

Abstract
Naturally occurring point mutations in apolipoprotein A-I (apoA-I), the major protein component of high-density lipoprotein (HDL), may affect plasma HDL-cholesterol levels and cardiovascular risk. Here, we evaluated the effect of human apoA-I mutations L144R (associated with low HDL-cholesterol), L178P (associated with low HDL-cholesterol and increased cardiovascular risk) and A164S (associated with increased cardiovascular risk and mortality without low HDL-cholesterol) on the structural integrity and functions of lipid-free and lipoprotein-associated apoA-I in an effort to explain the phenotypes of subjects carrying these mutations. All three mutants, in lipid-free form, presented structural and thermodynamic aberrations, with apoA-I[L178P] presenting the greatest thermodynamic destabilization. Additionally, apoA-I[L178P] displayed reduced ABCA1-mediated cholesterol efflux capacity. When in reconstituted HDL (rHDL), apoA-I[L144R] and apoA-I[L178P] were more thermodynamically destabilized compared to wild-type apoA-I, both displayed reduced SR-BI-mediated cholesterol efflux capacity and apoA-I[L144R] showed severe LCAT activation defect. ApoA-I[A164S] was thermodynamically unaffected when in rHDL, but exhibited a series of functional defects. Specifically, it had reduced ABCG1-mediated cholesterol and 7-ketocholesterol efflux capacity, failed to reduce ROS formation in endothelial cells and had reduced capacity to induce endothelial cell migration. Mechanistically, the latter was due to decreased capacity of rHDL-apoA-I[A164S] to activate Akt kinase possibly by interacting with endothelial LOX-1 receptor. The impaired capacity of rHDL-apoA-I[A164S] to preserve endothelial function may be related to the increased cardiovascular risk for this mutation. Overall, our structure–function analysis of L144R, A164S and L178P apoA-I mutants provides insights on how HDL-cholesterol levels and/or atheroprotective properties of apoA-I/HDL are impaired in carriers of these mutations.
Electronic supplementary material
The online version of this article (10.1007/s00018-020-03583-y) contains supplementary material, which is available to authorized users.
Keywords: Apolipoprotein A-I, High-density lipoprotein, Mutant, Protein conformation, ApoA-I/HDL functional properties, Coronary artery disease
Introduction
Apolipoprotein A-I (apoA-I), the main protein constituent of high-density lipoprotein (HDL), has a critical role in HDL biogenesis and structure and is involved in many of HDL atheroprotective properties [1, 2]. The biogenesis and catabolism of HDL take place via a complex pathway that involves several membrane bound and plasma proteins [1, 2]. Initially, lipid-free apoA-I interacts with ATP-binding cassette A1 (ABCA1) lipid transporter and this interaction leads to transfer of cellular phospholipids and cholesterol to apoA-I [1, 2]. As apoA-I acquires more lipids, it is gradually converted to discoidal particles enriched in unesterified cholesterol. Subsequently, lipidated apoA-I activates enzyme lecithin:cholesterol acyltransferase (LCAT), which esterifies cholesterol molecules resulting in the conversion of the discoidal particles to mature spherical HDL particles [1, 2]. Following synthesis, HDL can be remodeled by interactions with various cell receptors and transporters as well as plasma enzymes and lipid transfer proteins [1, 2]. HDL binds to scavenger receptor class B type I (SR-BI) which mediates selective delivery of cholesterol esters from HDL to cells or transfer of cholesterol from cells to HDL [1, 2]. HDL also interacts with ATP-binding cassette G1 (ABCG1) cholesterol transporter and this interaction leads to transfer of cellular cholesterol to HDL [1, 2]. It has been proposed that apoA-I plays an active role in interactions between HDL and SR-BI or ABCG1 [3, 4]. Additionally, HDL is remodeled following hydrolysis of lipids of HDL by hepatic lipase and endothelial lipase and exchange of lipids by phospholipid transfer protein and by cholesteryl ester transfer protein [1, 2]. Interactions between apoA-I and various proteins of the HDL metabolism pathway control plasma HDL concentration and also affect the antiatherogenic properties of HDL pertinent to the reverse cholesterol transport pathway [5–7]. Furthermore, apoA-I and HDL possess anti-inflammatory, antioxidant, antithrombolytic, endothelial-protective and other beneficial properties [5–7].
Human apoA-I has 243 amino acid residues and the amphipathic α-helix as its main structural motif [8]. X-ray crystallography studies combined with mutation analyses, biophysical and functional studies proposed that the N-terminal domain 1-184 of lipid-free apoA-I forms a helical bundle that is important for stability of protein and harbors the LCAT activation domain. The C-terminal domain 185–243 of the protein is considered to lack a well-defined structure, but plays a critical role in the initiation of lipid-binding and is involved in cholesterol efflux [9, 10]. Recently, a combination of distance constraints from cross-linking experiments, small-angle X-ray scattering, hydrogen–deuterium exchange and crystallography data resulted in a consensus model for the lipid-free full-length apoA-I structure [8]. This model postulates a two-domain structure for apoA-I with an N-terminal domain forming a helical bundle and a C-terminal domain assuming a mostly random-coil conformation [8]. In discoidal HDL, two molecules of apoA-I are located in an antiparallel double-belt configuration around the lipid molecules [11]. ApoA-I on spherical HDL also forms a double-belt, but it bents in a “trefoil/tetrafoil” fashion that allows packing of apoA-I and other proteins on the surface of spherical particles [12–14].
A large number of rare point mutations have been detected in the gene of human APOA-I, in most cases resulting in altered HDL-cholesterol (HDL-C) levels, but also in increased risk for cardiovascular disease (CVD) and total mortality, as well as in hereditary amyloidosis [15–18]. Amongst them, apoA-I[L144R] (also denoted as L168R based on the amino acid numbering of the entire translated protein) has been shown to reduce HDL-C levels in heterozygous carriers, but without any effect on ischemic heart disease (IHD) risk [18, 19]. ApoA-I[L178P] (also denoted as L202P based on the amino acid numbering of the entire translated protein) also results in low HDL-C, but in addition to increased carotid intima–media thickness (IMT) and a remarkable 24-fold increased CVD risk in heterozygous carriers [20]. Interestingly, apoA-I[A164S] (also denoted as A188S based on the amino acid numbering of the entire translated protein) has been shown to increase the risk of IHD and mortality in heterozygotes despite normal levels of HDL-C [18]. The molecular basis behind the impact of these apoA-I naturally occurring point mutations on HDL metabolism and function is still largely unexplored.
In this study, we aimed to evaluate the effect of L144R, A164S and L178P mutations on the structural integrity and functions of lipid-free and lipoprotein-associated apoA-I in an effort to explain the in vivo phenotypes of subjects carrying the aforementioned apoA-I mutations. To achieve this, we produced recombinant apoA-I[L144R], apoA-I[A164S] and apoA-I[L178P] and characterized their structural and thermodynamic integrity by biophysical techniques, as well as their functions related to HDL metabolism and atheroprotection by cell-based assays. Overall, our findings suggest that the L144R, A164S and L178P mutations affect the conformation and thermodynamic stability of apoA-I/HDL and can induce distinct functional defects that may impair HDL metabolism and/or atheroprotective properties of apoA-I/HDL.
Materials and methods
Construction of vectors for apoA-I forms expression
To produce WT human apoA-I we used the pET32a-apoAI3C vector, containing a thioredoxin (Trx)-tag, a 6x His-tag and a 3C-protease site at the fusion junction with the human cDNA for full-length apoA-I, that was generated as described previously [21]. The L144R, A164S and L178P mutations were introduced into the cDNA of apoA-I in pET32a-apoAI3C vector by site directed mutagenesis, using the QuickChange II XL site directed mutagenesis kit (Agilent; Santa Clara, CA, USA) according to the manufacturer’s instructions. The sequences of primers used for mutagenesis are: L144R primers 5′-AG AAG CTG AGC CCA CGG GGC GAG GAG ATG CGC-3′ and 5′-GCG CAT CTC CTC GCC CCG TGG GCT CAG CTT CT-3′; A164S primers 5′-G CTG CGC ACG CAT CTG AGC CCC TAC AGC GAC GAG-3′ and 5′-CTC GTC GCT GTA GGG GCT CAG ATG CGT GCG CAG C-3′; L178P primers GC TTG GCC GCG CGC CCT GAG GCT CTC AAG GAG AA and 5′-TT CTC CTT GAG AGC CTC AGG GCG CGC GGC CAA GC-3′. Successful mutagenesis was confirmed by DNA sequencing.
Expression in bacterial cells and purification of wild type (WT) and mutant apoA-I forms
The expression of WT or mutant apoA-I forms in bacterial cells was carried out as described previously [21]. Briefly, E. coli BL21-Gold (DE3) cells (Stratagene, Cedar Creek, TX, USA) were transformed with WT or mutant apoA-I expressing vectors and grown at 37 °C until the optical density of culture reached 0.6. At that point, protein expression was induced by isopropyl β-d-thiogalactopyranoside (0.5 mM final concentration) and the culture was incubated for 2.5 h at 37 °C.
All proteins were expressed in soluble form, fused with a Trx-tag, and purified by Ni2+-nitrilotriacetic acid (Ni–NTA) (Thermo Fisher Scientific, USA) affinity chromatography following elution by increasing concentrations of imidazole as described previously [21]. The Trx-tag was subsequently cleaved from apoA-I by His-tagged 3C protease (prepared as described previously [22] using the vector pET-24/His-3C that was generously supplied by Dr. Arie Geerlof (EMBL, Heidelberg, Germany), and the released apoA-I was isolated by a second Ni–NTA resin affinity chromatography step in the flow-through [21]. After purification, each apoA-I form was extensively dialyzed against 5 mM NH4HCO3, lyophilized and stored at − 80 °C.
Preparation of apoA-I samples
Before analyses, the lyophilized proteins were dissolved in 8 M guanidine hydrochloride (GdnHCl) in Dulbecco’s phosphate-buffered saline (DPBS) and refolded by extensive dialysis against the same buffer. The samples were then centrifuged at 10.000 × g for 10 min at 4 °C, to remove any precipitated protein. The refolded proteins were at least 95% pure, as estimated by SDS-PAGE. All analyses were performed on freshly refolded proteins.
Preparation of reconstituted HDL particles containing phosphatidylcholine, cholesterol and apoA-I
Reconstituted discoidal HDL (rHDL) particles containing WT or mutant apoA-I forms were prepared using 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) (Sigma-Aldrich, Germany), cholesterol (C) (Sigma-Aldrich), apoA-I and sodium cholate in a molar ratio 100:10:1:100 in 10 mM Tris–HCl buffer, pH 8, containing 0.01% EDTA, 150 mM NaCl, as described previously [4]. rHDL formation was verified by analysis with non-denaturing polyacrylamide gradient (4–20%) gel electrophoresis. The hydrodynamic diameter of the complexes was estimated from the gels using high-molecular mass protein markers [9]. The phospholipid, cholesterol and protein content of rHDL particles was determined using the Phospholipids reagent (Sentinel Diagnostics, Milan, Italy), Cholesterol-LS reagent (Chemelex S.A., Barcelona, Spain) and modified Lowry assay (DC™ Protein Assay, Bio-Rad, Hercules, CA, USA), respectively, according to the manufacturer’s instructions. For physicochemical analyses, rHDL particles were extensively dialyzed against 50 mM sodium phosphate buffer, pH 7.4. For each set of analyses the rHDL particles containing WT or mutant apoA-I forms were prepared using the same phospholipid-cholesterol suspension, and the procedure was performed in parallel. Particles were stored at 4 °C under nitrogen to prevent oxidation of lipids and used for biophysical studies within 2 days and for functional studies within a week.
Circular dichroism (CD) spectroscopy
Far-UV CD spectra of lipid-free WT and mutant apoA-I samples [0.1 mg/ml in DPBS (pH 7.4)] were recorded from 190 to 260 nm at 25 °C. Spectra were collected with a Jasco 715 (USA) spectropolarimeter, as described before [23]. Similarly, the far-UV CD spectra of WT and mutant apoA-I in rHDL [0.1 mg/ml of protein component of the particle in 50 mM sodium phosphate buffer (pH 7.4)] were recorded. Protein concentrations in samples were determined before and after CD experiments and the values agreed within 5%. Concentration of lipid-free proteins was quantitated by measuring their absorbance at 280 nm and of rHDL-associated proteins by a modified Lowry assay (DC™ Protein Assay). The helical content of apoA-I forms was calculated as described [24] using the equation: % α-helix = (− [Θ]222 + 3000)/(36,000 + 3000) × 100.
For thermal denaturation studies, the change in molar ellipticity at 222 nm of lipid-free WT or mutant apoA-I forms was monitored during variation of temperature from 20 to 80 °C at a rate of 1 °C/min. Boltzmann simple sigmoidal model curve was used to fit the data using the GraphPad Prism™ software (La Jolla, CA, USA), allowing the determination of the apparent melting temperature Tm, as the midpoint of the thermal transition. In addition, the relative enthalpy change ΔH was determined as described before [25].
For thermal denaturation analysis of rHDL-associated apoA-I, all measuring parameters were identical to that of the lipid-free apoA-I forms, with the exception of the temperature range used (20–100 °C). The midpoint of thermal unfolding T1/2, the temperature at which denaturation of rHDL-associated apoA-I was half-complete, was determined from the thermal unfolding curves.
Chemical denaturation experiments
To analyze the chemical profile of lipid-free WT and mutant apoA-I forms [0.1 mg/ml in DPBS (pH 7.4)], we monitored the change in wavelength of maximum intrinsic fluorescence of tryptophans by recording emission spectra from 310 to 420 nm using a 295 nm excitation wavelength, upon adding increasing amounts of 8.0 M GdnHCl, as described previously [23]. Fluorescence measurements were carried out using a Quantamaster 4 fluorescence spectrometer (Photon Technology International, USA). A Boltzmann simple sigmoidal model curve was used to fit the data using the GraphPad Prism™ software, allowing the determination of the midpoint of denaturation D1/2. The free energy difference between the native state and unfolded state of the protein in the absence of denaturant ΔGoD was determined using a linear extrapolation method, as described previously [25].
ANS fluorescence measurement
The fluorescence signal (excitation 395 nm, emission 425–600 nm) of WT and mutant apoA-I forms [0.062 mg/ml in DPBS (pH 7.4)] in the presence of 8-anilino-1-naphthalenesulfonic acid (ANS, Sigma-Aldrich) at a final ANS concentration of 310 μM was measured using an Infinite M200 microplate reader (Tecan group Ltd., Switzerland), as described previously [23].
Cholesterol and 7-ketocholesterol efflux assays
ABCA1-dependent efflux of cholesterol was measured in cultures of J774 macrophages following induction with a cAMP analog that leads to increased expression of ABCA1 and using WT and mutant apoA-I forms as cholesterol acceptors [26]. Briefly, J774 mouse macrophages were labeled with 0.25 µCi/ml [14C]-cholesterol ([4-14C]cholesterol, 0.1 mCi/ml of specific activity 55 mCi/mmol; ARC, USA) for 24 h and then treated in the presence or absence of 0.3 mM cpt-cAMP [8-(4-chloro phenylthio)-cAMP] for 18 h. At the end of this treatment period, the cells were incubated in the presence or absence of 1 μΜ lipid-free WT or mutant apoA-I forms for 4 h. The radioactivity of cell media and cell lysates were determined by liquid scintillation counting. The [14C]-cholesterol efflux was expressed as the percentage of radioactivity released in the medium relative to total radioactivity in cells and medium. To calculate the net cpt-dependent (ABCA1-mediated) cholesterol efflux, the cholesterol efflux of the untreated with cpt-cAMP cells was subtracted from the cholesterol efflux of the cells treated with cpt-cAMP.
SR-BI- and ABCG1-mediated efflux of cholesterol was measured in cultures of HEK293 cells following transfection with human SR-BI or ABCG1 expression plasmid using rHDL containing WT and mutant apoA-I forms as cholesterol acceptors [4, 27]. Briefly, HEK293 cells transiently transfected with a pcDNA3.1(+) plasmid encoding human SR-BI (a generous gift from Dr. Daisy Sahoo, Medical College of Wisconsin, Milwaukee, Wisconsin, USA) [28] or human ABCG1 [4] or with vector alone (mock) as described before [4, 27], were labeled with 0.22 µCi/ml [14C]-cholesterol for 24 h. At the end of this period, the cells were incubated in the presence or absence of 1 μΜ (concentration of the protein component) rHDL particles containing WT or mutant apoA-I for 4 h. The [14C]-cholesterol efflux was expressed as the percentage of radioactivity released in the medium relative to total radioactivity in cells and medium, determined by liquid scintillation counting. To calculate the net SR-BI- or ABCG1-mediated cholesterol efflux, the cholesterol efflux of cells transfected with the mock plasmid was subtracted from the cholesterol efflux of cells transfected with the SR-BI- or ABCG1-expressing plasmid, respectively. Untransfected HEK293 cells do not express detectable levels of SR-BI [29] or ABCG1 [30]. Furthermore, the subtraction of cholesterol efflux of cells transfected with the mock plasmid from cholesterol efflux of cells transfected with the SR-BI or ABCG1 plasmid minimizes any potential role of endogenous SR-BI or ABCG1 in cholesterol efflux.
ABCG1-mediated 7-ketocholesterol efflux was measured in HEK293 cells, transiently transfected with human ABCG1 expressing plasmid or vector alone, that were labeled with 1.6 µCi/ml [3H]-7-ketocholesterol (1,2,6-[3H]-7-ketocholesterol, 1 mCi/ml of specific activity 40 Ci/mmol; ARC, USA) using 1 µΜ (concentration of the protein component) rHDL containing WT and mutant apoA-I forms as 7-ketocholesterol acceptors [4]. The [3H]-7-ketocholesterol efflux was expressed as the percentage of radioactivity released in the medium relative to total radioactivity in cells and medium. To calculate the net ABCG1-mediated 7-ketocholesterol efflux, the 7-ketocholesterol efflux of transfected cells with the mock plasmid was subtracted from the 7-ketocholesterol efflux of cells transfected with ABCG1-expressing plasmid.
LCAT activation assay
LCAT was purified from the serum-free culture medium of adenovirus-infected HTB-13 cells and esterification reactions of cholesterol on [14C]-cholesterol-labeled rHDL particles containing WT or mutant apoA-I by LCAT were carried out as described previously [31]. The conversion of [14C]-cholesterol to [14C]-cholesteryl ester was determined by liquid scintillation counting after lipid extraction of the incubation mixture followed by thin-layer chromatography. The cholesterol esterification rate was expressed as nanomoles of cholesteryl ester formed per hour. To calculate the apparent Vmax and Km, the rate of cholesteryl ester formation was plotted versus the concentration of apoA-I. The data were fitted to a standard Michaelis–Menten model using the GraphPad Prism™ software.
Endothelial cell migration assay
EA.hy926 cells were cultured in DMEM supplemented with 10% FBS and antibiotics. Primary Human Coronary Artery Endothelial Cells (HCAEC) were cultured in Human Meso Endo Cell Growth medium (mix of Meso Endo Basal medium and Meso Endo supplement, Cell Applications, Inc., USA). For endothelial cell migration analysis [32, 33], EA.hy926 cells and HCAEC were plated in 12-well plates at a density of 1.8 × 105 and 1.5 × 105 cells/well, respectively. When EA.hy926 cells and HCAEC reached 90% confluency, they were synchronized in DMEM/antibiotics medium and Meso Endo Basal medium/0.5% FBS, respectively, for 16 h and then cell monolayers were scratched using a pipette tip (200 μl). Wounded cells were rinsed with warm DPBS to remove non-adherent cells and then EA.hy926 cells and HCAEC were incubated in the presence or absence of 100 µg/ml (protein content) rHDL particles containing WT or mutant apoA-I forms in DMEM/antibiotics medium and Meso Endo Basal medium/0.25% FBS, respectively, for 24 h. At the end of the incubation period cells were fixed with ice-cold methanol for 15 min, permeabilized in 0.1% Triton X-100/PBS for 15 min and stained with hematoxylin (Sigma Aldrich) for 5 min. Cells were photographed on marked positions at 0 and 24 h, using the 10x objective lens of a Zeiss Primovert Inverted microscope (Zeiss, Germany). Wound healing was quantified, following analysis of images with ImageJ analysis software (NIH, Bethesda, MD, USA) [34], by measuring the number of cells that migrated (for HCAEC) or as follows (for EA.hy926 cells): % Wound area closure = [(cell-free area(0 h) − cell-free area(24 h))/cell-free area(0 h)] × 100. In specific experiments, EA.hy926 cells and HCAEC were treated with 15 µg/ml blocking monoclonal antibody for endothelial lectin-type oxidized low-density lipoprotein receptor 1 (LOX-1) (ΜΑΒ1798, R&D Systems Inc., USA) in the presence or absence of 100 µg/ml rHDL particles. In specific experiments, EA.hy926 cells were also treated with 1.5 µg/ml oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (oxPAPC) (Avanti Polar Lipids, USA) in the presence or absence of 100 µg/ml rHDL particles.
Western blot analysis
EA.hy926 cells and HCAEC were grown to near confluence in 12-well plates and were synchronized in DMEM/antibiotics medium and Meso Endo Basal medium/0.5% FBS, respectively, for 16 h. Afterwards, cells were incubated in the presence or absence of 100 μg/ml (protein content) rHDL particles containing WT or mutant apoA-I forms for 24 h. Following incubation, the culture medium and cells were collected separately. Cells were lysed at 4 °C in RIPA buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.25% sodium deoxycholate, 1% Triton X-100, 0.1% SDS) containing protease and phosphatase inhibitors cocktails (Roche, Germany). The protein concentration in cell lysates was determined using the BCA protein kit (Thermo Fisher Scientific). The calculated total cell protein content was not found to vary significantly between wells in each experiment. 30 µg of total protein from each cell lysate were separated on 12% SDS-PAGE gels and then transferred onto nitrocellulose membranes for immunoblotting. Phospho-Akt was detected using the rabbit anti-phospho-Akt (Ser473) monoclonal antibody D9E (1:2000, Cell Signaling Technology, USA) and a goat anti-rabbit IgG coupled with horseradish peroxidase (1:2000, Merck Millipore, USA). The membranes were then stripped using stripping buffer (Thermo Fisher Scientific) and subsequently re-probed for the detection of total Akt using the rabbit anti-Akt (pan) monoclonal antibody C67E7 (1:2000, Cell Signaling Technology) followed by incubation with goat anti-rabbit IgG coupled with horseradish peroxidase (1:2000). In addition, β-actin was detected by the rabbit anti-β-actin monoclonal antibody 13E5 (1:2000, Cell Signaling Technology) and a goat anti-rabbit IgG coupled with horseradish peroxidase (1:2000). Quantification of protein levels was performed by densitometry of the immunoreactive bands by ImageJ software. Phospho-Akt levels were normalized to their respective total Akt levels.
Measurement of lipid peroxidation products
EA.hy926 cells synchronized in serum free DMEM for 16 h, were afterwards incubated in the presence or absence of 100 μg/ml (protein content) rHDL particles containing WT or mutant apoA-I forms in serum free DMEM for 24 h. Following incubation, the culture medium was collected and the amount of lipid peroxidation products in the cell medium was measured by a commercially available Thiobarbituric Acid Reactive Substances (TBARS) assay kit (Cayman Chemical, USA). By this assay, malondialdehyde (MDA), a common lipid peroxidation product, is detected in rHDL containing cell medium. We followed the manufacturer’s instructions, but we omitted the step of acid precipitation of lipoproteins from the procedure to avoid the loss of TBARS that are associated to rHDL.
MDA levels were also measured in rHDL particles before their incubation with EA.hy926 cells, in the absence or presence of oxPAPC.
Measurement of reactive oxygen species (ROS) generation
Intracellular ROS generation was measured as described previously [35], with some modifications. Briefly, EA.hy926 cells were synchronized in serum free DMEM for 16 h and then were incubated with 0.5 ml DMEM in the presence or absence of 1 μM (protein concentration) rHDL particles containing WT or mutant apoA-I forms for 24 h at 37 °C. In specific experiments, EA.hy926 cells were also treated with 1.5 µg/ml oxPAPC. After incubation, cells were washed with DMEM and further incubated in the dark for 45 min at 37 °C in DMEM containing 25 μΜ 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA, Molecular Probes/Thermo Fisher Scientific). Subsequently, cells were washed with DPBS and production of ROS was detected by recording the fluorescence of 2′,7′-dichlorofluorescein (DCF) with a Zeiss Axiovert 25 Inverted microscope equipped for fluorescence microscopy (excitation 450–490 nm, emission 520 nm). DCF fluorescent intensity was measured for at least 30 cells from the fluorescent images of each sample using the ImageJ analysis software and the relative fluorescence intensity was taken as average of values of at least 5 images for each experiment.
Statistical analysis
Quantitative data are presented as means ± SD. Statistical analyses were conducted using the GraphPad Prism™ software. Statistical comparisons between two groups were analyzed for significance by unpaired two-tailed Student’s t test. p values are indicated in each figure legend.
Results
Protein expression and purification of apoA-I[L144R], apoA-I[A164S], apoA-I[L178P] mutants
ApoA-I mutations L144R and A164S are located on helix 6 and L178P on helix 7 of the N-terminal moiety of apoA-I [9, 11]. Recombinant WT and mutant human apoA-I forms were expressed using a bacterial expression system. WT and mutant apoA-I forms migrated at the expected molecular mass of 28 kDa following SDS-PAGE analysis (Fig. 1). All proteins were ~ 95% pure as judged by SDS-PAGE analysis (Fig. 1). Before each analysis, WT and mutant apoA-I forms were subjected in parallel to a refolding step, as described in “Materials and methods”.
Fig. 1.
SDS-PAGE analysis of recombinant WT and mutant apoA-I forms. WT and mutant apoA-I forms, produced and purified as described in ‘‘Materials and methods”, were subjected to electrophoresis on 15% SDS polyacrylamide gels stained with Coomassie Brilliant Blue. M molecular mass marker (kDa)
Biophysical characterization of lipid-free WT apoA-I and apoA-I[L144R], apoA-I[A164S], apoA-I[L178P] mutants
Since all studied mutations are located within helical regions of apoA-I, we examined whether they may affect the secondary structure of the protein. To test this, we recorded the CD spectra of lipid-free WT apoA-I and mutants. The CD spectra of all lipid-free apoA-I mutants were found to overall have a similar shape to the spectrum of WT apoA-I (Fig. 2a). The intensity of the molar ellipticity, however, was reduced in all apoA-I mutants, showing an approximately 4% loss of helicity for L144R and A164S as compared to WT apoΑ-Ι and a pronounced helicity loss for L178P (~ 11% loss) (Fig. 2b). The severe reduction of α-helical content observed for apoA-I[L178P] is unlikely to be induced only by local perturbation of the helical structure due to the presence of the mutated residue and suggests that the mutation may induce more extensive perturbations to the secondary or even the tertiary structure of apoA-I.
Fig. 2.
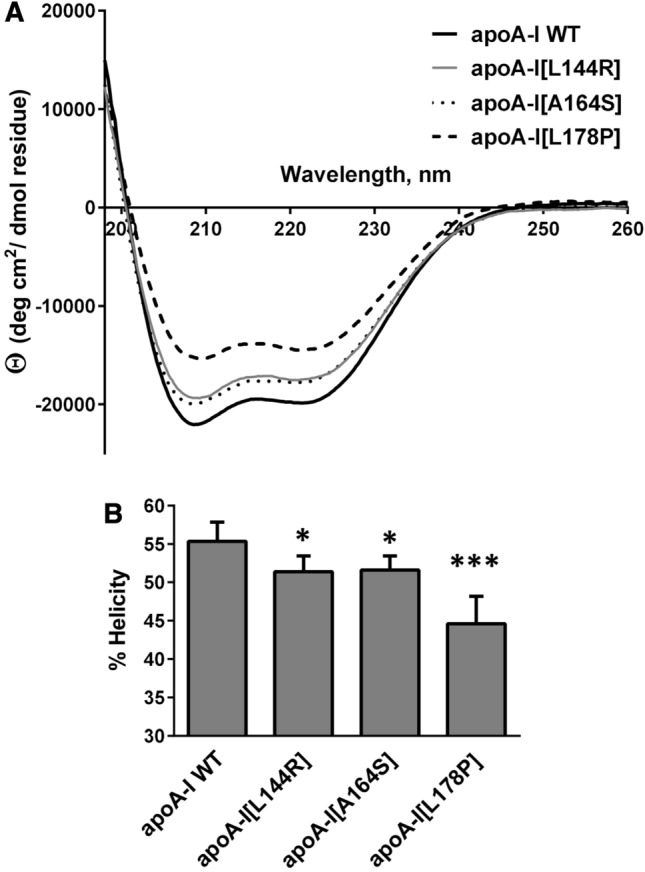
Effect of L144R, A164S and L178P mutations on the secondary structure of apoA-I. a Far-UV CD spectra of WT and mutant apoA-I forms. Spectra are averages of four independent analyses of separate protein preparations. b Percent α-helical content of WT and mutant apoA-I forms. Values represent the means ± SD (n = 4). *p < 0.05; ***p < 0.0001 versus apoA-I WT
Subsequently, we proceeded to examine the thermodynamic stability of apoA-I mutants. We compared the thermal denaturation profile of each mutant to that of WT apoA-I, following the CD signal at 222 nm, while the protein was gradually unfolded by increasing temperature. It has been proposed before that apoA-I thermal unfolding is reversible [36]. However, it is not known whether the apoA-I mutants share this property. Consecutive heating, cooling and re-heating of apoA-I samples was utilized to investigate the reversibility of thermal unfolding for apoA-I forms. Our analysis showed that WT apoA-I and mutant apoA-I forms demonstrated a predominantly reversible transition (Supplemental Figure 1). Analysis of thermal denaturation profiles of each mutant (Fig. 3a–c) showed that apoA-I[L178P] was the least stable toward thermal denaturation of the three apoA-I mutants, having a lower melting temperature midpoint Tm by 14.3 °C and a lower apparent enthalpy change of the transition ΔH by 15.6 kcal/mol compared to WT apoA-I (Fig. 3d, e). Furthermore, the thermal transition of apoA-I[L178P] showed significant loss of cooperativity as evidenced by the loss of sigmoidal shape, suggesting that this protein is significantly less thermodynamically stable than WT apoA-I. A much less pronounced change in thermodynamic stability was observed for apoA-I[L144R] and apoA-I[A164S]. These two apoA-I mutants unfolded at a temperature ~ 2.5 °C lower than WT apoA-I, while only apoA-I[L144R] displayed a decrease in apparent ΔH (Fig. 3d, e).
Fig. 3.

Effect of L144R, A164S and L178P mutations on thermal unfolding of apoA-I. a–c The thermal denaturation profile of each apoA-I mutant is presented in comparison to the WT protein. Solid line indicates the fit of data to Boltzmann sigmoidal model. The y axis has been normalized to correspond to the fraction of the protein in the unfolded state. For normalization, 0 is defined as the value of Boltzmann fit at 20 °C and 1 as the plateau value of Boltzmann fit. The fit is extrapolated to 90 °C to allow better visualization of the plateau. d, e Apparent Tm and ΔH values for the heating-induced transitions of WT and mutant apoA-I forms. Values represent the means ± SD (n = 3–4). *p < 0.05; ***p < 0.0001 versus apoA-I WT
To further examine possible changes in the thermodynamic stability of apoA-I due to the presence of mutations L144R, A164S and L178P, we compared the chemical denaturation profile of each apoA-I mutant to that of WT apoA-I. Fluorescence emission spectra of WT and mutant apoA-I forms were recorded in the presence of increasing concentrations of GdnHCl (Supplemental Figure 2). The wavelength of the maximum fluorescence signal was plotted versus the concentration of GdnHCl (Fig. 4a–c). Similar to the thermal denaturation measurements, apoA-I[L178P] was the least stable toward chemical denaturation of the three apoA-I mutants, exhibiting a decrease in the midpoint of denaturation D1/2 by 0.4 M and a decrease in the free energy change ΔGoD by 3.2 kcal/mol compared to WT apoA-I (Fig. 4d, e). ApoA-I[L144R] and apoA-I[A164S] displayed much smaller losses in stability versus chemical denaturation. ApoA-I[L144R] unfolded at a 0.1 M lower GdnHCl concentration compared to WT apoA-I and apoA-I[A164S] unfolded at a 0.12 M lower GdnHCl concentration, while it also displayed a decrease in ΔGoD (Fig. 4d, e).
Fig. 4.

Effect of L144R, A164S and L178P mutations on chemical denaturant unfolding of apoA-I. a–c The chemical denaturation profile of each apoA-I mutant is presented in comparison to the WT protein. Solid line indicates the fit of data to Boltzmann sigmoidal model. The y axis has been normalized to correspond to the fraction of the protein in the unfolded state. For normalization, 0 and 1 are defined as the bottom and plateau values, respectively, of the Boltzmann fit. d, e D1/2 and ΔGoD values for the denaturant-induced transitions of WT and mutant apoA-I forms. Values represent the means ± SD (n = 3–4). #p < 0.01; * < 0.05; **p < 0.005; ***p < 0.0001 versus apoA-I WT
Finally, to evaluate whether the introduction of L144R, A164S and L178P mutations in apoA-I affects the solvent-exposed hydrophobic surfaces of the protein we used the amphipathic probe ANS, whose fluorescence is enhanced upon binding to hydrophobic surfaces of apoA-I [23]. ANS binding analysis showed that only the L178P mutation resulted in an increase of solvent-accessible hydrophobic surface area in apoA-I (Fig. 5a, b). This finding is consistent with the severe thermodynamic destabilization of apoA-I[L178P] demonstrated by thermal and chemical denaturation analyses.
Fig. 5.
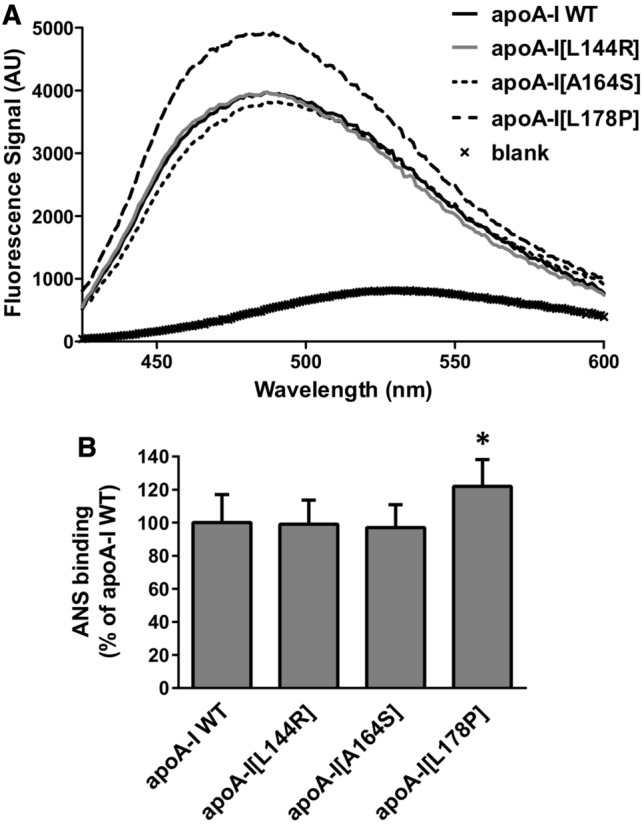
Effect of L144R, A164S and L178P mutations on solvent exposed hydrophobic sites of apoA-I. a ANS fluorescence spectra in the presence or absence of WT or mutant apoA-I forms. AU, arbitrary units. b The ANS fluorescence signal in the presence of WT or mutant apoA-I forms is divided by the fluorescence signal of free ANS in the same buffer and presented relative to ANS binding of WT apoA-I set to 100%. Values represent the means ± SD (n = 5–6). *p < 0.05 versus apoA-I WT
Overall, the biophysical analyses suggested that residues L144, A164 and L178 of apoA-I appear to have a role in the thermodynamic stability of lipid-free apoA-I and that the mutations of these residues studied here have negatively influenced the thermodynamic stability of apoA-I. Of the three mutations, L178P induced the most pronounced effects on the structure and folding of apoA-I, leading to significant thermodynamic destabilization and, possibly, local or generalized misfolding.
In an effort to gain atomic-level insight on the observed effects of the mutants on the structure and folding of apoA-I, we employed comparative all-atom molecular dynamics simulations (MDs). The mutations were introduced in silico to the crystal structure of a C-terminally truncated human apoA-I [9] and simulations were performed for 200 ns. Simulations showed high root-mean-square deviation (RMSD) values of 10–20 Å with respect to the initial crystallographic coordinates (Supplemental Figure 3), consistent with previous work showing a high degree of conformational plasticity of apoA-I [37]. Comparative analysis between WT structure and variants indicated that all three mutations induced perturbations in the root mean square atomic fluctuation (RMSF) values compared to the WT, but in distinct regions of the protein (Supplemental Figure 4). Overall, the MDs suggested strong local and global effects of the mutations and segregated L178P from L144R and A164S, similar to the biophysical analyses. This analysis suggested that the structural repercussions of each mutation can extend away from its local region and have more global effects on the structure of apoA-I, a result that probably explains the effects observed by the biophysical analyses.
Biophysical characterization of WT apoA-I and apoA-I[L144R], apoA-I[A164S], apoA-I[L178P] mutants in reconstituted HDL (rHDL) particles
ApoA-I in the circulation is found mostly in lipidated form in HDL particles. Therefore, we prepared rHDL consisting of WT apoA-I or apoA-I mutants, phosphatidylcholine and cholesterol and examined whether the L144R, A164S and L178P mutations also affect the structure and thermodynamic properties of apoA-I in rHDL particles. Non-denaturing polyacrylamide gradient gel electrophoresis verified the formation of lipoprotein particles with similar hydrodynamic diameter for each protein (Fig. 6a). Measurement of the lipid and protein levels showed that the phospholipid, cholesterol and protein content of the rHDL particles containing the WT and the three mutant apoA-I forms was comparable (Fig. 6a). Biophysical analyses showed that WT and mutant apoA-I forms displayed increased α-helical content and were significantly stabilized when associated in rHDL particles compared to lipid-free protein, as described before [38]. CD analysis suggested that compared to WT apoA-I, apoA-I[L144R] and apoA-I[L178P], but not apoA-I[A164S], had significantly lower α-helical content when in lipoprotein form (Fig. 6b, c). Thermal denaturation analysis showed that apoA-I[L144R] and apoA-I[L178P] mutants in rHDL undergo unfolding transition at lower temperatures, with the midpoint of the transition T1/2 being ~ 3 °C lower than WT apoA-I (Fig. 6d). ApoA-I[A164S] in rHDL, though, showed a similar value for the temperature at midpoint of thermal transition as WT apoA-I (Fig. 6d). Overall, these findings indicated that L144R and L178P mutations induced some thermodynamic perturbations in apoA-I when in lipoprotein particles.
Fig. 6.

Effect of L144R, A164S and L178P mutations on the secondary structure and thermal unfolding of apoA-I in rHDL. a Non-denaturing polyacrylamide gradient (4–20%) gel of rHDL particles containing WT or mutant apoA-I. In the first lane the high-molecular mass calibration proteins are shown [corresponding molecular masses (kDa) and Stokes diameters (nanometers) are indicated]. The molar ratio of phosphatidylcholine (POPC) : cholesterol (C): apoA-I (normalized against POPC) of rHDL particles containing WT or mutant apoA-I forms is shown at the bottom of the panel. Values are the means ± SD for four or five independent rHDL preparations. b Far-UV CD spectra of POPC/C/apoA-I particles containing WT or mutant apoA-I forms. Spectra are averages of four independent analyses of separate protein preparations. c Percent α-helical content of the protein component of lipoprotein particles. Values represent the means ± SD (n = 4). *p < 0.05; **p < 0.005 versus apoA-I WT. d Thermal denaturation profile of WT or mutant apoA-I forms in POPC/C/apoA-I particles. The y axis has been normalized to correspond to the fraction of the protein in the unfolded state. For normalization, 0 is defined as the smallest experiment value and 1 as largest value. Four independent analyses of separate protein preparations were performed
Effect of L144R, A164S and L178P mutations on the capacity of apoA-I/rHDL to promote ABCA1-, SR-BI- and ABCG1-mediated cholesterol efflux, ABCG1-mediated 7-ketocholesterol efflux and to activate LCAT
The observation of structural defects in apoA-I due to L144R, A164S and L178P mutations prompted us to examine whether the mutations also affect functional properties of apoA-I and HDL. We first evaluated the capacity of lipid-free or rHDL-associated WT and mutant apoA-I to promote cholesterol efflux (via ABCA1, SR-BI and ABCG1) from cells and to activate LCAT, processes that affect both HDL-C levels and atheroprotection.
For the measurement of ABCA1-mediated cholesterol efflux, we used a standard experimental approach in which macrophage cell lines, such as J774 cells, were labeled with radioactive cholesterol, pretreated with cAMP analogs, such as cpt-cAMP, and then the release of radiolabeled cellular cholesterol using lipid-free apoA-I as acceptor was quantified [26, 39–41]. cpt-cAMP increases the protein levels of ABCA1 in J774 cells as reported before [39–41] and as shown in Supplemental Figure 5. For the measurement of SR-BI- and ABCG1-mediated cholesterol efflux we used HEK293 cells following transfection with human SR-BI or ABCG1 expression plasmids or mock plasmids. The transfected cells were labeled with radioactive cholesterol and then the release of radiolabeled cellular cholesterol using lipoprotein-associated apoA-I as acceptor was quantified, as described before [4, 27, 28].
Amongst the three apoA-I mutants only apoA-I[L178P], that presents the most severe folding defects in lipid-free form, displayed a 33% reduced capacity to promote ABCA1-mediated cholesterol efflux (Fig. 7a), while only rHDL-associated apoA-I[L144R] was found to have defective LCAT activation capacity, showing a 70% reduction in LCAT activation compared to WT apoA-I (Fig. 7b). Furthermore, apoA-I[L144R] and apoA-I[L178P] in rHDL particles showed a reduced capacity, by 40 and 27%, respectively, to promote SR-BI-mediated cholesterol efflux (Fig. 7c), while apoA-I[A164S] had a 59% reduced capacity to promote ABCG1-mediated cholesterol efflux (Fig. 7d). In addition, rHDL-apoA-I[A164S] showed a 55% reduced capacity to promote ABCG1-mediated 7-ketocholesterol efflux (Fig. 7e).
Fig. 7.

ABCA1-, SR-BI- and ABCG1-mediated cholesterol efflux capacity, ABCG1-mediated 7-ketocholesterol efflux capacity, as well as LCAT activation capacity of WT apoA-I and apoA-I[L144R], apoA-I[A164S], apoA-I[L178P] mutants. a J774 mouse macrophages were labeled with [14C]-cholesterol, treated in the presence or absence of cpt-cAMP and incubated with lipid-free WT or mutant apoA-I forms for 4 h. The net ABCA1-mediated (cpt-cAMP-dependent) [14C]-cholesterol efflux is calculated as the difference in cholesterol efflux between cpt-cAMP-treated cells and untreated cells. ABCA1-mediated [14C]-cholesterol efflux is presented relative to cholesterol efflux in the presence of WT apoA-I (measured 2.6 ± 0.6% of total cholesterol) set to 100%. Values are the means ± SD for three independent experiments performed in triplicate. b LCAT activation by [14C]-cholesterol-labeled rHDL particles containing WT or mutant apoA-I forms shown as the ratio of values of the enzymatic reaction curves relative to the ratio measured for WT apoA-I (4.8 ± 0.1 nmol CE/h/µM apoA-I) set to 100%. The apparent Km and Vmax values of the enzyme are shown at the top of the panel. Values are the means ± SD for three independent experiments. c, d HEK293 cells were transfected with empty vector (mock), SR-BI- or ABCG1-expressing plasmid, labeled with [14C]-cholesterol and then incubated with rHDL containing WT or mutant apoA-I forms for 4 h. The net SR-BI- or ABCG1-mediated [14C]-cholesterol efflux is calculated as the difference in cholesterol efflux between SR-BI- or ABCG1-transfected and mock-transfected cells. SR-BI-mediated [14C]-cholesterol efflux is presented relative to cholesterol efflux in the presence of WT apoA-I (measured 10.6 ± 1.0% of total cholesterol) set to 100%. ABCG1-mediated [14C]-cholesterol efflux is presented relative to cholesterol efflux in the presence of WT apoA-I (measured 2.6 ± 0.4% of total cholesterol) set to 100%. Values are the means ± SD for four independent experiments performed in duplicate. e HEK293 cells were transfected with empty vector (mock) or ABCG1-expressing plasmid, labeled with [3H]-7-ketocholesterol and then incubated with rHDL containing WT or mutant apoA-I forms for 4 h. The net ABCG1-mediated [3H]-7-ketocholesterol efflux is calculated as the difference in 7-ketocholesterol efflux between ABCG1-transfected and mock-transfected cells. ABCG1-mediated [3H]-7-ketocholesterol efflux is presented relative to 7-ketocholesterol efflux in the presence of WT apoA-I (measured 5.0 ± 0.9% of total 7-ketocholesterol) set to 100%. Values are the means ± SD for three independent experiments performed in duplicate. #p < 0.01; **p < 0.005; ***p < 0.0001 versus apoA-I WT
Effect of L144R, A164S and L178P mutations on the capacity of rHDL to stimulate endothelial cell migration
The antiatherogenic functions of HDL involve its capacity to promote endothelial-protective effects including the stimulation of endothelial cell migration [32, 42]. Native HDL, as well as rHDL containing POPC, cholesterol and recombinant apoA-I, have been shown to stimulate endothelial cell migration at comparable levels [32]. However, to the best of our knowledge, the effect of disease-predisposing apoA-I mutations on the capacity of HDL particles to stimulate endothelial cell migration has not been studied before. Here, EA.hy926 endothelial cells were scratched and then incubated with rHDL containing WT or mutant apoA-I forms. Cells treated with rHDL-apoA-I[L144R] and rHDL-apoA-I[L178P] demonstrated similar migration compared to cells treated with rHDL-WT apoA-I (Fig. 8a, b). However, following treatment of cells with rHDL-apoA-I[A164S], migration was significantly reduced (by approximately 50% compared to rHDL-WT apoA-I).
Fig. 8.

Effect of L144R, A164S and L178P mutations on the capacity of lipoprotein-associated apoA-I to promote the migration of endothelial cells. a EA.hy926 cells or c HCAEC were wounded and either left untreated or treated with rHDL containing WT or mutant apoA-I forms for 24 h. Representative microscopic views at 0 h and 24 h are shown. Images were scanned and quantified by ImageJ software. b Quantitative analysis of wound area closure of EA.hy926 cells after 24 h. d Number of HCAEC migrated after 24 h. Values are the means ± SD for three independent experiments performed in duplicate. ***p < 0.0001 versus apoA-I WT
To test whether the reduced capacity of rHDL-containing apoA-I[A164S] to stimulate endothelial cell migration extends to a more physiologically relevant cellular system we performed the same analysis using human coronary artery endothelial cells (HCAEC). Similar to EA.hy926 cells, HCAEC treated with rHDL-apoA-I[L144R] and rHDL-apoA-I[L178P] demonstrated similar migration compared to cells treated with rHDL-WT apoA-I, while HCAEC treated with rHDL-apoA-I[A164S] displayed reduced migration (Fig. 8c, d). Interestingly, in this cellular system the A164S mutation appeared to inactivate completely the capacity of rHDL-associated apoA-I to stimulate cell migration, since apoA-I[A164S]-treated cells display similar migration pattern as untreated cells (Fig. 8c, d).
Understanding the mechanism by which rHDL-apoA-I[A164S] fails to stimulate endothelial cell migration
Previous studies have proposed that a main mechanism by which HDL promotes endothelial migration involves the activation of Akt kinase [32, 42]. Therefore, to understand how rHDL-apoA-I[A164S] fails to stimulate endothelial cell migration, we treated EA.hy926 endothelial cells with rHDL containing WT apoA-I or apoA-I[A164S] forms and assessed Akt activation. As expected, rHDL-WT apoA-I induced activation of Akt, as indicated by increased phosphorylation levels compared to untreated cells (Fig. 9a, b). rHDL-apoA-I[A164S] displayed reduced capacity of Akt activation (by approximately 60% compared to rHDL-WT apoA-I), in accordance to the reduced endothelial cell migration capacity that this mutant displayed.
Fig. 9.
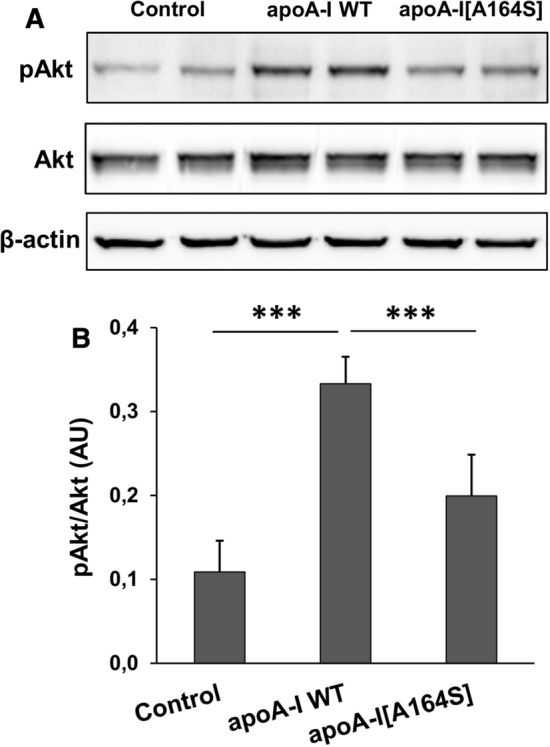
Activation of Akt kinase in EA.hy926 cells incubated with lipoprotein particles containing WT apoA-I or apoA-I[A164S]. Protein expression levels of pAkt, total Akt and β-actin in EA.hy926 cells left untreated or treated with rHDL containing WT apoA-I or apoA-I[A164S] for 24 h were measured by immunoblotting. a A representative set of immunoblots is shown. b Western blots were scanned and quantified by ImageJ software. The normalized levels of pAkt against total Akt are shown. Values are the means ± SD for four independent experiments performed in duplicate. AU arbitrary units. ***p < 0.0001
It has been suggested that Akt-mediated signaling by HDL in endothelial cells entails the SR-BI receptor [42], while the ABCG1 transporter is also involved in the protection of endothelial function by promoting cholesterol and oxysterols efflux [43]. Therefore, we examined whether rHDL-apoA-I[A164S] reduces Akt activation by reducing SR-BI or ABCG1 cellular levels. Our analysis showed no effect on SR-BI or ABCG1 levels of EA.hy926 treated in the absence or presence of rHDL containing WT apoA-I or apoA-I[A164S] (Supplemental Figure 6A, B).
Other studies have proposed that Akt activation can be blocked by dysfunctional HDL via a signaling pathway that involves the endothelial multiligand receptor LOX-1 [44]. Thus, we examined whether rHDL-apoA-I[A164S] reduces Akt activation by increasing LOX-1 cellular levels. We did not observe any difference in LOX-1 levels of EA.hy926 cells treated in the absence or presence of rHDL containing WT apoA-I or apoA-I[A164S] (Supplemental Figure 6C). Therefore, we examined whether rHDL-apoA-I[A164S] reduces Akt activation via interaction with LOX-1. To address this, we studied how EA.hy926 cells migration was affected when cells were treated with rHDL-apoA-I[A164S] in the presence of a LOX-1 blocking antibody. Notably, blocking of LOX-1 resulted in increased EA.hy926 cell migration in the presence of rHDL-apoA-I[A164S] similarly to what was observed for rHDL-WT apoA-I (Fig. 10a, b). In addition, blocking of LOX-1 in HCAEC treated with rHDL-apoA-I[A164S] also resulted in cell migration similar to that observed for rHDL-WT apoA-I (Fig. 10c, d). In line with these findings, blocking of LOX-1 resulted in restoration of Akt activation in EA.hy926 cells and HCAEC treated with rHDL-apoA-I[A164S] (Fig. 10e–h). These findings suggested that rHDL-apoA-I[A164S] fails to activate endothelial Akt and to stimulate migration of cells by interacting with endothelial LOX-1 receptor.
Fig. 10.

Migration of endothelial cells incubated with lipoprotein particles containing WT apoA-I or apoA-I[A164S] in the absence or presence of a LOX-1 receptor blocking antibody. a EA.hy926 cells were wounded and either left untreated or treated with rHDL containing WT apoA-I or apoA-I[A164S], in the absence or presence of a LOX-1 receptor blocking monoclonal antibody, for 24 h. Representative microscopic views at 0 h and 24 h are shown. Images were scanned and quantified by ImageJ software. b Quantitative analysis of wound area closure for EA.hy926 cells after 24 h. Values are the means ± SD for three independent experiments performed in duplicate. c HCAEC were wounded and treated with rHDL containing WT apoA-I or apoA-I[A164S], in the absence or presence of a LOX-1 receptor blocking monoclonal antibody, for 24 h. Representative microscopic views at 0 h and 24 h are shown. Images were scanned and quantified by ImageJ software. d Number of HCAEC migrated after 24 h. Values are the means ± SD for two independent experiments performed in triplicate. Protein expression levels of pAkt and total Akt in EA.hy926 cells (e) and HCAEC (g) left untreated or treated with rHDL containing apoA-I or apoA-I[A164S] for 24 h, in the absence or presence of a LOX-1 receptor blocking monoclonal antibody, were measured by immunoblotting. A representative set of immunoblots is shown. f, h Western blots were scanned and quantified by ImageJ software. The normalized levels of pAkt against total Akt are shown. Values are the means ± SD for four (f) or three (h) independent experiments performed in duplicate. AU arbitrary units. **p < 0.005; ##p < 0.001; ***p < 0.0001; ns not significant
It has been shown that accumulation of malondialdehyde (MDA), a common lipid peroxidation product, in HDL blocks the activation of Akt via the LOX-1 receptor [42, 44]. We, therefore, measured the MDA levels in rHDL-containing WT apoA-I or apoA-I[A164S] following their incubation with EA.hy926 cells. Before incubation with cells, MDA was undetectable in rHDL-containing WT apoA-I or apoA-I[A164S], but following incubation we found increased levels of MDA in rHDL-apoA-I[A164S] but not in rHDL-WT apoA-I (Fig. 11), suggesting that accumulation of MDA in rHDL-apoA-I[A164S] could contribute to its reduced capacity to stimulate Akt activation and endothelial cell migration.
Fig. 11.
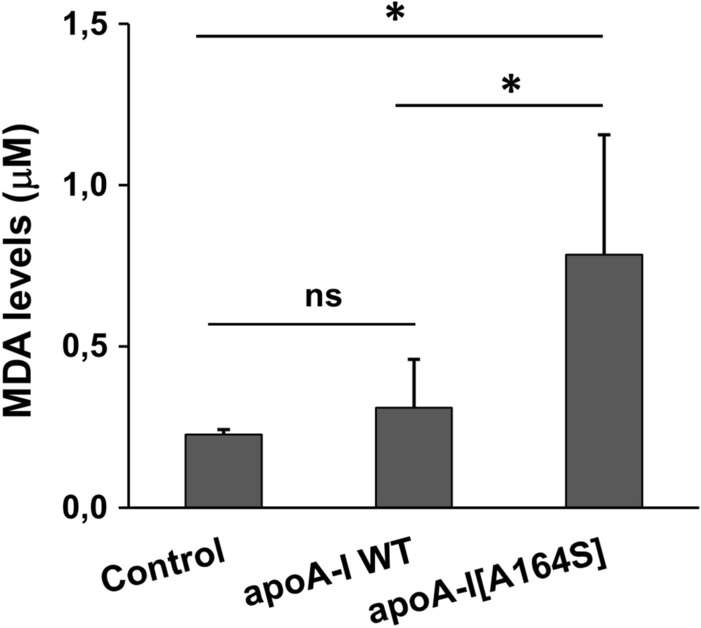
Lipid peroxidation products in the culture medium of EA.hy926 cells incubated with lipoprotein particles containing WT apoA-I or apoA-I[A164S]. MDA is detected in the rHDL containing cell medium of EA.hy926 cells by a commercially available TBARS assay kit. Values are the means ± SD (n = 6). *p < 0.05; ns not significant
It has been shown previously that incubation of endothelial cells with HDL prevents the production of ROS [43] and that MDA-containing HDL is associated with enhanced cellular oxidative stress and ROS formation [45]. Therefore, the increased levels of MDA in rHDL-apoA-I[A164S] following incubation with EA.hy926 cells prompted us to examine the formation of ROS in these cells. As shown in Fig. 12a, b, WT apoA-I in rHDL reduced the intracellular formation of ROS, while apoA-I[A164S] failed to reduce ROS formation. Collectively, our data suggested that HDL that contains apoA-I[A164S] can lose its capacity to preserve the endothelial function.
Fig. 12.
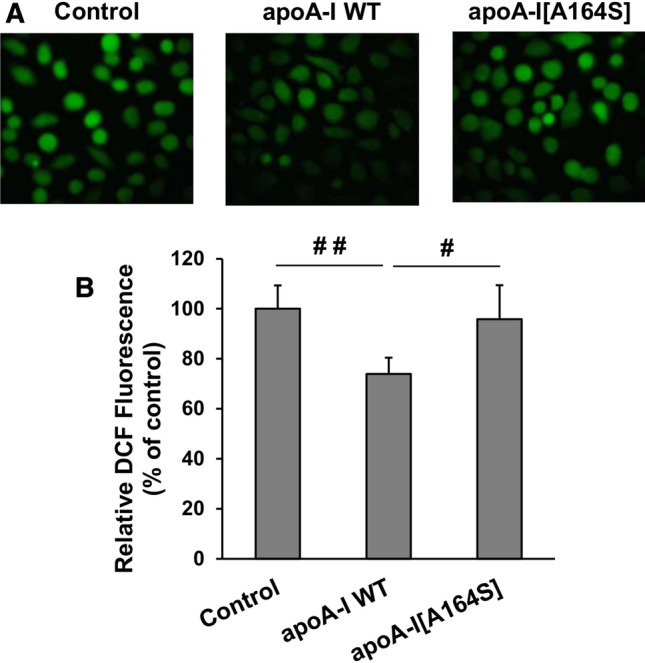
Effect of lipoprotein particles containing WT apoA-I or apoA-I[A164S] on ROS formation by EA.hy926 cells. a The formation of ROS by EA.hy926 cells treated with or without rHDL containing WT apoA-I or apoA-I[A164S] for 24 h was measured following incubation of cells with DCFH-DA and detection of fluorescent DCF emitted from cells using a fluorescence microscope. Representative microscopic views are shown. b Images were scanned and quantified by Image J software. The relative DCF fluorescence is shown. Values are the means ± SD (n = 6). #p < 0.01; ##p < 0.001
Terminal lipid peroxidation products, such as MDA, can be produced by oxidation of 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (PAPC) [46]. To examine the potential effect of lipid peroxidation products on the capacity of HDL to promote endothelial cell migration, we incubated EA.hy926 cells with rHDL-WT apoA-I in the presence of oxidized PAPC (oxPAPC). The concentration of MDA in cell medium containing rHDL-WT apoA-I/oxPAPC was 0.73 ± 0.06 µM, similar to the concentration of MDA found in cell medium containing rHDL-apoA-I[A164S] following incubation of lipoprotein particles with EA.hy926 cells for 24 h (Fig. 11). Our analysis showed that oxPAPC impaired the capacity of rHDL-WT apoA-I to promote endothelial cell migration (Fig. 13a, b), as well as to reduce ROS formation (Fig. 13c, d). These findings provide further support for the role of MDA in the dysfunctionality of HDL.
Fig. 13.

Migration and ROS formation by EA.hy926 cells incubated with lipoprotein particles containing WT apoA-I in the absence or presence of oxPAPC. a EA.hy926 cells were wounded and treated with or without rHDL containing WT apoA-I, in the absence or presence of oxPAPC, for 24 h. Representative microscopic views at 0 h and 24 h are shown. Images were scanned and quantified by ImageJ software. b Quantitative analysis of wound area closure for EA.hy926 cells after 24 h. Values are the means ± SD (n = 6). c EA.hy926 cells treated with or without rHDL containing WT apoA-I, in the absence or presence of oxPAPC, for 24 h, were then incubated with DCFH-DA. The formation of ROS by cells was measured following detection of fluorescent DCF emitted from cells using a fluorescence microscope. Representative microscopic views are shown. Images were scanned and quantified by Image J software. d The relative DCF fluorescence is shown. Values are the means ± SD (n = 10). ***p < 0.0001
Discussion
In an effort to gain mechanistic insight linking the presence of L144R, A164S and L178P mutations in apoA-I with HDL-C levels and CVD risk in carriers of these mutations we evaluated the effect of mutated residues on the structural integrity and functional properties of apoA-I in lipid-free and rHDL form. By combining the findings from the structural and functional analyses of the three apoA-I mutants we conclude that only severe conformational perturbations in lipid-free apoA-I, such as those found for apoA-I[L178P], are accompanied by reduced ABCA1-mediated cholesterol efflux which is the first step in HDL biogenesis pathway. ApoA-I[L144R] and apoA-I[L178P] in rHDL particles that display similar structural and thermodynamic perturbations of lipidated apoA-I, show a similarly reduced capacity to promote cellular cholesterol efflux by SR-BI and normal capacity for ABCG1-mediated cholesterol efflux and stimulation of endothelial cell migration. However, apoA-I[L144R] presents with a severe defect in LCAT activation. Inspection of the crystal structure of an N-terminal truncated apoA-I that has been proposed to mimic the apoA-I configuration on discoidal HDL [11] indicates that only L144, that is located in the external site of helix 6, is located in a position that would allow direct interactions leading to the activation of LCAT. ApoA-I[A164S] has only minimal structural perturbations in lipidated form and displays distinct functional defects that include reduced ABCG1-cholesterol efflux capacity and reduced stimulation of endothelial cell migration. Table 1 summarizes the effects of L144R, A164S and L178P mutations on biophysical and functional properties of apoA-I. How our findings could explain the observed phenotypes in humans is discussed below for each apoA-I mutant.
Table 1.
Summary of the observed changes in biophysical and functional properties of apoA-I L144R, A164S and L178P mutants as compared to WT apoA-I
| Change in property as compared to WT apoA-I | |||
|---|---|---|---|
| apoA-I[L144R] | apoA-I[A164S] | apoA-I[L178P] | |
| Biophysical properties (lipid-free apoA-I) | |||
| Helicity (%) | ~ 4 ↓ | ~ 4 ↓ | ~ 11 ↓ |
| Thermal unfolding | |||
| App Tm (οC) | ~ 2.5 ↓ | ~ 2.5 ↓ | 14.3 ↓ |
| App ΔH (kcal/mol) | 5.5 ↓ | ↔ | 15.6 ↓ |
| Chemical unfolding | |||
| D1/2 (M) | 0.1 ↓ | ~ 0.1 ↓ | 0.4 ↓ |
| ΔGoD (kcal/mol) | ↔ | 1.4 ↓ | 3.2 ↓ |
| ANS binding | ↔ | ↔ | 22% ↑ |
| Biophysical properties (rHDL-apoA-I) | |||
| Helicity (%) | ~ 8 ↓ | ↔ | ~ 6 ↓ |
| Thermal unfolding | |||
| T1/2 (οC) | ~ 3 ↓ | ↔ | ~ 3 ↓ |
| Functional properties (lipid-free apoA-I) | |||
| ABCA1-mediated cholesterol efflux | ↔ | ↔ | ~ 35% ↓ |
| Functional properties (rHDL-apoA-I) | |||
| LCAT activation | ~ 70% ↓ | ↔ | ↔ |
| SR-BI-mediated cholesterol efflux | ~ 40% ↓ | ↔ | ~ 25% ↓ |
| ABCG1-mediated cholesterol efflux | ↔ | ~ 60% ↓ | ↔ |
| Endothelial cell migration | |||
| EA.hy926 | ↔ | ~ 50% ↓ | ↔ |
| HCAEC | ↔ | ~ 100% ↓ | ↔ |
An upward-pointing arrow indicates that the property has increased, a downward-pointing arrow indicates that the property has decreased and a left–right arrow indicates no change
ApoA-I[L144R] Heterozygous carriers of apoA-I[L144R], identified at a frequency of 0.04% in the Danish population, had 47% reduced HDL-C levels, but none of the carriers had premature IHD [18]. Heterozygous carriers of apoA-I[L144R] with HDL-C levels below the 5th percentile and without evidence of CVD have also been reported in a Spanish family [47]. We show that rHDL-associated apoA-I[L144R] displays greatly reduced capacity to activate LCAT, which could be the underlying cause of reduced HDL-C levels in carriers of this mutation. Previous animal studies have shown that adenovirus-mediated gene transfer of apoA-I[L144R] in apoA-I-deficient mice results in low HDL-C levels and formation of discoidal HDL particles [18]. This phenotype is consistent with normal capacity for ABCA1-mediated cholesterol efflux by apoA-I[L144R] and reduced LCAT activation capacity. Indeed a decreased cholesteryl ester-to-total cholesterol ratio was measured in mice expressing apoA-I[L144R], while treatment of mice with adenoviruses expressing apoA-I[L144R] and human LCAT normalized HDL-C levels and the cholesteryl ester-to-total cholesterol ratio and promoted the formation of spherical HDL particles [18]. Our biophysical analyses demonstrate reduced helicity and thermodynamic perturbations in rHDL-associated apoA-I[L144R] that could underlie observed perturbations in functions, such as LCAT activation capacity and SR-BI-mediated cholesterol efflux. Furthermore, a previous study showed that carriers of apoA-I[L144R] had apoA-I fractional catabolic rate values more than two-fold higher than the fractional catabolic rate values of control subjects, suggesting enhanced catabolism of mutant apoA-I [47]. Overall, a combination of the findings from the current and previous studies suggest that the reduced HDL-C levels associated with apoA-I[L144R] may be caused by structural perturbations in apoA-I[L144R] that result in reduced activation of LCAT and enhanced catabolism of the mutant protein. The normal capacity of apoA-I[L144R] for ABCA1-mediated cholesterol efflux and stimulation of endothelial cell migration are consistent with the absence of CVD risk in carriers of this mutation.
ApoA-I[L178P] Heterozygous carriers of apoA-I[L178P] in Dutch families exhibited 63% reduced HDL-C levels, increased carotid IMT and 24-fold increased CVD risk [20]. The L178P mutation introduces a proline residue into the highly helical central region of apoA-I, and this could lead to structural perturbations due to the incompatibility of proline with helical secondary structure. Indeed, our biophysical studies show that lipid-free apoA-I[L178P] induces major defects in protein structure and stability. These may be due to the general helix-breaking properties of proline but could also indicate that the specific position 178 plays a key role in apoA-I structure and stability. In contrast to the lipid-free form, lipoprotein-associated apoA-I[L178P] displays milder conformational perturbations. The folding defects of lipid-free apoA-I[L178P] are accompanied with reduced capacity of the protein to promote ABCA1-mediated cholesterol efflux, something that may underlie the reduced HDL-C levels in carriers, as well as the increased CVD risk. Furthermore, the conformational perturbations in lipid-free and lipoprotein-associated apoA-I[L178P] are in accordance with a previous study from our lab showing that heterozygotes of this apoA-I mutant had HDL with disturbed apoA-I subpopulations profile and reduced antioxidant/anti-inflammatory capacity [48]. These two properties could also underlie the increased incidence of premature vascular events in these subjects.
ApoA-I[A164S] Among the three mutations we studied, A164S has the most striking pathological phenotype, since heterozygotes for this mutant have increased risk for IHD, myocardial infarction and total mortality compared to noncarriers, but normal plasma lipid and lipoprotein levels, including HDL-C and apoA-I [18]. Heterozygous carriers of apoA-I[A164S] were identified at a frequency of 0.23% in the Danish population and the median survival time of carriers was reduced by 10 years [18]. We show here that apoA-I[A164S] has normal capacity for ABCA1-mediated cholesterol efflux and LCAT activation, findings that are in accordance with the presence of normal HDL-C levels in carriers of this mutant. Adenovirus-mediated gene transfer of apoA-I[A164S] in apoA-I-deficient mice has confirmed the lack of effect of apoA-I[A164S] on plasma levels of HDL-C and apoA-I and led to formation of spherical HDL particles [18].
Analysis of clinical characteristics of A164S heterozygotes did not reveal any deposition of amyloid that could explain the increased risk of IHD, myocardial infarction and total mortality associated with this mutation [18]. However, due to the low sensitivity of the method used, intramural coronary vascular amyloidosis could not be excluded. Therefore, we examined the amyloidogenic propensity of apoA-I[A164S] by measuring binding to the amyloidophilic fluorescent probe Thioflavin T (ThT). Our analysis showed no difference in ThT binding between apoA-I[A164S] and WT apoA-I, following incubation of apoA-I forms for 24 and 48 h (Supplemental Figure 7). Thus, it appears that the apoA-I[A164S] mutant does not display a strong amyloidogenic propensity, in accordance with a previous study [49]. An alternative possibility is that the increased cardiovascular risk of apoA-I[A164S] may be related to dysfunctional HDL particles.
Our analyses show that lipidated apoA-I[A164S] displays minimal structural perturbations. However, apoA-I[A164S] in lipoprotein particles displays aberrant functions that could explain the association of this mutant with increased cardiovascular risk. Indeed, rHDL-containing apoA-I[A164S] has reduced capacity to stimulate endothelial cell migration following disruption of endothelial cells layer integrity, a process that has been related with vascular protection by HDL [5, 32, 42]. Several mechanisms have been proposed to explain the effect of HDL on endothelial cells migration, including the involvement of lipoprotein receptors and cholesterol transporters, such as SR-BI, ABCG1 and LOX-1. According to one proposed mechanism, HDL signaling through SR-BI (a process that requires SR-BI-mediated cholesterol efflux) results in Akt activation which promotes endothelial cell migration [5, 32, 42, 50]. We find, however, that although rHDL-apoA-I[A164S] has reduced capacity of Akt activation, neither SR-BI levels nor SR-BI-mediated cholesterol efflux are affected by this apoA-I mutant. As an alternative mechanism, it has been proposed that ABCG1 can preserve endothelial function by promoting efflux of cholesterol and oxysterols, such as 7-ketocholesterol, from endothelial cells [43]. Additionally, a recent study showed that the endothelial progenitor cells migration is stimulated following ABCG1 overexpression via activation of Akt pathway [51]. We do not observe any reduction in ABCG1 levels in rHDL-apoA-I[A164S] treated endothelial cells that could account for the observed reduction in activation of Akt. Nevertheless, the reduced capacity (by more than 55%) of rHDL-apoA-I[A164S] to promote ABCG1-mediated cholesterol and 7-ketocholesterol efflux could play at least a partial role in the reduced ability of this mutant to preserve endothelial function. Finally, the activation of endothelial multiligand receptor LOX-1 by dysfunctional HDL has been shown to promote the activation of PKCβII that subsequently blocks Akt activation [44, 52]. Our studies suggest that lipoprotein particles containing apoA-I[A164S] are prone to accumulation of lipid peroxidation products resulting in activation of LOX-1 which subsequently inactivates Akt leading to impaired stimulation of endothelial cell migration. When LOX-1 is blocked, then rHDL-apoA-I[A164S] particles are capable to promote endothelial cell migration probably through interactions with SR-BI.
HDL can protect endothelial cells from ROS [5, 42]. We show that apoA-I[A164S] containing rHDL failed to reduce ROS formation in endothelial cells. This finding may be related to the increased levels of MDA in these particles, since high concentrations of MDA in HDL have been shown to induce ROS generation [45]. Alternatively, this effect may be related to the reduced capacity of rHDL-apoA-I[A164S] to promote ABCG1-mediated 7-ketocholesterol efflux. Several oxysterols, most notably 7-ketocholesterol, have been shown to induce ROS generation and lower ABCG1-mediated 7-ketocholesterol efflux has been shown to induce ROS production [43].
Collectively, our studies propose that the apoA-I mutation A164S leads to the formation of dysfunctional lipoprotein particles. The impaired capacity of these particles to stimulate endothelial cells migration and to protect endothelial cells from ROS production may underlie the increased risk of IHD, myocardial infarction and total mortality observed in carriers of apoA-I[A164S] mutant.
In recent years, the association between HDL-C levels and cardiovascular risk has been challenged by genetic and clinical studies [6]. It has been suggested that the relationship between HDL-C levels and CHD risk is complex and HDL-C may not be the optimal therapeutic target. HDL possesses various atheroprotective functions and accumulating evidence supports that HDL functionality may be more relevant for atheroprotection than HDL-C levels [6]. In the current study we focus on the effect of naturally occurring apoA-I mutations on apoA-I and HDL functionality that may underlie the observed phenotypes in carriers of the mutations. Future animal studies using transgenic mice carrying the mutant apoA-I forms and modifications in genes of the HDL metabolism pathway (e.g., lipid transporters, lipoprotein receptors), that interact with apoA-I, could help us understand further the effect of apoA-I mutations on HDL functionality. In addition, future detailed compositional analyses of HDL particles from carriers of apoA-I mutations may provide information on potential changes in the proteome, lipidome and metabolome of HDL that may underlie functional changes.
In summary, we characterized the effect of three naturally occurring mutations of apoA-I (L144R, A164S and L178P) on the structure and function of the protein. We find that all mutations induce at least some disrupting structural and thermodynamic changes, which, although differ in specific effect and intensity, in all cases appear to affect a much larger part of the protein than the immediate location of the mutation. These structural and thermodynamic changes associate with specific functional defects that could underlie the reduction in HDL-C levels and/or increased cardiovascular risk observed in carriers of the mutations. Most notably, our results provide novel insights into the molecular basis for the pathological phenotype observed in carriers of apoA-I[A164S], which is the only apoA-I variant described to predict an increased risk of IHD, myocardial infarction and total mortality in the absence of low HDL-C levels.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Dr. Dimitris Kardassis (Medical School of the University of Crete, Greece) and members of his lab for providing valuable help with the endothelial cell migration assay. We acknowledge support of this work by the projects: (a) “An Open-Access Research Infrastructure of Chemical Biology and Target-Based Screening Technologies for Human and Animal Health, Agriculture and the Environment (OPENSCREEN-GR)” (MIS 5002691) which is implemented under the Action “Reinforcement of the Research and Innovation Infrastructure”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020) and co-financed by Greece and the European Union (European Regional Development Fund), (b) “The National Research Infrastructure on Integrated Structural Biology, Drug Screening Efforts and Drug target functional characterization (INSPIRED)” (MIS 5002550) which is implemented under the Action “Reinforcement of the Research and Innovation Infrastructure”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020) and co-financed by Greece and the European Union (European Regional Development Fund), (c) “A Greek Research Infrastructure for Visualizing and Monitoring Fundamental Biological Processes (BioImaging-GR)” (MIS 5002755) which is implemented under the Action “Reinforcement of the Research and Innovation Infrastructure”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020) and co-financed by Greece and the European Union (European Regional Development Fund) and (d) “CURE-PLaN” a grant from the Leducq Foundation for Cardiovascular Research.
Abbreviations
- ABCA1
ATP-binding cassette A1
- ABCG1
ATP-binding cassette G1
- ANS
8-anilino-1-naphthalenesulfonic acid
- apoA-I
Apolipoprotein A-I
- C
Cholesterol
- CD
Circular dichroism
- cpt-cAMP
8-(4-Chlorophenylthio)-cAMP
- CVD
Cardiovascular disease
- DCF
2′,7′-Dichlorofluorescein
- DCFH-DA
2′,7′-Dichlorodihydrofluorescein diacetate
- DPBS
Dulbecco’s phosphate-buffered saline
- GdnHCl
Guanidine hydrochloride
- HCAEC
Human coronary artery endothelial cells
- HDL
High-density lipoprotein
- HDL-C
HDL-cholesterol
- IHD
Ischemic heart disease
- IMT
Intima–media thickness
- LCAT
Lecithin:cholesterol acyltransferase
- LOX-1
Lectin-type oxidized low-density lipoprotein receptor 1
- MDA
Malondialdehyde
- Ni-NTA
Ni2+-nitrilotriacetic acid
- oxPAPC
Oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine
- POPC
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- rHDL
Reconstituted HDL
- ROS
Reactive oxygen species
- SR-BI
Scavenger receptor class B type I
- TBARS
Thiobarbituric acid reactive substances
- Trx
Thioredoxin
- WT
Wild type
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zannis VI, Fotakis P, Koukos G, Kardassis D, Ehnholm C, Jauhiainen M, Chroni A. HDL biogenesis, remodeling, and catabolism. Handb Exp Pharmacol. 2015;224:53–111. doi: 10.1007/978-3-319-09665-0_2. [DOI] [PubMed] [Google Scholar]
- 2.Siddiqi HK, Kiss D, Rader D. HDL-cholesterol and cardiovascular disease: rethinking our approach. Curr Opin Cardiol. 2015;30:536–542. doi: 10.1097/HCO.0000000000000211. [DOI] [PubMed] [Google Scholar]
- 3.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 4.Daniil G, Zannis VI, Chroni A. Effect of apoA-I mutations in the capacity of reconstituted HDL to promote ABCG1-mediated cholesterol efflux. PLoS One. 2013;8:e67993. doi: 10.1371/journal.pone.0067993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mineo C, Shaul PW. Novel biological functions of high-density lipoprotein cholesterol. Circ Res. 2012;111:1079–1090. doi: 10.1161/CIRCRESAHA.111.258673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marz W, Kleber ME, Scharnagl H, Speer T, Zewinger S, Ritsch A, Parhofer KG, et al. HDL cholesterol: reappraisal of its clinical relevance. Clin Res Cardiol. 2017;106:663–675. doi: 10.1007/s00392-017-1106-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Favari E, Thomas MJ, Sorci-Thomas MG. High-density lipoprotein functionality as a new pharmacological target on cardiovascular disease: unifying mechanism that explains high-density lipoprotein protection toward the progression of atherosclerosis. J Cardiovasc Pharmacol. 2018;71:325–331. doi: 10.1097/FJC.0000000000000573. [DOI] [PubMed] [Google Scholar]
- 8.Melchior JT, Walker RG, Cooke AL, Morris J, Castleberry M, Thompson TB, Jones MK, et al. A consensus model of human apolipoprotein A-I in its monomeric and lipid-free state. Nat Struct Mol Biol. 2017;24:1093–1099. doi: 10.1038/nsmb.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mei X, Atkinson D. Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J Biol Chem. 2011;286:38570–38582. doi: 10.1074/jbc.M111.260422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mei X, Atkinson D. Lipid-free apolipoprotein A-I structure: insights into HDL formation and atherosclerosis development. Arch Med Res. 2015;46:351–360. doi: 10.1016/j.arcmed.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borhani DW, Rogers DP, Engler JA, Brouillette CG. Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc Natl Acad Sci USA. 1997;94:12291–12296. doi: 10.1073/pnas.94.23.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silva RA, Huang R, Morris J, Fang J, Gracheva EO, Ren G, Kontush A, et al. Structure of apolipoprotein A-I in spherical high density lipoproteins of different sizes. Proc Natl Acad Sci USA. 2008;105:12176–12181. doi: 10.1073/pnas.0803626105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang R, Silva RA, Jerome WG, Kontush A, Chapman MJ, Curtiss LK, Hodges TJ, et al. Apolipoprotein A-I structural organization in high-density lipoproteins isolated from human plasma. Nat Struct Mol Biol. 2011;18:416–422. doi: 10.1038/nsmb.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gursky O. Crystal structure of Delta(185-243)ApoA-I suggests a mechanistic framework for the protein adaptation to the changing lipid load in good cholesterol: from flatland to sphereland via double belt, belt buckle, double hairpin and trefoil/tetrafoil. J Mol Biol. 2013;425:1–16. doi: 10.1016/j.jmb.2012.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sorci-Thomas MG, Thomas MJ. The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc Med. 2002;12:121–128. doi: 10.1016/s1050-1738(01)00163-3. [DOI] [PubMed] [Google Scholar]
- 16.Zannis VI, Chroni A, Krieger M. Role of apoA-I, ABCA1, LCAT, and SR-BI in the biogenesis of HDL. J Mol Med. 2006;84:276–294. doi: 10.1007/s00109-005-0030-4. [DOI] [PubMed] [Google Scholar]
- 17.Chroni A, Kardassis D. HDL dysfunction caused by mutations in apoA-I and other genes that are critical for HDL biogenesis and remodeling. Curr Med Chem. 2019;26:1544–1575. doi: 10.2174/0929867325666180313114950. [DOI] [PubMed] [Google Scholar]
- 18.Haase CL, Frikke-Schmidt R, Nordestgaard BG, Kateifides AK, Kardassis D, Nielsen LB, Andersen CB, et al. Mutation in APOA1 predicts increased risk of ischaemic heart disease and total mortality without low HDL cholesterol levels. J Intern Med. 2011;270:136–146. doi: 10.1111/j.1365-2796.2011.02381.x. [DOI] [PubMed] [Google Scholar]
- 19.Recalde D, Cenarro A, Garcia-Otin AL, Gomez-Coronado D, Civeira F, Pocovi M. Analysis of apolipoprotein A-I, lecithin:cholesterol acyltransferase and glucocerebrosidase genes in hypoalphalipoproteinemia. Atherosclerosis. 2002;163:49–58. doi: 10.1016/s0021-9150(01)00753-5. [DOI] [PubMed] [Google Scholar]
- 20.Hovingh GK, Brownlie A, Bisoendial RJ, Dube MP, Levels JH, Petersen W, Dullaart RP, et al. A novel apoA-I mutation (L178P) leads to endothelial dysfunction, increased arterial wall thickness, and premature coronary artery disease. J Am Coll Cardiol. 2004;44:1429–1435. doi: 10.1016/j.jacc.2004.06.070. [DOI] [PubMed] [Google Scholar]
- 21.Gkolfinopoulou C, Bourtsala A, Chroni A. Structural and functional basis for increased HDL-cholesterol levels due to the naturally occurring V19L mutation in human apolipoprotein A-I. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865:158593. doi: 10.1016/j.bbalip.2019.158593. [DOI] [PubMed] [Google Scholar]
- 22.Argyri L, Skamnaki V, Stratikos E, Chroni A. A simple approach for human recombinant apolipoprotein E4 expression and purification. Protein Expr Purif. 2011;79:251–257. doi: 10.1016/j.pep.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 23.Fotakis P, Tiniakou I, Kateifides AK, Gkolfinopoulou C, Chroni A, Stratikos E, Zannis VI, et al. Significance of the hydrophobic residues 225–230 of apoA-I for the biogenesis of HDL. J Lipid Res. 2013;54:3293–3302. doi: 10.1194/jlr.M043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrisett JD, David JS, Pownall HJ, Gotto AM., Jr Interaction of an apolipoprotein (apoLP-alanine) with phosphatidylcholine. Biochemistry. 1973;12:1290–1299. doi: 10.1021/bi00731a008. [DOI] [PubMed] [Google Scholar]
- 25.Gorshkova IN, Liadaki K, Gursky O, Atkinson D, Zannis VI. Probing the lipid-free structure and stability of apolipoprotein A-I by mutation. Biochemistry. 2000;39:15910–15919. doi: 10.1021/bi0014406. [DOI] [PubMed] [Google Scholar]
- 26.Chroni A, Liu T, Gorshkova I, Kan HY, Uehara Y, von Eckardstein A, Zannis VI. The central helices of apoA-I can promote ATP-binding cassette transporter A1 (ABCA1)-mediated lipid efflux. Amino acid residues 220-231 of the wild-type apoA-I are required for lipid efflux in vitro and high density lipoprotein formation in vivo. J Biol Chem. 2003;278:6719–6730. doi: 10.1074/jbc.M205232200. [DOI] [PubMed] [Google Scholar]
- 27.Dafnis I, Raftopoulou C, Mountaki C, Megalou E, Zannis VI, Chroni A. ApoE isoforms and carboxyl-terminal-truncated apoE4 forms affect neuronal BACE1 levels and Abeta production independently of their cholesterol efflux capacity. Biochem J. 2018;475:1839–1859. doi: 10.1042/BCJ20180068. [DOI] [PubMed] [Google Scholar]
- 28.Chadwick AC, Sahoo D. Functional characterization of newly-discovered mutations in human SR-BI. PLoS One. 2012;7:e45660. doi: 10.1371/journal.pone.0045660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambert G, Chase MB, Dugi K, Bensadoun A, Brewer HB, Jr, Santamarina-Fojo S. Hepatic lipase promotes the selective uptake of high density lipoprotein-cholesteryl esters via the scavenger receptor B1. J Lipid Res. 1999;40:1294–1303. [PubMed] [Google Scholar]
- 30.Kim WS, Rahmanto AS, Kamili A, Rye KA, Guillemin GJ, Gelissen IC, Jessup W, et al. Role of ABCG1 and ABCA1 in regulation of neuronal cholesterol efflux to apolipoprotein E discs and suppression of amyloid-beta peptide generation. J Biol Chem. 2007;282:2851–2861. doi: 10.1074/jbc.M607831200. [DOI] [PubMed] [Google Scholar]
- 31.Chroni A, Kan HY, Kypreos KE, Gorshkova IN, Shkodrani A, Zannis VI. Substitutions of glutamate 110 and 111 in the middle helix 4 of human apolipoprotein A-I (apoA-I) by alanine affect the structure and in vitro functions of apoA-I and induce severe hypertriglyceridemia in apoA-I-deficient mice. Biochemistry. 2004;43:10442–10457. doi: 10.1021/bi049782p. [DOI] [PubMed] [Google Scholar]
- 32.Seetharam D, Mineo C, Gormley AK, Gibson LL, Vongpatanasin W, Chambliss KL, Hahner LD, et al. High-density lipoprotein promotes endothelial cell migration and reendothelialization via scavenger receptor-B type I. Circ Res. 2006;98:63–72. doi: 10.1161/01.RES.0000199272.59432.5b. [DOI] [PubMed] [Google Scholar]
- 33.Jansen F, Yang X, Hoelscher M, Cattelan A, Schmitz T, Proebsting S, Wenzel D, et al. Endothelial microparticle-mediated transfer of MicroRNA-126 promotes vascular endothelial cell repair via SPRED1 and is abrogated in glucose-damaged endothelial microparticles. Circulation. 2013;128:2026–2038. doi: 10.1161/CIRCULATIONAHA.113.001720. [DOI] [PubMed] [Google Scholar]
- 34.Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 35.Dafnis I, Argyri L, Sagnou M, Tzinia A, Tsilibary EC, Stratikos E, Chroni A. The ability of apolipoprotein E fragments to promote intraneuronal accumulation of amyloid beta peptide 42 is both isoform and size-specific. Sci Rep. 2016;6:30654. doi: 10.1038/srep30654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gursky O, Atkinson D. Thermal unfolding of human high-density apolipoprotein A-1: implications for a lipid-free molten globular state. Proc Natl Acad Sci USA. 1996;93:2991–2995. doi: 10.1073/pnas.93.7.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X, Lei D, Zhang L, Rames M, Zhang S. A model of lipid-free apolipoprotein A-I revealed by iterative molecular dynamics simulation. PLoS One. 2015;10:e0120233. doi: 10.1371/journal.pone.0120233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gorshkova IN, Liu T, Kan HY, Chroni A, Zannis VI, Atkinson D. Structure and stability of apolipoprotein A-I in solution and in discoidal high-density lipoprotein probed by double charge ablation and deletion mutation. Biochemistry. 2006;45:1242–1254. doi: 10.1021/bi051669r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bortnick AE, Rothblat GH, Stoudt G, Hoppe KL, Royer LJ, McNeish J, Francone OL. The correlation of ATP-binding cassette 1 mRNA levels with cholesterol efflux from various cell lines. J Biol Chem. 2000;275:28634–28640. doi: 10.1074/jbc.M003407200. [DOI] [PubMed] [Google Scholar]
- 40.Favari E, Zanotti I, Zimetti F, Ronda N, Bernini F, Rothblat GH. Probucol inhibits ABCA1-mediated cellular lipid efflux. Arterioscler Thromb Vasc Biol. 2004;24:2345–2350. doi: 10.1161/01.ATV.0000148706.15947.8a. [DOI] [PubMed] [Google Scholar]
- 41.Hafiane A, Genest J. HDL-mediated cellular cholesterol efflux assay method. Ann Clin Lab Sci. 2015;45:659–668. [PubMed] [Google Scholar]
- 42.Besler C, Luscher TF, Landmesser U. Molecular mechanisms of vascular effects of High-density lipoprotein: alterations in cardiovascular disease. EMBO Mol Med. 2012;4:251–268. doi: 10.1002/emmm.201200224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terasaka N, Yu S, Yvan-Charvet L, Wang N, Mzhavia N, Langlois R, Pagler T, et al. ABCG1 and HDL protect against endothelial dysfunction in mice fed a high-cholesterol diet. J Clin Investig. 2008;118:3701–3713. doi: 10.1172/JCI35470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Besler C, Heinrich K, Rohrer L, Doerries C, Riwanto M, Shih DM, Chroni A, et al. Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease. J Clin Investig. 2011;121:2693–2708. doi: 10.1172/JCI42946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soumyarani VS, Jayakumari N. Oxidized HDL induces cytotoxic effects: implications for atherogenic mechanism. J Biochem Mol Toxicol. 2014;28:481–489. doi: 10.1002/jbt.21588. [DOI] [PubMed] [Google Scholar]
- 46.Solati Z, Ravandi A. Lipidomics of bioactive lipids in acute coronary syndromes. Int J Mol Sci. 2019;20:E1051. doi: 10.3390/ijms20051051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Recalde D, Velez-Carrasco W, Civeira F, Cenarro A, Gomez-Coronado D, Ordovas JM, Pocovi M. Enhanced fractional catabolic rate of apo A-I and apo A-II in heterozygous subjects for apo A-I(Zaragoza) (L144R) Atherosclerosis. 2001;154:613–623. doi: 10.1016/s0021-9150(00)00555-4. [DOI] [PubMed] [Google Scholar]
- 48.Daniil G, Phedonos AA, Holleboom AG, Motazacker MM, Argyri L, Kuivenhoven JA, Chroni A. Characterization of antioxidant/anti-inflammatory properties and apoA-I-containing subpopulations of HDL from family subjects with monogenic low HDL disorders. Clin Chim Acta. 2011;412:1213–1220. doi: 10.1016/j.cca.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 49.Dalla-Riva J, Lagerstedt JO, Petrlova J. Structural and functional analysis of the apolipoprotein A-I A164S variant. PLoS One. 2015;10:e0143915. doi: 10.1371/journal.pone.0143915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Assanasen C, Mineo C, Seetharam D, Yuhanna IS, Marcel YL, Connelly MA, Williams DL, et al. Cholesterol binding, efflux, and a PDZ-interacting domain of scavenger receptor-BI mediate HDL-initiated signaling. J Clin Investig. 2005;115:969–977. doi: 10.1172/JCI200523858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi Y, Lv X, Liu Y, Li B, Liu M, Yan M, Liu Y, et al. Elevating ATP-binding cassette transporter G1 improves re-endothelialization function of endothelial progenitor cells via Lyn/Akt/eNOS in diabetic mice. FASEB J. 2018;32:6525–6536. doi: 10.1096/fj.201800248RR. [DOI] [PubMed] [Google Scholar]
- 52.Marsche G, Levak-Frank S, Quehenberger O, Heller R, Sattler W, Malle E. Identification of the human analog of SR-BI and LOX-1 as receptors for hypochlorite-modified high density lipoprotein on human umbilical venous endothelial cells. FASEB J. 2001;15:1095–1097. doi: 10.1096/fj.00-0532fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.