

Abstract
Congenital heart disease (CHD) is the most common birth defect worldwide and a main cause of perinatal and infant mortality. Our previous genome-wide association study identified 53 SNPs that associated with CHD in the Han Chinese population. Here, we performed functional screening of 27 orthologous genes in zebrafish using injection of antisense morpholino oligos. From this screen, 5 genes were identified as essential for heart development, including iqgap2, ptprt, ptpn22, tbck and maml3. Presumptive roles of the novel CHD-related genes include heart chamber formation (iqgap2 and ptprt) and atrioventricular canal formation (ptpn22 and tbck). While deficiency of maml3 led to defective cardiac trabeculation and consequent heart failure in zebrafish embryos. Furthermore, we found that maml3 mutants showed decreased cardiomyocyte proliferation which caused a reduction in cardiac trabeculae due to inhibition of Notch signaling. Together, our study identifies 5 novel CHD-related genes that are essential for heart development in zebrafish and first demonstrates that maml3 is required for Notch signaling in vivo.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-022-04669-5.
Keywords: Congenital heart malformation, GWAS, Danio rerio, Cardiac development, Notch signaling
Introduction
Congenital heart disease (CHD) is the most common birth defect worldwide and a main cause of perinatal and infant mortality which affect 7–9 per 1000 newborns depending on the population, methods of diagnosis, and severity of the disease [1, 2]. Epidemiological and population-based studies suggest that approximately 20–30% of CHD occurs in the context of syndromes that encompass non-cardiac manifestations, such as Holt-Oram syndrome, DiGeorge syndrome and Noonan syndrome, and for many of these, the primary genetic defect has been identified [3, 4]. The remaining cases represent isolated non-syndromic CHD which refers to CHD with only cardiac abnormalities including simple and severe CHD [5]. Most non-syndromic CHD occur sporadically and may be explained by a multifactorial inheritance model, suggesting that malformations result from multiple susceptibility genes with low-penetrance mutations or intermediate-penetrance mutations in combination with adverse environmental factors [6, 7]. In recent years, several genes have been implicated in monogenic forms of non-syndromic CHD, including TBX5 and TBX1 [8]. To identify more common genetic variants associated with sporadic non-syndromic CHD in Han Chinese populations, we performed a multistage genome-wide association study (GWAS) previously [9] and identified 53 SNPs that were associated with CHD. We then identified candidate genes located within 200 kb of these SNPs and ascertained their potential function in CHD by a functional screen of orthologous genes in the zebrafish (Danio rerio) model. The zebrafish represents a well-studied model organism in biomedical research because of its rapid development, the large number of eggs produced by a single cross, and the ease of genetic manipulation [10–12]. Additionally, the optical clarity of zebrafish embryos allows for real-time in vivo observation of physiological or pathological events that occur during organ development, rendering zebrafish an optimal vertebrate model system for studying cardiovascular development and homeostasis [13, 14]. The zebrafish has a relatively simple cardiac and vascular system, and the molecular mechanisms underlying vessel formation and morphogenesis are very similar to those of higher vertebrates. In particular, the early stages of zebrafish and human heart development exhibit a remarkable degree of anatomical and functional conservation [15, 16].
Here, we used antisense morpholino oligos (MO) designed against small (~ 25 bp) regions of mRNA to knock down the genes associated with susceptibility loci identified from CHD GWAS during cardiovascular development. After two rounds of functional screening, we identified 5 novel CHD-related genes that were essential for heart development in zebrafish, including iqgap2, ptprt, ptpn22, tbck, and maml3. Furthermore, we found deficiency of maml3 led to abnormal heart morphology and heart failure due to inhibition of Notch signaling. Together, our study identified 5 novel CHD-related genes in regulating zebrafish heart development and demonstrated that maml3 is required for Notch signaling in vivo. In addition, our results indicated that functional analyses in model systems can effectively translate GWAS findings into relevant biological information in an objective and unbiased manner.
Results
Identification of candidate human CHD susceptibility genes and functional screening of orthologous genes in zebrafish
In our previous GWAS screen involving 4225 CHD cases and 5112 non-CHD controls [9], we identified 53 SNPs that were associated with CHD (P < 1 × 10−4). After 2 rounds of validation with 2160 cases and 3866 controls, we found strong evidence of genome-wide significance (P < 5 × 10−8) for CHD susceptibility for 2 SNPs, rs2474937 at 1p12 (P = 8.44 × 10−10) and rs1531070 at 4q31.1 (P = 4.99 × 10−12). The other SNPs did not reach genome-wide significance (P < 5 × 10−8) by multiple validation analysis; however, they may still mark surrounding genes essential for heart development that confer a risk for CHD. Using Drosophila as a model, we have previously shown that functional screening of genes surrounding SNPs that did not reach genome-wide significance can effectively identify disease-related genes and thereby control for false negatives in multiple-stage population data from GWAS [17]. Using a similar strategy, we evaluated genomic regions flanking these potential CHD susceptibility loci (Fig. S1). Outcomes from a recent study revealed that the majority (93%) of disease- and trait-associated SNPs identified by GWAS are located within noncoding sequences [18]. Approximately 32% of GWAS SNPs are located in regulatory DNA marked by deoxyribonuclease I hypersensitive sites (DHSs); these regulator regions may affect genes within 100 kb [18]. We therefore included genes within 200 kb of each SNP and classified these as Class I if the respective SNP was located within the gene body and as Class II if the gene represented an adjacent gene located within 200 kb upstream or downstream of the respective SNP. In total, we identified 56 candidate genes in the human genome that were associated with the 53 SNPs (Table S1 in Supporting Information). We next identified the corresponding zebrafish orthologues using the Ensembl and ZFIN databases and only considered genes with at least 20% sequence identity and of homology type ‘orthologue-one to one.’ We found 28 orthologous zebrafish genes corresponding to an equal number of human genes (Table S1 in Supporting Information). One orthologous zebrafish gene, tshz2, which corresponded to three human genes and three candidate SNPs, was excluded from the functional analysis because antisense morpholino oligos (MO) could not be designed. The remaining 27 genes were included in the next functional study (Fig. S1).
In vivo functional analysis in zebrafish identifies genes essential for heart development
To investigate the function of the 27 zebrafish orthologues during heart development, we conducted an in vivo cardiac development screen using a transgenic zebrafish line Tg(myl7:GFP) expressing enhanced green fluorescent protein (GFP) under the control of the cardiac myosin light chain 2 promoter [19]. In this strain, GFP is exclusively expressed in cardiomyocytes such that cardiac development and abnormalities can be clearly visualized during embryonic development. After injection of translation-blocking MO designed against the transcripts of each of the candidate genes (Table S2 in Supporting Information) into 1- to 2-cell stage zebrafish embryos, we assessed the viability and cardiac morphology of embryos at 48 h post-fertilization (hpf) (Fig. S2), when key aspects of heart development including cardiac chamber and atrioventricular canal (AVC) formation and looping are apparent [20]. For the vast majority (26/27) of candidate genes, knockdown was not associated with a reduction in embryo viability below 50%. Serp2 morphants exhibited malformation of the whole embryo and less than 50% of embryos were viable, suggesting that this gene plays an essential role during development. Serp2 was therefore excluded as a candidate CHD susceptibility gene (Fig. S2A). Of the remaining 26 morphants, 7 (29.17%) exhibited a cardiac phenotype with a significantly increased rate of cardiac abnormalities in embryos versus controls (Fig. S2B). The 7 novel genes associated with cardiac defects after MO-mediated knockdown were iqgap2, ptprt, tbck, ptpn22, grm4, slc24a3, and maml3 (Fig. S3). Among them, 82.80% (207/250) of iqgap2 morphants and 71.40% (200/280) ptprt morphants exhibited a smaller ventricle (Fig. S3B, C); knockdown of tbck, ptpn22 and grm4 caused an extended AVC and heart looping defects (Fig. S3D–F); these defects were present in 82.31% (228/277), 85.32% (279/327) and 76.77% (195/254), respectively; While 74.31% (188/253) of slc24a3 morphants and 73.95% (159/215) maml3 morphants showed heart looping disorders (Fig. S3G, H). In general appearance, iqgap2 and maml3 morphants displayed normal morphology except slight pericardial edema in iqgap2 morphants (Fig. S3B’, H’); knockdown of ptprt and ptpn22 exhibited a smaller head, smaller eyes, unabsorbed yolk, and an edematous hindbrain, while ptpn22 morphants also showed pericardial edema and curved tail (Fig. S3C′, E′); tbck, grm4, and slc24a3 morphants had a smaller head, unabsorbed yolk and pericardial edema (Fig. S3D′, F′, G′).
To exclude the possible off-target effects, splice blocking MOs were designed to verify these phenotypes as second round of validation (Table S2 in Supporting Information) and the effectiveness of these MOs in affecting their target transcripts was confirmed by RT-PCR or sequencing (Fig. S4). Embryos injected with splice blocking MOs of iqgap2, ptprt, tbck and ptpn22 exhibited similar phenotypes to translation-blocking morphants, confirming an important role of these 4 genes in zebrafish heart development (Fig. 1A–E). While, even the splice blocking morphants of maml3 presented a similar phenotype, the defects of heart looping were relatively slight and these defects were just present in 55.73% (107/192) (Fig. 1F). However, the phenotypes of slc24a3 and grm4 splice blocking morphants were not reproducible, thus we excluded them from further analysis. In summary, we identified 5 genes that had important roles in heart development via a medium-throughput genetic screen based on antisense MOs in zebrafish.
Fig. 1.

Cardiac defects induced by knockdown of splice-blocking MO of iqgap2, ptprt, tbck, ptpn22, and maml3. A–F Representative heart images of control and morphant embryos. Ventral view, anterior to the top. Scale bars: 50 μm. A’–F’ Representative bright-field images of whole embryos. Scale bars: 500 μm
Knockdown of iqgap2 and ptprt causes cardiac chamber defects in developing zebrafish embryos
Similar to translation-blocking morphants, 82.69% (215/260) of iqgap2 splice-blocking morphants exhibited a smaller ventricle and/or an enlarged, balloon-like atrium and mild pericardial edema (Fig. 1B, B’). Double FISH for ventricular and atrial myosin heavy chain (vmhc and amhc), markers of ventricular and atrial cardiomyocytes, respectively, confirmed this phenotype (Fig. S5A).
Knockdown of ptprt with splice-blocking MO was associated with smaller ventricle size, whereas atrium size was not affected. Morphants also had smaller heads and eyes, and an edematous hindbrain (Fig. 1C, C’). These defects were present in 83.56% (244/292) of ptprt morphant embryos at 48 hpf. Smaller ventricle size in ptprt morphants was also confirmed by double FISH for vmhc and amhc (Fig. S5A).
The tbck and ptpn22 morphants exhibit an extended AVC and heart looping defects
Consistent with translation-blocking morphants, knockdown of tbck and ptpn22 by splice-blocking MO caused an extended AVC accompanied with heart looping defects (Fig. 1D, E), and these defects were present in 73.33% (209/285), and 73.11% (223/305), respectively. In addition, the tbck morphants exhibited smaller head and pericardial edema (Fig. 1D’); And the ptpn22 morphants had a series of severe morphological defects, including tail curling, hindbrain edema, smaller head, smaller eyes, unabsorbed yolk, and pericardial edema (Fig. 1E’). These results suggest that tbck and ptpn22 play an important role in AVC formation.
To more clearly define the cardiac defects in these two morphants, we examined the expression of the AVC myocardial marker bmp4 and endocardial marker has2. Strikingly, expression of bmp4 was upregulated and expanded throughout the entire ventricle in both morphants, rather than restricted to AVC region as observed in control MO-injected embryos (Fig. S5B). Similarly, has2 was also upregulated in these two morphants, with ectopic expression beyond the AVC. However, the cardiac chamber specific markers vmhc and amhc appeared unaffected in both morphants (Fig. S5B). Overall, we conclude that tbck and ptpn22 do not affect cardiac cell fate specification and patterning, but have an important role in the morphogenesis of the AVC.
To further verify the specificity of the phenotype exhibited in morphants, phenotypic rescue experiments were performed. Knockdown of iqgap2 with iqgap2 MO alone caused a smaller ventricle phenotype in 71.3% (72/101) of embryos (Fig. S6B); however, this cardiac chamber defects decreased to 32.3% (31/96) when co-injected iqgap2 MO with full-length zebrafish iqgap2 mRNA (Fig. S6B′). Similarly, 65.1% (95/146) and 62.4% (103/165) of embryos showed an extended AVC accompanied with heart looping defects in tbck and ptpn22 MO-injected groups, respectively (Fig. S6C, D). In contrast, such defects were corrected in 34.6% (46/133) and 28.7% (29/101) of embryos when co-injected with full-length tbck and ptpn22 mRNA, respectively (Fig. S6C′, D′). However, ptprt mRNA was not able to rescue the cardiac chamber defects in ptprt morphants. We speculate that this is caused by the fact that the full-length mRNA of ptprt is incomplete in the current database. Together, mRNA rescue experiments further verify the specificity of the phenotype exhibited in iqgap2, tbck, and ptpn22 morphants.
Expression pattern of the 5 CHD-related genes in the zebrafish heart
Based on previously reported data [21] and our whole-mount in situ hybridization (WISH) results, with the exception of iqgap2, the other 4 novel CHD susceptibility genes were not expressed clearly in the heart region at 24 hpf and 48 hpf (Fig. S7B, C). Besides, these 4 genes were also not expressed in other tissues that may impact heart development indirectly such as bilateral heart fields at 15-somite (Fig. S7A). We assume the expression of these genes was too low to be detected by WISH, and we used the highly sensitive absolute quantitative PCR to measure the absolute copy number of these 5 CHD-related genes in heart tissue. Besides, we also measure the absolute copy number of myl7, ctsk, and cryaa which should have robust, low and no expression in the heart, respectively. The expression level of ctsk was lower than iqgap2, but higher than maml3, ptprt, tbck, and ptpn22 (Fig. 2A). Besides, all the 5 CHD-related genes were expressed much higher than cryaa, so we assume they were indeed expressed in the heart and could not be noise.
Fig. 2.

Expression of iqgap2, ptprt, tbck, ptpn22 and maml3 in zebrafish heart. A Absolute q-PCR analysis of the expression of myl7, ctsk, cryaa, and the 5 CHD-related genes in zebrafish heart at 24 hpf and 48 hpf. Data represent mean ± SEM. B. The expression of iggap2 was assessed in zebrafish embryos at 36 hpf and 48 hpf by WISH. Dotted lines outline the heart. Ventral view, anterior to the top. Scale bars: 50 μm. C FISH-antibody staining for iqgap2 in Tg(myl7:GFP) embryos at 36 hpf and 48 hpf
As for iqgap2, although it has been mentioned that weak expression of this gene was observed in zebrafish heart, the precise expression pattern of iqgap2 is not clear [21]. We next assessed the developmental specific expression of iqgap2 in zebrafish by WISH and detected a low level of iqgap2 mRNA in the ventricular region at 36 hpf and 48 hpf (Fig. 2B). FISH-antibody staining confirmed the expression of iqgap2 in the ventricle at 36 hpf and 48 hpf (Fig. 2C). Together, these data suggest that iqgap2 is expressed in ventricle during heart development, which strongly implies that iqgap2 plays a significant role in zebrafish ventricular development.
Regional association plots for 3 novel CHD associated loci
Our experimental strategy combining GWAS data with functional screening identified 5 novel CHD susceptibility genes, TBCK, PTPN22, IQGAP2, PTPRT, and MAML3 as essential for cardiac development. Figure S8A-C shows regional risk association plots for the 3 SNPs in the vicinity of TBCK, PTPN22, and IQGAP2, respectively. Regional risk association of rs1531070 with MAML3 and rs490514 with PTPRT has been reported previously [9, 22].
The maml3-deficient zebrafish is generated by CRISPR/Cas9 technology
As previously reported, non-syndromic CHD occurs just as a heart defect without other organs anomaly. The maml3 morphants displayed nearly normal morphology in general appearance (Fig. 1F’) that is consistent with non-syndromic CHD. So we generated a maml3 mutant zebrafish line (maml3cq131) using CRISPR/Cas9 technology to further study the biological function of maml3 in zebrafish heart development (Fig. S9). As indel mutations may not completely disrupt the function of target gene [23], we deleted part of exon 1 and exon 2 of the maml3 gene by Co-injection of two gRNAs together with Cas9 mRNA (Fig. S9A). PCR amplification of genomic DNA isolated from the microinjected or the wild-type embryos (Fig. S9B) and sequencing of the PCR products (Fig. S9C) indicated the genomic sequence between the two target sites was deleted accurately.
The maml3 deficiency leads to abnormal heart morphology, impaired cardiac function, and heart failure
To better analyze the heart morphology in maml3 mutants, we crossed the maml3cq131 line with the transgenic line Tg(myl7:GFP) expressing GFP in cardiomyocytes as mentioned above and the heart defects were analyzed carefully. About 60% of maml3 mutants presented with slight cardiac looping defects compared with siblings at 2–3 dpf, which is similar to maml3 morphants (Fig. 3A). While the heart looping defects of maml3 mutants became apparent at 4–5 dpf (Fig. 3A) and about 15% of maml3 mutants showed collapsed chambers at 5 dpf (Fig. S10A). In the bright-field view, maml3 mutant larvae displayed a significant decrease in body length from 3 dpf compared with siblings (Fig. 3B, D). Besides, nearly all the mutant larvae exhibited smaller lens and most mutant larvae have no swim bladder (Fig. 3B). In addition, about half of the maml3 mutants began to develop pericardial edema at 3 dpf (Fig. 3B), and about 10% of maml3 mutants showed body edema and decreased blood circulation at 5 dpf (Fig. S10B and Movies S1, S2). Then, we analyzed the heart rate of sibling and maml3 mutant embryos from 2 to 5 dpf. At 2–4 dpf, heart rate showed no significant difference between sibling and maml3 mutant larvae (Fig. 3E). While, maml3 mutants displayed a significantly depressed heart rate at 5 dpf compared with siblings, which indicated that maml3 mutant larvae developed abnormal cardiac function at 5 dpf (Fig. 3E). In addition, we examined the expression level of maml3 from 3 to 5 dpf by FISH-antibody staining and found that maml3 was lowly expressed in heart region at these stages (Fig. 3F). This result implies that maml3 plays a role in zebrafish heart development from 3 to 5 dpf.
Fig. 3.

The maml3 mutants display abnormal heart morphology, cardiac dysfunction, and heart failure. A Representative heart images of siblings and maml3 mutants from 2 to 5 dpf. Ventral view, anterior to the top. Scale bars: 50 μm. B Representative bright-field images of sibling and maml3 mutant larvae from 2 to 5 dpf. Arrows, arrowheads, and asterisks indicate the lens, pericardium, and swim bladders, respectively. Scale bars: 300 μm. C WISH analysis of nppa and nppb of sibling and maml3 mutant embryos at 5 dpf. Ventral view, anterior to the top. Scale bars: 50 μm. D Mean body length of sibling and maml3 mutant larvae from 2 to 5 dpf. Data represent mean ± SEM. ns, no significance; ****P < 0.0001. Student’s t test. E Comparison of the heart rate (beats per minute) in sibling and maml3 mutant embryos from 2 to 5 dpf. Data represent mean ± SEM. ns, no significance; ***P < 0.001. Student’s t test. F FISH-antibody staining for maml3 in Tg(myl7:GFP) embryos at 3 dpf, 4dpf, and 5 dpf. Ventral view, anterior to the top. Scale bars: 50 μm
It has been reported that heart failure in zebrafish should include the following components: increased ventricular size, impaired contractility indicated by blood flow, increased edema, cardiac arrhythmias, and increased expression of heart failure biomarkers [24]. As the maml3 mutants exhibited decreased blood circulation, pericardial edema, and depressed heart rates (Fig. 3 and Fig. S10), we suspect that the mutants developed heart failure. As we know, natriuretic peptides A and B (nppa and nppb) are well-known biomarkers of heart failure and are extensively induced in ventricular during heart failure [24–26]. So, we examined the expression of nppa and nppb and found significantly increased expression of these two markers in the ventricular region of some maml3 mutants compared with siblings at 5 dpf (Fig. 3C), which further indicated maml3 mutant embryos develop heart failure. Together, except normal ventricular size, maml3 mutants showed all symptoms of heart failure. These data suggested that maml3 plays an essential role in zebrafish heart development and loss-of-function mutation in maml3 gene leads to heart failure in zebrafish.
The maml3 mutants show defective trabeculation
Embryonic heart failure, which is a common cause of morbidity and mortality in CHD, can be induced by abnormal ventricular trabeculation [27–29]. Previous studies showed that a reduction in trabeculation commonly leads to heart failure and early embryonic lethality in both zebrafish and mouse [30, 31]. So we detect whether the trabeculation is perturbed in maml3 mutants. As cardiac trabeculae are detectable around 3 dpf in zebrafish [30, 32], we observed trabeculation from 3 to 5 dpf in maml3 mutants. Although a radially arranged protrusion was readily discernible in siblings at 3 dpf, there was less and/or short trabeculae in about 30% of the maml3 mutants (Fig. 4A, B). In siblings, trabeculae became more pronounced and complex at 4 dpf and formed an elaborate network at 5 dpf (Fig. 4C, E). While about 20% of the maml3 mutants still showed poorly developed trabeculae carneae at 4–5 dpf (Fig. 4D, F). 3D surface reconstructions of sibling and maml3 mutant ventricles from 3 to 5 dpf further confirmed the reduced trabecular complexity in maml3 mutant hearts (Fig. 4A’–F’). Taken together, we assume the phenotype of heart failure in maml3 mutants is caused by a significant reduction in trabeculation.
Fig. 4.
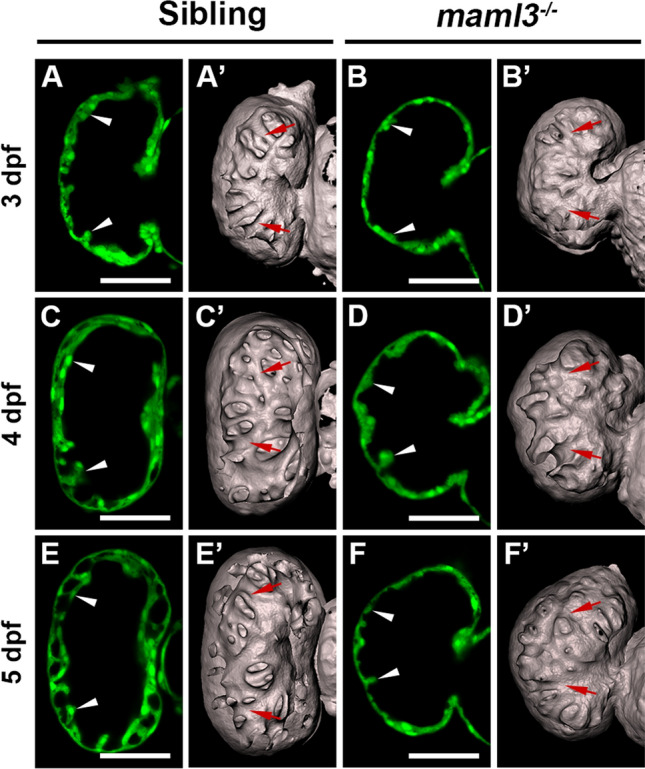
Knockout of maml3 leads to a reduction in trabeculae in zebrafish. A–F Representative ventricle images of siblings (A, C, E) and maml3 mutants (B, D, F). White arrowheads indicate the trabeculae of siblings and maml3 mutants. Ventral view, anterior to the top. Scale bars: 50 μm. A’–F’ Surface rendering of the ventricles of siblings (A’, C’, E’) and maml3 mutants (B’, D’, F’). Red arrows indicate the trabecular ridges of siblings and maml3 mutants
Notch signaling is down-regulated in maml3 mutants
To explore the regulatory mechanism of the maml3 gene involved in zebrafish heart development, we dissected the hearts from siblings or maml3 mutants at 3 dpf in Tg(myl7:GFP) background to perform RNA-Seq analysis. Transcriptional profile analyses revealed down-regulation of Notch signaling related gene in the mutant hearts (Fig. 5A). qPCR verified that the expression of Notch target genes (hey1, hey2, and her6) significantly decreased in the maml3 mutants (Fig. 5B). These results were surprising, as previous study showed that Maml3-null mice exhibited no obvious abnormalities and unchanged mRNA levels of Notch target genes [33]. To further confirm the change of Notch signaling in zebrafish maml3 mutant, we crossed this mutant line with the Notch reporter line Tg(Tp1:d2GFP) expressing a destabilized GFP upon Notch activation [34] and the Tg(myl7:CFPNTR)cq132 line expressing CFP under the cardiac myosin light chain 2 promoter. Then, Notch activity was assessed in this double transgenic fish at 3 dpf and 5 dpf, respectively. As previously reported [35], Notch-activated Tp1:d2GFP was prominent in a subset of ventricular cardiomyocytes and AV endocardial cells at 3 dpf while the Tp1:d2GFP+ signal gradually declined at 5 dpf in siblings (Fig. 5C, arrows). These Tp1:d2GFP+ cardiomyocytes in the myocardial wall form clusters across the surface of the ventricle in 3D images (Fig. 5C, arrows). However, maml3 mutant cardiomyocytes exhibited very weak Tp1:d2GFP+ signal in the ventricular cardiomyocytes at 3 or 5dpf (Fig. 5C, arrowheads), while the Tp1:d2GFP+ signal had no significant difference in AV region between siblings and mutants (Fig. 5C, asterisks). These results indicate that maml3 deficiency leads to a decrease in Notch signaling in the ventricular cardiomyocytes, not the AV region. As Notch activity in the ventricular zone is required for proper trabeculae development [36, 37], we assume trabeculation defect in the maml3 mutant is caused by decreased Notch signaling.
Fig. 5.

Notch signaling is down-regulated in maml3 mutant embryos. A Heatmap represents the expression analysis of genes related with Notch signaling pathway in siblings and maml3 mutant. Bar on the right represents the abundances of expression; color shading from blue to red indicates the expression abundances from low to high. B qPCR of representative Notch target genes for cardiac tissue from siblings and maml3 mutant embryos at 3 dpf. Data represent mean ± SEM. **P < 0.01. Student’s t test. C Confocal slices and 3D projections of the ventricles of siblings and maml3 mutants. Ventral view, anterior to the top. Arrows indicate the Tp1:d2GFP+ cardiomyocytes and asterisks indicate AV endocardial cells. Ventral view, anterior to the top. Scale bars: 50 μm
The maml3 mutants exhibit decreased cardiomyocyte proliferation
As Notch signaling is essential for regulating cardiomyocyte proliferation during ventricular trabeculation in mouse [36], we assumed the reduction in trabeculae in maml3 mutants caused by decreased ventricular cardiomyocyte proliferation. To assess if cardiomyocyte proliferation was impaired in maml3 mutants with defective trabeculae, we immunostained for proliferating cell nuclear antigen (PCNA), a marker of cell proliferation and expressed in the nuclei of cells during the DNA synthesis phase of the cell cycle. The maml3 mutant cardiomyocytes displayed a significantly reduced level of cell proliferation compared with siblings at 3 dpf (Fig. 6A, B), and cardiomyocyte proliferation was further reduced at 4 dpf. In addition, maml3 mutant larvae also showed a decrease in proliferative ventricular cells using another cell proliferation marker pH3 at 3 dpf(a marker of M phase) (Fig. 6C, D). Similarly, cardiomyocyte proliferation was more significantly suppressed at 4 dpf. These data indicate that the reduced trabeculae in maml3 mutants is induced by decreased ventricular cardiomyocyte proliferation with inhibition of Notch signaling.
Fig. 6.

Reduction in proliferative ventricular cardiac myocytes in maml3 mutants. A, C The PCNA and pH3 immunofluorescence staining in Tg(myl7:GFP) embryos showed decreased cell proliferative activity in maml3 cardiomyocytes. Ventral view, anterior to the top. Scale bars: 50 μm. B, D The number of PCNA and pH3 positive cells in maml3 mutant ventricles was significantly decreased when compared with siblings. Data represent mean ± SEM. *P < 0.05; **P < 0.01***; P < 0.001; ****P < 0.0001. Student’s t test
Inhibition of Notch signaling by DAPT in zebrafish shows a similar phenotype to maml3 mutants
If the cardiac defects in maml3 mutants were caused by inhibition of Notch signaling, we believed that blocking Notch signaling in wild-type embryos should display a similar phenotype to maml3 mutants. While, as previously reported, the obvious phenotype of zebrafish embryos treated with high concentrations at 50 μM of DAPT showed a curved tail [38] which does not exhibit in maml3 mutants. We assumed Notch signaling was just partially inhibited in maml3 mutants, as the Tp1:d2GFP+ signal was still present in the AV endocardial cells of maml3 mutant hearts (Fig. 5C, asterisks). To test this hypothesis, we inhibited Notch activity with different concentrations of DAPT starting at 24 hpf and found that most embryos did not show a curved tail when the concentration of DAPT was below 15 μM. So we treated zebrafish embryos with DAPT at 15 μM and observed the cardiac defect when Notch signaling was partially inhibited. The Notch-activated Tp1:d2GFP+ signals appeared in a subset of ventricular cardiomyocytes and AV endocardial cells in control group at 3 dpf and 5 dpf, whereas these Tp1:d2GFP+ signals almost disappeared in about half (10/18 in 3 dpf and 8/16 in 5 dpf) of the DAPT-treated embryo hearts (Fig. S11C). Meanwhile, some of DAPT-treated embryos showed similar cardiac defects to maml3 mutant, including abnormal cardiac looping, defective cardiac trabeculation, and heart failure (Fig. S11A, B). In addition, these embryos displayed pericardial edema and depressed heart rate at 3 and 5 days post-DAPT treatment compared with control group (Fig. S12A, B). Taken together, these data indicate that partial inhibition of Notch signaling with DAPT in zebrafish embryos exhibits a similar phenotype to maml3 mutant larvae.
Discussion
GWAS was a widely used approach for the identification of susceptibility loci of human disease [39]. However, the biological significance of these SNPs is largely unknown. Considering the highly heterogeneous human population structure and properties of SNPs, many SNPs surrounding causal genes may have been missed due to low association values and multiple validations. To assess the value of SNPs not reaching genome-wide significance, we developed a systematic screening strategy using zebrafish and our previous GWAS data in this study.
Herein, we identified 5 novel CHD-related genes that are essential for embryonic heart development in zebrafish. Previously, we had combined human disease-associated GWAS and animal model screening and identified genes essential for male fertility [17]. We now discovered 5 disease-related genes by selecting genes surrounding SNPs followed by functional screening in zebrafish in the same way. Our approach thus provides a very efficient way to discover risk genes for human disease; moreover, the outcomes of our study suggest GWAS stage data can provide useful information with and without population validation. In addition, different with our previous knockdown strategy on fruit fly of screening for male fertility [17], we perform a second round validation and 5 MOs were validated in the second round study. Two rounds of knockdown screening help us to discover valid CHD-related genes in a short cycle.
IQGAP2 is a member of the IQGAP family which is relevant to cell migration, cell proliferation, and other cellular processes in humans [40]. There is no report about IQGAP2 in cardiac development, while deficiency of IQGAP1, which is another member of IQGAP family, shows impaired heart function and increased cardiomyocyte apoptosis after pressure overload in mice [41]. Ptprt is a member of the protein tyrosine phosphatase (PTP) family, and interestingly, Ptpn22 is a member of the non-receptor of the PTP family. We found knockdown both of ptprt and ptpn22 exhibited a small head, small eyes, and an edematous hindbrain in zebrafish embryos (Fig. 1C’, E’), indicating that these two genes have similar functions. Ptprz, another member of the PTP family, was reported to play essential roles in cardiac development in mouse and zebrafish [42]. While mutation of PTPN11, another member of the non-receptor of the PTP family, caused CHD in newborns [43]. Together with our results, we speculated that the PTP family had a close relationship with CHD. Tbck contains a presumptive kinase domain, the TBC (Tre-2/Bub2/Cdc16) domain, and a rhodanese homology domain (RHOD) and is involved in cell proliferation and cell growth [44, 45], which may explain heart abnormalities in tbck morphants.
MAML3, a member of Mastermind-like protein family which contain MAML1, MAML2, and MAML3, is a transcriptional coactivator for Notch signaling [46] and there are no reports linking cardiac development with Mastermind-like family to our knowledge. Although Maml3 is predicted to play an important role in Notch pathway, the majority of research on this gene has been linked to tumors [47–49]. Herein, we found deficiency of maml3 led to inhibition of Notch signaling and consequent abnormal heart morphology and impaired cardiac function in zebrafish embryos for the first time. While, a previous study showed that Maml3-null mice exhibited no decrease in Notch signaling or apparent abnormalities [33]. The discrepancy for the different phenotype in zebrafish and mouse is not fully understood, and it is possible that we deleted partial sequences of maml3 exon 1 and exon 2 in zebrafish, while only exon 1 of Maml3 was knocked out in mouse [33]. In addition, although mice are more closely related to humans in evolutionary than zebrafish, for some diseases, the human symptoms can be recapitulated in zebrafish rather than mice. For example, the mutation in human SEC23B cause congenital dyserythropoietic anemia type II (CDAII) [50], while deficiency of Sec23b in mice has a markedly different phenotype exhibiting abnormalities in pancreas [51]. However, knockdown of zebrafish sec23b leads to aberrant erythrocyte development which recapitulates part of the human phenotype [50]. Combined with our previous report which identified the CHD susceptibility loci of rs1531070 at 4q31.1 for MAML3 in Han Chinese populations [9], we assume the zebrafish maml3 mutants recapitulate part of the symptoms in CHD patients.
Notch signaling plays important role in cardiac trabeculation, while different components of Notch pathway exhibit diverse functions in different species. For example, deletion of the Notch ligand Dll4 results in impaired trabeculation in mouse, whereas disruption of another Notch ligand Jag1 in mouse shows normal cardiac trabeculae [52]. In contrast, zebrafish jag2b mutant hearts exhibit increased trabeculation [35]. Herein, we found deficiency of maml3 led to decrease in ventricular cardiomyocyte proliferation due to inhibition of Notch signaling, which caused impaired cardiac trabeculation and heart failure in zebrafish.
In summary, integrating with our previous GWAS data, we identified 5 novel CHD-related genes that are essential for embryonic cardiac development in zebrafish, including iqgap2, ptprt, ptpn22, tbck, and maml3. Furthermore, we found that deficiency of maml3 led to abnormal heart morphology and heart failure due to inhibition of Notch signaling. In addition, our results demonstrate that functional analyses in zebrafish can effectively translate GWAS findings into relevant biological information. The 5 essential risk genes we found warrant further study and may help reveal mechanisms underlying CHD.
Materials and methods
Zebrafish strains
All animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee of Southwest University (Approval No. 20140920-01) and were consistent with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The transgenic or mutant zebrafish lines Tg(myl7:GFP) [19], Tg(Tp1:d2GFP) [34], Tg(myl7:CFPNTR)cq132 and maml3cq131 mutant were used in this study. Embryos were treated with 0.003% 1-phenyl-2-thiourea (PTU, Sigma) from 24 hpf to inhibit pigmentation.
Microinjection of MOs and synthetic mRNAs
MO against each of the 27 candidate genes and a negative control MO was designed by and obtained from Gene tools and injected into 1- to 2-cell embryos to ensure ubiquitous distribution described previously [53–55]. Each of MOs was injected into embryos from a low dose to a high dose (1 ng, 2 ng, 4 ng, 6 ng, 8 ng, and 10 ng per embryo) in MO screen experiment. If embryos exhibited cardiac abnormalities (one or more of the following: developmental delay in AVC formation, looping defects, a smaller or enlarged atrium or ventricle) when injected with MOs within these doses, we considered these MOs as candidates and established the optimal concentration of each MO according to the embryonic mortality and phenotypic severity in different doses. However, if embryos showed normal hearts even injected with 10 ng MO, we assume that the MO did not affect heart development. MOs used in this study are summarized in Table S2 in Supporting Information. The doses of translation-blocking MOs used in this study are as follows: iqgap2 MO = 3 ng; ptprt MO = 4.5 ng; tbck MO = 1.5 ng; ptpn22 MO = 6 ng; grm4 MO = 2 ng; slc24a3 MO = 2 ng; maml3 MO = 0.8 ng. The doses of splice-blocking MOs used in this study are as follows: iqgap2 MO = 6 ng; ptprt MO = 3 ng; tbck MO = 4.5 ng; ptpn22 MO = 3 ng; maml3 MO = 8 ng. The viability of zebrafish embryos was ascertained at 48 h post-fertilization (hpf), and morphology was assessed at this time point in all lines with more than 70% viability.
For rescue experiments, full-length cDNAs of iqgap2, ptprt, tbck, and ptpn22 were subcloned from the pGEMT vector into the pCS2 + vector. Capped mRNA transcripts were generated in vitro from the linearized plasmids using mMESSAGE mMACHINE kit (Ambion) according to the manufacturer’s protocol. Each of these mRNAs was co-injected with the corresponding MO at 1- to 2-cell embryos.
Morphology analysis
A total of 200–400 whole embryos were collected in each group, and heart morphology was observed using a fluorescent stereomicroscope (Leica). We graded abnormalities of heart morphology, size and atrioventricular valve as described previously [56–58]. Confocal images were captured by ZEN2010 software equipped on an LSM780 confocal microscope (Carl Zeiss). Bright-field images were captured using a SteREO Discovery V20 microscope equipped with Axio Vision Rel 4.8.2 software (Carl Zeiss).
In situ hybridization and antibody staining
Whole-mount in situ hybridization (WISH), fluorescent in situ hybridization (FISH), and antibody staining were performed as previously described [59–62]. The primers used for probe synthesis are listed in Table S3 in Supporting Information. Images of WISH were captured using a SteREO Discovery V20 microscope equipped with Axio Vision Rel 4.8.2 software (Carl Zeiss). Images of FISH were captured using ZEN2010 software equipped on an LSM780 or LSM880 confocal microscope (Carl Zeiss). Whole-mount antibody staining was performed as previously described [63]. Primary antibodies against PCNA (1:500; Abcam) and pH3 (1:500; Millipore) were applied. Images were captured using ZEN2010 software equipped on an LSM780 or LSM880 confocal microscope (Carl Zeiss).
Dissection of zebrafish heart
Zebrafish heart isolation was performed as previously described [64]. Briefly, zebrafish embryos were anaesthetized (about 800 in number for each sample) in PBS by treatment with tricaine (Sigma, USA). Manually dissect out GFP-positive hearts under a fluorescence stereomicroscope (Leica) with a pair of fine tweezers (WPI) and concentrate them in the center of the dish. Collect the hearts in the smallest possible volume and pipette into a 1.5 ml tube containing 0.75 ml PBS (on ice). Then, the hearts were sedimented by centrifuging at 2,000 × g for 5 min at 4 °C. Carefully remove the supernatant, then add 0.5 ml TriZol (Invitrogen) to the tube containing the sedimented hearts.
Transcriptome sequencing (RNA-Seq)
Three samples each for WT and maml3 mutant were prepared for sequencing, and mean values were calculated for the final analysis. The RNA library of each sample was constructed and sequenced using an Illumina HiSeq platform by Novogene following their standard procedures. Raw data of fastq format were firstly processed through in-house perl scripts. In this step, clean data were obtained by removing reads containing adapter, reads containing ploy-N and low quality reads from raw data. All the downstream analyses were based on the clean data with high quality. The expressed values of all genes were calculated and normalized to fragments per kilobase of transcript per million mapped fragments (FPKM). FPKM values were obtained from RNA-seq data to generate a heatmap by the Helm software. The color scale shown represents FPKM values which were log2 transformed. Raw data were deposited in the NCBI Sequence Read Archive database under accession number PRJNA900016.
Reverse transcription-polymerase chain reaction (RT-PCR)
To verify the effectiveness of MOs in affecting their target transcripts, RT-PCR was performed. Embryos were injected with MOs at the 1- to 2-cell stage, and total RNA was isolated from whole embryos at 48 hpf using TriZol reagent (Invitrogen). RNA was reverse transcribed into cDNA using OmniScript Reverse Transcription Kit (QIAGEN). The primers used for RT-PCR are listed in Table S3 in Supporting Information.
Absolute quantitative real-time PCR (qPCR)
RT-qPCR was carried out using the FastStart Universal SYBR Green Master (Roche). Total RNA was isolated from heart tissue at 24 hpf and 48 hpf using TriZol reagent (Invitrogen). RNA was reverse transcribed into cDNA using OmniScript Reverse Transcription Kit (QIAGEN). The primers used for RT-qPCR are listed in Table S3 in Supporting Information. For absolute quantification by PCR, iqgap2, ptprt, tbck, ptpn22, maml3, myl7 (high expression in heart), ctsk (low expression in heart), and cryaa (no expression in heart) cDNA fragments were amplified and cloned into pGEMT-Easy vector (Promega) as templates. Each plasmid DNA was tenfold serially diluted from 107 to 103 copies and used in subsequent experiments for generating standard curve. The qPCR reaction (15 μl) in triplicates comprised of PCR grade water (3.3 μl); SYBR Green (2x, 7.5 μl); Primer F and R (10 pmol/μl, 0.6 μl each) and plasmid DNA(3 μl) or cDNA (10 ng/μl, 3.0 μl). The obtained Ct values were plotted against the logarithm of their template copy numbers of each standard plasmid DNA, and the standard curve was generated by a linear regression of the plotted points. Absolute quantification of the sample of 24hpf and 48hpf was read from the standard curve [65].
Generation of the maml3cq131 mutant line
The maml3cq131 mutant line was generated by CRISPR/Cas9 technology as previously described [23]. In brief, two gRNAs (50 pg) together with Cas9 mRNA (300 pg) were co-injected into 1-cell stage wild-type embryos, and the lysate of about 10 embryos at 24 hpf was used as templates for PCR. The PCR products were sequenced to examine the deleted genomic sequences in maml3 target region, and embryos with effective genome editing were raised to adults (F0). Then, F0 fish were screened to identify the founder whose progeny carry the mutation, and offspring of identified F0 was raised up to get the candidate progeny (F1). Last, individual F1 adults were screened by PCR using the tail fin genomic DNA and the genotypes of mutants were determined by DNA sequencing. The target sequence of maml3 and PCR primers is depicted in Table S3 in Supporting Information.
Statistical methods
Differences between treatment and control groups after MO injection and assessment at 48 hpf were analyzed using one-way ANOVA and the Fisher’s exact test (Stata Software 11.0, Stata Corporation, College Station, TX). Other statistical calculations in this paper were performed using GraphPad Prism 8. Unpaired Student’s t test was used for statistical analysis. Values of P < 0.05 were considered statistically significant (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001); ns (P > 0.05) indicated not significant. All values were expressed as mean ± SEM.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Prof. Peidong Han for providing the Tg(Tp1:d2GFP) line.
Author contributions
ML, LL, ZH, JS, and ZZ designed the experiments. ML and LL designed the experiments. JM, YG, JL, JS, TZ, MJ, YW, XG, and JH performed most experiments and analysis. ML wrote the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was supported by the National Key Project of Research and Development Program (2018YFC1004200, 2018YFC1004202), the Program of the National Natural Science Foundation of China (81830100), and the National Key R&D Program of China (2021YFA0805000).
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics approval
All animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee of Southwest University (Approval No. 20140920-01).
Consent to participate
This article does not contain any studies involving human participants performed by any of the authors.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jianlong Ma, Yayun Gu, Juanjuan Liu and Jingmei Song contributed equally to this work.
Contributor Information
Lingfei Luo, Email: lluo@swu.edu.cn.
Mingxi Liu, Email: mingxi.liu@njmu.edu.cn.
References
- 1.van der Linde D, Konings EE, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJ, Roos-Hesselink JW. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. 2011;58:2241–2247. doi: 10.1016/j.jacc.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 2.Liu Y, Chen S, Zuhlke L, Black GC, Choy MK, Li N, Keavney BD. Global birth prevalence of congenital heart defects 1970–2017: updated systematic review and meta-analysis of 260 studies. Int J Epidemiol. 2019;48:455–463. doi: 10.1093/ije/dyz009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landis BJ, Cooper DS, Hinton RB. CHD associated with syndromic diagnoses: peri-operative risk factors and early outcomes. Cardiol Young. 2016;26:30–52. doi: 10.1017/S1047951115001389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Øyen N, Poulsen G, Boyd HA, Wohlfahrt J, Jensen PKA, Melbye M. Recurrence of congenital heart defects in families. Circulation. 2009;120:295–301. doi: 10.1161/CIRCULATIONAHA.109.857987. [DOI] [PubMed] [Google Scholar]
- 5.Yang L, Liu X, Chen Y, Shen B. An update on the CHDGKB for the systematic understanding of risk factors associated with non-syndromic congenital heart disease. Comput Struct Biotechnol J. 2021;19:5741–5751. doi: 10.1016/j.csbj.2021.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ware S, Jefferies J (2012) New genetic insights into congenital heart disease. J Clin Exp Cardiolog S8:003 [DOI] [PMC free article] [PubMed]
- 7.Yasuhara J, Garg V. Genetics of congenital heart disease: a narrative review of recent advances and clinical implications. Transl Pediatr. 2021;10:2366–2386. doi: 10.21037/tp-21-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wessels M, Willems P. Genetic factors in non-syndromic congenital heart malformations. Clin Genet. 2010;78:103–123. doi: 10.1111/j.1399-0004.2010.01435.x. [DOI] [PubMed] [Google Scholar]
- 9.Hu Z, Shi Y, Mo X, Xu J, Zhao B, Lin Y, Yang S, Xu Z, Dai J, Pan S, et al. A genome-wide association study identifies two risk loci for congenital heart malformations in Han Chinese populations. Nat Genet. 2013;45:818–821. doi: 10.1038/ng.2636. [DOI] [PubMed] [Google Scholar]
- 10.Lieschke GJ, Currie PD. Animal models of human disease: zebrafish swim into view. Nat Rev Genet. 2007;8:353–367. doi: 10.1038/nrg2091. [DOI] [PubMed] [Google Scholar]
- 11.Francoeur N, Sen R. Advances in cardiac development and regeneration using zebrafish as a model system for high-throughput research. J Dev Biol. 2021;9:40. doi: 10.3390/jdb9040040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brittijn SA, Duivesteijn SJ, Belmamoune M, Bertens LFM, Bitter W, de Bruijn JD, Champagne DL, Cuppen E, Flik G, Vandenbroucke-Grauls CM, et al. Zebrafish development and regeneration: new tools for biomedical research. Int J Dev Biol. 2009;53:835–850. doi: 10.1387/ijdb.082615sb. [DOI] [PubMed] [Google Scholar]
- 13.Gays D, Santoro MM. The admiR-able advances in cardiovascular biology through the zebrafish model system. Cell Mol Life Sci CMLS. 2013;70:2489–2503. doi: 10.1007/s00018-012-1181-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bowley G, Kugler E, Wilkinson R, Lawrie A, van Eeden F, Chico TJA, Evans PC, Noël ES, Serbanovic-Canic J. Zebrafish as a tractable model of human cardiovascular disease. Br J Pharmacol. 2021;179:900–917. doi: 10.1111/bph.15473. [DOI] [PubMed] [Google Scholar]
- 15.Isogai S, Horiguchi M, Weinstein B. The vascular anatomy of the developing zebrafish: an atlas of embryonic and early larval development. Dev Biol. 2001;230:278–301. doi: 10.1006/dbio.2000.9995. [DOI] [PubMed] [Google Scholar]
- 16.Brown DR, Samsa LA, Qian L, Liu J. Advances in the study of heart development and disease using zebrafish. J Cardiovasc Dev Dis. 2016;3:13. doi: 10.3390/jcdd3020013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu J, Wu H, Wen Y, Liu Y, Zhou T, Ni B, Lin Y, Dong J, Zhou Z, Hu Z, et al. Identification of seven genes essential for male fertility through a genome-wide association study of non-obstructive azoospermia and RNA interference-mediated large-scale functional screening in Drosophila. Hum Mol Genet. 2015;24:1493–1503. doi: 10.1093/hmg/ddu557. [DOI] [PubMed] [Google Scholar]
- 18.Maurano M, Humbert R, Rynes E, Thurman R, Haugen E, Wang H, Reynolds A, Sandstrom R, Qu H, Brody J, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burns CG, Milan DJ, Grande EJ, Rottbauer W, MacRae CA, Fishman MC. High-throughput assay for small molecules that modulate zebrafish embryonic heart rate. Nat Chem Biol. 2005;1:263–264. doi: 10.1038/nchembio732. [DOI] [PubMed] [Google Scholar]
- 20.Miura G, Yelon D. A guide to analysis of cardiac phenotypes in the zebrafish embryo. Methods Cell Biol. 2011;101:161–180. doi: 10.1016/B978-0-12-387036-0.00007-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fang X, Zhang B, Thisse B, Bloom GS, Thisse C. IQGAP3 is essential for cell proliferation and motility during zebrafish embryonic development. Cytoskeleton. 2015;72:422–433. doi: 10.1002/cm.21237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin Y, Guo X, Zhao B, Liu J, Da M, Wen Y, Hu Y, Ni B, Zhang K, Yang S, et al. Association analysis identifies new risk loci for congenital heart disease in Chinese populations. Nat Commun. 2015;6:8082. doi: 10.1038/ncomms9082. [DOI] [PubMed] [Google Scholar]
- 23.Xiao A, Wang Z, Hu Y, Wu Y, Luo Z, Yang Z, Zu Y, Li W, Huang P, Tong X, et al. Chromosomal deletions and inversions mediated by TALENs and CRISPR/Cas in zebrafish. Nucleic Acids Res. 2013;41:e141. doi: 10.1093/nar/gkt464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Narumanchi S, Wang H, Perttunen S, Tikkanen I, Lakkisto P, Paavola J. Zebrafish heart failure models. Front Cell Dev Biol. 2021;9:662583. doi: 10.3389/fcell.2021.662583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Becker JR, Robinson TY, Sachidanandan C, Kelly AE, Coy S, Peterson RT, MacRae CA. In vivo natriuretic peptide reporter assay identifies chemical modifiers of hypertrophic cardiomyopathy signalling. Cardiovasc Res. 2012;93:463–470. doi: 10.1093/cvr/cvr350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi X, Verma S, Yun J, Brand-Arzamendi K, Singh KK, Liu X, Garg A, Quan A, Wen XY. Effect of empagliflozin on cardiac biomarkers in a zebrafish model of heart failure: clues to the EMPA-REG OUTCOME trial? Mol Cell Biochem. 2017;433:97–102. doi: 10.1007/s11010-017-3018-9. [DOI] [PubMed] [Google Scholar]
- 27.Sedmera D. Pathways to embryonic heart failure. Am J Physiol Heart Circ Physiol. 2009;297:H1578–1579. doi: 10.1152/ajpheart.00873.2009. [DOI] [PubMed] [Google Scholar]
- 28.Zhang W, Chen H, Qu X, Chang C-P, Shou W. Molecular mechanism of ventricular trabeculation/compaction and the pathogenesis of the left ventricular noncompaction cardiomyopathy (LVNC) Am J Med Genet C Semin Med Genet. 2013;163:144–156. doi: 10.1002/ajmg.c.31369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyer HV, Dawes TJW, Serrani M, Bai W, Tokarczuk P, Cai J, de Marvao A, Henry A, Lumbers RT, Gierten J, et al. Genetic and functional insights into the fractal structure of the heart. Nature. 2020;584:589–594. doi: 10.1038/s41586-020-2635-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu J, Bressan M, Hassel D, Huisken J, Staudt D, Kikuchi K, Poss KD, Mikawa T, Stainier DY. A dual role for ErbB2 signaling in cardiac trabeculation. Development. 2010;137:3867–3875. doi: 10.1242/dev.053736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gassmann M, Casagranda F, Orioli D, Simon H, Lai C, Klein R, Lemke G. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature. 1995;378:390–394. doi: 10.1038/378390a0. [DOI] [PubMed] [Google Scholar]
- 32.Peshkovsky C, Totong R, Yelon D. Dependence of cardiac trabeculation on neuregulin signaling and blood flow in zebrafish. Dev Dyn. 2011;240:446–456. doi: 10.1002/dvdy.22526. [DOI] [PubMed] [Google Scholar]
- 33.Oyama T, Harigaya K, Sasaki N, Okamura Y, Kokubo H, Saga Y, Hozumi K, Suganami A, Tamura Y, Nagase T, et al. Mastermind-like 1 (MamL1) and mastermind-like 3 (MamL3) are essential for Notch signaling in vivo. Development. 2011;138:5235–5246. doi: 10.1242/dev.062802. [DOI] [PubMed] [Google Scholar]
- 34.Clark BS, Cui S, Miesfeld JB, Klezovitch O, Vasioukhin V, Link BA. Loss of Llgl1 in retinal neuroepithelia reveals links between apical domain size, Notch activity and neurogenesis. Development (Cambridge, England) 2012;139:1599–1610. doi: 10.1242/dev.078097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han P, Bloomekatz J, Ren J, Zhang R, Grinstein JD, Zhao L, Burns CG, Burns CE, Anderson RM, Chi NC. Coordinating cardiomyocyte interactions to direct ventricular chamber morphogenesis. Nature. 2016;534:700–704. doi: 10.1038/nature18310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grego-Bessa J, Luna-Zurita L, del Monte G, Bolos V, Melgar P, Arandilla A, Garratt AN, Zang H, Mukouyama YS, Chen H, et al. Notch signaling is essential for ventricular chamber development. Dev Cell. 2007;12:415–429. doi: 10.1016/j.devcel.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Samsa LA, Givens C, Tzima E, Stainier DYR, Qian L, Liu J. Cardiac contraction activates endocardial Notch signaling to modulate chamber maturation in zebrafish. Development. 2015;142:4080–4091. doi: 10.1242/dev.125724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang T, Arslanova D, Gu Y, Augelli-Szafran C, Xia W. Quantification of gamma-secretase modulation differentiates inhibitor compound selectivity between two substrates Notch and amyloid precursor protein. Mol Brain. 2008;1:15. doi: 10.1186/1756-6606-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, Yang J. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101:5–22. doi: 10.1016/j.ajhg.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hedman AC, Smith JM, Sacks DB. The biology of IQGAP proteins: beyond the cytoskeleton. EMBO Rep. 2015;16:427–446. doi: 10.15252/embr.201439834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sbroggio M, Carnevale D, Bertero A, Cifelli G, De Blasio E, Mascio G, Hirsch E, Bahou WF, Turco E, Silengo L, et al. IQGAP1 regulates ERK1/2 and AKT signalling in the heart and sustains functional remodelling upon pressure overload. Cardiovasc Res. 2011;91:456–464. doi: 10.1093/cvr/cvr103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Katraki-Pavlou S, Kastana P, Bousis D, Ntenekou D, Varela A, Davos CH, Nikou S, Papadaki E, Tsigkas G, Athanasiadis E, et al. Protein tyrosine phosphatase receptor-zeta1 deletion triggers defective heart morphogenesis in mice and zebrafish. Am J Physiol Heart Circ Physiol. 2022;322:H8–H24. doi: 10.1152/ajpheart.00400.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarkozy A, Conti E, Seripa D, Digilio MC, Grifone N, Tandoi C, Fazio VM, Di Ciommo V, Marino B, Pizzuti A, et al. Correlation between PTPN11 gene mutations and congenital heart defects in Noonan and LEOPARD syndromes. J Med Genet. 2003;40:704–708. doi: 10.1136/jmg.40.9.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Y, Yan X, Zhou T. TBCK influences cell proliferation, cell size and mTOR signaling pathway. PLoS ONE. 2013;8:e71349. doi: 10.1371/journal.pone.0071349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu J, Lu G. Multiple functions of TBCK protein in neurodevelopment disorders and tumors. Oncol Lett. 2021;21:17. doi: 10.3892/ol.2020.12278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kitagawa M. Notch signalling in the nucleus: roles of Mastermind-like (MAML) transcriptional coactivators. J Biochem. 2016;159:287–294. doi: 10.1093/jb/mvv123. [DOI] [PubMed] [Google Scholar]
- 47.Alzofon N, Koc K, Panwell K, Pozdeyev N, Marshall CB, Albuja-Cruz M, Raeburn CD, Nathanson KL, Cohen DL, Wierman ME, et al. Mastermind like transcriptional coactivator 3 (MAML3) drives neuroendocrine tumor progression. Mol Cancer Res. 2021;19:1476–1485. doi: 10.1158/1541-7786.MCR-20-0992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang X, Bledsoe KL, Graham RP, Asmann YW, Viswanatha DS, Lewis JE, Lewis JT, Chou MM, Yaszemski MJ, Jen J, et al. Recurrent PAX3-MAML3 fusion in biphenotypic sinonasal sarcoma. Nat Genet. 2014;46:666–668. doi: 10.1038/ng.2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li J, Li T, Lu Y, Shen G, Guo H, Wu J, Lei C, Du F, Zhou F, Zhao X, et al. MiR-2392 suppresses metastasis and epithelial-mesenchymal transition by targeting and in gastric cancer. FASEB J. 2017;31:3774–3786. doi: 10.1096/fj.201601140RR. [DOI] [PubMed] [Google Scholar]
- 50.Schwarz K, Iolascon A, Verissimo F, Trede NS, Horsley W, Chen W, Paw BH, Hopfner KP, Holzmann K, Russo R, et al. Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat Genet. 2009;41:936–940. doi: 10.1038/ng.405. [DOI] [PubMed] [Google Scholar]
- 51.Tao J, Zhu M, Wang H, Afelik S, Vasievich MP, Chen XW, Zhu G, Jensen J, Ginsburg D, Zhang B. SEC23B is required for the maintenance of murine professional secretory tissues. Proc Natl Acad Sci USA. 2012;109:E2001–2009. doi: 10.1073/pnas.1209207109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.D'Amato G, Luxan G, del Monte-Nieto G, Martinez-Poveda B, Torroja C, Walter W, Bochter MS, Benedito R, Cole S, Martinez F, et al. Sequential Notch activation regulates ventricular chamber development. Nat Cell Biol. 2016;18:7–20. doi: 10.1038/ncb3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nasevicius A, Ekker SC. Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet. 2000;26:216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- 54.Lu H, Ma J, Yang Y, Shi W, Luo L. EpCAM is an endoderm-specific Wnt derepressor that licenses hepatic development. Dev Cell. 2013;24:543–553. doi: 10.1016/j.devcel.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 55.Yang Y, Wang H, He J, Shi W, Jiang Z, Gao L, Jiang Y, Ni R, Yang Q, Luo L (2021) A single-cell-resolution fate map of endoderm reveals demarcation of pancreatic progenitors by cell cycle. Proc Nat Acad Sci USA 118:e2025793118 [DOI] [PMC free article] [PubMed]
- 56.Bakkers J. Zebrafish as a model to study cardiac development and human cardiac disease. Cardiovasc Res. 2011;91:279–288. doi: 10.1093/cvr/cvr098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tu S, Chi NC (2012) Zebrafish models in cardiac development and congenital heart birth defects. Differentiation 84:4–16 [DOI] [PMC free article] [PubMed]
- 58.Staudt D, Stainier D. Uncovering the molecular and cellular mechanisms of heart development using the zebrafish. Annu Rev Genet. 2012;46:397–418. doi: 10.1146/annurev-genet-110711-155646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He J, Lu H, Zou Q, Luo L. Regeneration of liver after extreme hepatocyte loss occurs mainly via biliary transdifferentiation in zebrafish. Gastroenterology. 2014;146(789–800):e788. doi: 10.1053/j.gastro.2013.11.045. [DOI] [PubMed] [Google Scholar]
- 60.Liu C, Wu C, Yang Q, Gao J, Li L, Yang D, Luo L. Macrophages mediate the repair of brain vascular rupture through direct physical adhesion and mechanical traction. Immunity. 2016;44:1162–1176. doi: 10.1016/j.immuni.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 61.He J, Chen J, Wei X, Leng H, Mu H, Cai P, Luo L. Mammalian target of rapamycin complex 1 signaling is required for the dedifferentiation from biliary cell to bipotential progenitor cell in zebrafish liver regeneration. Hepatology. 2019;70:2092–2106. doi: 10.1002/hep.30790. [DOI] [PubMed] [Google Scholar]
- 62.Cai P, Mao X, Zhao J, Nie L, Jiang Y, Yang Q, Ni R, He J, Luo L. Farnesoid X receptor is required for the redifferentiation of bipotential progenitor cells during biliary-mediated zebrafish liver regeneration. Hepatology. 2021;74:3345–3361. doi: 10.1002/hep.32076. [DOI] [PubMed] [Google Scholar]
- 63.Chen J, He J, Ni R, Yang Q, Zhang Y, Luo L. Cerebrovascular injuries induce lymphatic invasion into brain parenchyma to guide vascular regeneration in zebrafish. Dev Cell. 2019;49:697. doi: 10.1016/j.devcel.2019.03.022. [DOI] [PubMed] [Google Scholar]
- 64.Lombardo VA, Otten C, Abdelilah-Seyfried S (2015) Large-scale zebrafish embryonic heart dissection for transcriptional analysis. J Vis Exp 95:52087 [DOI] [PMC free article] [PubMed]
- 65.Whelan JA, Russell NB, Whelan MA. A method for the absolute quantification of cDNA using real-time PCR. J Immunol Methods. 2003;278:261–269. doi: 10.1016/S0022-1759(03)00223-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.