

Abstract
Cortical bone structure is a crucial determinant of bone strength, yet for many years studies of novel genes and cell signalling pathways regulating bone strength have focused on the control of trabecular bone mass. Here we focus on mechanisms responsible for cortical bone development, growth, and degeneration, and describe some recently described genetic-driven modifications in humans and mice that reveal how these processes may be controlled. We start with embryonic osteogenesis of preliminary bone structures preceding the cortex and describe how this structure consolidates then matures to a dense, vascularised cortex containing an increasing proportion of lamellar bone. These processes include modelling-induced, and load-dependent, asymmetric cortical expansion, which enables the cortex’s transition from a highly porous woven structure to a consolidated and thickened highly mineralised lamellar bone structure, infiltrated by vascular channels. Sex-specific differences emerge during this process. With aging, the process of consolidation reverses: cortical pores enlarge, leading to greater cortical porosity, trabecularisation and loss of bone strength. Each process requires co-ordination between bone formation, bone mineralisation, vascularisation, and bone resorption, with a need for locational-, spatial- and cell-specific signalling pathways to mediate this co-ordination. We will discuss these processes, and a number of cell-signalling pathways identified in both murine and human genetic studies to regulate cortical bone mass, including signalling through gp130, STAT3, PTHR1, WNT16, NOTCH, NOTUM and sFRP4.
Keywords: Cortical bone, Cortical porosity, Bone strength, Bone growth, sFRP4, Notum
Introduction
Cortical bone is a toughened compact structure, infiltrated with blood vessels, that forms the outer boundary of every element of the skeleton. It houses the bone marrow, and its structure both enables movement and provides protection to internal organs. Surprisingly, despite its importance, the cellular and genetic control mechanisms underlying the development, growth and maintenance of cortical bone have received far less attention than mechanisms contributing to the internal honeycomb-like (trabecular) structure of bone. Over many years, detailed study of trabecular structure and how it is disrupted in genetically altered mice and human mutations has been instrumental in the development of current therapies for skeletal fragility, but similar studies of cortical bone have been rare. In this review, we describe a framework for cortical bone by describing the cellular mechanisms controlling its genesis during embryonic development, its consolidation, expansion and shaping during growth, and its degeneration with age. For each of these stages, we provide examples of experimental studies in rodent models (Table 1), and descriptions of human syndromes (Table 2) with genetic or other modifications associated with disrupted cortical structure. We discuss how these studies might shed light on the intercellular and intracellular signals influencing the cortex. A better understanding of the control of cortical structure is likely to inform future approaches to strengthen cortical bone in conditions of skeletal fragility.
Table 1.
Examples of genetically altered mouse models with defective cortical structure; see text for further details
| Condition | Gene responsible | Bone width, length | Cortical thickness, porosity and maturity | Trabecular bone mass | References |
|---|---|---|---|---|---|
| Embryonic and early postnatal cortical defects | |||||
| Delayed chondrocyte differentiation | Pth1r | Width not recorded, short | Thick and immature | Low | [8, 136] |
| Accelerated chondrocyte differentiation | Pthlh | Width not recorded, short | Normal thickness, more mature | Low | [8, 136–138] |
| Osteopetrosis | Src | Erlenmeyer defect, short | Thin, immature | High | [10] |
| Osteopetrosis | Ctsk | Erlenmeyer defect, short | Thin, immature, porous later in life | High | [11, 72, 139] |
| Impaired angiogenesis | Vegf | Narrow, short | Thin | Low | [12] |
| Muscle paralysis in utero | Myf5:MyoD | Round cross-section | Normal thickness | Not reported | [13] |
| Maternal uterine muscle paralysis | Cacng1 | Round cross-section | Normal thickness | Not reported | [5] |
| Maternal PTHrP | Pthlh | Wide, normal length | Normal thickness | High (males) | [14] |
| Osteoblast-targeted G protein deletion | Gnas | Narrow, normal length | Thin, porous, immature | Low | [9] |
| Defective cortical consolidation | |||||
| Elevated STAT3 signalling in osteocytes | Socs3 | Normal width and length | Thick and porous in metaphysis | High early, low in adulthood | [32] |
| Constitutively active PTH receptor | Pth1r | Short, bowed limbs | Thin | High | [35] |
| Osteocyte-targeted constitutively active PTH receptor | Pth1r | Wide, normal length | Thick and porous | High early | [36, 37] |
| Hajdu–Cheney gene mutation in Notch2 | Notch2 | Short, narrow (initially, then normalises) | Thin and porous | Low | [42] |
| Osteoblast/osteocyte targeted Notch1 activation | Notch1 | Short, wide (osteocyte-targeted only) | Thick and porous | High | [43] |
| Wnt16 deletion | Wnt16 | Normal length, narrow | Thin | Normal | [44, 46] |
| Osteoblast and osteocyte targeted Wnt16 deletion | Wnt16 | Normal length, narrow (osteoblast-targeted only) | Thin, porous (osteoblast-targeted only) | Normal | [44] |
| Osteoblast-targeted Wnt16 overexpression | Wnt16 | Slightly shorter (female only), width not reported | Thick | High | [48] |
| Chondrocyte-targeted VEGFA overexpression | Vegfa | Not reported | Porous | High | [49] |
| Mineral defect with no defect in cortical thickness | |||||
| Hypermineralised cortex | EphrinB2 | Normal length and width | Normal, more mineralised | Normal | [58] |
| Impaired periosteal expansion | |||||
| Osteoblast progenitor-targeted BMP2 deletion | Bmp2 | Slender, slight reduction in length | Normal, porous, immature | Normal | [70] |
| Greater bone width | |||||
| Osteoblast- and osteocyte-targeted gp130 deletion | Il6st | Wide, normal length | Normal, excess woven bone | Low | [31, 78] |
| Osteocyte-targeted STAT3 deletion | Stat3 | Wide, length not reported | Normal thickness | Normal | [81] |
| Osteoblast-targeted connexin 43 deletion | Gja1 | Wide | Thin, excess woven bone [84]; normal [83] | Normal | [83, 84] |
| Osteocyte-targeted connexin 43 deletion, or deletion of the connexin 43 C-terminus | Gja1 | Wide | Normal, greater crystallinity and D spacing | Normal | [83, 85, 87] |
| Notum+/− | Notum | Wide, normal length | Thick | Normal | [91] |
| Osteoblast lineage NOTUM deletion | Notum | Wide, normal length | Thick | Normal | [90] |
| Induced systemic NOTUM deletion | Notum | Wide, normal length | Thick | Normal | [90] |
| Osteoblast lineage FGFR1/2 deletion | Fgfr1 / Fgfr2 | Wide, length not reported | Thick | High | [92] |
| Germline sFRP4 deletion | Sfrp4 | Wide, length not reported | Thin | High | [102, 103] |
| Estrogen receptor alpha deletion early in osteoblast lineage | Esr1 | Short, narrow | Thin | Normal | [118] |
| Increased cortical porosity developed after initial formation of cortex | |||||
| Decreased osteoblast and osteocyte apoptosis | Bak, Bax | Width not reported, normal length | Thick, porous | High | [128] |
| Adenine-induced kidney disease | Diet-induced | Not reported | Porous | Not reported | [130] |
| Osteoblast-targeted VEGF-C overexpression | Vegfc | Not reported | Porous | Not reported | [135] |
n/a indicates that data have not been recorded. Note that some papers do not record bone width, but record cross-sectional area, or periosteal circumference. Mice are listed in the same order as in the text
aEarly postnatal phenotype not reported
Table 2.
Some examples of human conditions with defective cortical structure
| Condition | Cause | Cortical defect | Trabecular defect | References |
|---|---|---|---|---|
| Caffey disease | COL1A1 | Hyperostosis. Local inflammation with subperiosteal new lamellar bone formation | Unknown | [15] |
| Jansen’s metaphyseal chondrodysplasia | PTH1R | Cortical bone erosion | Diffuse demineralization, rickets-like metaphyseal changes | [140] |
| Hajdu–Cheney syndrome | NOTCH2 | Thin cortical bone | Osteoporosis, focal bone lysis of distal phalanges | [41] |
| Camurati–Engelman disease | TGFB1 | Bilateral and symmetrical hyperostosis at the diaphysis | Unknown | [93] |
| Kenny–Caffey syndrome | TBCE (type 1), FAM111A (type 2) | Cortical thickness | Unknown | [96, 98] |
| Pyle’s disease | SFRP4 | Wide and thin cortices | Wide metaphysis with increased trabecular bone | [102] |
| Gorham Stout disease | Unknown | Osteolysis | Osteolysis | [134] |
Conditions are listed in the same order as in the text
Embryonic osteogenesis: initial formation of cortical and trabecular components
Osteogenesis (the formation of bone) begins during embryogenesis. Two distinct processes are responsible for forming the preliminary structures of cortical and trabecular bone: intramembranous ossification and endochondral ossification.
Flat bones, such as the skull, mandible, maxilla, and clavicles form by intramembranous ossification. In this process mesenchymal progenitors accumulate and differentiate directly into osteoblasts; these deposit collagen I-containing osteoid that becomes mineralised to form bone. Flat bones then grow by osteoblast differentiation and bone deposition at the periphery (periosteum) of the newly forming bone.
Most skeletal elements form by endochondral ossification. This is illustrated from two perspectives in Fig. 1, with the lower panel highlighting the appearance of a cross section of the cortex. Endochondral ossification, like intramembranous ossification, is also initiated by mesenchymal condensation, but a cartilaginous (chondrocyte) template is formed before bone formation commences. The small cartilage template of the future bone enlarges through chondrocyte proliferation and hypertrophy and their production of cartilage and gradually transforms to model into a larger mineralized bone by two processes: formation of the bone collar (see below), and formation of the primary ossification centre. The primary ossification centre forms when chondrocytes at the centre of the diaphysis become hypoxic, their lacunae become enlarged (hypertrophic), and the surrounding cartilage matrix accumulates mineral. This mineralized cartilage is invaded by blood vessels, which bring osteoclast and osteoblast precursors [1]. Subsequent osteoclast formation leads to resorption of the mineralized cartilage, thereby making space for blood vessels and expanding the marrow cavity; the first region of vascular invasion and marrow formation within each mineralized template is the “primary ossification centre”. Differentiated osteoblasts form osteoid on the remnant cartilage templates, which mineralizes to form bone.
Fig. 1.

Bone development, viewed as a longitudinal section (A) or a cross-section (B). During embryogenesis, mesenchymal stem cells condensate (A) and differentiate to form a cartilage model (anlagen) of the bone that is to form. As the bone grows, chondrocytes in the centre become hypertrophic and hypoxic, and release mineral, which accumulates within the cartilage; at the same time, new bone is deposited by osteoblasts on the perichondrium forming a porous “bone collar”. Blood vessels are drawn to the primary ossification centre, bringing osteoclast precursors, which resorb a space into which marrow forms. As the marrow expands, remnants of mineralized cartilage remain (shown in orange), and woven bone continues to form at the bone collar, making a porous pre-cortical structure, with infiltrating vasculature. A Shows the formation of the secondary ossification centres, and the regions of the immature long bone, including the growth plate (epiphysis), diaphysis and metaphysis. Shown in B: under the influence of uterine and embryonic muscle activity, the bone grows asymmetrically through the formation of struts and rings of new bone. As the bone continues to grow asymmetrically after birth (modelling), this preliminary bone structure is gradually reshaped through bone resorption and bone formation. Remnants of mineralized cartilage and woven bone are gradually removed and replaced with lamellar bone (remodelling), but some remain
Collagen is deposited rapidly by osteoblasts during these early stages of development, both in the primary ossification centre and in the bone collar. During this rapid phase, collagen fibres are deposited with an irregular orientation leading to the term “woven bone”. Over time, through remodelling, the woven bone is replaced with a more mechanically competent layered structure, termed lamellar bone, with parallel collagen fibres oriented in perpendicular planes in adjacent lamellae [2]. Replacement of woven bone with lamellar bone occurs in all bones as they mature, and although the cellular signals determining whether osteoblasts deposit woven or lamellar bone are unknown, cellular projections of osteoblasts on the bone surface appear to be involved [3].
Cortical and trabecular structures begin to emerge during this earliest stage of bone development. The precursor to diaphyseal cortex, termed the “bone collar” or “ring of Lacroix”, is formed by osteoblasts during vascular invasion of the perichondrium, a cellular condensation that emerges to surround the cartilage template [4]. The bone collar is initially a highly porous material. The diaphyseal cortex grows through periosteal bone formation; at these early stages, cortical growth occurs by formation of alternating struts and rings of woven bone, interspersed with blood vessels and marrow [5]. After birth, the highly porous woven bone precursor to cortical bone is remodelled to form a dense lamellar structure, which lacks the larger marrow containing spaces observed in neonates but retains an infiltrating vascular structure [6]. This process will be discussed below.
As the cartilaginous growth plates move apart from each other during longitudinal growth driven by hypertrophic enlargement of chondrocytes [7], remnants of mineralized cartilage remain (see Fig. 1). These are used as templates on which both cortical and trabecular bone form. As the bones grow, through cycles of bone remodelling, mineralized cartilage is gradually replaced with woven bone, which is then replaced by lamellar bone. In the embryo, particularly the murine embryo, the small amount of trabecular bone present is largely mineralized cartilage and woven bone. In the murine cortex, remnant mineralized cartilage and woven bone remain until at least early adulthood (Fig. 1).
Defects in embryonic cortical bone development
The dependence of embryonic cortical structure on appropriate chondrocyte differentiation is one explanation for how embryonic cortical structure is modified in genetically altered mouse models. One early example of this is the difference between embryonic mice lacking parathyroid-hormone-related protein (PTHrP), and those lacking its receptor (PTH1R), which is also used by parathyroid hormone (PTH) [8]. Both PTHrP and PTH1R null mice exhibit neonatal lethal phenotypes with short bones. In PTHrP null mice, chondrocyte differentiation is accelerated, leading to early mineralization, vascular invasion and early maturation of cortical bone compared with controls. In contrast, mice lacking PTH1R have delayed chondrocyte differentiation, later vascular invasion and a porous, poorly mineralized cortical structure, which is thicker than usual, likely due to defective calcium and phosphorus supply.
A change in cortical bone maturity in the early postnatal period was also noted in mice lacking the signalling G protein Gsα in osteoblasts, which mediates intracellular signalling by a range of receptors, including PTH1R. Mice lacking Gsα in osteoblasts exhibited narrow cortical bone, which was immature since it contained a greater quantity of woven bone than controls [9]. Osteoblasts therefore control the thickness and maturation of cortical bone even during early development.
Osteoclasts are also important for cortical development: if osteoclast-mediated invasion and destruction of the cartilage model is defective, cortical bone development is delayed. An example of this is in the syndrome of osteopetrosis, caused either by a lack of osteoclasts or by defective osteoclast activity. Severe osteopetrotic mouse models exhibit thin cortical bone soon after birth, and a marrow space filled with abundant trabecular bone mainly comprising mineralized cartilage. This is particularly evident in the src null mouse, which has extensive marrow spaces, even in the diaphyseal cortex, indicating a lack of cortical consolidation [10]. Even in conditions with mild osteoclast defects, such as the Cathepsin K null mouse, high cortical porosity is observed in early adulthood [11]. Initially this seems confusing: we would expect osteoblasts to mediate consolidation of the cortical structure. The effect of osteoclast suppression suggests cortical consolidation is driven by remodelling: the osteoblasts responsible for closing cortical pores may depend on prior resorption of the cortex by osteoclasts. Such a concept is supported by delayed formation of the bone collar and cortical consolidation in VEGF-deficient mice which have reduced osteoclast invasion due to defective vascular formation [12]. In contrast to the intracortical environment, src null mice exhibit normal periosteal diameter; this indicates bone width during embryogenesis is determined by modelling and is independent of osteoclast function [5].
Bone shape is at least partially determined by muscle forces experienced by the developing bone in utero. Muscle contractions of the developing embryo produce signals responsible for the formation of attachment sites, and bone curvature, as revealed by studies of mice lacking striated muscle (Myf5−/−: MyoD−/−) [13]. The maternal musculature is also important; in the absence of intrauterine muscle forces, embryos form bones with normal periosteal diameter, but the normal pattern of oval development, caused by formation of struts in a preferential direction is disrupted and the bone retain a circular cross-section [5].
Few studies of embryonic bone have assessed the cross-sectional area of skeletal elements, so very little is known about how radial bone growth is controlled in utero. We recently described a mouse model that develops wide bones of normal length during embryonic development [14]. Bone width can therefore be controlled independently of bone length in utero. While the communication pathway underlying this phenotype remains unknown, it is associated with accelerated mineralization due to impaired PTHrP production by the maternal decidua (the interface between the placenta and the uterine wall).
Few human syndromes have been described with specific defects in cortical development during embryogenesis. One possible example is infantile cortical hyperostosis (OMIM 114000), also known as Caffey disease, a rare disease affecting various skeletal elements and contiguous connective tissue [15]. Although a lethal prenatal form has been reported, the disease is usually self-limiting, and periosteal new bone formation leads to hyperostosis of the affected bones and swelling of the overlying soft tissue. Histologically, local inflammation along with subperiosteal new lamellar bone formation was observed. Autosomal dominant form of the disease is caused by a heterozygous missense mutation (R836C) in the type I collagen (COL1A1) gene. There are several type I collagen disorders, including osteogenesis imperfecta, but only Caffey disease is characterized by increased cortical bone formation. The underlying cause remains unknown.
Thus, bone width, cortical shape, and cortical bone porosity are controlled by multiple mechanisms during embryogenesis, from both the embryo and the maternal environment. Within the developing embryo, these include influences of chondrocyte development, osteoclast formation, vascularization, and muscle activity. The mother also influences embryonic development through intrauterine muscle contractions, by controlling the supply of calcium to the developing embryo, and preliminary studies now suggest some level of control of embryonic bone width through the decidua.
Consolidation of cortical bone during bone growth
During postnatal bone growth, cortical structure continues to evolve through three processes: consolidation of trabecular bone (i.e., corticalisation), transformation of the cortical structure from woven to lamellar bone, and accumulation of mineral by secondary mineralization.
During corticalisation, thin bony trabeculae arising from the growing cartilaginous growth plate coalesce at the periphery of the metaphysis to form the thickening cortical shell (Fig. 2) [16, 17]. At the junction of the growth plate and the nascent cortical bone is the groove of Ranvier, which provides both osteoblasts to the growing cortex and chondrocytes to the expanding growth plate. Thus, trabecular development at the metaphysis during childhood, rather than metaphyseal cortical morphology, is likely to determine cortical morphology in adulthood, thereby determining fragility and risk of fracture. For example, healthy girls and pre- and postmenopausal women with thinner or fewer trabeculae exhibit lower cortical area and higher cortical porosity [18, 19]. Cortical morphology during growth is generally correlated with longitudinal growth velocity, and differs according to the stage of puberty [20].
Fig. 2.
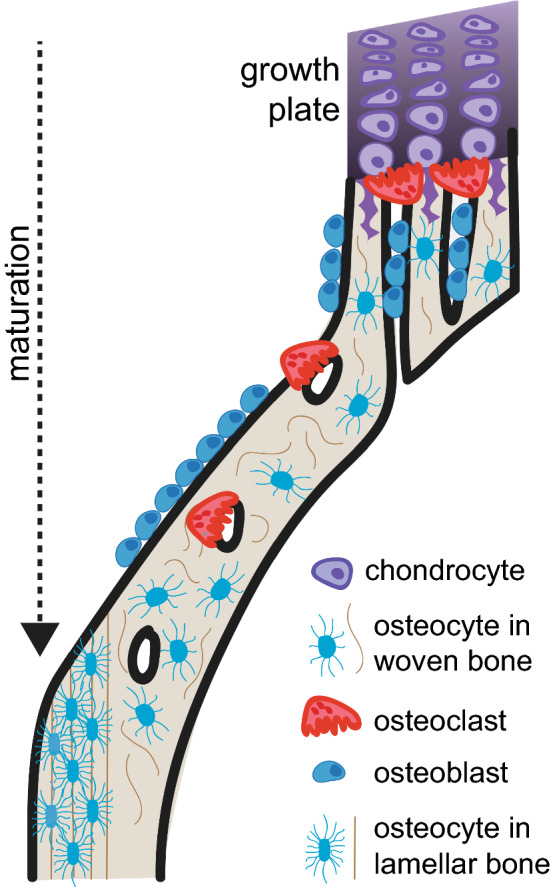
Metaphyseal consolidation of cortical bone. During long bone maturation, chondrocyte differentiation and hypertrophy occurs at the epiphyseal growth plates. During hypertrophy, osteoclasts begin to resorb the mineralised cartilage, leaving remnants of mineralised cartilage, on which osteoblasts form woven bone, thereby making trabeculae in the primary spongiosa. At the periphery of the bone, these trabeculae become consolidated to form thickened woven bone, with vascular pores, which may contain both osteoblasts and osteoclasts. The activity of osteoclasts within these pores must be suppressed for continued consolidation of the woven bone. Over time, the woven bone is replaced with lamellar bone, which contains flattened, aligned osteocytes; this is an asymmetric process. Here it is shown occurring on the endocortical surface
The replacement of woven bone with lamellar bone occurs by both modelling and remodelling in the cortex [16]. As the cortical bone becomes consolidated, it passes through a transitional state of “coarse, irregular whorls or convolutions” of compact bone, described by Enlow in 1962 [16]. This bone is gradually replaced with lamellar bone during asymmetric growth: new lamellar bone is deposited on both inner (endocortical) and outer (periosteal) surfaces of the cortex, depending on mechanical forces. On the opposite side of the cortex, woven endochondral bone and mineralized cartilage are resorbed by osteoclasts, leaving a central core of woven bone and cartilage (see Fig. 1). This asymmetric process is termed “modelling drift”, and is responsible for both bone enlargement and changes in bone shape during growth [21, 22] as well as age-associated changes in bone shape [23]. There may be multiple mechanisms of control for this, including: (1) signals from osteoclasts transmitted to osteoblasts on the other side of the cortical structure, termed osteotransmitters [24, 25], (2) independent responses of osteoblasts and osteoclasts to the mechanical loads they are experiencing, or (3) responses of mechanosensitive osteocytes sending specific signals in different directions to induce formation and resorption where it is needed.
It should be noted that large mammals, including humans, develop Haversian systems after the initial period of cortical consolidation. Haversian systems are an organised structure within the cortex of larger bones, characterised by bundles of longitudinal cylinders each containing central blood vessels surrounded by concentric layers of lamellar bone. During development, human metaphyseal bone, like murine bone, is composed of thin, porous, non-coalesced bone [26]. This is one reason why the border with the growth plate is a site of greater bone fragility in children [26]. The key difference in cortical development between small (non-Haversian) and large (Haversian) bone occurs after the transitional “coarse compact” bone phase. This suggests large and small mammals share similar initial steps in cortical bone development, and there may be similar signalling pathways at play.
After the woven and “coarse compact” bone phase, remodelling into lamellar Haversian systems occurs on intracortical surfaces (i.e., within the cortical structure). In adulthood, these Haversian systems continue to be remodelled, with intracortical blood vessels providing osteoclast and osteoblast precursors in osteonal cutting cones [16, 27]. It is often surmised that rodent bone is stagnant, and does not remodel after cortical shape is established, since it lacks Haversian systems. However, rodent cortical bone is heavily vascularised with trans-osteal (or transcortical) vessels [28, 29]. Since these vessels house both osteoclasts [29] and osteoblast progenitors [30], the murine cortex may also be remodelled on intracortical surfaces, even after consolidation. Consistent with this suggestion, murine bone continues to accrue mineral after the lamellar structure has formed [31].
Defects in cortical consolidation in the postnatal skeleton
How is cortical consolidation controlled? Certainly, osteoclast activity is required, since, as discussed above, osteopetrotic models exhibit delayed cortical consolidation. Chondrocytes may also play a role, since chondrocyte differentiation at the growth plate determines the generation of new bone at the metaphysis. Another contributor is the osteocyte. Delayed formation of dense cortical bone in the presence of normal chondrocyte differentiation was noted in mice lacking a cytokine suppressor (SOCS3-suppressor of cytokine signalling 3) in osteocytes and late osteoblasts [32]. In these mice, a consolidated cortical structure did not form in the metaphyseal region until after 12 weeks of age. SOCS3 is an intracellular protein which attenuates STAT3 intracellular signalling, and in its absence, STAT3 phosphorylation downstream of locally acting cytokines, including IL-6, IL-11, and oncostatin M, is hyperactivated. Cortical consolidation therefore requires suppression of STAT3 signalling in osteocytes and late osteoblasts. In the mice with hyperactive STAT3 signalling in osteocytes, osteoclast formation was elevated specifically within the cortical structure; this prevented cortical consolidation and delayed the accrual of lamellar bone, leading, ultimately to reduced bone strength [31, 32]. Thus, osteocyte signalling, particularly through STAT3, must be suppressed for cortical bone to mature normally. The finding that osteoclast formation was normal in the trabecular compartment indicates separate mechanisms of control between the cortical and trabecular bone compartments.
In contrast to the impairment in cortical consolidation when STAT3 signalling in osteocytes was elevated, cortical bone was unaffected when STAT3 was hyperactivated in the osteoclast lineage (again by deleting SOCS3), but trabecular bone mass was reduced due to osteoclast formation in that compartment [33]. This reinforces the concept that osteoclast formation is controlled separately in trabecular and cortical bone, and that cortical consolidation requires a sufficiently low level of remodelling for it to progress in a timely manner.
A high level of cortical porosity was also observed in an earlier study of Jansen’s metaphyseal chondrodysplasia (OMIM 156400), a rare bone skeletal disease caused by activating mutations of parathyroid hormone 1 receptor (PTH1R). It is characterized by severe short stature, short, bowed limbs with cortical bone erosion, and rickets-like metaphyseal dysplasia, clinodactyly, prominent upper face and small mandible [34]. When a Jansen’s-type mutation was targeted to osteoblasts in a genetically altered mouse model, porous, thin cortical bone, and high trabecular bone mass was observed [35]. This was explained by high levels of both bone formation and bone resorption on both trabecular and endocortical surfaces. Surprisingly, when this mutation was introduced to osteocytes, using Cre-targeting, the phenotype was exaggerated to the extent that the marrow space was full of unremodelled woven bone, and there was no clear distinction between trabecular and cortical bone, a phenotype that persisted until 26 weeks of age [36]. Further cortical analysis revealed that PTHR1 activation increased cortical remodelling, and increased periosteal bone formation, leading to thickened, unconsolidated woven cortical bone in 12-week-old mice, and increased bone width [37]. This phenotype is, in some ways, similar to the mice with osteocyte targeted SOCS3 deletion; both exhibit immature woven cortical bone, suggesting both STAT3 and PTH1R signalling must be suppressed for cortical bone to mature fully, but of the two, only PTH1R activation increases periosteal bone formation.
A third pathway in osteocytes that must be suppressed for full cortical consolidation in both mice and humans is Notch signalling. Hajdu-Cheney syndrome (OMIM 102500) is caused by gain-of function mutations in exon 34 of the NOTCH2 gene [38, 39]. It is a rare multisystem disorder characterized by acro-osteolysis, osteoporosis, short stature, specific craniofacial features, neurologic symptoms, cardiovascular defects and polycystic kidneys [40]. Osteoporosis in these individuals is characterised by a highly porous cortex early in life, with no clear distinction between the cancellous and cortical compartments [41]. This human syndrome was recapitulated by a mouse model with a heterozygous Q2319X Notch2 mutation, which showed trabecular and cortical bone osteopenia, enhanced osteoclastogenesis, and increased bone resorption [42]. Notably, poor cortical consolidation was also observed in three mouse models with activated Notch1 signalling targeted to early stages of osteoblast development (Col2.3-Cre; RosaNotch), mature osteoblasts and osteocytes (Oc-Cre; RosaNotch) and osteocytes (Dmp1-Cre; RosaNotch) [43]. As observed in the two models discussed above with elevated STAT3 and PTHR1 signalling, the mice with elevated Notch1 signalling exhibited a high level of woven bone, but unlike those other models, and Hajdu–Cheney syndrome, bone resorption was suppressed [43]; it is unclear how this could lead to increased cortical porosity, and histomorphometry within the cortical region was not reported.
Impaired cortical consolidation due to elevated osteoclast activity is also likely to contribute to the highly porous and thin cortical bone observed in mice with global deletion of Wnt16 [44, 45]. Indeed, increased osteoclast formation was observed in these mice on their endocortical surface, a site of abundant Wnt16 protein localization [44]. The thin cortical bone was observed at 5 weeks of age and maintained until at least 24 weeks of age, indicating that this pathway is important both during cortical development and maintenance [44]. Wnt16 null mice exhibited no significant bone length phenotype, nor any difference in trabecular bone mass, suggesting Wnt16 regulates cortical bone mass independently of trabecular mass. Indeed, this was confirmed in adult mice, when Wnt16 deletion induced in adult (10 week old) and aged (47 week old) mice selectively reduced cortical bone mass [46]. Porosity was not reported in this later study. Wnt16 is also important in determining human cortical structure: in human population-based GWAS studies, WNT16 was a determinant of cortical structure and shape, including forearm cortical thickness and BMD [45], femoral neck BMD and buckling ratio [47]. Although Wnt16 may be required to maintain cortical bone independently of trabecular bone in a physiological setting, osteoblast-specific overexpression of human WNT16 in mice increased both cortical and trabecular bone mass [48]. Pharmacological approaches to increase WNT16 levels, if feasible, might therefore be expected to increase both cortical and trabecular bone mass. Although increased porosity was observed in the Wnt16 deficient mice, no description of cortical bone composition, intracortical osteoclast formation, vascularization, or distribution of lamellar and cortical bone has been published.
Wnt16 is a member of the WNT (Wingless-type MMTV integration site) family, and regulates bone homeostasis through two pathways, termed canonical (β-catenin-dependent) and non-canonical (β-catenin-independent) signalling. Wnt16 appears to inhibit osteoclastogenesis through both mechanisms: it acts indirectly through canonical Wnt signalling in osteoblasts to increase expression of osteoprotegerin (an endogenous RANKL inhibitor), and acts directly via an inhibitory β-catenin-independent action on osteoclast precursors [44]. The importance of osteoblast-derived Wnt16 was highlighted by targeted deletion of Wnt16 in the osteoblast lineage, using a Runx2-directed Cre recombinase, which also resulted in increased osteoclast formation and thin cortical bone [44]. In contrast, when Wnt16 recombination was targeted to late osteoblasts and osteocytes (with Dmp1Cre), only a modest decrease in cortical thickness was observed, and only with aging [44], suggesting early stages in the osteoblast lineage are most important in regulating osteoclastogenesis through these pathways.
The interplay between the vascular network and the developing cortical structure is also important to consider, although there is little data yet concerning how this is controlled. In mice with increased vascularization due to chondrocyte-targeted overexpression of vascular endothelial growth factor A (VEGFA), cortical porosity and trabecular bone mass were both significantly elevated, even when overexpression was induced in the young adult skeleton [49]. However, this was a complex phenotype since VEGFA also influences osteoblast differentiation and caused significant marrow fibrosis. It remains unknown whether and how the vascular network per se exerts control over the cortical structure once the skeletal structure has been formed.
Defects in cortical mineralisation in the postnatal skeleton
While gradual accrual of mineral during maturation of the cortex may be partially mediated by cortical remodelling, the gradual increase in mineral content may also reflect secondary mineralization of bone. Mineralisation of bone's ground substance (osteoid) occurs in two stages: 70% of the mineral is deposited during an initial rapid phase of mineral accumulation, termed “primary mineralisation” [50]. This is followed by a slower phase termed “secondary mineralisation” which can continue for years [51], either until that region is resorbed during bone remodelling, or until it reaches maximal mineral levels. Multiple processes occur during both primary and secondary mineralization: mineral crystal size and shape increase, the bioapatite structure becomes more ordered, carbonate is incorporated into the bioapatite, collagen fibres become more compact and crosslinked, and water content reduces (these processes are described in detail in [52]). Such “bone-age” differences in composition can be observed within the same tissue, either along the radius of the Haversian osteon, or across the lamellar bone deposited on the periosteum [51, 53–55].
Remodelling of cortical bone, particularly within Haversian systems, leads to regional variations in the degree of mineralization at the tissue level; some osteons are younger, and therefore less mineralized than others. Younger bone, including immature cortical bone close to the growth plate is less mineralized than older bone, since more rapid activation of remodelling and osteoclast-mediated resorption truncates the time available for secondary mineralization [16, 56, 57]. In contrast, when bone remodelling cycles are activated less frequently, for example, when anti-resorptive therapies like bisphosphonates are administered, more bone completes secondary mineralization uninterrupted and reaches a greater mineralization level [53].
The strength of cortical bone can also be influenced, independently of bone structure, by the level of mineralisation achieved. We observed this in a recent study of a mouse with osteocyte-targeted deletion of the protein EphrinB2, a ligand for the EphB4 receptor tyrosine kinase. Although these mice had bones of normal size, shape and cortical thickness, we found their cortical bone strength was severely compromised due to a higher level of cortical bone mineralisation [58]. Although bone formation occurred at a normal rate in these mice, mineral accrual was accelerated due to a RhoA-ROCK dependent defect in osteocyte autophagy, which is an intracellular recycling process. This suggests osteocytes can control the mechanical competency of bone independently of bone mass by modifying the composition of their surrounding matrix.
While no human genetic defects of hypermineralisation with normal bone mass have been described, such defects could explain why some patients experience fragility fractures even though they do not have clinically diagnosed osteoporosis. While areal BMD is used as a standard screening tool for osteoporosis, it is becoming clear that it has low sensitivity as a predictor of fracture risk, with recent estimates suggesting most patients with fragility fracture do not have clinically defined osteoporosis (T score < − 2.5 by areal BMD) [59]. This suggests there may be a diversity of bone fragility phenotypes—some can be detected by areal BMD scans, but others may relate to alterations in mineral content [60], mineral crystal composition [61], or mild changes in collagen content or orientation.
It has recently been noted that atypical femoral fracture (AFF), a rare condition experienced by a subset of patients receiving bisphosphonate-based therapies for osteoporosis may also have an intrinsic defect in bone composition without a change in bone shape. When bone from fracture sites of patients with AFF was assessed by Fourier-transform infra-red microspectroscopy (FTIRM) it was found to have a higher mineral:matrix ratio than bone from women with typical osteoporotic fractures [62]. Additionally, long-term bisphosphonate treatment leads to a higher degree of mineralisation in iliac crest biopsies of patients with AFF compared to patients who did not suffer from AFF [63]. The underlying cellular defect leading to high mineral deposition in AFF has not been defined, although a number of patients suffer AFF in conjunction with undiagnosed osteogenesis imperfecta, reinforcing the concept that this is a material defect [64]. These studies raise the questions of whether AFF is caused by a defect in mineral deposition secondary to a genetic change like that observed in Dmp1Cre.Efnb2f/f mice or caused by an effect of bisphosphonates on osteocyte intracellular dynamics and RhoA-ROCK signalling. Understanding how this pathway regulates bone mineralization may help provide ways to predict susceptible patients. Recent family studies have identified a number of putative genetic changes, including differences in collagen type I, GGPS1, ATRAID and OFD1, that may give rise to this defect. Mechanistic studies are still in the early stages, and this has been very recently reviewed in detail [64].
Continuing cortical expansion and development of bone width
After the embryonic period, as bones growth in length, they also growth in width (radial growth), to support the increase in body weight during growth. Bone width is controlled by multiple processes [26] including: (1) periosteal bone formation at the diaphysis, (2) periosteal bone resorption at the metaphysis, and at least in theory, (3) radial cartilage expansion at the epiphysis (Fig. 3).
Fig. 3.
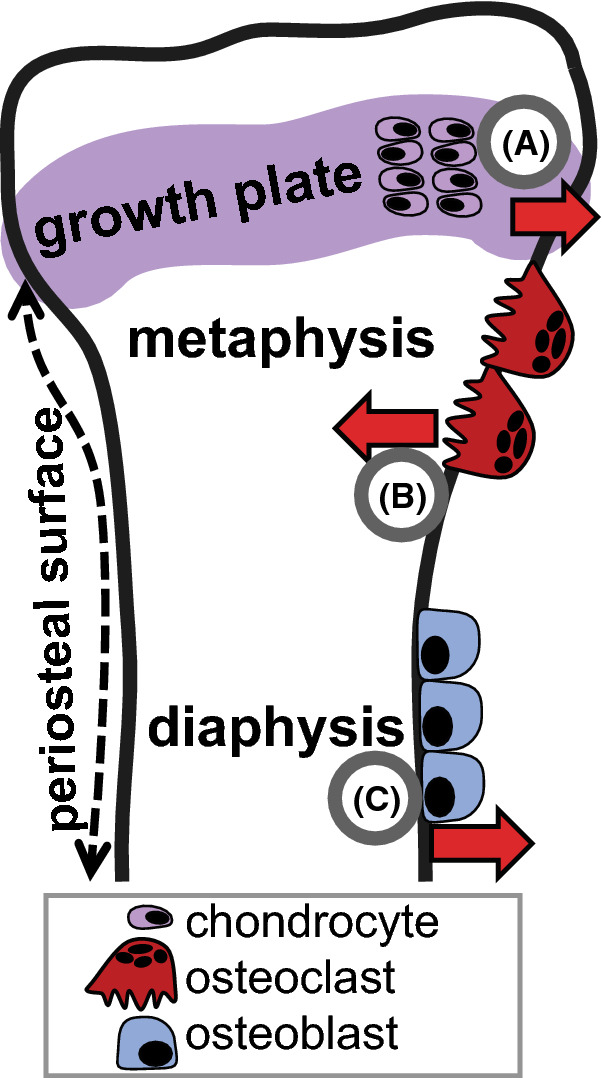
Mechanisms of cortical expansion. During bone growth there are multiple processes that determine bone width: A radial expansion at the epiphyseal growth plate increases bone width, B periosteal resorption by osteoclasts at the metaphysis narrow the bone to fit the wide growth plate to the narrower diaphysis, and C osteoblast-mediated periosteal bone formation at the diaphysis
Radial expansion at the epiphysis
To date, although there are variations in bone width at the epiphyseal growth plate, mechanisms controlling postnatal widening of bone at this site remain undefined. Lineage tracing studies have led to the suggestion that there is a Wnt-responsive population of fibrocartilage cells that proliferate at the periochondrial edge of the growth plate (termed the Groove of Ranvier) [65], but the mechanisms controlling their proliferation have not been identified. We will therefore focus much of this discussion on diaphyseal periosteal expansion and metaphyseal periosteal resorption.
Periosteal expansion at the diaphysis
In very general terms, periosteal expansion at the diaphysis involves bone formation on bone’s outer surface (the periosteum) to increase bone width. The cellular layer of the periosteum (cambium) contains osteoblast progenitors and osteoblasts in contact with the bone surface; the thickness of this layer reduces after development, becoming very thin in adult bone [66]. The cellular layer, and the ability of a subpopulation of periosteal cells to provide a source of osteoblast progenitors has been studied extensively in the context of fracture healing [67–69], but far less so in the context of bone growth. Periosteal progenitors, like their endosteal (bone marrow residing) counterparts are self-renewing and have a high capacity to differentiate to osteoblasts. Recent work has shown that both periosteal and bone marrow-residing osteoblast progenitors are derived during bone development from the local mesenchymal lineage within each element, and are not transported from the vasculature to the periosteum when the primary ossification centre is established [69].
A recent study revealed that BMP signalling in the earliest osteoblast progenitors at the periosteum is required to establish normal postnatal bone width in mice [70]. A narrow bone phenotype observed when BMP2 deletion was targeted by Prrx1Cre was not reproduced if more mature osteoblast progenitors or mature osteoblasts were targeted for the same deletion. This suggests that BMP2 action at the periosteum stimulates osteoblast differentiation, and commits progenitors in the periosteal niche to differentiate and expand bone width by periosteal bone formation. The cortical bone formed in these Prrx1Cre.Bmp2f/f mice was not only narrower (more slender), but was also immature, retaining cartilage bone remnants [70]. This provides further evidence that cortical expansion is required for the maturation of cortical bone matrix.
During bone growth, periosteal expansion is usually co-ordinated with concurrent expansion of the marrow space through bone resorption on the endocortical surface. If these processes are matched, cortical thickness remains constant. During growth, asymmetry of these processes leads to shape change (termed “modelling drift”). Even though bone formation and resorption occur on different surfaces, the co-ordinated change in bone shape suggests that activities on the periosteal and endocortical surfaces are linked. Some years ago, we proposed a conceptual framework for this communication in the context of a mouse model with osteoclast-targeted deletion of gp130 which we were surprised to find had reduced bone formation on the periosteum, a site lacking osteoclasts. We suggested that osteoclasts on the endocortical surface might release signals termed “osteotransmitters” which are transmitted through the cortical osteocyte network (either by lacunocanalicular transport, or by a cellular signal relay) to influence osteoblasts on the periosteal surface [25]. An influence of endocortical osteoclast-derived osteotransmitters is also evident in RANKL-deficient mice (which lack osteoclasts), when cortical morphology was restored by marrow transfer from CD4-driven RANKL-transgenic mice which restored osteoclasts to endocortical, but not periosteal surfaces [71]. Such a communication mechanism may also occur in the other direction, for example, when osteoclasts are stimulated to resorb the periosteum, and bone formation is stimulated on the endocortical surface [24]. Any communication across the cortex, whether emanating from the periosteum or the endocortical surface, could be modified by osteocytes as they sense the changes in strain across the bone during growth and activity, so that bone shape adapts, as needed, to forces experienced by the bone in the growing individual.
In most mouse models described to date, when cortical bone formation has been assessed, it is usually regulated in the same direction as trabecular bone formation. For example, bone formation is stimulated on both trabecular and cortical surfaces in mice with global cathepsin K deletion [72], global sclerostin deletion [73], or osteocyte-targeted constitutively active PTH1R [37], as discussed above. Other models exhibit suppressed bone formation on both trabecular and periosteal bone surfaces, such as mice lacking receptors for the related cytokines interleukin-11, cardiotrophin-1 or oncostatin M [74–76]. Similarly, mice lacking periostin, a protein expressed at high levels in the periosteum, exhibit suppressed bone formation both on periosteal and trabecular bone surfaces [77]; although periostin may regulate periosteal growth with experimental loading [77] and in bone regeneration [69], the null mouse phenotype suggests periostin is not a specific stimulus of the periosteum in the physiological setting.
One example where periosteal and trabecular bone formation exhibit different phenotypes are mice with osteoblast- and osteocyte-targeted deletions of gp130 (the receptor for IL-6 family cytokines); three different models have been described, which all exhibit low levels of bone formation on trabecular surfaces, but greater than normal bone formation on the periosteum, leading to increased bone width [31, 78]. These differences in endocortical and periosteal bone cell activities might relate to differences in the periosteal and trabecular microenvironments: trabecular bone is adjacent to bone marrow, but the periosteal bone surface is surrounded by the periosteum and is adjacent to muscles. Cytokines released by muscle adjacent to cortical bone (myokines) may control the widening of cortical bone [79] by passing through the cellular periosteum to act on osteoblasts residing on the periosteal surface [80], but probably lack such access to trabecular osteoblasts. Modifications in muscle size can modify bone growth and shape, suggesting that periosteal cells experience different compressive forces than trabecular cells [24]. Increased periosteal growth could also occur in response to poor bone material strength; this was observed in the two mouse models lacking gp130 in the osteoblast lineage, since they also exhibited a high level of woven bone in the cortex, and low collagen production [31, 78]. A more recent study provides confirmation that gp130/IL-6 cytokines have different actions in trabecular and cortical compartments. Osteocyte-targeted deletion of STAT3, one of the intracellular molecules through which gp130/IL-6 cytokines act, also resulted in a greater periosteal circumference (indicated by increased bone width), with reduced bone formation on trabecular surfaces [81]. These mice also exhibited a reduction in bone strength, suggesting they may also have a defect in bone quality.
Another example of wide bones associated with poor quality bone matrix is mice with targeted deletion of the gap junction protein connexin 43 in osteoblasts [82, 83] in osteoblast-chondrocyte progenitors [84] or in osteocytes [83, 85]. Mice with connexin 43 suppression due to expression of a dominant negative form in osteocytes also showed greater bone width than controls [86]. The observations in these mice varied depending on the stage of osteoblast differentiation in which connexin 43 was deleted, but there was no defect in trabecular bone mass. In osteoblasts or their progenitors, connexin 43 deletion was associated with more endocortical resorption, leading also to thin cortical bone, even though the targeted cells were osteoblasts [82, 84]. In those works, the cortical bone was highly porous and contained both a high proportion of woven bone, and low mineral content, this is a phenotype of immature cortical bone, due to impaired bone forming ability of connexin 43-deficient osteoblasts. In contrast, when connexin 43 was deleted in osteocytes, the increase in bone width was not associated with any change in cortical thickness [83], but a material defect was observed: increased mineral crystallinity and collagen D-spacing [85]. The cortical size defect was rescued when the C-terminus of connexin 43 was re-expressed in osteocytes [87], but this intervention did not rescue the material defect, suggesting that, in these mice at least, greater bone width was not secondary to a defect in bone matrix quality.
Although there is great interest in identifying specific modifiers of cortical thickness independent of actions in trabecular bone, there are few mouse models reported to date with a specific increase in cortical thickness. One that has been described is mice heterozygous for Notum, a secreted lipase that inactivates WNTs by removing their attached lipid group essential for activating the Frizzled receptor [88, 89]. Notum+/− mice exhibit greater bone width and cortical thickness than controls without a change in trabecular bone mass [90, 91]. When osteoblast-lineage-specific deletion of Notum led to a similar phenotype, this led to the conclusion that osteoblast lineage cells are the principal source of NOTUM that limits cortical thickness [90]. The mechanism by which cortical thickness is elevated in Notum+/− mice seems to involve increased endocortical bone formation, since bone cross-sectional area was normal. This action is likely to involve canonical, rather than non-canonical Wnt signalling, since no disruption in bone resorption was reported. In the human, two genetic variants in the NOTUM locus were identified, and although the changes in signalling induced by these variants are not yet defined, but one was associated with greater bone mineral density, and one with less [90]. When Notum was genetically inactivated in adult mice, whether this was initiated in osteoblast lineage or in all cells, periosteal bone formation was increased [90]. Pharmacological inhibition of NOTUM in ovariectomized rats increased cortical thickness by stimulating endocortical bone formation [91]. Such a pharmacological approach to thicken the cortex is appealing as a therapeutic approach to strengthen cortical bone in osteoporosis.
Another recent mouse model has been described with a progressive phenotype of increasing bone width, however, unlike Notum+/− mice, the newly deposited bone is highly porous [92]. This was observed in two mouse models lacking both fibroblast growth factor receptors (FGFR) 1 and 2 in mature osteoblasts and osteocytes. These mice exhibited increased cortical bone remodelling, with abundant bone resorption, and lamellar bone formation on the endocortical surfaces, leading to infilling of the tibial cortex, as well as periosteal bone formation. The authors noted that osteocyte lacunae within the thickened cortex were frequently empty, and observed a significant increase in osteocyte apoptosis, which may be the cause of the increased remodelling of the cortex. This suggests that both osteocyte activation (through STAT3, PTH1R and NOTCH pathways) and osteocyte cell death can cause cortical porosity and cortical thickening.
Cortical thickening of the diaphysis is a characteristic of Camurati-Engelmann disease (OMIN 131300: CED) or progressive diaphyseal dysplasia. In these individuals there is bilateral and symmetrical hyperostosis, which usually starts at the diaphyses of the femora and tibiae and expands to the fibulae, humeri, ulnae and radii [93]. Although the pathology has not been fully elucidated, the narrowing of the medullary canal with periosteal expansion suggest bone formation may be increased on both surfaces, and a defect in osteoclast activity may also play a role. CED is caused by a mutation in the transforming growth factor (TGF) β1 gene (TGFB) [94] which leads to increased TGF-beta signalling [95]. This adds to the growing list of locally acting factors that, when hyperactive, lead to increased bone formation and cortical thickening.
Another clinical syndrome with cortical thickening is Kenny–Caffey syndrome (OMIN 127000, 24460: KCS). This is quite different to Caffey’s disease, and is a very rare dysmorphologic syndrome characterized by proportionate short stature, cortical thickening on the endocortical surface, with medullary stenosis of tubular bones, delayed closure of anterior fontanelle, eye abnormalities, and hypoparathyroidism [96–98]. There is no evidence that KCS patients are susceptible to fractures although the causative gene for KCS type 2 (FAM111A) mutation also causes gracial bone dysplasia, which is neonatal lethal with brittle bones [97]; no mouse models have yet been developed to explore the causation of this phenotype.
Periosteal resorption at the metaphysis
The metaphyseal periosteum is also a site of bone resorption during growth. Such action is responsible for the even narrowing of the metaphyseal cortex that leads to the formation of a concave curve meeting the narrow diaphysis [26]. This morphological process is notably disrupted in human and murine osteopetroses. The absence of osteoclasts leads to a characteristic change in bone shape, termed an Erlenmeyer flask morphology where the metaphysis is uncharacteristically wide and lacks the concave connection to the diaphysis [99–101].
An Erlenmeyer flask morphology has also been described in Pyle’s disease (OMIM 265900), which is characterized by wide metaphyses, significant thinning of cortical bone, and fragility fractures. Surprisingly though, Pyle’s disease is not an osteopetrosis. Pyle’s disease is associated with loss-of-function mutations in the human SFRP4 gene, and patients exhibit elevated serum bone formation markers, but normal serum markers of bone resorption [102]. High trabecular bone mass, and widened metaphyses were also described in mice rendered null for the murine Sfrp4 gene [102, 103]. In the null mice, histomorphometry of trabecular, periosteal and endocortical surfaces indicated a difference in osteoclast generation between trabecular and endocortical bone. Osteoclast numbers were only elevated on endocortical bone, thereby explaining the marrow expansion and cortical thinning. Pyle’s disease and Sfrp4 null mice provide further clear evidence that cortical and trabecular bone are regulated by different mechanisms in the metaphysis.
However, the explanation for the bone widening in the Sfrp4 null mice remains obscure since bone formation was, in contrast to the patients, lower on periosteal surfaces than in control mice. The Sfrp4 null mouse phenotype of widened metaphyses was not observed at birth, but developed by 2 weeks of age [103], so this phenotype may involve radial expansion of the chondrocyte population (see above). However, at this stage, there is no evidence yet that the Erlenmeyer flask morphology of patients with Pyle’s disease or Sfrp4 null mice is caused by reduced periosteal resorption at the metaphysis. sFRP4 is a soluble Wnt decoy receptor and inhibits both canonical and noncanonical Wnt signalling, and this phenotype therefore strengthens the hypothesis made on the basis of the Wnt16 null mouse phenotype: canonical Wnt signalling may promote trabecular bone formation while non-canonical Wnt signalling erodes cortical bone. Consistent with this distinction between trabecular and cortical influences, Wnt16 null bones, in contrast to Sfrp4 null bones, exhibit smaller cross-sectional area [45], and lower periosteal bone formation rate [104]. A recent report, using cell culture, demonstrated that both osteoblast-expressed and osteoclast-expressed sFRP4 regulate osteoclastogenesis, and the increased endocortical resorption in sFRP4-deficient mice was caused by noncanonical Wnt/Ror2/Jnk signalling activation in osteoclasts [105]. In short, sFRP4 inhibits bone formation on trabecular bone surfaces through canonical signalling while inhibiting osteoclastogenesis on the endocortical surface of bone through noncanonical Wnt signalling.
The above examples show that widening of cortical bone can stem from chondrocytes at the epiphysis, osteoblasts at the periosteal metaphysis and diaphysis, and by osteoclasts at the metaphysis (Fig. 3). The degree to which osteoblast activity is matched by osteoclasts at the endocortical surface determines whether cortical bone is thick or thin, and therefore whether the bone is strong or weak.
Sexual dimorphism in bone width, shape, and cortical development
Men have wider bones than women even after correcting for bone length; this leads to exponentially greater bone strength in men. Clinical data have identified very small sex differences in bone width in utero [106] and during the pre-pubertal period [107], but major sex differences appear at the peri-pubertal period [108]. Young adult men have a 25–33% larger cross-sectional bone area and 18–21% more bone mass than young adult women, and this is maintained throughout life [109]. Notably, trabecular volumetric bone mineral density (vBMD) was greater in men at all sites measured, while despite the difference in cross-sectional area, cortical vBMD was similar between sexes. A major difference contributing to bone strength was identified using peripheral quantitative CT (pQCT): although cortical thickness at the age of peak bone mass was similar, bone cross-sectional area and medullary area were larger in young men than young women [110]. This wider distribution of mineral is a mechanically superior arrangement of bone. Such sex differences arise mainly during puberty when cortical bone expansion is greater in men than women [111] due to the later pubertal timing and therefore, extended longitudinal growth in boys before puberty [107]. Thus, puberty is a critical period for establishing sexual dimorphism in bone mass and geometry.
Rodent models also have sexual dimorphism in cortical dimensions. Whether this is also due to a difference in pubertal timing has not been established, but since female mice and rats have narrower bones than males due to a shorter growth period [112–114], the use of these models to study this sexual difference is warranted. Several mechanism of sex steroid regulation in cortical bone have been revealed by the use of knock out (KO) mice for estrogen receptor (ER) isoforms α, β and the androgen receptor (AR), but these studies were complicated by compensatory elevations in serum testosterone and estradiol [115]. Nevertheless, gonadectomy and sex-steroid treatment studies showed that ERα is the dominant mediator of estrogen actions in bone and the role of ERβ is limited [116]. Androgens such as testosterone can be converted to estrogens through aromatization; this means they can act through both AR and ERα, and periosteal expansion in male mice depends both on both receptors. This was suggested by partially reduced testosterone action on periosteal bone formation when administrating an aromatase inhibitor in orchiectomized wild mice [117]. Cortical bone in male mice became thinner when ERα was deleted in the osteoblast lineage, including in precursors using Prrx1-Cre, but there was no cortical phenotype when this deletion was targeted to mature osteoblasts and osteocytes using Osteocalcin-Cre, Col1a1-Cre or Dmp1-Cre [118]. This indicates that ERα in osteoblast precursors increases cortical bone thickness in young male mice. On the other hand, the AR-expressing cell through which non-aromatised testosterone stimulates cortical bone expansion in male mice remains largely unknown [111]. In females, ERα in osteoblast progenitors may be responsible for cortical bone mass accretion by inhibiting endocortical bone resorption and stimulating periosteal bone formation; this is supported by the report that ERα deletion in osteoblast progenitors did not show the expected increase in endocortical osteoclast numbers nor loss of endocortical bone following ovariectomy [118].
There are several reports that growth hormone (GH) and insulin-like growth factor (IGF1) may play an important role in sexual dimorphism in both longitudinal growth and periosteal bone expansion [111, 119, 120]. When puberty arises, the GH pulse amplitude rises and IGF-1 increases, so both may contribute to longitudinal and transverse bone growth. This happens later but continues longer in men than in women, which results in sexual dimorphism (as discussed above). This GH secretion pattern may be important for sex differences in periosteal expansion since male mice with conditional GH receptor knockout mice in osteoblasts had narrower bones, which may be interpreted as a female-like bone geometry [121]. Whether GH and IGF1 influence bone width independent of their effects on longitudinal growth has not been specifically explored.
Maturation of the cortical structure also differs between male and female mice. Since female mice have a higher level of trabecular bone remodelling than males throughout life, it seems reasonable that cortical bone remodelling would also be maintained at a higher level in female mice. This has not been studied extensively, but our work in Dmp1Cre.Socs3f/f mice showed a more severe phenotype in female animals, which exhibited a further delay in cortical consolidation than males [31, 32]. This difference was sex-steroid dependent, and the sex difference was, at least in part, due to elevated IL-6 signalling in male mice, since the male skeleton took on the appearance of female bone if IL-6 was deleted [32].
Trabecularisation of the cortex with age and pathological examples of trabecularisation
Approximately 80% of fractures in older individuals are non-vertebral, and mainly occur in cortical bone [122]. Approximately 70% of all age-related appendicular bone loss is cortical, and most of this occurs by intracortical remodelling (i.e., bone remodelling within the cortical bone). Such intracortical remodelling with ageing forms cavities in the cortex producing porosity. As the resorption progresses with age, cortical bone becomes more porous [123], eventually taking on the appearance of trabecular bone [122], leading to the term “trabecularisation”. This increased porosity is regional, with most bone loss occurring along the neutral axis of the cortex, where bending stress, and the need to retain a strong cortex is lowest [124]. Increased porosity is caused by an imbalance in the normal process of intra-cortical bone remodelling that renews the cortical bone within cutting cones. With ageing, bone resorption exceeds formation, leading to bone loss within intracortical pores [125].
Increased cortical porosity also occurs in ageing mice. In this case, it occurs at the same time as cortical thinning, caused by endocortical resorption [126]. The number and interconnectivity of vascular channels increases with age, and closely spaced clusters of pores develop and merge from capillary branches of the vascular channel [127]. These events also lead the cortex adjacent to the bone marrow to take on a trabecular bone-like structure, again, similar to observations in human bone.
Although the underlying cellular and molecular mechanisms responsible for the emergence of age-related cortical porosity are unknown, some of the pathways required for cortical consolidation may be involved, such as increased osteocyte signalling (STAT3, PTH1R) or increased osteocyte apoptosis (downstream of FGFR1/2 deletion). Another possibility is that cortical porosity emerges when the normal process of osteocyte cell death is disrupted. This was suggested when Bak and Bax, two pro-apoptotic members of the Bcl2-family of proteins, were conditionally deleted in the osteoblast lineage (using Osx1-Cre). This led to greater trabecular bone mass and increasing cortical porosity with age due to increased intracortical remodelling [128]. These mice exhibited decreased apoptosis of osteoblasts, and increased numbers of “dysmorphic” osteocytes characterised by condensed cytoplasm and darkened nuclei without clear nucleoli; surprisingly these dysmorphic osteocytes were observed in the periosteal zone of the cortex, which lacked the high level of bone remodelling [128]. Both RANKL and VEGFA mRNA levels were high in bones from these mice, possibly due to higher numbers of blood vessels and osteoblasts within the cortical samples. The mechanism responsible for the increased porosity therefore remains obscure.
The emergence of cortical porosity also occurs in pathological conditions, including chronic kidney disease (CKD). CKD leads to rapid cortical bone loss, including cortical thinning and increased porosity, which has been attributed mainly to secondary hyperparathyroidism and subsequent elevated remodelling [129]. When experimental CKD was induced in young mice, using an adenine-rich diet, cortical porosity also developed [130] providing a model by which to study the pathogenesis of this condition. Using this model, the authors suggested elevated porosity was caused by reduced osteocyte apoptosis and increased RANKL expression by osteocytes. However, although this is an appealing model, osteoclast precursors must make direct contact with RANKL-expressing cells, which functions primarily as a membrane-bound protein [131], and it is difficult to see how the RANKL-expressing osteocytes would make contact with osteoclast precursors. Furthermore, osteocytes, even when cultured directly with osteoclast precursors do not fully support osteoclast formation [132, 133].
A rare instance in which cortical porosity emerges is Gorham-Stout disease (GSD, also known as vanishing bone disease). This is characterized by the presence of lymphatics in bone, with bone loss and its replacement with fibrous tissue [134]. GSD is mostly diagnosed in children and young adults. It can affect any bone in the body but most frequently affects the maxilla, mandible, clavicle, ribs, cervical vertebrae, pelvis and femur. Bone of patients with GSD gradually disappears. Recently, to recapitulate GSD, a mouse with osteoblast-lineage targeted overexpression of vascular endothelial growth factor-C (VEGF-C), a principal driver of lymphangiogenesis, was generated [135]. These mice developed pathological lymphatics within in their bones (where they are not normally located). This resulted in significantly more porous and trabecularised cortical bone than control mice [135]. Again, this upholds the model that increased signalling by the osteoblast/osteocyte lineage an stimulate cortical remodelling and initiate cortical pore expansion through multiple mechanisms.
Concluding statements
To conclude, cortical bone structure is determined by distinct processes at specific stages of life. The cortical structure is first shaped by embryo- and maternal- specific influences during embryogenesis. During growth, cortical composition changes as it consolidates, expands, and is shaped in the metaphyseal region. Cortical consolidation, both during embryogenesis and in growth involves closure of cortical pores by osteoblasts, and this process requires prior resorption. Periosteal expansion occurs by osteoblast-mediated bone formation, but is also influenced by cortical remodelling: if the material strength of the bone is compromised, as in mice retaining a high proportion of woven bone, cortical width expansion may occur to compensate. Such control of cortical thickness and shape requires co-ordination of osteoblast and osteoclast activities across the cortical bone itself, and osteocytes may be involved in this. With age, cortical porosity emerges in both rodents and humans, but the major signals that cause this remain unknown. Further study of each of these stages of cortical development, growth, and degeneration are needed to understand how this multicellular process is controlled and can be manipulated to improve human health.
Acknowledgements
The authors thank Emma C. Walker for assistance in preparing the figures and tables for this manuscript.
Author contributions
NAS conceived of the article, TI performed the literature search, both NAS and TI drafted sections of the review, and both critically revised the work; both authors approve of the final submission.
Funding
Tsuyoshi Isojima was supported by Travel Grants from Mochida Memorial Foundation for Medical and Pharmacological Research and The Foundation for Growth Science, Japan. Natalie Sims is supported by an NHMRC Senior Research Fellowship. St. Vincent’s Institute acknowledges the support of the Victorian State Government’s Operational Infrastructure Support program.
Availability of data and material
Not applicable.
Code availability
Not applicable.
Declarations
Conflict of interest
The author(s) declare(s) that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Maes C, Kobayashi T, Selig MK, Torrekens S, Roth SI, Mackem S, et al. Osteoblast precursors, but not mature osteoblasts, move into developing and fractured bones along with invading blood vessels. Dev Cell. 2010;19(2):329–344. doi: 10.1016/j.devcel.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giraud-Guille MM. Twisted plywood architecture of collagen fibrils in human compact bone osteons. Calcif Tissue Int. 1988;42(3):167–180. doi: 10.1007/BF02556330. [DOI] [PubMed] [Google Scholar]
- 3.Yamamoto T, Hasegawa T, Sasaki M, Hongo H, Tabata C, Liu Z, et al. Structure and formation of the twisted plywood pattern of collagen fibrils in rat lamellar bone. J Electron Microsc. 2012;61(2):113–121. doi: 10.1093/jmicro/dfs033. [DOI] [PubMed] [Google Scholar]
- 4.Kronenberg HM. The role of the perichondrium in fetal bone development. Ann NY Acad Sci. 2007;1116(1):59–64. doi: 10.1196/annals.1402.059. [DOI] [PubMed] [Google Scholar]
- 5.Sharir A, Stern T, Rot C, Shahar R, Zelzer E. Muscle force regulates bone shaping for optimal load-bearing capacity during embryogenesis. Development. 2011;138(15):3247–3259. doi: 10.1242/dev.063768. [DOI] [PubMed] [Google Scholar]
- 6.Zimmermann EA, Riedel C, Schmidt FN, Stockhausen KE, Chushkin Y, Schaible E, et al. Mechanical competence and bone quality develop during skeletal growth. J Bone Miner Res. 2019;34(8):1461–1472. doi: 10.1002/jbmr.3730. [DOI] [PubMed] [Google Scholar]
- 7.Cooper KL, Oh S, Sung Y, Dasari RR, Kirschner MW, Tabin CJ. Multiple phases of chondrocyte enlargement underlie differences in skeletal proportions. Nature. 2013;495:375. doi: 10.1038/nature11940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lanske B, Amling M, Neff L, Guiducci J, Baron R, Kronenberg HM. Ablation of the PTHrP gene or the PTH/PTHrP receptor gene leads to distinct abnormalities in bone development. J Clin Invest. 1999;104(4):399–407. doi: 10.1172/JCI6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu JY, Aarnisalo P, Bastepe M, Sinha P, Fulzele K, Selig MK, et al. Gsalpha enhances commitment of mesenchymal progenitors to the osteoblast lineage but restrains osteoblast differentiation in mice. J Clin Invest. 2011;121(9):3492–3504. doi: 10.1172/JCI46406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soriano P, Montgomery C, Geske R, Bradley A. Targeted disruption of the c-src proto-oncogene leads to osteopetrosis in mice. Cell. 1991;64(4):693–702. doi: 10.1016/0092-8674(91)90499-O. [DOI] [PubMed] [Google Scholar]
- 11.Li CY, Jepsen KJ, Majeska RJ, Zhang J, Ni R, Gelb BD, et al. Mice lacking cathepsin K maintain bone remodeling but develop bone fragility despite high bone mass. J Bone Miner Res. 2006;21(6):865–875. doi: 10.1359/jbmr.060313. [DOI] [PubMed] [Google Scholar]
- 12.Maes C, Carmeliet P, Moermans K, Stockmans I, Smets N, Collen D, et al. Impaired angiogenesis and endochondral bone formation in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Mech Dev. 2002;111(1–2):61–73. doi: 10.1016/S0925-4773(01)00601-3. [DOI] [PubMed] [Google Scholar]
- 13.Rot-Nikcevic I, Reddy T, Downing KJ, Belliveau AC, Hallgrímsson B, Hall BK, et al. Myf5−/−:MyoD−/− amyogenic fetuses reveal the importance of early contraction and static loading by striated muscle in mouse skeletogenesis. Dev Genes Evol. 2006;216(1):1–9. doi: 10.1007/s00427-005-0024-9. [DOI] [PubMed] [Google Scholar]
- 14.Ansari N, Isojima T, Crimeen-Irwin B, Poulton IJ, McGregor NE, Ho PWM, et al. Dmp1Cre-directed knockdown of PTHrP in murine decidua is associated with increased bone width and a life-long increase in strength specific to male progeny. J Bone Miner Res. 2021 doi: 10.1002/jbmr.4388. [DOI] [PubMed] [Google Scholar]
- 15.Nistala H, Makitie O, Juppner H. Caffey disease: new perspectives on old questions. Bone. 2014;60:246–251. doi: 10.1016/j.bone.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Enlow DH. A study of the post-natal growth and remodeling of bone. Am J Anat. 1962;110:79–101. doi: 10.1002/aja.1001100202. [DOI] [PubMed] [Google Scholar]
- 17.Cadet ER, Gafni RI, McCarthy EF, McCray DR, Bacher JD, Barnes KM, et al. Mechanisms responsible for longitudinal growth of the cortex: coalescence of trabecular bone into cortical bone. J Bone Jt Surg Am. 2003;85-A(9):1739–1748. doi: 10.2106/00004623-200309000-00013. [DOI] [PubMed] [Google Scholar]
- 18.Bala Y, Bui QM, Wang XF, Iuliano S, Wang Q, Ghasem-Zadeh A, et al. Trabecular and cortical microstructure and fragility of the distal radius in women. J Bone Miner Res. 2015;30(4):621–629. doi: 10.1002/jbmr.2388. [DOI] [PubMed] [Google Scholar]
- 19.Wang Q, Ghasem-Zadeh A, Wang X-F, Iuliano-Burns S, Seeman E. Trabecular bone of growth plate origin influences both trabecular and cortical morphology in adulthood. J Bone Miner Res. 2011;26(7):1577–1583. doi: 10.1002/jbmr.360. [DOI] [PubMed] [Google Scholar]
- 20.Pritchett JW. Longitudinal growth and growth-plate activity in the lower extremity. Clin Orthop Relat Res. 1992;275:274–279. doi: 10.1097/00003086-199202000-00041. [DOI] [PubMed] [Google Scholar]
- 21.Shipov A, Zaslansky P, Riesemeier H, Segev G, Atkins A, Shahar R. Unremodeled endochondral bone is a major architectural component of the cortical bone of the rat (Rattus norvegicus) J Struct Biol. 2013;183(2):132–140. doi: 10.1016/j.jsb.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 22.Maggiano IS, Maggiano CM, Tiesler VG, Chi-Keb JR, Stout SD. Drifting diaphyses: asymmetry in diametric growth and adaptation along the humeral and femoral length. Anat Rec (Hoboken) 2015;298(10):1689–1699. doi: 10.1002/ar.23201. [DOI] [PubMed] [Google Scholar]
- 23.Ferguson VL, Ayers RA, Bateman TA, Simske SJ. Bone development and age-related bone loss in male C57BL/6J mice. Bone. 2003;33(3):387–398. doi: 10.1016/S8756-3282(03)00199-6. [DOI] [PubMed] [Google Scholar]
- 24.Chan ASM, McGregor NE, Poulton IJ, Hardee JP, Cho EH, Martin TJ, et al. Bone geometry is altered by follistatin-induced muscle growth in young adult male mice. JBMR Plus. 2021;5(4):e10477. doi: 10.1002/jbm4.10477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson RW, McGregor NE, Brennan HJ, Crimeen-Irwin B, Poulton IJ, Martin TJ, et al. Glycoprotein130 (Gp130)/interleukin-6 (IL-6) signalling in osteoclasts promotes bone formation in periosteal and trabecular bone. Bone. 2015;81:343–351. doi: 10.1016/j.bone.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 26.Rauch F. The dynamics of bone structure development during pubertal growth. J Musculoskelet Neuronal Interact. 2012;12(1):1–6. [PubMed] [Google Scholar]
- 27.Sims NA, Martin TJ. Osteoclasts provide coupling signals to osteoblast lineage cells through multiple mechanisms. Annu Rev Physiol. 2020;82:507–529. doi: 10.1146/annurev-physiol-021119-034425. [DOI] [PubMed] [Google Scholar]
- 28.de Saint-Georges L, Miller SC. The microcirculation of bone and marrow in the diaphysis of the rat hemopoietic long bones. Anat Rec. 1992;233(2):169–177. doi: 10.1002/ar.1092330202. [DOI] [PubMed] [Google Scholar]
- 29.Grüneboom A, Hawwari I, Weidner D, Culemann S, Müller S, Henneberg S, et al. A network of trans-cortical capillaries as mainstay for blood circulation in long bones. Nat Metab. 2019;1(2):236–250. doi: 10.1038/s42255-018-0016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Root SH, Wee NKY, Novak S, Rosen CJ, Baron R, Matthews BG, et al. Perivascular osteoprogenitors are associated with transcortical channels of long bones. STEM CELLS. 2020;38(6):769–781. doi: 10.1002/stem.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walker EC, Truong K, McGregor NE, Poulton IJ, Isojima T, Gooi JH, et al. Cortical bone consolidation requires local SOCS3-dependent suppression of osteoclasts through gp130/STAT3 signalling in osteocytes. Elife. 2020;9:e56666. doi: 10.7554/eLife.56666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cho DC, Brennan HJ, Johnson RW, Poulton IJ, Gooi JH, Tonkin BA, et al. Bone corticalization requires local SOCS3 activity and is promoted by androgen action via interleukin-6. Nat Commun. 2017;8(1):806. doi: 10.1038/s41467-017-00920-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong PK, Egan PJ, Croker BA, O'Donnell K, Sims NA, Drake S, et al. SOCS-3 negatively regulates innate and adaptive immune mechanisms in acute IL-1-dependent inflammatory arthritis. J Clin Invest. 2006;116(6):1571–1581. doi: 10.1172/JCI25660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schipani E, Langman CB, Parfitt AM, Jensen GS, Kikuchi S, Kooh SW, et al. Constitutively activated receptors for parathyroid hormone and parathyroid hormone-related peptide in Jansen's metaphyseal chondrodysplasia. N Engl J Med. 1996;335(10):708–714. doi: 10.1056/NEJM199609053351004. [DOI] [PubMed] [Google Scholar]
- 35.Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, et al. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. J Clin Invest. 2001;107(3):277–286. doi: 10.1172/JCI11296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O'Brien CA, Plotkin LI, Galli C, Goellner JJ, Gortazar AR, Allen MR, et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE. 2008;3(8):e2942. doi: 10.1371/journal.pone.0002942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rhee Y, Allen MR, Condon K, Lezcano V, Ronda AC, Galli C, et al. PTH receptor signaling in osteocytes governs periosteal bone formation and intracortical remodeling. J Bone Miner Res. 2011;26(5):1035–1046. doi: 10.1002/jbmr.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Isidor B, Lindenbaum P, Pichon O, Bezieau S, Dina C, Jacquemont S, et al. Truncating mutations in the last exon of NOTCH2 cause a rare skeletal disorder with osteoporosis. Nat Genet. 2011;43(4):306–308. doi: 10.1038/ng.778. [DOI] [PubMed] [Google Scholar]
- 39.Simpson MA, Irving MD, Asilmaz E, Gray MJ, Dafou D, Elmslie FV, et al. Mutations in NOTCH2 cause Hajdu–Cheney syndrome, a disorder of severe and progressive bone loss. Nat Genet. 2011;43(4):303–305. doi: 10.1038/ng.779. [DOI] [PubMed] [Google Scholar]
- 40.Canalis E, Zanotti S. Hajdu–Cheney syndrome, a disease associated with NOTCH2 mutations. Curr Osteoporos Rep. 2016;14(4):126–131. doi: 10.1007/s11914-016-0311-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sakka S, Gafni RI, Davies JH, Clarke B, Tebben P, Samuels M, et al. Bone structural characteristics and response to bisphosphonate treatment in children with Hajdu–Cheney syndrome. J Clin Endocrinol Metab. 2017;102(11):4163–4172. doi: 10.1210/jc.2017-01102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Canalis E, Schilling L, Yee SP, Lee SK, Zanotti S. Hajdu Cheney mouse mutants exhibit osteopenia, increased osteoclastogenesis, and bone resorption. J Biol Chem. 2016;291(4):1538–1551. doi: 10.1074/jbc.M115.685453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Canalis E, Parker K, Feng JQ, Zanotti S. Osteoblast lineage-specific effects of notch activation in the skeleton. Endocrinology. 2013;154(2):623–634. doi: 10.1210/en.2012-1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moverare-Skrtic S, Henning P, Liu X, Nagano K, Saito H, Borjesson AE, et al. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat Med. 2014;11:1279–1288. doi: 10.1038/nm.3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng HF, Tobias JH, Duncan E, Evans DM, Eriksson J, Paternoster L, et al. WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PLoS Genet. 2012;8(7):e1002745. doi: 10.1371/journal.pgen.1002745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ohlsson C, Henning P, Nilsson KH, Wu J, Gustafsson KL, Sjogren K, et al. Inducible Wnt16 inactivation: WNT16 regulates cortical bone thickness in adult mice. J Endocrinol. 2018;237(2):113–122. doi: 10.1530/JOE-18-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia-Ibarbia C, Perez-Nunez MI, Olmos JM, Valero C, Perez-Aguilar MD, Hernandez JL, et al. Missense polymorphisms of the WNT16 gene are associated with bone mass, hip geometry and fractures. Osteoporos Int. 2013;24(9):2449–2454. doi: 10.1007/s00198-013-2302-0. [DOI] [PubMed] [Google Scholar]
- 48.Alam I, Alkhouli M, Gerard-O'Riley RL, Wright WB, Acton D, Gray AK, et al. Osteoblast-specific overexpression of human WNT16 increases both cortical and trabecular bone mass and structure in mice. Endocrinology. 2016;157(2):722–736. doi: 10.1210/en.2015-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maes C, Goossens S, Bartunkova S, Drogat B, Coenegrachts L, Stockmans I, et al. Increased skeletal VEGF enhances beta-catenin activity and results in excessively ossified bones. Embo J. 2010;29(2):424–441. doi: 10.1038/emboj.2009.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tonna S, Takyar FM, Vrahnas C, Crimeen-Irwin B, Ho PW, Poulton IJ, et al. EphrinB2 signaling in osteoblasts promotes bone mineralization by preventing apoptosis. FASEB J. 2014;28(10):4482–4496. doi: 10.1096/fj.14-254300. [DOI] [PubMed] [Google Scholar]
- 51.Fuchs RK, Allen MR, Ruppel ME, Diab T, Phipps RJ, Miller LM, et al. In situ examination of the time-course for secondary mineralization of Haversian bone using synchrotron Fourier transform infrared microspectroscopy. Matrix Biol. 2008;27(1):34–41. doi: 10.1016/j.matbio.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 52.Blank M, Sims NA. Cellular processes by which osteoblasts and osteocytes control bone mineral deposition and maturation revealed by stage-specific EphrinB2 knockdown. Curr Osteoporos Rep. 2019;17(5):270–280. doi: 10.1007/s11914-019-00524-y. [DOI] [PubMed] [Google Scholar]
- 53.Fuchs RK, Faillace ME, Allen MR, Phipps RJ, Miller LM, Burr DB. Bisphosphonates do not alter the rate of secondary mineralization. Bone. 2011;49(4):701–705. doi: 10.1016/j.bone.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 54.Vrahnas C, Buenzli PR, Pearson TA, Pennypacker BL, Tobin MJ, Bambery KR, et al. Differing effects of parathyroid hormone, alendronate, and odanacatib on bone formation and on the mineralization process in intracortical and endocortical bone of ovariectomized rabbits. Calcif Tissue Int. 2018;103(6):625–637. doi: 10.1007/s00223-018-0455-8. [DOI] [PubMed] [Google Scholar]
- 55.Vrahnas C, Pearson TA, Brunt AR, Forwood MR, Bambery KR, Tobin MJ, et al. Anabolic action of parathyroid hormone (PTH) does not compromise bone matrix mineral composition or maturation. Bone. 2016;93:146–154. doi: 10.1016/j.bone.2016.09.022. [DOI] [PubMed] [Google Scholar]
- 56.Boivin G, Meunier PJ. Changes in bone remodeling rate influence the degree of mineralization of bone. Connect Tissue Res. 2002;43(2–3):535–537. doi: 10.1080/03008200290000934. [DOI] [PubMed] [Google Scholar]
- 57.Paschalis EP, Betts F, DiCarlo E, Mendelsohn R, Boskey AL. FTIR microspectroscopic analysis of normal human cortical and trabecular bone. Calcif Tissue Int. 1997;61(6):480–486. doi: 10.1007/s002239900371. [DOI] [PubMed] [Google Scholar]
- 58.Vrahnas C, Blank M, Dite TA, Tatarczuch L, Ansari N, Crimeen-Irwin B, et al. Increased autophagy in EphrinB2-deficient osteocytes is associated with elevated secondary mineralization and brittle bone. Nat Commun. 2019;10(1):3436. doi: 10.1038/s41467-019-11373-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lespessailles E, Cortet B, Legrand E, Guggenbuhl P, Roux C. Low-trauma fractures without osteoporosis. Osteoporos Int. 2017;28(6):1771–1778. doi: 10.1007/s00198-017-3921-7. [DOI] [PubMed] [Google Scholar]
- 60.Roschger P, Misof B, Paschalis E, Fratzl P, Klaushofer K. Changes in the degree of mineralization with osteoporosis and its treatment. Curr Osteoporos Rep. 2014;12(3):338–350. doi: 10.1007/s11914-014-0218-z. [DOI] [PubMed] [Google Scholar]
- 61.McCreadie BR, Morris MD, Chen TC, Sudhaker Rao D, Finney WF, Widjaja E, et al. Bone tissue compositional differences in women with and without osteoporotic fracture. Bone. 2006;39(6):1190–1195. doi: 10.1016/j.bone.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 62.Lloyd AA, Gludovatz B, Riedel C, Luengo EA, Saiyed R, Marty E, et al. Atypical fracture with long-term bisphosphonate therapy is associated with altered cortical composition and reduced fracture resistance. Proc Natl Acad Sci USA. 2017;114(33):8722–8727. doi: 10.1073/pnas.1704460114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Farlay D, Rizzo S, Ste-Marie LG, Michou L, Morin SN, Qiu S, et al. Duration-dependent increase of human bone matrix mineralization in long-term bisphosphonate users with atypical femur fracture. J Bone Miner Res. 2021;36:1031–1041. doi: 10.1002/jbmr.4244. [DOI] [PubMed] [Google Scholar]
- 64.Zhou W, van Rooij JGJ, Ebeling PR, Verkerk A, Zillikens MC. The genetics of atypical femur fractures—a systematic review. Curr Osteoporos Rep. 2021;19(2):123–130. doi: 10.1007/s11914-021-00658-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Usami Y, Gunawardena AT, Francois NB, Otsuru S, Takano H, Hirose K, et al. Possible contribution of Wnt-responsive chondroprogenitors to the postnatal murine growth plate. J Bone Miner Res. 2019;34(5):964–974. doi: 10.1002/jbmr.3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dwek JR. The periosteum: what is it, where is it, and what mimics it in its absence? Skeletal Radiol. 2010;39(4):319–323. doi: 10.1007/s00256-009-0849-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grcevic D, Pejda S, Matthews BG, Repic D, Wang L, Li H, et al. In vivo fate mapping identifies mesenchymal progenitor cells. STEM CELLS. 2012;30(2):187–196. doi: 10.1002/stem.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Colnot C. Skeletal cell fate decisions within periosteum and bone marrow during bone regeneration. J Bone Miner Res. 2009;24(2):274–282. doi: 10.1359/jbmr.081003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Duchamp de Lageneste O, Julien A, Abou-Khalil R, Frangi G, Carvalho C, Cagnard N, et al. Periosteum contains skeletal stem cells with high bone regenerative potential controlled by Periostin. Nat Commun. 2018;9(1):773. doi: 10.1038/s41467-018-03124-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Salazar VS, Capelo LP, Cantu C, Zimmerli D, Gosalia N, Pregizer S, et al. Reactivation of a developmental Bmp2 signaling center is required for therapeutic control of the murine periosteal niche. Elife. 2019;8:e42386. doi: 10.7554/eLife.42386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim N, Odgren PR, Kim DK, Marks SC, Jr, Choi Y. Diverse roles of the tumor necrosis factor family member TRANCE in skeletal physiology revealed by TRANCE deficiency and partial rescue by a lymphocyte-expressed TRANCE transgene. Proc Natl Acad Sci USA. 2000;97(20):10905–10910. doi: 10.1073/pnas.200294797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pennypacker B, Shea M, Liu Q, Masarachia P, Saftig P, Rodan S, et al. Bone density, strength, and formation in adult cathepsin K(−/−) mice. Bone. 2009;44(2):199–207. doi: 10.1016/j.bone.2008.08.130. [DOI] [PubMed] [Google Scholar]
- 73.Li X, Ominsky MS, Niu QT, Sun N, Daugherty B, D'Agostin D, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23(6):860–869. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 74.Sims NA, Jenkins BJ, Nakamura A, Quinn JM, Li R, Gillespie MT, et al. Interleukin-11 receptor signaling is required for normal bone remodeling. J Bone Miner Res. 2005;20(7):1093–1102. doi: 10.1359/JBMR.050209. [DOI] [PubMed] [Google Scholar]
- 75.Walker EC, McGregor NE, Poulton IJ, Pompolo S, Allan EH, Quinn JM, et al. Cardiotrophin-1 is an osteoclast-derived stimulus of bone formation required for normal bone remodeling. J Bone Miner Res. 2008;23(12):2025–2032. doi: 10.1359/jbmr.080706. [DOI] [PubMed] [Google Scholar]
- 76.Walker EC, McGregor NE, Poulton IJ, Solano M, Pompolo S, Fernandes TJ, et al. Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J Clin Invest. 2010;120(2):582–592. doi: 10.1172/JCI40568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bonnet N, Standley KN, Bianchi EN, Stadelmann V, Foti M, Conway SJ, et al. The matricellular protein periostin is required for sost inhibition and the anabolic response to mechanical loading and physical activity. J Biol Chem. 2009;284(51):35939–35950. doi: 10.1074/jbc.M109.060335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Johnson RW, Brennan HJ, Vrahnas C, Poulton IJ, McGregor NE, Standal T, et al. The primary function of gp130 signaling in osteoblasts is to maintain bone formation and strength, rather than promote osteoclast formation. J Bone Miner Res. 2014;29(6):1492–1505. doi: 10.1002/jbmr.2159. [DOI] [PubMed] [Google Scholar]
- 79.Johnson RW, White JD, Walker EC, Martin TJ, Sims NA. Myokines (muscle-derived cytokines and chemokines) including ciliary neurotrophic factor (CNTF) inhibit osteoblast differentiation. Bone. 2014;64C:47–56. doi: 10.1016/j.bone.2014.03.053. [DOI] [PubMed] [Google Scholar]
- 80.Evans SF, Parent JB, Lasko CE, Zhen X, Knothe UR, Lemaire T, et al. Periosteum, bone's "smart" bounding membrane, exhibits direction-dependent permeability. J Bone Miner Res. 2013;28(3):608–617. doi: 10.1002/jbmr.1777. [DOI] [PubMed] [Google Scholar]
- 81.Corry KA, Zhou H, Brustovetsky T, Himes ER, Bivi N, Horn MR, et al. Stat3 in osteocytes mediates osteogenic response to loading. Bone Rep. 2019;11:100218. doi: 10.1016/j.bonr.2019.100218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grimston SK, Brodt MD, Silva MJ, Civitelli R. Attenuated response to in vivo mechanical loading in mice with conditional osteoblast ablation of the connexin43 gene (Gja1) J Bone Miner Res. 2008;23(6):879–886. doi: 10.1359/jbmr.080222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bivi N, Nelson MT, Faillace ME, Li J, Miller LM, Plotkin LI. Deletion of Cx43 from osteocytes results in defective bone material properties but does not decrease extrinsic strength in cortical bone. Calcif Tissue Int. 2012;91(3):215–224. doi: 10.1007/s00223-012-9628-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Watkins M, Grimston SK, Norris JY, Guillotin B, Shaw A, Beniash E, et al. Osteoblast connexin43 modulates skeletal architecture by regulating both arms of bone remodeling. Mol Biol Cell. 2011;22(8):1240–1251. doi: 10.1091/mbc.e10-07-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hammond MA, Berman AG, Pacheco-Costa R, Davis HM, Plotkin LI, Wallace JM. Removing or truncating connexin 43 in murine osteocytes alters cortical geometry, nanoscale morphology, and tissue mechanics in the tibia. Bone. 2016;88:85–91. doi: 10.1016/j.bone.2016.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu H, Gu S, Riquelme MA, Burra S, Callaway D, Cheng H, et al. Connexin 43 channels are essential for normal bone structure and osteocyte viability. J Bone Miner Res. 2015;30(3):436–448. doi: 10.1002/jbmr.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pacheco-Costa R, Davis HM, Sorenson C, Hon MC, Hassan I, Reginato RD, et al. Defective cancellous bone structure and abnormal response to PTH in cortical bone of mice lacking Cx43 cytoplasmic C-terminus domain. Bone. 2015;81:632–643. doi: 10.1016/j.bone.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kakugawa S, Langton PF, Zebisch M, Howell S, Chang TH, Liu Y, et al. Notum deacylates Wnt proteins to suppress signalling activity. Nature. 2015;519(7542):187–192. doi: 10.1038/nature14259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nusse R. Cell signalling: disarming Wnt. Nature. 2015;519(7542):163–164. doi: 10.1038/nature14208. [DOI] [PubMed] [Google Scholar]
- 90.Moverare-Skrtic S, Nilsson KH, Henning P, Funck-Brentano T, Nethander M, Rivadeneira F, et al. Osteoblast-derived NOTUM reduces cortical bone mass in mice and the NOTUM locus is associated with bone mineral density in humans. FASEB J. 2019;33(10):11163–11179. doi: 10.1096/fj.201900707R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brommage R, Liu J, Vogel P, Mseeh F, Thompson AY, Potter DG, et al. NOTUM inhibition increases endocortical bone formation and bone strength. Bone Res. 2019;7:2. doi: 10.1038/s41413-018-0038-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.McKenzie J, Smith C, Karuppaiah K, Langberg J, Silva MJ, Ornitz DM. Osteocyte death and bone overgrowth in mice lacking fibroblast growth factor receptors 1 and 2 in mature osteoblasts and osteocytes. J Bone Miner Res. 2019;34(9):1660–1675. doi: 10.1002/jbmr.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Janssens K, Vanhoenacker F, Bonduelle M, Verbruggen L, Van Maldergem L, Ralston S, et al. Camurati–Engelmann disease: review of the clinical, radiological, and molecular data of 24 families and implications for diagnosis and treatment. J Med Genet. 2006;43(1):1–11. doi: 10.1136/jmg.2005.033522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Janssens K, Gershoni-Baruch R, Guanabens N, Migone N, Ralston S, Bonduelle M, et al. Mutations in the gene encoding the latency-associated peptide of TGF-beta 1 cause Camurati–Engelmann disease. Nat Genet. 2000;26(3):273–275. doi: 10.1038/81563. [DOI] [PubMed] [Google Scholar]
- 95.Janssens K, ten Dijke P, Ralston SH, Bergmann C, Van Hul W. Transforming growth factor-β1 mutations in Camurati–Engelmann disease lead to increased signaling by altering either activation or secretion of the mutant protein*. J Biol Chem. 2003;278(9):7718–7724. doi: 10.1074/jbc.M208857200. [DOI] [PubMed] [Google Scholar]
- 96.Parvari R, Hershkovitz E, Grossman N, Gorodischer R, Loeys B, Zecic A, et al. Mutation of TBCE causes hypoparathyroidism–retardation–dysmorphism and autosomal recessive Kenny–Caffey syndrome. Nat Genet. 2002;32(3):448–452. doi: 10.1038/ng1012. [DOI] [PubMed] [Google Scholar]
- 97.Unger S, Gorna MW, Le Bechec A, Do Vale-Pereira S, Bedeschi MF, Geiberger S, et al. FAM111A mutations result in hypoparathyroidism and impaired skeletal development. Am J Hum Genet. 2013;92(6):990–995. doi: 10.1016/j.ajhg.2013.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Isojima T, Doi K, Mitsui J, Oda Y, Tokuhiro E, Yasoda A, et al. A recurrent de novo FAM111A mutation causes Kenny–Caffey syndrome type 2. J Bone Miner Res. 2014;29(4):992–998. doi: 10.1002/jbmr.2091. [DOI] [PubMed] [Google Scholar]
- 99.Sobacchi C, Schulz A, Coxon FP, Villa A, Helfrich MH. Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol. 2013;9(9):522–536. doi: 10.1038/nrendo.2013.137. [DOI] [PubMed] [Google Scholar]
- 100.Faden MA, Krakow D, Ezgu F, Rimoin DL, Lachman RS. The Erlenmeyer flask bone deformity in the skeletal dysplasias. Am J Med Genet A. 2009;149A(6):1334–1345. doi: 10.1002/ajmg.a.32253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Stein M, Barnea-Zohar M, Shalev M, Arman E, Brenner O, Winograd-Katz S, et al. Massive osteopetrosis caused by non-functional osteoclasts in R51Q SNX10 mutant mice. Bone. 2020;136:115360. doi: 10.1016/j.bone.2020.115360. [DOI] [PubMed] [Google Scholar]
- 102.Kiper POS, Saito H, Gori F, Unger S, Hesse E, Yamana K, et al. Cortical-bone fragility-insights from sFRP4 deficiency in Pyle's disease. N Engl J Med. 2016;374(26):2553–2562. doi: 10.1056/NEJMoa1509342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Haraguchi R, Kitazawa R, Mori K, Tachibana R, Kiyonari H, Imai Y, et al. sFRP4-dependent Wnt signal modulation is critical for bone remodeling during postnatal development and age-related bone loss. Sci Rep. 2016;6(1):25198. doi: 10.1038/srep25198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wergedal JE, Kesavan C, Brommage R, Das S, Mohan S. Role of WNT16 in the regulation of periosteal bone formation in female mice. Endocrinology. 2015;156(3):1023–1032. doi: 10.1210/en.2014-1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen K, Ng PY, Chen R, Hu D, Berry S, Baron R, et al. Sfrp4 repression of the Ror2/Jnk cascade in osteoclasts protects cortical bone from excessive endosteal resorption. Proc Natl Acad Sci USA. 2019;116(28):14138–14143. doi: 10.1073/pnas.1900881116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang Q, Alen M, Lyytikainen A, Xu L, Tylavsky FA, Kujala UM, et al. Familial resemblance and diversity in bone mass and strength in the population are established during the first year of postnatal life. J Bone Miner Res. 2010;25(7):1512–1520. doi: 10.1002/jbmr.45. [DOI] [PubMed] [Google Scholar]
- 107.Iuliano-Burns S, Hopper J, Seeman E. The age of puberty determines sexual dimorphism in bone structure: a male/female co-twin control study. J Clin Endocrinol Metab. 2009;94(5):1638–1643. doi: 10.1210/jc.2008-1522. [DOI] [PubMed] [Google Scholar]
- 108.Almeida M, Laurent MR, Dubois V, Claessens F, O'Brien CA, Bouillon R, et al. Estrogens and Androgens in Skeletal Physiology and Pathophysiology. Physiol Rev. 2017;97(1):135–187. doi: 10.1152/physrev.00033.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Riggs BL, Melton LJ, III, Robb RA, Camp JJ, Atkinson EJ, Peterson JM, et al. Population-based study of age and sex differences in bone volumetric density, size, geometry, and structure at different skeletal sites. J Bone Miner Res. 2004;19(12):1945–1954. doi: 10.1359/jbmr.040916. [DOI] [PubMed] [Google Scholar]
- 110.Khosla S, Riggs BL, Atkinson EJ, Oberg AL, McDaniel LJ, Holets M, et al. Effects of sex and age on bone microstructure at the ultradistal radius: a population-based noninvasive in vivo assessment. J Bone Miner Res. 2006;21(1):124–131. doi: 10.1359/JBMR.050916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vanderschueren D, Laurent MR, Claessens F, Gielen E, Lagerquist MK, Vandenput L, et al. Sex steroid actions in male bone. Endocr Rev. 2014;35(6):906–960. doi: 10.1210/er.2014-1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sontag W. Quantitative measurements of periosteal and cortical-endosteal bone formation and resorption in the midshaft of male rat femur. Bone. 1986;7(1):63–70. doi: 10.1016/8756-3282(86)90153-5. [DOI] [PubMed] [Google Scholar]
- 113.Sontag W. Quantitative measurement of periosteal and cortical-endosteal bone formation and resorption in the midshaft of female rat femur. Bone. 1986;7(1):55–62. doi: 10.1016/8756-3282(86)90152-3. [DOI] [PubMed] [Google Scholar]
- 114.Glatt V, Canalis E, Stadmeyer L, Bouxsein ML. Age-related changes in trabecular architecture differ in female and male C57BL/6J mice. J Bone Miner Res. 2007;22(8):1197–1207. doi: 10.1359/jbmr.070507. [DOI] [PubMed] [Google Scholar]
- 115.Sims NA, Dupont S, Krust A, Clement-Lacroix P, Minet D, Resche-Rigon M, et al. Deletion of estrogen receptors reveals a regulatory role for estrogen receptors-beta in bone remodeling in females but not in males. Bone. 2002;30(1):18–25. doi: 10.1016/S8756-3282(01)00643-3. [DOI] [PubMed] [Google Scholar]
- 116.Sims NA, Clement-Lacroix P, Minet D, Fraslon-Vanhulle C, Gaillard-Kelly M, Resche-Rigon M, et al. A functional androgen receptor is not sufficient to allow estradiol to protect bone after gonadectomy in estradiol receptor-deficient mice. J Clin Invest. 2003;111(9):1319–1327. doi: 10.1172/JCI200317246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Venken K, De Gendt K, Boonen S, Ophoff J, Bouillon R, Swinnen JV, et al. Relative impact of androgen and estrogen receptor activation in the effects of androgens on trabecular and cortical bone in growing male mice: a study in the androgen receptor knockout mouse model. J Bone Miner Res. 2006;21(4):576–585. doi: 10.1359/jbmr.060103. [DOI] [PubMed] [Google Scholar]
- 118.Almeida M, Iyer S, Martin-Millan M, Bartell SM, Han L, Ambrogini E, et al. Estrogen receptor-alpha signaling in osteoblast progenitors stimulates cortical bone accrual. J Clin Invest. 2013;123(1):394–404. doi: 10.1172/JCI65910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Liu Z, Mohan S, Yakar S. Does the GH/IGF-1 axis contribute to skeletal sexual dimorphism? Evidence from mouse studies. Growth Horm IGF Res. 2016;27:7–17. doi: 10.1016/j.ghir.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sims NA, Clement-Lacroix P, Da Ponte F, Bouali Y, Binart N, Moriggl R, et al. Bone homeostasis in growth hormone receptor-null mice is restored by IGF-I but independent of Stat5. J Clin Invest. 2000;106(9):1095–1103. doi: 10.1172/JCI10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Singhal V, Goh BC, Bouxsein ML, Faugere MC, DiGirolamo DJ. Osteoblast-restricted disruption of the growth hormone receptor in mice results in sexually dimorphic skeletal phenotypes. Bone Res. 2013;1(1):85–97. doi: 10.4248/BR201301006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zebaze RM, Ghasem-Zadeh A, Bohte A, Iuliano-Burns S, Mirams M, Price RI, et al. Intracortical remodelling and porosity in the distal radius and post-mortem femurs of women: a cross-sectional study. Lancet. 2010;375(9727):1729–1736. doi: 10.1016/S0140-6736(10)60320-0. [DOI] [PubMed] [Google Scholar]
- 123.Feik SA, Thomas CD, Clement JG. Age-related changes in cortical porosity of the midshaft of the human femur. J Anat. 1997;191(Pt 3):407–416. doi: 10.1046/j.1469-7580.1997.19130407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Thomas CD, Feik SA, Clement JG. Regional variation of intracortical porosity in the midshaft of the human femur: age and sex differences. J Anat. 2005;206(2):115–125. doi: 10.1111/j.1469-7580.2005.00384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sims NA, Martin TJ. Chapter 10—coupling of bone formation and resorption. In: Bilezikian JP, Martin TJ, Clemens TL, Rosen CJ, editors. Principles of bone biology. 4. Academic Press; 2020. pp. 219–243. [Google Scholar]
- 126.Piemontese M, Almeida M, Robling AG, Kim HN, Xiong J, Thostenson JD, et al. Old age causes de novo intracortical bone remodeling and porosity in mice. JCI Insight. 2017;2(17):e93771. doi: 10.1172/jci.insight.93771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cooper DM, Thomas CD, Clement JG, Turinsky AL, Sensen CW, Hallgrimsson B. Age-dependent change in the 3D structure of cortical porosity at the human femoral midshaft. Bone. 2007;40(4):957–965. doi: 10.1016/j.bone.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 128.Jilka RL, O'Brien CA, Roberson PK, Bonewald LF, Weinstein RS, Manolagas SC. Dysapoptosis of osteoblasts and osteocytes increases cancellous bone formation but exaggerates cortical porosity with age. J Bone Miner Res. 2014;29(1):103–117. doi: 10.1002/jbmr.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nickolas TL, Stein EM, Dworakowski E, Nishiyama KK, Komandah-Kosseh M, Zhang CA, et al. Rapid cortical bone loss in patients with chronic kidney disease. J Bone Miner Res. 2013;28(8):1811–1820. doi: 10.1002/jbmr.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Metzger CE, Swallow EA, Allen MR. Elevations in cortical porosity occur prior to significant rise in serum parathyroid hormone in young female mice with adenine-induced CKD. Calcif Tissue Int. 2020;106(4):392–400. doi: 10.1007/s00223-019-00642-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Martin TJ, Sims NA. RANKL/OPG; Critical role in bone physiology. Rev Endocr Metab Disord. 2015;16(2):131–139. doi: 10.1007/s11154-014-9308-6. [DOI] [PubMed] [Google Scholar]
- 132.Chia LY, Walsh NC, Martin TJ, Sims NA. Isolation and gene expression of haematopoietic-cell-free preparations of highly purified murine osteocytes. Bone. 2015;72:34–42. doi: 10.1016/j.bone.2014.11.005. [DOI] [PubMed] [Google Scholar]
- 133.McGregor NE, Murat M, Elango J, Poulton IJ, Walker EC, Crimeen-Irwin B, et al. IL-6 exhibits both cis- and trans-signaling in osteocytes and osteoblasts, but only trans-signaling promotes bone formation and osteoclastogenesis. J Biol Chem. 2019;294(19):7850–7863. doi: 10.1074/jbc.RA119.008074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dellinger MT, Garg N, Olsen BR. Viewpoints on vessels and vanishing bones in Gorham–Stout disease. Bone. 2014;63:47–52. doi: 10.1016/j.bone.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 135.Hominick D, Silva A, Khurana N, Liu Y, Dechow PC, Feng JQ, et al. VEGF-C promotes the development of lymphatics in bone and bone loss. Elife. 2018;7:e34323. doi: 10.7554/eLife.34323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lanske B, Divieti P, Kovacs CS, Pirro A, Landis WJ, Krane SM, et al. The parathyroid hormone (PTH)/PTH-related peptide receptor mediates actions of both ligands in murine bone. Endocrinology. 1998;139(12):5194–5204. doi: 10.1210/endo.139.12.6361. [DOI] [PubMed] [Google Scholar]
- 137.Karaplis AC, Luz A, Glowacki J, Bronson RT, Tybulewicz VL, Kronenberg HM, et al. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes Dev. 1994;8(3):277–289. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]
- 138.Amizuka N, Warshawsky H, Henderson JE, Goltzman D, Karaplis AC. Parathyroid hormone-related peptide-depleted mice show abnormal epiphyseal cartilage development and altered endochondral bone formation. J Cell Biol. 1994;126(6):1611–1623. doi: 10.1083/jcb.126.6.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gowen M, Lazner F, Dodds R, Kapadia R, Feild J, Tavaria M, et al. Cathepsin K knockout mice develop osteopetrosis due to a deficit in matrix degradation but not demineralization. J Bone Miner Res. 1999;14(10):1654–1663. doi: 10.1359/jbmr.1999.14.10.1654. [DOI] [PubMed] [Google Scholar]
- 140.Nampoothiri S, Fernandez-Rebollo E, Yesodharan D, Gardella TJ, Rush ET, Langman CB, et al. Jansen metaphyseal chondrodysplasia due to heterozygous H223R-PTH1R mutations with or without overt hypercalcemia. J Clin Endocrinol Metab. 2016;101(11):4283–4289. doi: 10.1210/jc.2016-2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.
Not applicable.