

Abstract
We describe the unique structural features of a large telomere repeat DNA complex (TRDC) of >20 kb generated by a simple PCR using (TTAGGG)4 and (CCCTAA)4 as both primers and templates. Although large, as determined by conventional agarose gel electrophoresis, the TRDC was found to consist of short single-stranded DNA telomere repeat units of between several hundred and 3000 bases, indicating that it is a non-covalent complex comprising short cohesive telomere repeat units. S1 nuclease digestion showed that the TRDC contains both single- and double-stranded portions stable enough to survive glycerol density gradient centrifugation, precipitation with ethanol and gel electrophoresis. Sedimentation analysis suggests that a part of the TRDC is non-linear and consists of a three-dimensional network structure. After treatment with Werner DNA helicase the TRDC dissociated into smaller fragments, provided that human replication protein A was present, indicating that: (i) the TRDC is a new substrate for the Werner syndrome helicase; (ii) the telomere repeat sequence re-anneals rapidly unless unwound single-stranded regions are protected by replication protein A; (iii) the TRDC may provide a new clue to understanding deleterious telomere–totelomere interactions that can lead to genomic instability. Some properties of the TRDC account for the extra-chromosomal telomere repeat (ECTR) DNA that exists in telomerase-negative immortalized cell lines and may be involved in maintaining telomeres.
INTRODUCTION
Telomeres are special structures at the end of eukaryotic chromosomes and have been thought to protect the chromosome ends from degradation and fusion to other chromosomes (1). In most eukaryotes telomeric DNA consists of tandem repeats of G-rich sequences, for example TTAGGG in mammals and other vertebrates (2).
In humans all chromosome ends contain ∼5 kb of telomeric DNA (3) and telomeres shorten with each cell division in most somatic cells, resulting in genomic instability (4). However, cells that divide indefinitely, such as germline cells and tumor cells, maintain the length of their telomeres, suggesting that a telomere maintenance pathway is activated in these immotalized cells (5). Immortalized cells overcome telomere shortening by using telomerase, which is a kind of reverse transcriptase and adds TTAGGG repeats to the 3′-ends of chromosomes (6). Some immortalized cell lines, however, are devoid of telomerase and they maintain telomeres by an alternative mechanism (7,8). Recently, we and others found an extra-chromosomal telomere repeat (ECTR) DNA in telomerase-negative immortalized cell lines (9,10). Cloned ECTR DNA is composed of linear, double-stranded telomere repeat sequences alone. We argued that this unique DNA may have an important role in the mechanism of maintenance of telomeres without telomerase.
We also showed that the telomere dynamics of cells from patients with Werner syndrome (WS), a genomic instability disease caused by defective mutations in a DNA helicase gene (11), are abnormal (12). For instance, the telomeres of some B lymphoblastoid cell lines obtained from WS patients and transformed with Epstein–Barr virus (EBV) show a marked shortening or lengthening of telomeres compared with normal cell lines. Accordingly, we hypothesized that both ECTR DNA and Werner syndrome helicase (WRNp), the product of the causative WRN gene, are partly responsible for maintaining telomeres of EBV-transformed lymphoblasts (13).
WRNp belongs to a DNA helicase family of which the prototypical member is Escherichia coli RecQ (14). WRNp is a member of a family of five RecQ helicases in humans; two other members are the BLM and RTS helicases deficient in Bloom’s syndrome (15) and Rothmund-Thomson syndrome (16–18), respectively, both of which cause genomic instability. The remaining two human RecQ helicases are RecQL1 (19) and RecQL5 (20), whose correlation with human disease is unknown. All RecQ helicases are considered to be involved in maintaining genomic integrity, although their physiological role(s) remains to be clarified. Among the other eukaryotic helicases in the RecQ family are Saccharomyces cerevisiae Sgs1 (21) and Schizosaccharomyces pombe Rqh1 (22), helicases that are important for genomic stability in yeast cells. The RecQ family helicases are to some extent functional homologs, as both human BLM helicase and WRNp can suppress the hyper-recombination phenotype of an Sgs1 mutant (23).
As a model of ECTR DNA, we here propose a non-covalent large telomere repeat DNA complex (TRDC) that can be generated by simple PCR using (TTAGGG)4 and (CCCTAA)4. The TRDC shows large molecular sizes in the conventional agarose gel electrophoresis used to determine telomere size, but consists of short single-stranded (ss)DNA telomere repeat units. We also show that the TRDC can be dissociated into smaller units by incubation with WRNp provided that replication protein A (RPA) is also present, indicating that telomere repeat sequences in the TRDC are re-annealed rapidly unless unwound single-stranded regions are simultaneously protected by RPA. From the unique nature of the G-rich TRDC we speculate possible roles of telomeres, ECTR DNA and WRNp in the maintenance of genomic stability.
MATERIALS AND METHODS
Preparation of ECTR-like DNA
ECTR-like DNA, i.e. telomere-like DNA, was made by PCR using (TTAGGG)4 and (CCCTAA)4 as both primers and templates, essentially by the method described by Ijdo et al. (24). Unless otherwise mentioned, the standard reaction mixture (50 µl) contained 5 pmol (TTAGGG)4 and (CCCTAA)4, 5 µl of 10× Pfu buffer (Stratagene, La Jolla, CA), 4 µl of 2.5 mM dNTP, 2.5 µl of 80% glycerol and 0.5 µl of Pfu polymerase (2.5 U/ml). The 10× Pfu buffer contained 200 mM Tris–HCl, pH 8.0, 20 mM MgCl2, 100 mM KCl, 60 mM (NH4)2SO4, 1% Triton X-100 and 0.1% nuclease-free bovine serum albumin (BSA). The thermal cycle conditions were initial denaturation at 94°C for 1 min followed by 10–60 cycles of denaturation at 94°C for 30 s, annealing at 60°C for 30 s and extension at 72°C for 90 s. The resulting DNAs were purified by phenol/chloroform extraction and precipitated using ethanol.
Gel electrophoresis
The PCR reaction products were separated by neutral agarose gel electrophoresis to separate double-stranded (ds)DNA or alkaline agarose gel electrophoresis to separate ssDNA. For neutral agarose gel electrophoresis DNA samples were applied to a 0.8% agarose gel containing TAE buffer (40 mM Tris base, 20 mM glacial acetic acid and 1 mM EDTA) and electrophoresed at 8 V/cm. After electrophoresis the gel was stained with ethidium bromide and photographed using UV illumination. For alkaline agarose gel electrophoresis DNA samples were applied to a 0.8% agarose gel containing 50 mM NaOH and 1 mM EDTA and electrophoresed in this solution at 8 V/cm (25). After electrophoresis the gel was soaked for 45 min in 1 M Tris–HCl, pH 7.6, 1.5 M NaCl, stained with SYBR Green II (Takara) and detected with a FluoroImager SI (Molecular Dynamics).
Glycerol density gradient centrifugation
The PCR product was separated using density gradient centrifugation at 36 000 r.p.m. for 18 h using a SW41 rotor (Beckman). We used a linear gradient of 5–30% glycerol in 50 mM Tris–HCl buffer, pH 7.6, containing 0.15 M NaCl and 5 mM MgCl2. A total of 10 aliquots each containing 1.2 ml were fractionated. To monitor the sedimentation coefficient, yeast tRNA (Gibco BRL), 16S rRNA and 23S rRNA were separated using the same conditions as for PCR product separation.
S1 nuclease digestion
The TRDC purified by glycerol density gradient centrifugation was digested with S1 nuclease in 30 mM sodium acetate buffer, pH 4.6, containing 280 mM NaCl and 1 mM ZnSO4. Between 200 ng and 1 µg DNA was digested at 37°C for 30 min with 1–100 U S1 nuclease (Takara). The PCR product, as well as SmaI digests of plasmid pUC18 (Takara) as a reference for double-stranded DNA and plasmid M13mp18 (Takara) as a reference for single-stranded DNA, were used as DNA substrates.
Unwinding of the TRDC by purified WRN helicase and RPA
Recombinant WRNp (26) and human RPA (27) were purified as described previously. The substrate DNA including the TRDC (1 µg) was incubated at 37°C for 60 min with or without 25 ng WRNp, 1 µg human RPA or both in a reaction mixture (20 µl) containing 50 mM Tris–HCl, pH 7.5, 1 mM MgCl2, 2 mM ATP, 0.5 mg/ml BSA and 2 mM 2-mercaptoethanol. As a control, the TRDC was boiled for 5 min and immediately chilled on ice with and without human RPA.
RESULTS
The TRDC formed by simple PCR consists of small telomere DNA repeat units
ECTR DNA exists in telomerase-negative immortalized cell lines (9,10). To study the physicochemical properties of ECTR DNA, we synthesized ECTR-like DNA by PCR. The sequences (TTAGGG)4 and (CCCTAA)4 acted as both primers and templates and telomere repeats were elongated by PCR. Figure 1 shows the effect of increasing the number of PCR cycles on the length of the elongated telomere repeat sequences. The sizes of the products measured by 0.8% neutral agarose gel electrophoresis increased as the number of PCR cycles increased. After 30 cycles most elongated DNAs were >12 kb in size, and after 50 cycles the majority of the product DNAs appeared to be too large to penetrate the agarose gel matrix (Fig. 1A). However, the ssDNA component in the same DNA products was surprisingly short, from several hundred to 3000 bases, after treatment with alkali and 0.8% alkaline agarose gel electrophoresis to measure the size (Fig. 1B). The data clearly indicate that the high molecular weight DNAs found by neutral agarose gel electrophoresis consisted of complex non-covalent pairings of short ssDNA pieces; we refer to this high molecular weight DNA complex as the TRDC.
Figure 1.
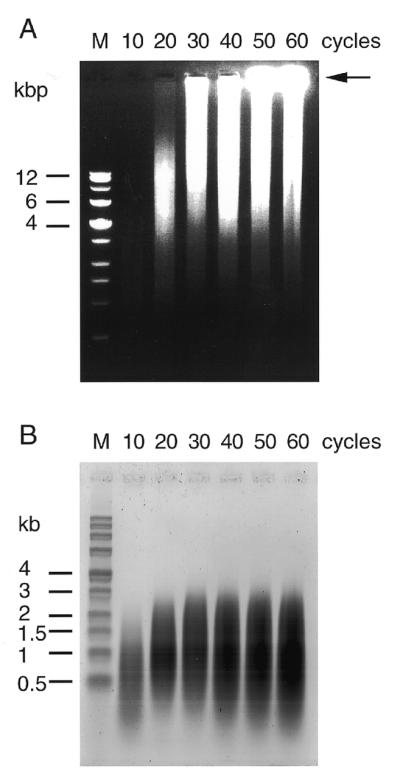
Analysis of PCR products by agarose gel electrophoresis. The PCR products were separated by 0.8% agarose gel electrophoresis in (A) a Tris/acetate/EDTA buffer (pH 7.0) (neutral conditions) or (B) an alkaline buffer containing 60 mM NaOH and 1 mM EDTA (alkaline conditions).
Sedimentation analysis of the TRDC
We examined the molecular sizes of the TRDC produced after 50 PCR cycles by glycerol density gradient centrifugation (Fig. 2A). The size of the TRDC ranged widely, from ∼4 to 23 S, as shown by tRNA and rRNA markers used as references, indicating that the size of the TRDC was heterogeneous; some of the TRDC was extremely large and precipitated at the bottom of the centrifugation tube. When the size of each TRDC fraction was measured by neutral agarose gel electrophoresis they ranged widely from 1 to 8 kb. In particular, the TRDC in fractions 8–10 contained large DNA complexes remaining at the origin of the gel, which was most pronounced in fraction 10 in which most DNA components were huge, were precipitated during centrifugation in the glycerol density gradient and could not penetrate the 0.8% agarose gel matrix (Fig. 2B, fraction 10, arrowhead). Regardless of the large variations in the sizes of the TRDC molecules, the sizes of the ssDNA telomere repeats were almost the same (Fig. 2C). These results clearly indicate that large TRDCs comprise smaller telomere unit sequences aggregating with each other to form a large DNA complex. The results also showed that the TRDC is stable and can survive ultracentrifugation at physiological salt concentrations, although the TRDC consisted of non-covalently linked multiple ssDNA telomere repeat components.
Figure 2.

Analysis of PCR products separated by glycerol density gradient ultracentrifugation. (A) Separation of PCR products by glycerol density gradient ultracentrifugation. The numbers 1–10 are fractions each containing 1.2 ml corresponding to the gradient from 5 to 30% glycerol. (B) The PCR products were separated under neutral or (C) alkaline conditions as in the legend to Figure 1.
Optimal conditions for large TRDC formation
To determine conditions by which to manipulate the sizes of the TRDC and component ssDNAs, PCR was carried out with varying concentrations of (TTAGGG)4/(CCCTAA)4 while keeping the dNTP concentration constant. The lower the concentration of (TTAGGG)4/(CCCTAA)4, the larger the sizes of both the TRDC and component ssDNA as measured by neutral agarose gel electrophoresis (Fig. 3A) and alkaline agarose gel electrophoresis (Fig. 3B), respectively; the highest efficiency for formation of a large TRDC was obtained with ≤0.4 µM each (TTAGGG)4 and (CCCTAA)4 per 0.2 mM each of the four dNTPs. These results indicate that longer component ssDNAs are necessary to form a larger TRDC. Moreover, a lower concentration of (TTAGGG)4/(CCCTAA)4, which is both the primer and template for the reaction, produced longer component ssDNA products, because of the greater frequency of elongated primer/template in the mutiple cycles of the PCR reaction. In contrast, a high concentration of (TTAGGG)4/(CCCTAA)4 apparently competes for annealing between elongated products, resulting in the generation of smaller component ssDNA units. Although biologically unimportant, similar large and stable DNA complexes can be formed artificially by PCR with GC-rich or tandem GC-containing primers, such as (AGCGCTA)4 and (TTCGAA)4, but not with GC-less primers (data not shown), because of strong GC base pairing and G-G interactions.
Figure 3.

Correlation between the size of PCR products and their telomere repeat units. The correlation was determined when the concentration of (TTAGGG)4/(CCCTAA)4 was increased while the concentrations of dNTPs were kept constant. The numbers (0.4–400 µM) at the top indicate molar concentration ratios of (TTAGGG)4/(CCCTAA)4 per dNTP: the concentrations of dNTP were kept at 0.2 mM. (A) The PCR products were separated under neutral or (B) alkaline conditions as in the legend to Figure 1.
TRDC is partially sensitive to S1 nuclease
To understand the overall structure, purified TRDC from glycerol gradient centrifugation fraction 5 (Fig. 2A) was incubated with 1–100 U S1 nuclease and the reaction mixtures were analyzed by neutral agarose gel electrophoresis (Fig. 4). As controls, pUC18 dsDNA and M13mp18 ssDNA were similarly treated. Under these conditions the TRDC was digested slightly at 1 U, but showed nearly 50 and 90% digestion at 10 and 100 U, respectively. Under the same conditions, linear duplex pUC18 DNA, obtained by SmaI digestion, was completely resistant to 1 and 10 U S1 nuclease, while M13mp18 ssDNA was completely digested with 1 U S1 nuclease. These results suggest that the TRDC contains both single- and double-stranded portions in its structure.
Figure 4.

The effect of S1 nuclease on the PCR product. The PCR product, as well as M13mp18 (single-stranded) DNA and SmaI digests of pUC18 DNA, were digested with S1 nuclease. The top numbers indicate the amounts of S1 nuclease added per tube. DNAs were separated by 0.8% agarose gel electrophoresis under neutral conditions as in the legend to Figure 1.
Dissociation of the TRDC into smaller fragments after treatment with WRNp
To examine whether WRNp, which efficiently unwinds duplex DNAs and RNA/DNA heteroduplexes containing single-stranded regions (26), can dissociate the TRDC into smaller fragments, we incubated the TRDC with purified WRNp together with or without human RPA, which binds to the dissociated ssDNA and prevents re-annealing. After the treatments the reaction products were analyzed by agarose gel electrophoresis under both neutral and alkaline conditions. Denaturation of the TRDC with an apparent molecular size of 23 kb by boiling and subsequent chilling alone did not affect the migration profile in the neutral gel, perhaps because the dissociated telomere repeat ssDNAs re-annealed during chilling (Fig. 5A). In contrast, when the TRDC was boiled and chilled in the presence of RPA, the migration profile changed dramatically. Here, much of the TRDC were dissociated into ssDNAs that stained weakly with ethidium bromide and were dispersed in the gel, shifting part of the TRDC to a fraction of diminished molecular size (6.6 kb). Co-incubation of the TRDC with RPA alone yielded no mobility change in the TRDC: the major part of the TRDC stayed at the origin of the neutral agarose gel. Also, no mobility change was found when the TRDC was incubated with WRNp alone. Lastly, when the TRDC was incubated with WRNp in the presence of RPA, most of the DNA in the original TRDC dispersed in the gel as faint smears, leaving small TRDC repeats (6.6 kb), but no TRDC at the origin of the gel. The mobility change of the TRDC after co-incubation with WRNp and RPA was essentially the same as when the TRDC was treated by heating and chilling in the presence of RPA. These findings prompted us to speculate that the major part of the TRDC was dissociated by WRNp into component ssDNAs that then were bound by RPA proteins and migrated differentially in the gels depending on the size of the telomere ssDNA–RPA complex. The results are also consistent with the observation by Brosh et al. (28) that WRNp can catalyze the unwinding of long duplex DNA substrates in a reaction dependent on human RPA. Most component ssDNAs remained intact during these studies (Fig. 5B), in which the same amount of TRDC was analyzed by alkaline agarose gel electrophoresis. Is this dissociation of TRDC to ssDNA specific for WRNp? To answer this question we tested E.coli DNA helicase II (Wako) and SV40 large T antigen helicase (HIMERx; Madison), which do not belong to the RecQ helicase family, under the same reaction conditions in vitro and found that they could also dissociate the TRDC in the presence of RPA or gene 32 protein of λ phage, but less efficiently than WRNp (data not shown).
Figure 5.
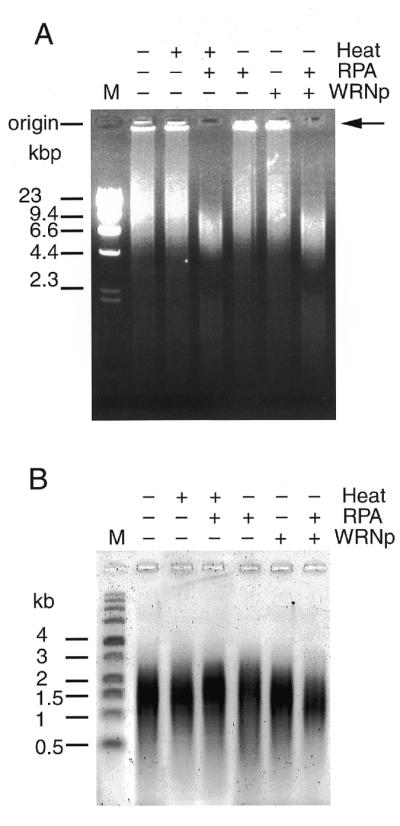
Unwinding of the TRDC by WRN helicase and RPA. (A) TRDC was incubated with WRNp with or without RPA. + indicates boiling, RPA treatment or WRNp treatment, while – indicates none of these. The products were analyzed by 0.8% neutral agarose gel electrophoresis. (B) The same amounts of incubation mixture were analyzed for ssDNA by alkaline agarose gel electrophoresis.
DISCUSSION
We have shown in this study that telomere repeat DNA fragments produced by PCR using (TTAGGG)4/(CCCTAA)4 as primers and templates form large DNA complexes, referred to here as the TRDC. Although the TRDC is stable under the low salt conditions (40 mM Tris–acetate buffer, pH 8.5) used with agarose gel electrophoresis at room temperature or at the physiological salt concentrations (0.15 M NaCl) used in ultracentrifugation, it unwound and dissociated into smaller component units when treated with WRNp together with RPA or under alkaline conditions (50 mM NaOH). Figure 6 shows two models for the possible structure of the TRDC, a network structure (Fig. 6A) and a linear sturucture (Fig. 6B). The following observations and speculations support the view that the TRDC forms a network structure: (i) significant portions of the TRDC precipitate on ultracentrifugation in a glycerol gradient (Fig. 2A) or on centrifugation in a tabletop centrifuge (∼20 000 g for 15 min), conditions that do not precipitate linear DNAs; (ii) the TRDC always clogged the fine needles (≤0.1 µm diameter) used for microinjection into cells (our unpublished observation), while linear DNAs do not clog the needles. The linear structure (Fig. 6B), however, can explain some of the structural features of the TRDC, but it is assumed to have many short single-stranded side chains which it may be difficult to prevent from forming intra- or inter-molecular links with side chains of other single-stranded portions of the TRDC, resulting in a network structure similar to that shown in Figure 6A.
Figure 6.

Network (A) and linear (B) models of the TRDC. Two extreme model structures are shown for the TRDC, consisting of multiple single-strand telomere components.
Previously we and others found that the N-terminal portion of WRNp contains an integral exonuclease activity (29–31). We also showed that the 5′→3′ exonuclease activity is dependent on duplex unwinding and that it digests unwound strands efficiently when the free 5′-end is close to the unwound region, but less efficiently when the free 5′-end is distal from the unwound region (29). In unwinding of the TRDC no apparent decrease in ssDNA component units was noticeable (Fig. 5B), indicating that the TRDC is not a good substrate for WRN exonuclease. Perhaps WRNp predominantly unwinds the TRDC from internal single-stranded loop regions, which were confirmed by sensitivity to S1 nuclease.
The G-rich sequences contained in telomere and rDNA repeats have the unique physicochemical property of forming guanine–guanine (G-G) interactions, distinguishing them from other cellular nucleotide sequences. These G-G interactions promote the association of DNA or RNA strands that contain runs of three or more consecutive guanine residues (32). The structures formed by G-G pairing are distinguished by strand stoichiometry, strand orientation and conformation of the glycosidic bonds, including four-stranded parallel G4 DNA or antiparallel two-stranded G2′ DNA (reviewed in 32).
Recently Sun et al. (33) showed that both human BLM helicase and yeast Sgs1 helicase can efficiently unwind DNAs in which G-G pairing stabilizes interstrand interactions. Extended studies by Sun et al. (34) demonstrated that Sgs1 helicase unwinds G-G paired yeast telomere-like sequences at least 10-fold more efficiently than it unwinds standard Watson–Crick duplex DNA, suggesting that one function of Sgs1 (and BLM helicase) may be to resolve the G4 DNA formed by G-G pairing in telomeres and eventually to prevent telomere–telomere interactions that can lead to chromosome non-disjunction in vivo. Similar G-G pairings and G4 DNA formation are predicted to exist in the G-rich TDRC, consisting of human telomeric sequences, described in this study. The capability of WRNp to dissociate the G-rich TDRC implies that WRNp may have a similar function to that deduced for Sgs1 and BLM helicases. Related to this is the fact that human WRNp unwinds tetrahelical structures of the fragile X syndrome repeat sequence d(CGG)n (35). Our previous findings that WRNp and BLM helicase complement the defects of an Sgs1 mutation in yeast cells (23), which results in hyper-recombination, also support this view.
The physicochemical nature of the telomere repeat sequence that forms the TRDC described in this paper may resolve other enigmas associated with the dynamics of telomeres that may relate to the intriguing biological meaning of telomeres at the ends of chromosomes. Previously we reported that immortalized cell lines without detectable levels of telomerase activity have ECTR DNA. These cell lines are also characterized by having apparently long telomere restriction fragments (TRFs), as measured by neutral agarose gel electrophoresis (8,9). The telomeres of some of these cell lines, surprisingly, did not stain well by cycling oligonucleotide-primed in situ hybridization. This discrepancy could now be resolved in the light of the findings of this study; the apparent long TRF detected previously could have been the result of overestimating the telomere size due to formation of a TRDC-like complex between short TRFs and ECTR DNAs during electrophoresis. This could be due to the cohesive nature of telomere repeat DNA fragments that form larger complexes by self-annealing, as shown in this study. The findings of this study also raise the possibility that ECTR DNA is not simply linear, but more likely forms a DNA network, like the TRDC. These facts and speculation also support the idea that ECTR DNA may have an important role in maintaining the length of telomeres in telomerase-negative immortalized cell lines.
EBV-transformed lymphoblastoid cell lines (LCL) from WS patients show abnormal telomere dynamics (12). Most LCL cells have no or weak telomerase activity, although they have relatively long lifespans (on average 90 population doublings), and often have ECTR DNA (13). From these facts we previously argued that both ECTR DNA and WRNp may be involved in maintaining telomeres in LCLs obtained from normal individuals, but not in LCLs from WS patients that lack WRNp. From this study an obvious possibility is that short telomere sequences are released from ECTR DNA in the TRDC-like complex, which contribute to elongation of telomeres by homologous recombination. This speculation is supported by this study showing that WRNp together with RPA unwinds the TDRC and dissociates it into smaller TRFs and by our recent immunocytochemical study indicating that WRNp and RPA co-localize in the ECTR DNA complex in cells (our unpublished results).
Acknowledgments
ACKNOWLEDGEMENTS
We thank Drs A. Shimamoto, S.-J. Heo, S. Kitao and S. Sakamoto of the AGENE Research Institute for valuable discussions and encouragement. This work was supported by The Organization for Drug ADR Relief (R and D promotion and Product Review) of the Japanese Government.
REFERENCES
- 1.Blackburn E.H. (1991) Nature, 350, 569–573. [DOI] [PubMed] [Google Scholar]
- 2.Greider C.W. (1996) Annu. Rev. Biochem., 65, 337–365. [DOI] [PubMed] [Google Scholar]
- 3.Martens U.M., Zijlmans,J.M., Poon,S.S., Dragowska,W., Yui,J., Chavez,E.A., Ward,R.K. and Lansdorp,P.M. (1998) Nature Genet., 18, 76–80. [DOI] [PubMed] [Google Scholar]
- 4.Harley C.B., Futcher,A.B. and Greider,C.W. (1990) Nature, 345, 458–460. [DOI] [PubMed] [Google Scholar]
- 5.de Lange T. (1995) In Greider,I.E.H.B. and Greider,C.W. (eds), Telomeres. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, pp. 69–106.
- 6.Counter C.M., Avilion,A.A., LeFeuvre,C.E., Stewart,N.G., Greider,C.W., Harley,C.B. and Bacchetti,S. (1992) EMBO J., 11, 1921–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bryan T.M. and Reddel,R.R. (1997) Eur. J. Cancer, 33, 767–773. [DOI] [PubMed] [Google Scholar]
- 8.Yeager T.R., Neumann,A.A., Englezou,A., Huschtscha,L.I., Noble,J.R. and Reddel,R.R. (1999) Cancer Res., 59, 4175–4179. [PubMed] [Google Scholar]
- 9.Tokutake Y., Matsumoto,T., Maeda,S., Watanabe,T., Tahara,H., Sakamoto,S., Niida,H., Sugimoto,M., Ide,T. and Furuichi,Y. (1998) Biochem. Biophys. Res. Commun., 247, 765–772. [DOI] [PubMed] [Google Scholar]
- 10.Ogino H., Nakabayashi,K., Suzuki,M., Takahashi,E., Fujii,M., Suzuki,T. and Ayusawa,D. (1998) Biochem. Biophys. Res. Commun., 248, 223–227. [DOI] [PubMed] [Google Scholar]
- 11.Yu C.E., Oshima,J., Fu,Y.H., Wijsman,E.M., Hisama,F., Alisch,R., Matthews,S., Nakura,J., Miki,T., Ouais,S., Martin,G.M., Mulligan,J. and Schellenberg,G.D. (1996) Science, 272, 258–262. [DOI] [PubMed] [Google Scholar]
- 12.Tahara H., Tokutake,Y., Maeda,S., Kataoka,H., Watanabe,T., Satoh,M., Matsumoto,T., Sugawara,M., Ide,T., Goto,M., Furuichi,Y. and Sugimoto,M. (1997) Oncogene, 15, 1911–1920. [DOI] [PubMed] [Google Scholar]
- 13.Sugimoto M., Ide,T., Goto,M. and Furuichi,Y. (1999) Mech. Ageing Dev., 107, 51–60. [DOI] [PubMed] [Google Scholar]
- 14.Nakayama H., Nakayama,K., Nakayama,R., Irino,N., Nakayama,Y. and Hanawalt,P.C. (1984) Mol. Gen. Genet., 195, 474–480. [DOI] [PubMed] [Google Scholar]
- 15.Ellis N.A., Groden,J., Ye,T.Z., Straughen,J., Lennon,D.J., Ciocci,S., Proytcheva,M. and German,J. (1995) Cell, 83, 655–666. [DOI] [PubMed] [Google Scholar]
- 16.Kitao S., Shimamoto,A., Goto,M., Miller,R.W., Smithson,W.A., Lindor,N.M. and Furuichi,Y. (1999) Nature Genet., 22, 82–84. [DOI] [PubMed] [Google Scholar]
- 17.Kitao S., Lindor,N.M., Shiratori,M., Furuichi,Y. and Shimamoto,A. (1999) Genomics, 61, 268–276. [DOI] [PubMed] [Google Scholar]
- 18.Lindor N.M., Furuichi,Y., Kitao,S., Shimamoto,A., Arndt,C. and Jalal,S. (2000) Am. J. Med. Genet., 90, 223–228. [DOI] [PubMed] [Google Scholar]
- 19.Seki M., Miyazawa,H., Tada,S., Yanagisawa,J., Yamaoka,T., Hoshino,S., Ozawa,K., Eki,T., Nogami,M., Okumura,K., Taguchi,H., Hanaoka,F. and Enomoto,T. (1994) Nucleic Acids Res., 22, 4566–4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimamoto A., Nishikawa,K., Kitao,S. and Furuichi,Y. (2000) Nucleic Acids Res., 28, 1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gangloff S., McDonald,J.P., Bendixen,C., Arthur,L. and Rothstein,R. (1994) Mol. Cell. Biol., 14, 8391–8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart E., Chapman,C.R., Al-Khodairy,F., Carr,A.M. and Enoch,T. (1997) EMBO J., 16, 2682–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamagata K., Kato,J., Shimamoto,A., Goto,M., Furuichi,Y. and Ikeda,H. (1998) Proc. Natl Acad. Sci. USA, 95, 8733–8738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ijdo J.W., Wells,R.A., Baldini,A. and Reeders,S.T. (1991) Nucleic Acids Res., 19, 4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
- 26.Suzuki N., Shimamoto,A., Imamura,O., Kuromitsu,J., Kitao,S., Goto,M. and Furuichi,Y. (1997) Nucleic Acids Res., 25, 2973–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brush G.S., Kelly,T.J. and Stillman,B. (1995) Methods Enzymol., 262, 522–546. [DOI] [PubMed] [Google Scholar]
- 28.Brosh R.M. Jr, Orren,D.K., Nehlin,J.O., Ravn,P.H., Kenny,M.K., Machwe,A. and Bohr,V.A. (1999) J. Biol. Chem., 274, 18341–18350. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki N., Shiratori,M., Goto,M. and Furuichi,Y. (1999) Nucleic Acids Res., 27, 2361–2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang S., Li,B., Gray,M.D., Oshima,J., Mian,I.S. and Campisi,J. (1998) Nature Genet., 20, 114–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen J.C., Gray,M.D., Oshima,J., Kamath-Loeb,A.S., Fry,M. and Loeb,L.A. (1998) J. Biol. Chem., 273, 34139–34144. [DOI] [PubMed] [Google Scholar]
- 32.Williamson J.R. (1994) Annu. Rev. Biophys. Biomol. Struct., 23, 703–730. [DOI] [PubMed] [Google Scholar]
- 33.Sun H., Karow,J.K., Hickson,I.D. and Maizels,N. (1998) J. Biol. Chem., 273, 27587–27592. [DOI] [PubMed] [Google Scholar]
- 34.Sun H., Bennett,R.J. and Maizels,N. (1999) Nucleic Acids Res., 27, 1978–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fry M. and Loeb,L.A. (1999) J. Biol. Chem., 274, 12797–12802. [DOI] [PubMed] [Google Scholar]