbe K, Adamczyk A, Buer J, Geffers R, Hansen W, Pastille E, Jendrossek V. Hypoxia Enhances Immunosuppression by Inhibiting CD4+ Effector T Cell Function and Promoting Treg Activity. Cell Physiol Biochem. 2017;41(4):1271–1284. doi: 10.1159/000464429. [DOI] [PubMed] [Google Scholar]
- 56.Ge J, Wang J, Wang H, Jiang X, Liao Q, Gong Q, Mo Y, Li X, Li G, Xiong W, Zhao J, Zeng Z. The BRAF V600E mutation is a predictor of the effect of radioiodine therapy in papillary thyroid cancer. J Cancer. 2020;11(4):932–939. doi: 10.7150/jca.33105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu Q, Zhou W, Yin S, Zhou Y, Chen T, Qian J, Su R, Hong L, Lu H, Zhang F, Xie H, Zhou L, Zheng S. Blocking triggering receptor expressed on myeloid cells-1-positive tumor-associated macrophages induced by hypoxia reverses immunosuppression and anti-programmed cell death ligand 1 resistance in liver cancer. Hepatology. 2019;70(1):198–214. doi: 10.1002/hep.30593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lequeux A, Noman MZ, Xiao M, Sauvage D, Van Moer K, Viry E, Bocci I, Hasmim M, Bosseler M, Berchem G, Janji B. Impact of hypoxic tumor microenvironment and tumor cell plasticity on the expression of immune checkpoints. Cancer Lett. 2019;458:13–20. doi: 10.1016/j.canlet.2019.05.021. [DOI] [PubMed] [Google Scholar]
- 59.Chouaib S, Noman MZ, Kosmatopoulos K, Curran MA. Hypoxic stress: obstacles and opportunities for innovative immunotherapy of cancer. Oncogene. 2017;36(4):439–445. doi: 10.1038/onc.2016.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211(5):781–790. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miao Y, Yang H, Levorse J, Yuan S, Polak L, Sribour M, Singh B, Rosenblum MD, Fuchs E. Adaptive immune resistance emerges from tumor-initiating stem cells. Cell. 2019;177(5):1172–1186. doi: 10.1016/j.cell.2019.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, He X, Semenza GL. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113(14):E2047–2056. doi: 10.1073/pnas.1602883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thienpont B, Steinbacher J, Zhao H, D'Anna F, Kuchnio A, Ploumakis A, Ghesquiere B, Van Dyck L, Boeckx B, Schoonjans L, Hermans E, Amant F, Kristensen VN, Peng Koh K, Mazzone M, Coleman M, Carell T, Carmeliet P, Lambrechts D. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature. 2016;537(7618):63–68. doi: 10.1038/nature19081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bhandari V, Hoey C, Liu LY, Lalonde E, Ray J, Livingstone J, Lesurf R, Shiah YJ, Vujcic T, Huang X, Espiritu SMG, Heisler LE, Yousif F, Huang V, Yamaguchi TN, Yao CQ, Sabelnykova VY, Fraser M, Chua MLK, van der Kwast T, Liu SK, Boutros PC, Bristow RG. Molecular landmarks of tumor hypoxia across cancer types. Nat Genet. 2019;51(2):308–318. doi: 10.1038/s41588-018-0318-2. [DOI] [PubMed] [Google Scholar]
- 65.Greiner EF, Guppy M, Brand K. Glucose is essential for proliferation and the glycolytic enzyme induction that provokes a transition to glycolytic energy production. J Biol Chem. 1994;269(50):31484–31490. doi: 10.1016/S0021-9258(18)31720-4. [DOI] [PubMed] [Google Scholar]
- 66.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 68.Tasselli L, Chua KF. Cancer: Metabolism in 'the driver's seat. Nature. 2012;492(7429):362–363. doi: 10.1038/492362a. [DOI] [PubMed] [Google Scholar]
- 69.Fan C, Tu C, Qi P, Guo C, Xiang B, Zhou M, Li X, Wu X, Li X, Li G, Xiong W, Zeng Z. GPC6 promotes cell proliferation, migration, and invasion in nasopharyngeal carcinoma. J Cancer. 2019;10(17):3926–3932. doi: 10.7150/jca.31345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xiao L, Wei F, Liang F, Li Q, Deng H, Tan S, Chen S, Xiong F, Guo C, Liao Q, Li X, Zhang W, Wu M, Zhou Y, Xiang B, Zhou M, Li X, Xiong W, Zeng Z, Li G. TSC22D2 identified as a candidate susceptibility gene of multi-cancer pedigree using genome-wide linkage analysis and whole-exome sequencing. Carcinogenesis. 2019;40(7):819–827. doi: 10.1093/carcin/bgz095. [DOI] [PubMed] [Google Scholar]
- 71.Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, Williams LJ, Wang Z, Bristow CA, Carugo A, Peoples MD, Li L, Karpinets T, Huang L, Malu S, Creasy C, Leahey SE, Chen J, Chen Y, Pelicano H, Bernatchez C, Gopal YNV, Heffernan TP, Hu J, Wang J, Amaria RN, Garraway LA, Huang P, Yang P, Wistuba II, Woodman SE, Roszik J, Davis RE, Davies MA, Heymach JV, Hwu P, Peng W. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 2018;27(5):977–987. doi: 10.1016/j.cmet.2018.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, Renner K, Timischl B, Mackensen A, Kunz-Schughart L, Andreesen R, Krause SW, Kreutz M. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109(9):3812–3819. doi: 10.1182/blood-2006-07-035972. [DOI] [PubMed] [Google Scholar]
- 73.Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C, Hellerbrand C, Kastenberger M, Kunz-Schughart LA, Oefner PJ, Andreesen R, Gottfried E, Kreutz MP. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J Immunol. 2010;184(3):1200–1209. doi: 10.4049/jimmunol.0902584. [DOI] [PubMed] [Google Scholar]
- 74.Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11(5):325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 75.Qian X, Li X, Shi Z, Xia Y, Cai Q, Xu D, Tan L, Du L, Zheng Y, Zhao D, Zhang C, Lorenzi PL, You Y, Jiang BH, Jiang T, Li H, Lu Z. PTEN Suppresses Glycolysis by Dephosphorylating and Inhibiting Autophosphorylated PGK1. Mol Cell. 2019;76(3):516–527. doi: 10.1016/j.molcel.2019.08.006. [DOI] [PubMed] [Google Scholar]
- 76.Cheung EC, Vousden KH. The role of p53 in glucose metabolism. Curr Opin Cell Biol. 2010;22(2):186–191. doi: 10.1016/j.ceb.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 77.Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275(29):21797–21800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- 78.Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;330(6009):1340–1344. doi: 10.1126/science.1193494. [DOI] [PubMed] [Google Scholar]
- 79.Ancey PB, Contat C, Meylan E. Glucose transporters in cancer - from tumor cells to the tumor microenvironment. FEBS J. 2018;285(16):2926–2943. doi: 10.1111/febs.14577. [DOI] [PubMed] [Google Scholar]
- 80.Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Molecular Cancer. 2013;12:152. doi: 10.1186/1476-4598-12-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tang L, Wei F, Wu Y, He Y, Shi L, Xiong F, Gong Z, Guo C, Li X, Deng H, Cao K, Zhou M, Xiang B, Li X, Li Y, Li G, Xiong W, Zeng Z. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J Exp Clin Cancer Res. 2018;37(1):87. doi: 10.1186/s13046-018-0758-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25(34):4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 83.Oronsky BT, Oronsky N, Fanger GR, Parker CW, Caroen SZ, Lybeck M, Scicinski JJ. Follow the ATP: tumor energy production: a perspective. Anticancer Agents Med Chem. 2014;14(9):1187–1198. doi: 10.2174/1871520614666140804224637. [DOI] [PubMed] [Google Scholar]
- 84.Zhang J, Pavlova NN, Thompson CB. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. 2017;36(10):1302–1315. doi: 10.15252/embj.201696151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.DeBerardinis RJ, Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29(3):313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123(9):3678–3684. doi: 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yue M, Jiang J, Gao P, Liu H, Qing G. Oncogenic MYC Activates a feedforward regulatory loop promoting essential amino acid metabolism and tumorigenesis. Cell Rep. 2017;21(13):3819–3832. doi: 10.1016/j.celrep.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 88.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, DePinho RA, Cantley LC, Kimmelman AC. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jin K, Wang S, Zhang Y, Xia M, Mo Y, Li X, Li G, Zeng Z, Xiong W, He Y. Long non-coding RNA PVT1 interacts with MYC and its downstream molecules to synergistically promote tumorigenesis. Cellular Molecular Life Sci. 2019;76(21):4275–4289. doi: 10.1007/s00018-019-03222-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang W, Zhou R, Wu Y, Liu Y, Su W, Xiong W, Zeng Z. PVT1 Promotes Cancer Progression via MicroRNAs. Front Oncol. 2019;9:609. doi: 10.3389/fonc.2019.00609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tajan M, Hock AK, Blagih J, Robertson NA, Labuschagne CF, Kruiswijk F, Humpton TJ, Adams PD, Vousden KH. A Role for p53 in the adaptation to glutamine starvation through the expression of SLC1A3. Cell Metab. 2018;28(5):721–736. doi: 10.1016/j.cmet.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jeon YJ, Khelifa S, Ratnikov B, Scott DA, Feng Y, Parisi F, Ruller C, Lau E, Kim H, Brill LM, Jiang T, Rimm DL, Cardiff RD, Mills GB, Smith JW, Osterman AL, Kluger Y, Ronai ZA. Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress-inducing chemotherapies. Cancer Cell. 2015;27(3):354–369. doi: 10.1016/j.ccell.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fu S, Li Z, Xiao L, Hu W, Zhang L, Xie B, Zhou Q, He J, Qiu Y, Wen M, Peng Y, Gao J, Tan R, Deng Y, Weng L, Sun LQ. Glutamine synthetase promotes radiation resistance via facilitating nucleotide metabolism and subsequent DNA damage repair. Cell Rep. 2019;28(5):1136–1143. doi: 10.1016/j.celrep.2019.07.002. [DOI] [PubMed] [Google Scholar]
- 94.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619–634. doi: 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dias MM, Adamoski D, Dos Reis LM, Ascencao CFR, de Oliveira KRS, Mafra ACP, da Silva Bastos AC, Quintero M, de GCC, Ferreira IM, Fidelis CHV, Rocco SA, Bajgelman MC, Stine Z, Berindan-Neagoe I, Calin GA, Ambrosio ALB, Dias SMG, (2019) GLS2 is protumorigenic in breast cancers. Oncogene. 10.1038/s41388-019-1007-z [DOI] [PubMed]
- 96.Mohamed A, Deng X, Khuri FR, Owonikoko TK. Altered glutamine metabolism and therapeutic opportunities for lung cancer. Clin Lung Cancer. 2014;15(1):7–15. doi: 10.1016/j.cllc.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Biancur DE, Paulo JA, Malachowska B, Quiles Del Rey M, Sousa CM, Wang X, Sohn ASW, Chu GC, Gygi SP, Harper JW, Fendler W, Mancias JD, Kimmelman AC. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun. 2017;8:15965. doi: 10.1038/ncomms15965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mullard A. Cancer metabolism pipeline breaks new ground. Nat Rev Drug Discov. 2016;15(11):735–737. doi: 10.1038/nrd.2016.223. [DOI] [PubMed] [Google Scholar]
- 99.Song M, Kim SH, Im CY, Hwang HJ. Recent development of small molecule glutaminase inhibitors. Curr Top Med Chem. 2018;18(6):432–443. doi: 10.2174/1568026618666180525100830. [DOI] [PubMed] [Google Scholar]
- 100.Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, Arwood ML, Bettencourt IA, Patel CH, Wen J, Tam A, Blosser RL, Prchalova E, Alt J, Rais R, Slusher BS, Powell JD. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. 2019;366(6468):1013–1021. doi: 10.1126/science.aav2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.DeBerardinis RJ. Tumor microenvironment, metabolism, and immunotherapy. N Engl J Med. 2020;382(9):869–871. doi: 10.1056/NEJMcibr1914890. [DOI] [PubMed] [Google Scholar]
- 102.Pavlova NN, Hui S, Ghergurovich JM, Fan J, Intlekofer AM, White RM, Rabinowitz JD, Thompson CB, Zhang J. As extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metab. 2018;27(2):428–438. doi: 10.1016/j.cmet.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kus K, Kij A, Zakrzewska A, Jasztal A, Stojak M, Walczak M, Chlopicki S. Alterations in arginine and energy metabolism, structural and signalling lipids in metastatic breast cancer in mice detected in plasma by targeted metabolomics and lipidomics. Breast Cancer Res. 2018;20(1):148. doi: 10.1186/s13058-018-1075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Venkateswaran N, Lafita-Navarro MC, Hao YH, Kilgore JA, Perez-Castro L, Braverman J, Borenstein-Auerbach N, Kim M, Lesner NP, Mishra P, Brabletz T, Shay JW, DeBerardinis RJ, Williams NS, Yilmaz OH, Conacci-Sorrell M. MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes Dev. 2019;33(17–18):1236–1251. doi: 10.1101/gad.327056.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang Y, Xia M, Jin K, Wang S, Wei H, Fan C, Wu Y, Li X, Li X, Li G, Zeng Z, Xiong W. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Molecular Cancer. 2018;17(1):45. doi: 10.1186/s12943-018-0796-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang H, Wang Y, Li J, Chen H, He X, Zhang H, Liang H, Lu J. Biosynthetic energy cost for amino acids decreases in cancer evolution. Nat Commun. 2018;9(1):4124. doi: 10.1038/s41467-018-06461-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Currie E, Schulze A, Zechner R, Walther TC, Farese RV., Jr Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18(2):153–161. doi: 10.1016/j.cmet.2013.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hopperton KE, Duncan RE, Bazinet RP, Archer MC. Fatty acid synthase plays a role in cancer metabolism beyond providing fatty acids for phospholipid synthesis or sustaining elevations in glycolytic activity. Exp Cell Res. 2014;320(2):302–310. doi: 10.1016/j.yexcr.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 109.Mo Y, Wang Y, Xiong F, Ge X, Li Z, Li X, Li Y, Li X, Xiong W, Li G, Zeng Z, Guo C. Proteomic analysis of the molecular mechanism of lovastatin inhibiting the growth of nasopharyngeal carcinoma cells. J Cancer. 2019;10(10):2342–2349. doi: 10.7150/jca.30454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, McGarry L, James D, Shanks E, Kalna G, Saunders RE, Jiang M, Howell M, Lassailly F, Thin MZ, Spencer-Dene B, Stamp G, van den Broek NJ, Mackay G, Bulusu V, Kamphorst JJ, Tardito S, Strachan D, Harris AL, Aboagye EO, Critchlow SE, Wakelam MJ, Schulze A, Gottlieb E. Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell. 2015;27(1):57–71. doi: 10.1016/j.ccell.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yi M, Li J, Chen S, Cai J, Ban Y, Peng Q, Zhou Y, Zeng Z, Peng S, Li X, Xiong W, Li G, Xiang B. Emerging role of lipid metabolism alterations in Cancer stem cells. J Exp Clin Cancer Res. 2018;37(1):118. doi: 10.1186/s13046-018-0784-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7(10):763–777. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 113.Rohrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer. 2016;16(11):732–749. doi: 10.1038/nrc.2016.89. [DOI] [PubMed] [Google Scholar]
- 114.Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, Lombardo PS, Van Nostrand JL, Hutchins A, Vera L, Gerken L, Greenwood J, Bhat S, Harriman G, Westlin WF, Harwood HJ, Jr, Saghatelian A, Kapeller R, Metallo CM, Shaw RJ. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med. 2016;22(10):1108–1119. doi: 10.1038/nm.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vriens K, Christen S, Parik S, Broekaert D, Yoshinaga K, Talebi A, Dehairs J, Escalona-Noguero C, Schmieder R, Cornfield T, Charlton C, Romero-Perez L, Rossi M, Rinaldi G, Orth MF, Boon R, Kerstens A, Kwan SY, Faubert B, Mendez-Lucas A, Kopitz CC, Chen T, Fernandez-Garcia J, Duarte JAG, Schmitz AA, Steigemann P, Najimi M, Hagebarth A, Van Ginderachter JA, Sokal E, Gotoh N, Wong KK, Verfaillie C, Derua R, Munck S, Yuneva M, Beretta L, DeBerardinis RJ, Swinnen JV, Hodson L, Cassiman D, Verslype C, Christian S, Grunewald S, Grunewald TGP, Fendt SM. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature. 2019;566(7744):403–406. doi: 10.1038/s41586-019-0904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang YA, Li XL, Mo YZ, Fan CM, Tang L, Xiong F, Guo C, Xiang B, Zhou M, Ma J, Huang X, Wu X, Li Y, Li GY, Zeng ZY, Xiong W. Effects of tumor metabolic microenvironment on regulatory T cells. Molecular Cancer. 2018;17(1):168. doi: 10.1186/s12943-018-0913-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 118.Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342(6155):1242454. doi: 10.1126/science.1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Palmer CS, Ostrowski M, Balderson B, Christian N, Crowe SM. Glucose metabolism regulates T cell activation, differentiation, and functions. Front Immunol. 2015;6:1. doi: 10.3389/fimmu.2015.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Siska PJ, Rathmell JC. T cell metabolic fitness in antitumor immunity. Trends Immunol. 2015;36(4):257–264. doi: 10.1016/j.it.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pearce EL. Metabolism in T cell activation and differentiation. Curr Opin Immunol. 2010;22(3):314–320. doi: 10.1016/j.coi.2010.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hu Z, Qu G, Yu X, Jiang H, Teng X, Ding L, Hu Q, Guo X, Zhou Y, Wang F, Li H, Chen L, Jiang J, Su B, Liu J, Zou Q. Acylglycerol kinase maintains metabolic state and immune responses of CD8+ T cells. Cell Metab. 2019;30(2):290–302. doi: 10.1016/j.cmet.2019.05.016. [DOI] [PubMed] [Google Scholar]
- 123.Le Bourgeois T, Strauss L, Aksoylar HI, Daneshmandi S, Seth P, Patsoukis N, Boussiotis VA. Targeting T cell metabolism for improvement of cancer immunotherapy. Front Oncol. 2018;8:237. doi: 10.3389/fonc.2018.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, Worley PF, Kozma SC, Powell JD. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30(6):832–844. doi: 10.1016/j.immuni.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460(7251):108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Palmer CS, Hussain T, Duette G, Weller TJ, Ostrowski M, Sada-Ovalle I, Crowe SM. Regulators of glucose metabolism in CD4(+) and CD8(+) T cells. Int Rev Immunol. 2016;35(6):477–488. doi: 10.3109/08830185.2015.1082178. [DOI] [PubMed] [Google Scholar]
- 127.Jones N, Cronin JG, Dolton G, Panetti S, Schauenburg AJ, Galloway SAE, Sewell AK, Cole DK, Thornton CA, Francis NJ. Metabolic adaptation of human CD4(+) and CD8(+) T-cells to T-cell receptor-mediated stimulation. Front Immunol. 2017;8:1516. doi: 10.3389/fimmu.2017.01516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, Rathmell JC. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20(1):61–72. doi: 10.1016/j.cmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cao Y, Rathmell JC, Macintyre AN. Metabolic reprogramming towards aerobic glycolysis correlates with greater proliferative ability and resistance to metabolic inhibition in CD8 versus CD4 T cells. PLoS ONE. 2014;9(8):e104104. doi: 10.1371/journal.pone.0104104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, Maseda D, Liberti MV, Paz K, Kishton RJ, Johnson ME, de Cubas AA, Wu P, Li G, Zhang Y, Newcomb DC, Wells AD, Restifo NP, Rathmell WK, Locasale JW, Davila ML, Blazar BR, Rathmell JC. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell. 2018;175(7):1780–1795. doi: 10.1016/j.cell.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Xiao Z, Dai Z, Locasale JW. Metabolic landscape of the tumor microenvironment at single cell resolution. Nat Commun. 2019;10(1):3763. doi: 10.1038/s41467-019-11738-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, Seifi B, Mohammadi A, Afshari JT, Sahebkar A. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233(9):6425–6440. doi: 10.1002/jcp.26429. [DOI] [PubMed] [Google Scholar]
- 133.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–896. doi: 10.1038/ni.1937. [DOI] [PubMed] [Google Scholar]
- 134.Komohara Y, Fujiwara Y, Ohnishi K, Takeya M. Tumor-associated macrophages: Potential therapeutic targets for anti-cancer therapy. Adv Drug Deliv Rev. 2016;99(Pt B):180–185. doi: 10.1016/j.addr.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 135.Mills CD. M1 and M2 Macrophages: Oracles of Health and Disease. Crit Rev Immunol. 2012;32(6):463–488. doi: 10.1615/CritRevImmunol.v32.i6.10. [DOI] [PubMed] [Google Scholar]
- 136.Rath M, Muller I, Kropf P, Closs EI, Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. 2014;5:532. doi: 10.3389/fimmu.2014.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38(4):633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mills EL, O'Neill LA. Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur J Immunol. 2016;46(1):13–21. doi: 10.1002/eji.201445427. [DOI] [PubMed] [Google Scholar]
- 139.Liu D, Chang C, Lu N, Wang X, Lu Q, Ren X, Ren P, Zhao D, Wang L, Zhu Y, He F, Tang L. Comprehensive proteomics analysis reveals metabolic reprogramming of tumor-associated macrophages stimulated by the tumor microenvironment. J Proteome Res. 2017;16(1):288–297. doi: 10.1021/acs.jproteome.6b00604. [DOI] [PubMed] [Google Scholar]
- 140.Netea-Maier RT, Smit JWA, Netea MG. Metabolic changes in tumor cells and tumor-associated macrophages: A mutual relationship. Cancer Lett. 2018;413:102–109. doi: 10.1016/j.canlet.2017.10.037. [DOI] [PubMed] [Google Scholar]
- 141.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116(11):3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhang L, Sun S, Wang Y, Mo Y, Xiong F, Zhang S, Zeng Z, Xiong W, Li G, Chen H, Guo C. Gossypol induces apoptosis of multiple myeloma cells through the JUN-JNK pathway. Am J Cancer Res. 2020;10(3):870–883. [PMC free article] [PubMed] [Google Scholar]
- 143.Xiang W, Shi R, Kang X, Zhang X, Chen P, Zhang L, Hou A, Wang R, Zhao Y, Zhao K, Liu Y, Ma Y, Luo H, Shang S, Zhang J, He F, Yu S, Gan L, Shi C, Li Y, Yang W, Liang H, Miao H. Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat Commun. 2018;9(1):2574. doi: 10.1038/s41467-018-04999-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Binenbaum Y, Fridman E, Yaari Z, Milman N, Schroeder A, Ben David G, Shlomi T, Gil Z. Transfer of miRNA in macrophage-derived exosomes induces drug resistance in pancreatic adenocarcinoma. Cancer Res. 2018;78(18):5287–5299. doi: 10.1158/0008-5472.CAN-18-0124. [DOI] [PubMed] [Google Scholar]
- 145.Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee HJ, Thurston G, Zhang Y, Lazarus J, Sajjakulnukit P, Hong HS, Kremer DM, Nelson BS, Kemp S, Zhang L, Chang D, Biankin A, Shi J, Frankel TL, Crawford HC, Morton JP, Pasca di Magliano M, Lyssiotis CA. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. 2019;29(6):1390–1399. doi: 10.1016/j.cmet.2019.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Di Pilato M, Kim EY, Cadilha BL, Prussmann JN, Nasrallah MN, Seruggia D, Usmani SM, Misale S, Zappulli V, Carrizosa E, Mani V, Ligorio M, Warner RD, Medoff BD, Marangoni F, Villani AC, Mempel TR. Targeting the CBM complex causes Treg cells to prime tumours for immune checkpoint therapy. Nature. 2019;570(7759):112–116. doi: 10.1038/s41586-019-1215-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature. 2013;499(7459):485–490. doi: 10.1038/nature12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kishore M, Cheung KCP, Fu H, Bonacina F, Wang G, Coe D, Ward EJ, Colamatteo A, Jangani M, Baragetti A, Matarese G, Smith DM, Haas R, Mauro C, Wraith DC, Okkenhaug K, Catapano AL, De Rosa V, Norata GD, Marelli-Berg FM. Regulatory T cell migration is dependent on glucokinase-mediated glycolysis. Immunity. 2017;47(5):875–889. doi: 10.1016/j.immuni.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, Zhao L, Vatan L, Shao I, Szeliga W, Lyssiotis C, Liu JR, Kryczek I, Zou W. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol. 2017;18(12):1332–1341. doi: 10.1038/ni.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 151.Zou W. Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol. 2006;6(4):295–307. doi: 10.1038/nri1806. [DOI] [PubMed] [Google Scholar]
- 152.Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27(1):109–118. doi: 10.1038/cr.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Parker KH, Beury DW, Ostrand-Rosenberg S. Myeloid-derived suppressor cells: critical cells driving immune suppression in the tumor microenvironment. Adv Cancer Res. 2015;128:95–139. doi: 10.1016/bs.acr.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ostrand-Rosenberg S. Myeloid derived-suppressor cells: their role in cancer and obesity. Curr Opin Immunol. 2018;51:68–75. doi: 10.1016/j.coi.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, Valle LD, Trillo-Tinoco J, Maj T, Zou W, Rodriguez PC, Ochoa AC. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. 2015;3(11):1236–1247. doi: 10.1158/2326-6066.CIR-15-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, Sanchez MD, Dean MJ, Rodriguez PC, Ochoa AC. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology. 2017;6(10):e1344804. doi: 10.1080/2162402X.2017.1344804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yan D, Adeshakin AO, Xu M, Afolabi LO, Zhang G, Chen YH, Wan X. Lipid metabolic pathways confer the immunosuppressive function of myeloid-derived suppressor cells in Tumor. Front Immunol. 2019;10:1399. doi: 10.3389/fimmu.2019.01399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37(3):208–220. doi: 10.1016/j.it.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, To TKJ, Schug Z, Basu S, Wang F, Ricciotti E, DiRusso C, Murphy ME, Vonderheide RH, Lieberman PM, Mulligan C, Nam B, Hockstein N, Masters G, Guarino M, Lin C, Nefedova Y, Black P, Kagan VE, Gabrilovich DI. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019;569(7754):73–78. doi: 10.1038/s41586-019-1118-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Du X, Wen J, Wang Y, Karmaus PWF, Khatamian A, Tan H, Li Y, Guy C, Nguyen TM, Dhungana Y, Neale G, Peng J, Yu J, Chi H. Hippo/Mst signalling couples metabolic state and immune function of CD8alpha(+) dendritic cells. Nature. 2018;558(7708):141–145. doi: 10.1038/s41586-018-0177-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wculek SK, Khouili SC, Priego E, Heras-Murillo I, Sancho D. Metabolic control of dendritic cell functions: digesting information. Front Immunol. 2019;10:775. doi: 10.3389/fimmu.2019.00775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Merad M, Salmon H. Cancer: A dendritic-cell brake on antitumour immunity. Nature. 2015;523(7560):294–295. doi: 10.1038/523294a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, Ellenson LH, Caputo T, Lee AH, Conejo-Garcia JR, Glimcher LH. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161(7):1527–1538. doi: 10.1016/j.cell.2015.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Giovanelli P, Sandoval TA, Cubillos-Ruiz JR. Dendritic cell metabolism and function in tumors. Trends Immunol. 2019;40(8):699–718. doi: 10.1016/j.it.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 166.Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017;542(7640):177–185. doi: 10.1038/nature21363. [DOI] [PubMed] [Google Scholar]
- 167.Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, van der Windt GJ, Tonc E, Schreiber RD, Pearce EJ, Pearce EL. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162(6):1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chang CH, Curtis JD, Maggi LB, Jr, Faubert B, Villarino AV, O'Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, Weber JD, Pearce EJ, Jones RG, Pearce EL. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153(6):1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, Wei S, Crespo J, Wan S, Vatan L, Szeliga W, Shao I, Wang Y, Liu Y, Varambally S, Chinnaiyan AM, Welling TH, Marquez V, Kotarski J, Wang H, Wang Z, Zhang Y, Liu R, Wang G, Zou W. Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol. 2016;17(1):95–103. doi: 10.1038/ni.3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Bo H, Fan L, Gong Z, Liu Z, Shi L, Guo C, Li X, Liao Q, Zhang W, Zhou M, Xiang B, Li X, Li G, Xiong W, Zeng Z, Cao K, Zhang S, Xiong F. Upregulation and hypomethylation of lncRNA AFAP1AS1 predicts a poor prognosis and promotes the migration and invasion of cervical cancer. Oncol Rep. 2019;41(4):2431–2439. doi: 10.3892/or.2019.7027. [DOI] [PubMed] [Google Scholar]
- 171.Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, Kleinstein SH, Abel ED, Insogna KL, Feske S, Locasale JW, Bosenberg MW, Rathmell JC, Kaech SM. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor t cell responses. Cell. 2015;162(6):1217–1228. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, Filisio F, Giles-Davis W, Xu X, Karakousis GC, Schuchter LM, Xu W, Amaravadi R, Xiao M, Sadek N, Krepler C, Herlyn M, Freeman GJ, Rabinowitz JD, Ertl HCJ. Enhancing CD8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell. 2017;32(3):377–391. doi: 10.1016/j.ccell.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Huang W, Choi W, Chen Y, Zhang Q, Deng H, He W, Shi Y. A proposed role for glutamine in cancer cell growth through acid resistance. Cell Res. 2013;23(5):724–727. doi: 10.1038/cr.2013.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Patil MD, Bhaumik J, Babykutty S, Banerjee UC, Fukumura D. Arginine dependence of tumor cells: targeting a chink in cancer's armor. Oncogene. 2016;35(38):4957–4972. doi: 10.1038/onc.2016.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Raber P, Ochoa AC, Rodriguez PC. Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: mechanisms of T cell suppression and therapeutic perspectives. Immunol Invest. 2012;41(6–7):614–634. doi: 10.3109/08820139.2012.680634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rodriguez PC, Ochoa AC. Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev. 2008;222:180–191. doi: 10.1111/j.1600-065X.2008.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Fletcher M, Ramirez ME, Sierra RA, Raber P, Thevenot P, Al-Khami AA, Sanchez-Pino D, Hernandez C, Wyczechowska DD, Ochoa AC, Rodriguez PC. l-Arginine depletion blunts antitumor T-cell responses by inducing myeloid-derived suppressor cells. Cancer Res. 2015;75(2):275–283. doi: 10.1158/0008-5472.CAN-14-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfor K, Rofstad EK, Mueller-Klieser W. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000;60(4):916–921. [PubMed] [Google Scholar]
- 179.Haas R, Smith J, Rocher-Ros V, Nadkarni S, Montero-Melendez T, D'Acquisto F, Bland EJ, Bombardieri M, Pitzalis C, Perretti M, Marelli-Berg FM, Mauro C. Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PLoS Biol. 2015;13(7):e1002202. doi: 10.1371/journal.pbio.1002202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, Matos C, Bruss C, Klobuch S, Peter K, Kastenberger M, Bogdan C, Schleicher U, Mackensen A, Ullrich E, Fichtner-Feigl S, Kesselring R, Mack M, Ritter U, Schmid M, Blank C, Dettmer K, Oefner PJ, Hoffmann P, Walenta S, Geissler EK, Pouyssegur J, Villunger A, Steven A, Seliger B, Schreml S, Haferkamp S, Kohl E, Karrer S, Berneburg M, Herr W, Mueller-Klieser W, Renner K, Kreutz M. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016;24(5):657–671. doi: 10.1016/j.cmet.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 181.Ippolito L, Morandi A, Giannoni E, Chiarugi P. Lactate: A metabolic driver in the tumour landscape. Trends Biochem Sci. 2019;44(2):153–166. doi: 10.1016/j.tibs.2018.10.011. [DOI] [PubMed] [Google Scholar]
- 182.Liu N, Luo J, Kuang D, Xu S, Duan Y, Xia Y, Wei Z, Xie X, Yin B, Chen F, Luo S, Liu H, Wang J, Jiang K, Gong F, Tang ZH, Cheng X, Li H, Li Z, Laurence A, Wang G, Yang XP. Lactate inhibits ATP6V0d2 expression in tumor-associated macrophages to promote HIF-2alpha-mediated tumor progression. J Clin Invest. 2019;129(2):631–646. doi: 10.1172/JCI123027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. 2012;188(1):21–28. doi: 10.4049/jimmunol.1101029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Luan B, Yoon YS, Le Lay J, Kaestner KH, Hedrick S, Montminy M. CREB pathway links PGE2 signaling with macrophage polarization. Proc Natl Acad Sci U S A. 2015;112(51):15642–15647. doi: 10.1073/pnas.1519644112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Wondimu A, Liu Y, Su Y, Bobb D, Ma JS, Chakrabarti L, Radoja S, Ladisch S. Gangliosides drive the tumor infiltration and function of myeloid-derived suppressor cells. Cancer Res. 2014;74(19):5449–5457. doi: 10.1158/0008-5472.CAN-14-0927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117(5):1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185(6):3190–3198. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, Palmer DC, Gros A, Yamamoto TN, Patel SJ, Guittard GC, Yu Z, Carbonaro V, Okkenhaug K, Schrump DS, Linehan WM, Roychoudhuri R, Restifo NP. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature. 2016;537(7621):539–543. doi: 10.1038/nature19364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Vodnala SK, Eil R, Kishton RJ, Sukumar M, Yamamoto TN, Ha NH, Lee PH, Shin M, Patel SJ, Yu Z, Palmer DC, Kruhlak MJ, Liu X, Locasale JW, Huang J, Roychoudhuri R, Finkel T, Klebanoff CA, Restifo NP. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science. 2019 doi: 10.1126/science.aau0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ye J, Ma C, Hsueh EC, Dou J, Mo W, Liu S, Han B, Huang Y, Zhang Y, Varvares MA, Hoft DF, Peng G. TLR8 signaling enhances tumor immunity by preventing tumor-induced T-cell senescence. EMBO Mol Med. 2014;6(10):1294–1311. doi: 10.15252/emmm.201403918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ye J, Peng G. Controlling T cell senescence in the tumor microenvironment for tumor immunotherapy. Oncoimmunology. 2015;4(3):e994398. doi: 10.4161/2162402X.2014.994398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Goossens P, Rodriguez-Vita J, Etzerodt A, Masse M, Rastoin O, Gouirand V, Ulas T, Papantonopoulou O, Van Eck M, Auphan-Anezin N, Bebien M, Verthuy C, Vu Manh TP, Turner M, Dalod M, Schultze JL, Lawrence T. Membrane cholesterol efflux drives tumor-associated macrophage reprogramming and tumor progression. Cell Metab. 2019;29(6):1376–1389. doi: 10.1016/j.cmet.2019.02.016. [DOI] [PubMed] [Google Scholar]
- 193.Li X, Wenes M, Romero P, Huang SC, Fendt SM, Ho PC. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat Rev Clin Oncol. 2019;16(7):425–441. doi: 10.1038/s41571-019-0203-7. [DOI] [PubMed] [Google Scholar]
- 194.Feng J, Yang H, Zhang Y, Wei H, Zhu Z, Zhu B, Yang M, Cao W, Wang L, Wu Z. Tumor cell-derived lactate induces TAZ-dependent upregulation of PD-L1 through GPR81 in human lung cancer cells. Oncogene. 2017;36(42):5829–5839. doi: 10.1038/onc.2017.188. [DOI] [PubMed] [Google Scholar]
- 195.Feichtinger RG, Lang R. Targeting L-lactate metabolism to overcome resistance to immune therapy of melanoma and other tumor entities. J Oncol. 2019;2019:2084195. doi: 10.1155/2019/2084195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Kleinfeld AM, Okada C. Free fatty acid release from human breast cancer tissue inhibits cytotoxic T-lymphocyte-mediated killing. J Lipid Res. 2005;46(9):1983–1990. doi: 10.1194/jlr.M500151-JLR200. [DOI] [PubMed] [Google Scholar]
- 197.Torisu H, Ono M, Kiryu H, Furue M, Ohmoto Y, Nakayama J, Nishioka Y, Sone S, Kuwano M. Macrophage infiltration correlates with tumor stage and angiogenesis in human malignant melanoma: possible involvement of TNFalpha and IL-1alpha. Int J Cancer. 2000;85(2):182–188. doi: 10.1002/(SICI)1097-0215(20000115)85:2<182::AID-IJC6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 198.Zhang Y, Yu G, Chu H, Wang X, Xiong L, Cai G, Liu R, Gao H, Tao B, Li W, Li G, Liang J, Yang W. Macrophage-associated PGK1 phosphorylation promotes aerobic glycolysis and tumorigenesis. Mol Cell. 2018;71(2):201–215. doi: 10.1016/j.molcel.2018.06.023. [DOI] [PubMed] [Google Scholar]
- 199.Chen F, Chen J, Yang L, Liu J, Zhang X, Zhang Y, Tu Q, Yin D, Lin D, Wong PP, Huang D, Xing Y, Zhao J, Li M, Liu Q, Su F, Su S, Song E. Extracellular vesicle-packaged HIF-1alpha-stabilizing lncRNA from tumour-associated macrophages regulates aerobic glycolysis of breast cancer cells. Nat Cell Biol. 2019;21(4):498–510. doi: 10.1038/s41556-019-0299-0. [DOI] [PubMed] [Google Scholar]
- 200.Sanmamed MF, Chen L. A Paradigm shift in cancer immunotherapy: from enhancement to normalization. Cell. 2018;175(2):313–326. doi: 10.1016/j.cell.2018.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hegde PS, Chen DS. Top 10 challenges in cancer immunotherapy. Immunity. 2020;52(1):17–35. doi: 10.1016/j.immuni.2019.12.011. [DOI] [PubMed] [Google Scholar]
- 202.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9(10):1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 203.Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. 2012;72(21):5435–5440. doi: 10.1158/0008-5472.CAN-12-0569. [DOI] [PubMed] [Google Scholar]
- 204.Munn DH, Mellor AL. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. 2016;37(3):193–207. doi: 10.1016/j.it.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Mbongue JC, Nicholas DA, Torrez TW, Kim NS, Firek AF, Langridge WH. The role of indoleamine 2, 3-dioxygenase in immune suppression and autoimmunity. Vaccines (Basel) 2015;3(3):703–729. doi: 10.3390/vaccines3030703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Li H, Bullock K, Gurjao C, Braun D, Shukla SA, Bosse D, Lalani AA, Gopal S, Jin C, Horak C, Wind-Rotolo M, Signoretti S, McDermott DF, Freeman GJ, Van Allen EM, Schreiber SL, Stephen Hodi F, Sellers WR, Garraway LA, Clish CB, Choueiri TK, Giannakis M. Metabolomic adaptations and correlates of survival to immune checkpoint blockade. Nat Commun. 2019;10(1):4346. doi: 10.1038/s41467-019-12361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhai L, Spranger S, Binder DC, Gritsina G, Lauing KL, Giles FJ, Wainwright DA. Molecular pathways: targeting IDO1 and other tryptophan dioxygenases for cancer immunotherapy. Clin Cancer Res. 2015;21(24):5427–5433. doi: 10.1158/1078-0432.CCR-15-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yue EW, Sparks R, Polam P, Modi D, Douty B, Wayland B, Glass B, Takvorian A, Glenn J, Zhu W, Bower M, Liu X, Leffet L, Wang Q, Bowman KJ, Hansbury MJ, Wei M, Li Y, Wynn R, Burn TC, Koblish HK, Fridman JS, Emm T, Scherle PA, Metcalf B, Combs AP. INCB24360 (Epacadostat), a highly potent and selective indoleamine-2,3-dioxygenase 1 (IDO1) inhibitor for immuno-oncology. ACS Med Chem Lett. 2017;8(5):486–491. doi: 10.1021/acsmedchemlett.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Zakharia Y, Drabick J, Khleif S, Munn D, Link C, Vahanian N, Kennedy E, Rixe O, Milhem M. 514 Results of Phase 1b trial of the Indoleamine 2,3-dioxygenase (IDO) Pathway Inhibitor Indoximod plus Ipilimumab for the treatment of unresectable stage III or IV melanoma. Eur J Cancer. 2015;51:S108–S108. doi: 10.1016/S0959-8049(16)30315-X. [DOI] [Google Scholar]
- 210.Mullard A. IDO takes a blow. Nat Rev Drug Discov. 2018;17(5):307. doi: 10.1038/nrd.2018.67. [DOI] [PubMed] [Google Scholar]
- 211.Wu W, Shi X, Xu C. Regulation of T cell signalling by membrane lipids. Nat Rev Immunol. 2018;18(3):219. doi: 10.1038/nri.2018.9. [DOI] [PubMed] [Google Scholar]
- 212.Lochner M, Berod L, Sparwasser T. Fatty acid metabolism in the regulation of T cell function. Trends Immunol. 2015;36(2):81–91. doi: 10.1016/j.it.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 213.Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, Meng X, Li L, Wang J, Xu C, Yan C, Wang L, Chang CC, Chang TY, Zhang T, Zhou P, Song BL, Liu W, Sun SC, Liu X, Li BL, Xu C. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature. 2016;531(7596):651–655. doi: 10.1038/nature17412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Pal P, Gandhi H, Giridhar R, Yadav MR. ACAT inhibitors: the search for novel cholesterol lowering agents. Mini Rev Med Chem. 2013;13(8):1195–1219. doi: 10.2174/1389557511313080007. [DOI] [PubMed] [Google Scholar]
- 215.Jiang Y, Sun A, Zhao Y, Ying W, Sun H, Yang X, Xing B, Sun W, Ren L, Hu B, Li C, Zhang L, Qin G, Zhang M, Chen N, Zhang M, Huang Y, Zhou J, Zhao Y, Liu M, Zhu X, Qiu Y, Sun Y, Huang C, Yan M, Wang M, Liu W, Tian F, Xu H, Zhou J, Wu Z, Shi T, Zhu W, Qin J, Xie L, Fan J, Qian X, He F, Chinese Human Proteome Project C Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature. 2019;567(7747):257–261. doi: 10.1038/s41586-019-0987-8. [DOI] [PubMed] [Google Scholar]
- 216.Ohta A. A metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol. 2016;7:109. doi: 10.3389/fimmu.2016.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Cai XY, Wang XF, Li J, Dong JN, Liu JQ, Li NP, Yun B, Xia RL, Qin J, Sun YH. High expression of CD39 in gastric cancer reduces patient outcome following radical resection. Oncol Lett. 2016;12(5):4080–4086. doi: 10.3892/ol.2016.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Perrot I, Michaud HA, Giraudon-Paoli M, Augier S, Docquier A, Gros L, Courtois R, Dejou C, Jecko D, Becquart O, Rispaud-Blanc H, Gauthier L, Rossi B, Chanteux S, Gourdin N, Amigues B, Roussel A, Bensussan A, Eliaou JF, Bastid J, Romagne F, Morel Y, Narni-Mancinelli E, Vivier E, Paturel C, Bonnefoy N. Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep. 2019;27(8):2411–2425. doi: 10.1016/j.celrep.2019.04.091. [DOI] [PubMed] [Google Scholar]
- 219.Hammami A, Allard D, Allard B, Stagg J. Targeting the adenosine pathway for cancer immunotherapy. Semin Immunol. 2019;42:101304. doi: 10.1016/j.smim.2019.101304. [DOI] [PubMed] [Google Scholar]
- 220.Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, Huang X, Caldwell S, Liu K, Smith P, Chen JF, Jackson EK, Apasov S, Abrams S, Sitkovsky M. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A. 2006;103(35):13132–13137. doi: 10.1073/pnas.0605251103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Willingham SB, Ho PY, Hotson A, Hill C, Piccione EC, Hsieh J, Liu L, Buggy JJ, McCaffery I, Miller RA. A2AR antagonism with CPI-444 induces antitumor responses and augments efficacy to Anti-PD-(L)1 and Anti-CTLA-4 in preclinical models. Cancer Immunol Res. 2018;6(10):1136–1149. doi: 10.1158/2326-6066.CIR-18-0056. [DOI] [PubMed] [Google Scholar]
- 222.Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene. 2010;29(39):5346–5358. doi: 10.1038/onc.2010.292. [DOI] [PubMed] [Google Scholar]
- 223.Adinolfi E, Capece M, Amoroso F, De Marchi E, Franceschini A. Emerging roles of P2X receptors in cancer. Curr Med Chem. 2015;22(7):878–890. doi: 10.2174/0929867321666141012172913. [DOI] [PubMed] [Google Scholar]
- 224.Burnstock G, Knight GE. The potential of P2X7 receptors as a therapeutic target, including inflammation and tumour progression. Purinergic Signal. 2018;14(1):1–18. doi: 10.1007/s11302-017-9593-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.De Marchi E, Orioli E, Pegoraro A, Sangaletti S, Portararo P, Curti A, Colombo MP, Di Virgilio F, Adinolfi E. The P2X7 receptor modulates immune cells infiltration, ectonucleotidases expression and extracellular ATP levels in the tumor microenvironment. Oncogene. 2019;38(19):3636–3650. doi: 10.1038/s41388-019-0684-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zhou B, Yuan Y, Zhang S, Guo C, Li X, Li G, Xiong W, Zeng Z. Intestinal flora and disease mutually shape the regional immune system in the intestinal tract. Front Immunol. 2020;11:575. doi: 10.3389/fimmu.2020.00575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Routy B, Gopalakrishnan V, Daillere R, Zitvogel L, Wargo JA, Kroemer G. The gut microbiota influences anticancer immunosurveillance and general health. Nat Rev Clin Oncol. 2018;15(6):382–396. doi: 10.1038/s41571-018-0006-2. [DOI] [PubMed] [Google Scholar]
- 228.Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CP, Poirier-Colame V, Roux A, Becharef S, Formenti S, Golden E, Cording S, Eberl G, Schlitzer A, Ginhoux F, Mani S, Yamazaki T, Jacquelot N, Enot DP, Berard M, Nigou J, Opolon P, Eggermont A, Woerther PL, Chachaty E, Chaput N, Robert C, Mateus C, Kroemer G, Raoult D, Boneca IG, Carbonnel F, Chamaillard M, Zitvogel L. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350(6264):1079–1084. doi: 10.1126/science.aad1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Lei YM, Jabri B, Alegre ML, Chang EB, Gajewski TF. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350(6264):1084–1089. doi: 10.1126/science.aac4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, Fidelle M, Flament C, Poirier-Colame V, Opolon P, Klein C, Iribarren K, Mondragon L, Jacquelot N, Qu B, Ferrere G, Clemenson C, Mezquita L, Masip JR, Naltet C, Brosseau S, Kaderbhai C, Richard C, Rizvi H, Levenez F, Galleron N, Quinquis B, Pons N, Ryffel B, Minard-Colin V, Gonin P, Soria JC, Deutsch E, Loriot Y, Ghiringhelli F, Zalcman G, Goldwasser F, Escudier B, Hellmann MD, Eggermont A, Raoult D, Albiges L, Kroemer G, Zitvogel L. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359(6371):91–97. doi: 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 231.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, Prieto PA, Vicente D, Hoffman K, Wei SC, Cogdill AP, Zhao L, Hudgens CW, Hutchinson DS, Manzo T, Petaccia de Macedo M, Cotechini T, Kumar T, Chen WS, Reddy SM, Szczepaniak Sloane R, Galloway-Pena J, Jiang H, Chen PL, Shpall EJ, Rezvani K, Alousi AM, Chemaly RF, Shelburne S, Vence LM, Okhuysen PC, Jensen VB, Swennes AG, McAllister F, Marcelo Riquelme Sanchez E, Zhang Y, Le Chatelier E, Zitvogel L, Pons N, Austin-Breneman JL, Haydu LE, Burton EM, Gardner JM, Sirmans E, Hu J, Lazar AJ, Tsujikawa T, Diab A, Tawbi H, Glitza IC, Hwu WJ, Patel SP, Woodman SE, Amaria RN, Davies MA, Gershenwald JE, Hwu P, Lee JE, Zhang J, Coussens LM, Cooper ZA, Futreal PA, Daniel CR, Ajami NJ, Petrosino JF, Tetzlaff MT, Sharma P, Allison JP, Jenq RR, Wargo JA. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359(6371):97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, Luke JJ, Gajewski TF. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359(6371):104–108. doi: 10.1126/science.aao3290. [DOI] [PMC free article] [PubMed] [Google Scholar]