

Abstract
The iconic Tasmanian devil (Sarcophilus harrisii) is endangered due to the transmissible cancer Devil Facial Tumour Disease (DFTD), of which there are two genetically independent subtypes (DFT1 and DFT2). While DFT1 and DFT2 can be differentially diagnosed using tumour biopsies, there is an urgent need to develop less-invasive biomarkers that can detect DFTD and distinguish between subtypes. Extracellular vesicles (EVs), the nano-sized membrane-enclosed vesicles present in most biofluids, represent a valuable resource for biomarker discovery. Here, we characterized the proteome of EVs from cultured DFTD cells using data-independent acquisition–mass spectrometry and an in-house spectral library of > 1500 proteins. EVs from both DFT1 and DFT2 cell lines expressed higher levels of proteins associated with focal adhesion functions. Furthermore, hallmark proteins of epithelial–mesenchymal transition were enriched in DFT2 EVs relative to DFT1 EVs. These findings were validated in EVs derived from serum samples, revealing that the mesenchymal marker tenascin-C was also enriched in EVs derived from the serum of devils infected with DFT2 relative to those infected with DFT1 and healthy controls. This first EV-based investigation of DFTD increases our understanding of the cancers’ EVs and their possible involvement in DFTD progression, such as metastasis. Finally, we demonstrated the potential of EVs to differentiate between DFT1 and DFT2, highlighting their potential use as less-invasive liquid biopsies for the Tasmanian devil.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-021-03955-y.
Keywords: Exosomes, Microvesicles, Marsupials, Size exclusion chromatography, Proteomics, Cancer diagnostics
Introduction
The Tasmanian devil (Sarcophilus harrisii) is the world’s largest extant carnivorous marsupial, currently endangered due to two transmissible cancers, both of which cause devil facial tumour disease (DFTD). The first described DFTD (referred to as DFT1) has caused an 82% decline in the wild since its discovery in 1996 [1]. DFT1 is caused by a clonal cell of Schwann cell origin [2], transmitted among devils as an allograft through biting during social interactions, in which the tumour cells are hypothesised to be inoculated into penetrating injuries in the skin and oral mucosa to establish and proliferate [3]. The second genetically independent transmissible cancer (DFT2) was first reported in 2014 and is also of Schwann cell origin and transmitted as an allograft [4]. In contrast to DFT1, which arose in a female devil [5], DFT2 appears to have originated in a male individual [4]. As the majority of documented DFT2 cases to date are among males [6], it is thought that the Y chromosome present in the DFT2 karyotype results in antigenicity, and thus resistance to infection, among female hosts [7]. On the other hand, females hosts have been demonstrated to present higher tolerance to DFT1 infection [8].While DFT1 has widely spread across Tasmania since its discovery [9], DFT2 is currently confined to the Channel region of southeast Tasmania [6]. Fatal in almost 100% of cases, DFT1 and DFT2 present with tumour masses on facial, oral, and neck regions [10].
Both of these non-virally induced cancers exploit the antigen presentation pathway to avoid immune recognition. For instance, DFT1 epigenetically downregulates the major histocompatibility complex class I (MHC-I) molecules to escape the devil immune system [11]. In contrast to DFT1, DFT2 tumour cells express MHC-I molecules. However, the MHC-I molecules expressed in DFT2 cells share allotype identity with devils in the region in which DFT2 was first identified, reducing the hosts’ capacity to detect DFT2 tumours as foreign tissues [12]. As the devil immune system is unable to “see” and respond to the grafted tumour, DFTD progresses and kills its host within 6–12 months after the presentation of clinical signs [13]. For DFT1, death of the host has been proposed to be a consequence of three main mechanisms: secondary infection, starvation, and metastasis [14]. Large DFT1 tumours often present necrotic areas in contact with the environment, and therefore, bacterial infections are common in later stages of the disease. These infections may invade healthy tissue and induce sepsis [14]. Starvation occurs as a consequence of mechanical obstruction of feeding or through the secretion of by-products from the tumour, inducing anorexia [14]. With DFT1, metastasis occurs in 65% of cases, primarily affecting regional lymph nodes, lungs, and spleen [10]. However, the timing and mechanisms of DFT1 metastasis require further investigation to provide a better understanding of the pathogenesis and progression of the cancer. Information on DFT2 pathogenesis, such as metastasis or time until host death, is not available due to the relatively recent discovery of DFT2 [4] and the limited number of DFT2 infected animals (n = 25) in the literature thus far [6].
DFT1 and DFT2 are grossly indistinguishable, but can be differentially diagnosed using histopathology, immunohistochemistry, cytogenetics, and PCR techniques [4, 15, 16]. These laboratory techniques require the collection of a tumour biopsy from the infected animal, a procedure which is avoided unless tumours exhibit ulceration (especially inside of the oral cavity) to reduce possibilities of secondary infections, tumour spread, and/or potential risk of facilitating rapid tumour growth [17]. Discovery of a biomarker that can lead to the development of a rapid and less-invasive differential diagnostic test for DFT1 and DFT2 would significantly improve the potential scope and scale of disease surveillance across Tasmania. Most cells, including cancer cells in humans and animal models, secrete extracellular vesicles (EVs) into their extracellular environment. EVs are lipid bilayer structures which contain bioactive cargo, such as proteins, genetic material, and lipids [18]. EV cargo is enriched in a subset of common EV marker proteins related to their biogenesis [19], but also may represent the proteome of the parent cell. EVs have been proposed to be major players in the process of intercellular communication, through the transfer of their cargo, both locally and systemically [20]. Using transcellular and paracellular routes, EVs can enter a variety of body fluids, including blood; therefore, EVs can be readily isolated from serum or plasma [21]. The EV phospholipid bilayer membrane protects cargo against degradation by serum proteases and nucleases [22]. These properties have made EVs an increasingly attractive tool for biomarker discovery in cancer research and other diseases [23].
A major hallmark of cancer is the need for communication with other cells to survive, invade, and progress. As EVs play a crucial role in intercellular communication, they are active participants in the process of tumorigenesis and cancer progression [24]. Thus, EVs originating from tumour cells, or the tumour microenvironment, can support tumour cell growth and promote successful colonization of local and distant organs [25–27]. EVs facilitate the process of metastasis through a variety of mechanisms, including immune modulation, microenvironment remodelling, angiogenesis, intravasation and extravasation of tumour cells, and preparation of the metastasis niche at the target organs [28, 29]. As metastasis is the leading cause of death in cancer patients [30], and as EVs express molecules which are associated with cancer progression, they have been investigated as a source of prognostic biomarkers for cancer [31].
Here, we provide the first analysis of the proteome of EVs from DFTD cells. We identified cell and focal adhesion proteins related to metastasis and validated the use of an EV-associated protein to identify DFTD subtypes in clinical serum samples, demonstrating the potential application of EVs as a liquid biopsy for the differential diagnosis of DFTD.
Materials and methods
Cell lines and cell culture
Three tumour cell lines (C5065, 1426, and 4906) were established from three devils with confirmed DFT1 by personnel of the Tasmanian Government Department of Primary Industries, Parks, Water and Environment (DPIPWE). DFT2 cell lines (JARVIS, SNUG and RED VELVET) and devil fibroblasts cells lines were originally established at Menzies Institute for Medical Research from three devils with confirmed DFT2 [4] and three healthy captive devils, respectively. DFTD and fibroblast cell lines were obtained from tumour and ear biopsies, respectively. Both tumour and ear biopsies were collected from devils using a 4 mm disposable punch (Kai Medical, Singen) under a Standard Operating Procedure approved by the General Manager, Natural and Cultural Heritage Division, Tasmanian Government Department of Primary Industries, Parks, Water and the Environment. Cell line sources are detailed in Online Resource 1. Frozen stocks of DFT1, DFT2, and devil fibroblasts cell lines were thawed to perform the study experiments.
Cell lines were cultured in RPMI medium (Gibco), supplemented with 10% heat-inactivated foetal bovine serum (FBS), and 1% antibiotic–antimycotic (Thermo Fisher Scientific). Each cell line was cultured in duplicates in a 175 cm2 culture flask (Corning) at 35 °C (Tasmanian devil normal body temperature) in a fully humidified atmosphere of 5% CO2.
Tasmanian devil serum samples
Frozen serum samples from 12 advanced-stage DFT1 infected devils, 5 advanced-stage DFT2 infected devils, and 10 healthy controls were used for this study. DFT1 infected devils were considered to be in advanced stage (mid or late) of the disease based on large tumour volumes (15 ml–161 ml). Tumour volumes of DFT2 infected devils were not recorded; however, all these animals were euthanised at the time of sampling due to poor physiological conditions associated with advanced DFT2. Blood was obtained from conscious or anesthetised devils by venepuncture from either jugular or marginal ear vein (between 0.3 and 1 ml) and transferred into empty or clot activating tubes. Samples were centrifuged at 1000 g for 10 min, and the serum was pipetted off and stored frozen at − 20 °C (short term storage; up to 3 months) or − 80 °C (long-term storage, up to 6 years) until further use. All animal procedures were performed under a Standard Operating Procedure approved by the General Manager, Natural and Cultural Heritage Division, Tasmanian Government Department of Primary Industries, Parks, Water and the Environment and under the auspices of the University of Tasmania Animal Ethics Committee (permit numbers: A0017550, A0013326, A0015835).
Isolation of extracellular vesicles
Cell culture
Cell cultures were used at 60%–70% confluence. Culture medium was discarded, and cells were washed twice with phosphate-buffered saline (PBS) and then incubated in culture medium, supplemented with 5% of heat-inactivated FBS, for 48 h. This medium had been previously subjected to centrifugation at 100,000 g for 18 h at 4 °C to deplete bovine EVs from FBS [32]. After 48 h, the cultured medium of each cell line was collected and centrifuged at 1500 g for 10 min at 4 °C to remove cells and debris. The supernatant was further centrifuged at 10,000 g for 10 min at 4 °C to remove larger extracellular vesicles. Centrifuged supernatants were then concentrated using Amicon Ultra-15 centrifugal filters (MWCO 100 kDa) to a final volume of 2 ml. Then, following the manufacturer’s instructions, the concentrated supernatant was subjected to size exclusion chromatography on qEV2-35 columns (IZON; recovery range: 35 nm–350 nm). Briefly, EVs were eluted in PBS containing 0.05% sodium azide in eight fractions of 1 ml each after a 14 ml void volume and pooled. The EV samples were concentrated with Amicon Ultra-15 centrifugal filters (MWCO 100 kDa) to a final volume of 500 µl and stored at − 80 °C until further use.
Serum samples
A similar methodology used to isolate EVs derived from cultured DFTD and devil fibroblast cell lines was used to isolate EVs derived from devil serum samples. Briefly, serum samples were thawed on ice, and 500 μl of each sample was aliquoted into an Eppendorf tube and centrifuged at 1,500 g for 10 min at 4 °C to remove debris. The samples were further centrifuged at 10,000 g for 10 min at 4 °C to pellet larger extracellular vesicles. The supernatants were taken and subjected immediately to size exclusion chromatography on qEV2-35 columns (IZON) following the manufacturer’s instructions. Briefly, EVs were eluted in PBS containing 0.05% sodium azide in eight fractions of 1 ml each after the collection of 14 ml of void volume and pooled. The EV samples were concentrated with Amicon Ultra-15 centrifugal filters (MWCO 100 kDa) to a final volume of 1 ml and stored in aliquots of 500 μl at − 80 °C until future use.
Electron microscopy
The EVs derived from cultured DFT1, DFT2, devil fibroblasts cells and devil serum were imaged using a JEM-2100 transmission electron microscope (JEOL Tokyo, Japan) equipped with a LaB6 filament operating at 200 kV. Images were recorded using a Gatan Orius SC200 2 k × 2 k charge-coupled device (CCD) camera at a range of magnifications. 400-mesh carbon-coated copper grids (ProSciTech) which had been glow-discharged in the air to render them hydrophilic using an Emitech 950X equipped with a K350 glow discharge unit (Quorum Technologies) were used. 10 μl of the EV samples derived from cultured cells and devil serum obtained with the SEC columns were dropped onto the prepared grids and left for at least 30 s. Excess fluid was drawn off with filter paper, and two drops of 2% Uranyl Acetate were added for approximately 10 s each before being drawn off with filter paper. The grids were then dried and transferred into transmission electron microscopy for viewing.
Nano-particle tracking analysis
EV size distribution and concentration of EVs derived from cultured cells (DFT1, DFT2, and devil fibroblast cells) were determined using a Nanosight NS300 nanoparticle analyser (Malvern Panalytical, Malvern, UK) equipped with a 405 nm laser and Nanosight NTA 3.2 software. Samples were measured in PBS, and camera level was set at 14 for all recordings. Camera focus was adjusted to make the particles appear as sharp individual dots, and three 30-s videos were recorded for each sample. For post-acquisition analysis, all functions were set to automatic except detection threshold, which was set at 5. EV size data were normalised to the equal area under the curve for comparison between samples. EV size distribution and concentration of devil serum EVs were determined using a ZetaView PMX-120 nanoparticle analyser (Particle Metrix, Inning am Ammersee, Germany) equipped with Zetaview Analyse Software version 8.05.12. Prior to measurement, the system was calibrated as per the manufacturer’s instructions with 100 nm Nanospheres 3100A (Thermo Fisher Scientific). Measurements were performed in scatter mode, and for all measurements, the cell temperature was maintained at 25 °C. Each sample was diluted in PBS to a final volume of 1 ml. Capture settings were sensitivity 80, shutter 100, and frame rate 30. Post-acquisition settings were minimum trace length 10, min brightness 30, min area 5, and max area 1000.
Protein preparation
EV proteins derived from cultured DFT1, DFT2, devil fibroblast cells, and devil serum were extracted according to the method of Abramowicz et al. [33]. EV samples were mixed with acetonitrile to a final concentration of 50% (v/v) and evaporated using a centrifugal vacuum concentrator. Protein samples were resuspended in 100 µl and 150 µl of denaturation buffer (7 M urea and 2 M thiourea in 40 mM Tris, pH 8.0) for EV samples derived from cultured cells and serum samples, respectively. Duplicate samples of the cell lines were washed twice in PBS after collecting cultured supernatants for EVs’ isolation. A cell count of 1 × 107 cells per ml was determined using a hemocytometer. Pelleted cells were carefully lysed in 700 µl of denaturation buffer supplemented with 1% (w/v) of Halt protease inhibitor cocktail (100X, Thermo Fisher Scientific) and then incubated on a tube rotator for 2 h at 4 °C. The lysate was centrifuged at 13,000 g for 10 min, and the supernatant finally cleaned up by precipitation using 9 × volumes of 100% ethanol overnight at − 20 °C. Precipitated proteins were pelleted by centrifugation at 13,000 g for 10 min. Protein pellets were briefly air-dried and then reconstituted in 100 µl of denaturation buffer. Protein concentration from lysates and EV samples derived from cultured cells and devil serum was determined using EZQ protein quantification Kit (Thermo Fisher Scientific). For mass spectrometry analysis, aliquots of 30 μg of protein from each sample (n = 18 EV samples derived from cultured cells and 18 lysates samples; n = 27 EV samples derived from devil serum) were sequentially reduced using 10 mM DTT overnight at 4 °C, alkylated using 50 mM iodoacetamide for 2 h at ambient temperature, and then digested with 1.2 μg proteomics-grade trypsin/LysC (Promega) according to the SP3 protocol [34]. Digests were acidified by the addition of TFA to 0.1%, and peptides were collected by centrifugation at 21,000 × g for 20 min. Samples were further cleaned up by offline desalting using ZipTips (Merck) according to the manufacturer’s instructions.
SDS-PAGE and western blotting
EVs from cultured cells and lysate samples (DFT1, DFT2, and devil fibroblast cells) resuspended in denaturation buffer were mixed with freshly prepared β-mercaptoethanol (5% (v/v) Sigma-Aldrich) and heated for 10 min at 95 °C. Protein samples (20 μg in each lane) were separated on a Bolt™ 4%–12%, Bis–Tris, 1.0 mm, Mini Protein Gel (Thermo Fisher Scientific Invitrogen™) in NuPAGE™ MES SDS Running Buffer, alongside a molecular weight marker (SeeBlue™ Plus2 Pre-stained Protein Standard, Thermo Fisher Scientific Invitrogen™). Blotting was performed on an Immobilin®-P PVDF membrane with 0.45 µm pore size (Merck Millipore) using a Mini Blot Module (Thermo Fisher Scientific Invitrogen™). Membranes were blocked in 5% skimmed milk in TBS containing 0.1% Tween-20 (TBS-T) for 1 h at room temperature, primary antibodies rabbit anti-Syntenin-ab19903 (Abcam) at 1:1000, mouse anti-Flotillin-1-BD610821 (BD Transduction Laboratories) at 1:1000, and mouse anti-Golgi matrix protein—BD610822 at 1:1000 (BD Transduction Laboratories) were incubated overnight in 5% skimmed milk in TBS-T at 4 °C. After incubation, membranes were washed three times in TBS-T for 10 min at room temperature. Membranes were subsequently probed with Amersham ECL Mouse IgG, HRP-linked whole Ab (from sheep) (NA931) or Amersham ECL Rabbit IgG, HRP-linked whole Ab (from donkey) (NA934) at 1:5000 in TBS-T for 1 h at room temperature. After incubation, membranes were washed three times in TBS-T for 10 min at room temperature and subsequently visualised using Clarity Western ECL Substrate (Bio-Rad). Images were acquired on a ChemiDoc™ Touch Imaging System (Bio-Rad) and analysed with Image Lab Software (Bio‐Rad).
Liquid chromatography and mass spectrometry analysis
High-pH peptide fractionation
Experiment-specific peptide spectral libraries were generated for DFT1, DFT2, and devil fibroblast cell lysates (n = 18, 3 biological replicates and 2 technical replicates of each cell line), EVs derived from cultured cells (DFT1, DFT2 and devil fibroblast cells; n = 18, 3 biological replicates and 2 technical replicates of each cell line), and devil serum (n = 27; 10 captive healthy devils, 12 devils infected with DFT1 and 5 devils infected with DFT2) using offline high-pH fractionation. For each library, a pooled digest comprising aliquots of each sample set (180 μg) was desalted using Pierce desalting spin columns (Thermo Fisher Scientific) according to the manufacturer’s guidelines. Each sample was evaporated to dryness and then resuspended in 25 μl HPLC loading buffer (2% acetonitrile containing 0.05% TFA) and injected onto a 100 × 1 mm Hypersil GOLD (particle size 1.9 μm) HPLC column. Peptides were separated using an Ultimate 3000 RSLC system equipped with microfractionation and automated sample concatenation, operated at 30 μl/minute using a 40 min linear gradient of 96% mobile phase A (water containing 1% triethylamine, adjusted to pH 9.6 using acetic acid) to 50% mobile phase B (80% acetonitrile containing 1% triethylamine), followed by 6 min washing in 90% B and re-equilibration in 96% A for 8 min. Sixteen concatenated fractions were collected into 0.5 ml low-bind Eppendorf tubes, evaporated to dryness, and then reconstituted in 12 μl HPLC loading buffer.
Mass spectrometry–data-dependent acquisition (DDA)
Peptide fractions of the pooled digest samples coming from cell lysates, EVs derived from cultured cells, and devil serum were analysed by nanoflow HPLC–MS/MS using an ultimate 3000 nano-RSLC system (Thermo Fisher Scientific) coupled with a Q-Exactive HF mass spectrometer fitted with a nano-spray flex ion source (Thermo Fisher Scientific), and controlled using Xcalibur software (ver 4.3). Approximately 1 μg of each fraction was injected and separated using a 90-min segmented gradient by preconcentration onto a 20 mm×75 μm PepMap 100 C18 trapping column and then separation on a 250 mm×75 μm PepMap 100 C18 analytical column at a flow rate of 300 nL/min and held at 45 °C. MS Tune software (version 2.9) parameters used for data acquisition were: 2.0 kV spray voltage, S-lens RF level of 60, and heated capillary set to 250 °C. MS1 spectra (390–1500 m/z) were acquired at a scan resolution of 600,000 followed by MS2 scans using a Top15 DDA method, with 20-s dynamic exclusion of fragmented peptides. MS2 spectra were acquired at a resolution of 15,000 using an AGC target of 2e5, maximum IT of 28 ms, and normalised collision energy of 30.
Mass spectrometry–data-independent acquisition (DIA)
Individual peptide samples derived from cell lysates (DFT1, DFT2, and devil fibroblast cells; n = 18, 3 biological replicates and 2 technical replicates of each cell line) and EVs derived from cultured cells (DFT1, DFT2, and devil fibroblast cells; n = 18, 3 biological replicates and 2 technical replicates of each cell line) and devil serum (n = 27; 10 captive healthy devils, 12 infected devils with DFT1 and 5 devils infected with DFT2) were analysed by nanoflow HPLC–MS/MS using the instrumentation and LC gradient conditions described above but using DIA mode. The sequence of sample injections was randomised by blinding the MS operator to the sample codes. MS1 spectra (390–1240 m/z) were acquired at 60,000 k resolution, followed by sequential MS2 scans across 26 DIA × 25 amu windows over the range of 397.5–1027.5 m/z, with 1 amu overlap between sequential windows. MS2 spectra were acquired at a resolution of 30,000 using an AGC target of 1e6, maximum IT of 55 ms, and normalised collision energy of 27.
Raw data processing
Both DDA–MS and DIA–MS raw files were processed using Spectronaut software (version 13.12, Biognosys AB). Each project-specific library was generated using the Pulsar search engine to search DDA MS2 spectra against the Sarcophilus harrisii UniProt reference proteome (comprising 22,388 entries, last modified in August 2020). With the exception that single-hit proteins were excluded, default (BGS factory) settings were used for both spectral library generation and DIA data extraction. For library generation, these included N-terminal acetylation and methionine oxidation as variable modifications and cysteine carbamidomethylation as a fixed modification, up to two missed cleavages allowed and peptide, protein, and PSM thresholds set to 0.01. Mass tolerances were based on first-pass calibration and extensive calibration for the calibration and main searches, respectively, with correction factors set to 1 at the MS1 and MS2 levels. Briefly, XIC extraction deployed dynamic retention time alignment with a correction factor of 1. Protein identification deployed a 1% q value cut-off at precursor and protein levels, automatic generation of mutated peptide decoys based on 10% of the library and dynamic decoy limitation for protein identification. MS2-level data were used for relative peptide quantitation between experimental samples, using the intensity values for the Top3 peptides (stripped sequences) and cross-run normalization based on median peptide intensity.
Statistical analysis
Spectronaut protein quantitation pivot reports, including protein description, gene names, and UniProt accession numbers, were uploaded into Perseus software (version 1.6.10.50) for further data processing and statistical analysis. Quantitative values were log2 transformed and proteins filtered according to the number of valid values. Proteins detected in at least 70% of one group were considered for the analysis. Remaining missing values were imputed with random intensity values for low-abundance proteins based on a normal abundance distribution using default Perseus settings. Principal component analysis (PCA) was carried out using the filtered proteome for the cultured cell EVs and lysate samples to reduce the dimensionality of the dataset using Perseus software (version 1.6.10.50). Differential abundance of proteins between sample groups was determined using Student’s t test with a permutation-based false discovery rate (FDR) controlled at 0.05 and s0 (minimum fold change) values set to 0.1 to exclude proteins with very small differences between means. Significantly upregulated proteins (FDR-corrected p value < 0.05) of EVs derived from serum samples of devils with DFT2 (n = 5) relative to those infected with DFT1 (n = 12) and healthy controls (n = 10) were compared with significantly upregulated proteins (FDR-corrected p value < 0.05) of EVs derived from DFT2 relative to EVs derived from DFT1 cultured cells. One protein was found to overlap between groups. This protein was evaluated as a DFT2 status classifier by subjecting DFT2/healthy and DFT2/DFT1 cohort protein intensity values of each to receiver-operating characteristic (ROC) curve analysis to calculate their area under the curve, sensitivity, and specificity with bootstrapped confidence intervals using R 3.6.2 [35]. The classification cut-off values were determined using Youden’s index.
Bioinformatics analysis
FUNRICH version 3.1.3 [36] was used to compare the EV proteins derived from the Tasmanian devil cell lines with the human Vesiclepedia, and with the top-100 EV proteins reported in the Exocarta database [37]. We compared the proteome of cell lysates and EVs and used DAVID software for functional enrichment analysis of the proteins present only in EVs [38, 39]. DAVID software was also used for bioinformatic analysis of proteins that were upregulated in EVs derived from DFT1 and DFT2 cultured cells compared to fibroblasts EVs and also upregulated in DFT1 and DFT2 lysates relative to fibroblasts’ lysates. These analyses were carried out to identify whether DFT1 and DFT2 EVs maintain key features of their cell of origin. To investigate general biological patterns from DFT1 and DFT2 EVs relative to a healthy control, we used two different bioinformatics approaches. Over Representation Analysis (ORA) is used to determine whether particular biological functions or processes are over-represented in a subset of genes or proteins when compared to a background proteome [40]. We used ORA to analyse the subset of proteins demonstrating a pattern of upregulation in EVs derived from DFT1 and DFT2 compared to EVs derived from devil fibroblasts and EVs derived from DFT1 cells relative to those released from DFT2 cells. UniProt accessions for these proteins were uploaded to DAVID [38, 39] to identify clusters of GO terms, protein families, and pathways. Functional terms with p values < 0.05 after Benjamini–Hochberg correction for multiple hypothesis testing were considered significant [38]. The protein list was analysed using the Sarcophilus harrisii species proteome UniProt database. To complement the ORA, we performed Gene Set Enrichment Analysis (GSEA) to analyse patterns across the entire post-filtering EV proteome. GSEA tests for non-random patterns of enrichment of functional protein groups with quantitative expression data rather than just the presence of proteins names within a subset, and thus can detect subtle changes in biological functions evidenced in a coordinated way in a set of related genes [40]. GSEA software ver. 4.0.3 [41, 42] was used for the GSEA analyses, according to the method of Reimand et al. [43]. GSEA and ORA approaches were also used to determine whether the functional pathways enriched in EVs released by DFT1 and DFT2 cells can be detectable in the whole lysate proteome.
Results
Characterization of EVs from DFTD and devil fibroblast cells.
The experimental workflow including EV isolation and the approach for mass spectrometry analysis of EV proteins and cell lysates is illustrated in Fig. 1. Transmission electron microscopy confirmed the presence of EV structures, revealing a typical EV morphology as small closed vesicles with a cup-shaped structure, consistent with previous analysis [44] (Fig. 2A). Nano-particle tracking analysis (NTA) demonstrated successful isolation of particles with a typical small/medium EV size distribution. The mean ± standard deviation diameters were 185.9 ± 78.6 nm for DFT1 EVs, 217 ± 91.2 nm for DFT2 EVs, and 183.2 ± 78.6 nm for devil fibroblast EVs (Fig. 2B). In addition, NTA demonstrated that DFT2 EVs were secreted in significantly greater numbers than EVs derived from DFT1 and devil fibroblast cell lines (Fig. 2C).
Fig. 1.

Characterization of EVs from DFTD and devil fibroblast cells. Schematic representation of the isolation of EVs derived from cell cultures and the EV proteomic workflow analyses. Briefly, three biological replicates of each cell line (DFT1, DFT2, and devil fibroblast cells) were cultured in duplicates for 48 h at 35 °C (Tasmanian devil normal body temperature) in a fully humidified atmosphere of 5% CO2. After 48 h, the cultured medium of each cell line was collected, centrifuged, concentrated, and subjected to size exclusion chromatography to obtain extracellular vesicles. Additionally, lysate samples were prepared from each cell line. A pooled EV protein sample for EVs and cell lysates was prepared to generate specific-spectral libraries using data-dependent acquisition–mass spectrometry techniques (DDA–MS). Each fraction (16 in total) of the EV and lysate pooled samples were run in the mass spectrometer for a total of 90 min for each fraction. Individual EV and lysate protein samples (n = 36) were run using data-independent acquisition–mass spectrometry techniques (DIA–MS). Each DIA sample was run on the mass spectrometer machine for a total of 90 min
Fig. 2.

Characterization of EVs from DFTD and devil fibroblast cells. A. Transmission electron microscopy images of isolated EVs from cell cultures. Red arrows indicate EV structures. B. Particle-size distributions of cell culture-derived EVs measured by nanoparticle tracking analysis (NTA), shaded areas represent 95% confidence intervals. C. EV concentration obtained by NTA. The letters “a” and “b” indicate significant pairwise differences between groups (i.e., groups denoted with the same letters are not significantly different; one-way ANOVA, Tukey post hoc test, p < 0.05); error bars represent 95% confidence intervals. D Venn diagram of overlapping genes identified in EVs derived from cell cultures with Vesiclepedia, and the top hundred exosomal genes reported in the Exocarta database. E EV and cell lysate Western blots of a purity (Golgi matrix protein ~ 130 kDa) and two cytosolic EV markers (Flotillin-1 ~ 48 kDa and Syntenin-1 ~ 32 kDa). F Heat map of expression patterns of mass spectrometry intensities of membrane and cytosolic EV markers presented in the cell culture-derived EV proteome
The post-filtering EV proteome from the DIA–MS analysis was converted to gene symbols for comparison with the human Vesiclepedia database. Gene symbols were retrieved for 1,315 proteins, while 185 proteins were uncharacterised, with no gene symbol reported. The EV proteome of devil samples shared 1283 genes with the Vesiclepedia database (Fig. 2D). Of these Vesiclepedia-devil EV shared proteins, 75 proteins were also in the ExoCarta top-100 proteins reported from EV preparations (Fig. 2D). In addition to the proteins identified by mass spectrometry, western blotting was used for targeted analysis of the cytoplasmic EV markers Flotillin-1 (FLOT1) and Syntenin-1 (SDCBP) (Fig. 2E). These EV markers showed expression patterns that aligned with the corresponding proteomic data for these two proteins displayed in Fig. 2F. The marker Golgi matrix protein (GM130) was detected in cell lysates but absent from corresponding EV samples, indicating purity in the EV preparation (Fig. 2E). Other commonly recovered proteins in EV preparations identified in the filtered proteome dataset (1,500 proteins) are represented as a heat map in Fig. 2F. These proteins are recommended as protein content-based EV characterisation by the minimal information of extracellular vesicles 2018 ISEV guidelines [45].
EVs derived from DFTD cells represent their cell of origin
To investigate whether EVs from DFT1, DFT2, and fibroblast cells bear hallmarks of their respective cell types of origin, we compared proteomic datasets for EVs and cell lysates using principal component analysis (PCA) (Fig. 3A). Based on a filtered DIA–MS dataset (see methods), variances of the 4437 cell lysate proteins and 1500 cell-cultured EV proteins were explained by the first two principal components in each plot, respectively. For both the lysate and EV samples, PC 1 explained 45.6% and 50.1%, while PC 2 explained 14.7% and 18%, respectively. DFT1, DFT2 and fibroblast cell lysates and their respective EV samples shared a similar pattern, with biological and technical replicates of each cell and EV type distinctly clustered in the PCA plot.
Fig. 3.

EVs derived from DFTD cells represent their cell of origin. A Principal component analysis (PCA) biplots of cell lysate and cell culture-derived EV filtered proteomes. B Venn diagram of cell lysate and cell culture-derived EV protein overlap. D Venn diagram comparing significantly upregulated proteins of DFT2 EVs relative to devil fibroblasts EVs and significantly upregulated proteins of DFT2 lysates relative to fibroblast lysates. E Over-representation analysis (ORA) of cellular component gene ontology (GO) terms associated with proteins that are both significantly upregulated in DFT1 cells and their released EVs (relative to fibroblast cells and their released EVs; 129 proteins; FDR-corrected p ≤ 0.05). F ORA of cellular component GO terms associated with proteins that are both significantly upregulated in DFT2 cells and their released EVs (relative to fibroblast cells and their released EVs; 141 proteins; FDR-corrected p ≤ 0.05). For E and F, the total number of proteins included in each functional term is denoted by a number on the edge of each bar. G List of proteins that formed part of the enriched GO term myelin sheath in DFT1 cells and their EVs. H List of proteins that formed part of the enriched GO term myelin sheath in DFT2 cells and their EVs
To further understand the relationship between sample types, we identified the proteins detected in EVs only, lysates only, and those detected in both datasets. Over 80% of the EV proteins (1,221/1,500) were also detected in cell lysates (Fig. 3B). However, about one-fifth of the EV proteins (279/1,500) were exclusively present in EVs (Fig. 3B). Functional enrichment analysis of these only EV proteins revealed that the majority of them are annotated by the term extracellular exosome (Online resource 2).
To investigate whether EVs from DFTD cells maintain the same pattern of protein upregulation as their parental cells, we compared subsets of significant proteins. First, of the 604 EV proteins significantly upregulated in DFT1 relative to fibroblasts, 129 proteins were also significantly upregulated in DFT1 lysates relative to fibroblast lysates (Fig. 3C). Second, of the 517 EV proteins upregulated in DFT2 compared to fibroblasts, 141 proteins were also significantly upregulated in DFT2 lysates relative to fibroblast lysates (Fig. 3D). Both overlapping protein groups (129 proteins for DFT1 and 141 proteins for DFT2) showed enrichment of the gene ontology (GO) term myelin sheath (Fig. 3E, F), previously reported as a significant functional term associated with DFTD cells and biopsies [46]. Eight and seven proteins formed part of the GO term myelin sheath for DFT1 and DFT2, respectively (Fig. 3G, H). Interestingly, myelin protein zero (MPZ) is the protein most significantly upregulated in DFT1 EVs compared to fibroblasts EVs with a log2 increase of 11-fold (Fig. 3G). MPZ has been previously reported as highly expressed on DFT1 cells and tumour biopsies [2, 46].
DFTD-derived EVs’ enriched cell and focal adhesion proteins relative to fibroblasts derived EVs
Next, to define the protein signature of DFTD and devil fibroblast EVs, differentially abundant proteins between groups were subjected to functional enrichment analyses. We also performed functional enrichment analyses in the differentially expressed proteins obtained from the cell lysates database to investigate whether the EV functional signatures were also found in their cell of origin.
Over-representation analysis (ORA)
Relative to fibroblast EVs, 604 and 517 proteins were significantly upregulated in DFT1 (log2 0.4—11-fold) and DFT2 (log2 0.4—ninefold) derived EVs, respectively (Online Resource 3). Over-representation analysis (ORA) of DFT1 and DFT2 upregulated EV proteins revealed that many of the enriched functional terms were linked to focal adhesion (Fig. 4C), such as integrin, PI3K–Akt signalling pathway, ECM–receptor interaction, and cytoskeleton (Fig. 4A, B and Online Resource 4). The most significant enriched GO term for DFT1 and DFT2 EVs was GTP binding (Fig. 4A, B), including RAS oncogenes and G-proteins alpha subunit, which are involved in the RAP1 signalling pathway (Fig. 4D), also a significant KEGG pathway in both DFT1 and DFT2 EVs (Fig. 4A, B). In contrast, proteins that were significantly more abundant in devil fibroblast EVs were enriched in functional terms related to protein biosynthesis such as ribosome, amino acyl tRNA biosynthesis, and translation elongation, as well as protein folding (TCP-1) and disposal (proteasome) (Fig. 4A, B and Online Resource 4).
Fig. 4.

DFTD-derived EVs enriched cell and focal adhesion proteins relative to fibroblasts derived EVs. A. Over-representing analysis (ORA) for gene ontology (GO) terms, protein families, and pathways of the EV proteins upregulated in DFT1 (604) relative to fibroblast (569), and B. of the EV proteins significantly upregulated in DFT2 (517) compared to fibroblasts (634). Only functional terms with p ≤ 0.001 are illustrated in panels A and B (see Online resource 3 for all significant terms). The total number of proteins included in each functional term is denoted by a number on the edge of each bar. C. KEGG pathway map (shr04510) of the focal adhesion signalling pathway. Interconnected signalling pathways significantly upregulated in both DFT1 and DFT2 EVs are outlined in black borders. Proteins that are significantly upregulated in DFT1 EVs, DFT2 EVs or both are highlighted by red stars and labelled with gene names and with colours corresponding to the key shown. D. Heat map of expression patterns of mass spectrometry intensities of proteins belonging to the GTP binding GO term
Relative to fibroblast EVs, 1588 and 1319 proteins were significantly upregulated in DFT1 (log2 0.3—ninefold) and DFT2 cells (log2 0.3—eightfold), respectively (Online Resource 5). Surprisingly, ORA analyses of DFT1 and DFT2 upregulated proteins did not reveal a significant enrichment of functional terms related to focal adhesion pathways as found in the EV ORA analyses, suggesting that proteins related to focal adhesion pathways are enriched in their released EVs (Online Resource 6).
Gene enrichment set analysis (GSEA)
To further explore biological information from our proteomics data, the complementary GSEA approach identified those protein groups in both DFT1 and DFT2 derived EVs relative to fibroblast EVs that were specifically associated with cell adhesion and cell signalling. Additionally, enrichment of protein groups related to the Schwann cell origin of DFTD was identified according to their annotated GO terms such as myelin sheath, glial cell development and ensheathment of neurons, and axon and neuron development (see others in Online Resource 7). Consistent with the ORA analyses, EVs derived from devil fibroblasts enriched protein groups related with protein synthesis, including the GO terms ribonucleoprotein complex, amide and peptide biosynthetic process, and ribosome (see others in Online Resource 7). This complementary approach therefore confirmed that in contrast to whole-cell lysates, DFTD EVs are specifically enriched in cell and focal adhesion proteins relative to fibroblast EVs (Online Resource 8).
DFT2-derived EVs enriched the epithelial–mesenchymal transition hallmark relative to DFT1-derived EVs
In addition to using the DFT1, DFT2, and fibroblast data to identify tumour EV-associated signatures, we could also identify differentially abundant proteins between EVs derived from DFT2 and DFT1 cells. Based on t tests, 138 proteins were significantly upregulated in DFT2 EVs by log2 0.6–8.2-fold, and 294 proteins were significantly upregulated in DFT1 EVs by log2 0.6–9.2-fold, respectively (FDR-corrected p < 0.05, Online Resource 3). ORA analyses revealed that the EV proteins upregulated in DFT2 were associated with significant functional terms such as calcium binding, extracellular matrix (ECM) receptor interaction, focal adhesion, laminin G domain, fibrinogen, and immunoglobulin-like fold (Fig. 5A). In contrast, proteins upregulated in EVs derived from DFT1 cells showed enrichment of a very distinct set of significant functional terms such as nucleotide binding, GTP binding, biosynthesis of antibiotics, and epidermal growth factor (Fig. 5A).
Fig. 5.
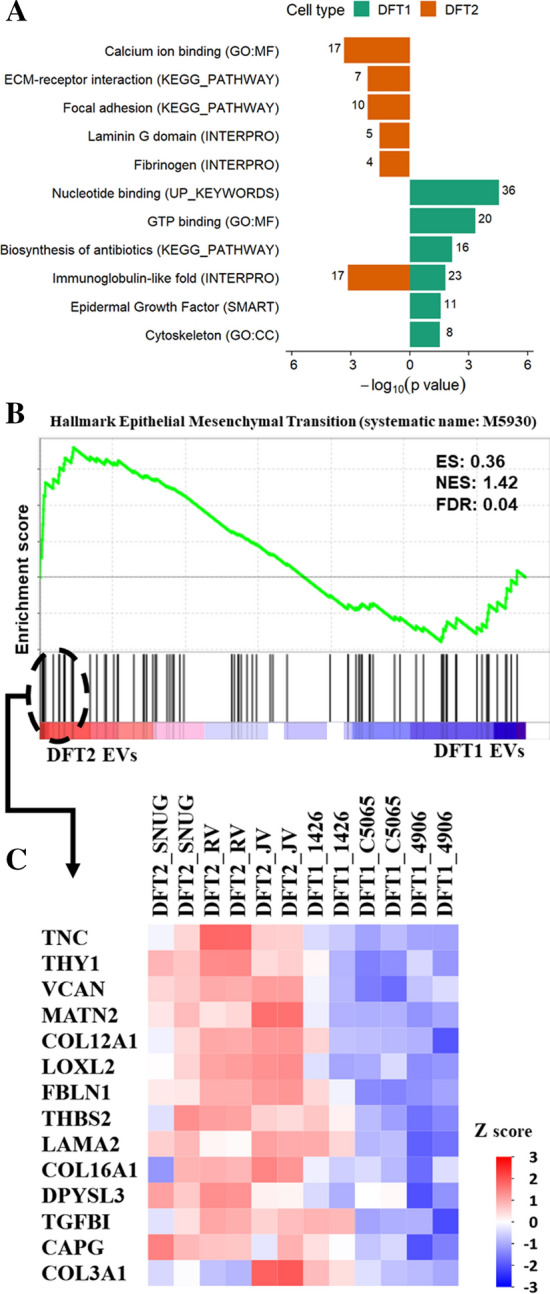
DFT2-derived EVs enriched the epithelial–mesenchymal transition hallmark relative to DFT1-derived EVs. A. Over-representing analysis (ORA) for gene ontology (GO) terms, protein families, and pathways of the EV proteins significantly upregulated in DFT1 (138) relative to DFT2 (294) (FDR-corrected p ≤ 0.05). The total number of proteins included in each functional term is denoted by a number on the edge of each bar. B. Enrichment plot contains enrichment score (ES), normalized enrichment score (NES), and FDR-corrected p value. The bottom portion of the plot shows the genes belonging to the hallmark, and they are ranked according to their differential expression. Higher and lower expressions are represented by red and blue colour, respectively. C Heat map showing the core list of proteins that contribute the most to the Epithelial–Mesenchymal Transition Hallmark
Comparison of the EV datasets from DFT1 and DFT2 by GSEA showed only one significant term that was enriched in DFT2 derived EVs, which corresponded to the epithelial–mesenchymal transition hallmark (EMT; systematic name: M5930; Fig. 5B). The EMT hallmark core list is composed of 14 mesenchymal proteins that contribute the most to the EMT hallmark enrichment (Fig. 5C). Notably, the EMT hallmark enriched in DFT2 was specific to DFT2 EVs and was not evident among the set of significant lysate proteins in DFT2 vs DFT1, either by ORA (Online Resource 6) or GSEA (Online Resource 8).
The mesenchymal protein TNC as potential biomarker for DFT2
To explore whether our proteomic comparison of EV proteins derived from cultured DFT1 and DFT2 cells could be translated into a potential clinical application, we prepared EVs from the serum of DFT1, DFT2, and non-infected animals. In this preliminary analysis, our goal was to first investigate whether the mesenchymal markers enriched in DFT2 EVs in vitro could serve as potential DFT2 biomarkers.
EVs derived from devil serum samples were characterised by TEM and NTA to evaluate the morphology and size of the isolated extracellular vesicles. TEM images confirmed the presence of EV structures in DFT1- and DFT2-infected devils as well as healthy controls, showing a typical EV morphology as closed vesicles with a cup-shaped structure [44] (Fig. 6A). NTA demonstrated the presence of a heterogeneous nanoparticle population with a small-to-medium-sized distribution (Fig. 6B). The mean (± standard deviation) diameters of the EVs were 134.4 ± 2.7 nm for captive healthy devils, 142.8 ± 17.1 nm for DFT1-infected devils, and 139.8 ± 18.4 nm for DFT2-infected devils (Fig. 6B). NTA indicated that DFT2-infected devils have a significantly greater concentration of EVs relative to those infected with DFT1 (Fig. 6C). Using DIA–MS, 370 proteins were quantified across our cohort of DFT2 infected devils (n = 5), DFT1-infected devils (n = 12) and healthy controls (n = 10), and the 350 that passed filtering criteria were considered for analyses (Online Resource 9). Among the filtered proteins, 24 established EV markers were identified, including CD9, annexins, heat shock, and major histocompatibility complex proteins [45, 46] (Fig. 6D). Serum-derived contaminants, including albumin and lipoproteins, are also reported in a heat map in Fig. 6E.
Fig. 6.

Characterisation of extracellular vesicles (EVs) derived from Tasmanian devil serum. A. Transmission electron microscopy images for EVs isolated from serum of healthy control captive devils (n = 4), DFT1-infected devils (n = 4), and DFT2-infected devils (n = 5). Red arrows indicate EV structures. B. Size distribution profiles determined by nanoparticle tracking analysis (NTA) of EVs isolated from serum of captive healthy control devils (n = 4), DFT1-infected devils (n = 4), and DFT2-infected devils (n = 5). Shaded areas represent 95% confidence intervals. C EV concentrations of the same NTA groups. The letters “a” and “b” indicate significant pairwise differences among groups (i.e., groups denoted with the same letter are not significantly different; one-way ANOVA, Tukey post hoc test, p < 0.05). Error bars represent 95% confidence intervals. D. Heat map of intensity values of commonly recovered EV proteins, and E. serum contaminants found in EV samples derived from captive healthy controls devils (n = 10), DFT1-infected devils (n = 12), and devils infected with DFT2 (n = 5)
Among the mesenchymal proteins that contribute the most to the EMT hallmark significantly enriched in DFT2 EVs relative to DFT1 EVs (Fig. 5C), tenascin-C (TNC) was the only protein significantly upregulated by log2 1.6 and 1.5-fold in serum EVs derived from DFT2-infected devils, relative to both DFT1 infected devils and healthy controls, respectively (Fig. 7A, B; see other upregulated proteins and their fold changes in Online Resource 9).
Fig. 7.

The mesenchymal protein TNC as a potential biomarker for DFT2. A. Venn diagram of overlapping proteins identified as: (a) upregulated in EVs derived from devils infected with DFT2 relative to devils infected with DFT1; (b) upregulated in EVs derived from devils infected with DFT2 relative to healthy controls; (c) upregulated in EVs derived from DFT2 cultured cells relative to DFT1 cultured cells; and (d) the core list of mesenchymal proteins that contribute the most to the epithelial–mesenchymal transition hallmark enrichment in EVs derived from DFT2 cultured cells. Note that one protein in the core enrichment list was present, but not significantly upregulated in DFT2 cultured cell EVs. B. Dot plot showing the relative abundance of EV TNC detected in 10 healthy, 12 DFT1-infected devils, and 5 DFT2-infected devils, different letters “a” and “b” indicate significant pairwise differences between groups (i.e., groups denoted with the same letter are not significantly different; one-way ANOVA and Tukey post hoc test, p < 0.05). C. Receiver-operating characteristic (ROC) curve analysis for TNC EVs (5 DFT2-infected devils relative to 12 DFT1-infected devils). D. ROC curve analysis for TNC EVs (5 DFT2 infected devils relative to 10 healthy controls). For both C and D, the dashed red line indicates random performance. The cut-off values were determined using Youden’s index and are indicated in blue at the left top corner of the ROC curve, and specificity and sensitivity are indicated in brackets, respectively
Receiver-operating characteristic (ROC) curve analysis confirmed that TNC differentiated devils with DFT2 from DFT1-infected ones with 100% sensitivity and 91.7% specificity (Fig. 7C). Moreover, TNC distinguished devils with DFT2 from healthy controls with 100% sensitivity and 90% specificity (Fig. 7D), which indicated that TNC is not only a potential EV biomarker candidate to differentiate DFTD tumours, but also is a potential liquid biopsy for DFT2 when tumours cannot be sampled.
Discussion
Despite the value of non-invasive liquid biopsies, and in particular the potential for extracellular vesicles (EVs) to provide insights about cancer signalling mechanisms, attempts to characterise EV molecular cargo in wild animals have not been extensively exploited and it is currently a growing field [47–49]. This is largely due to challenges inherent to collecting samples from wild animals, such as the logistical difficulty of capturing animals and sample storage in remote areas. Here, we have demonstrated that the proteomic characterisation of EVs derived from devil facial tumour cells can provide insights about DFTD mechanisms, including metastasis and phenotypic signatures of the cancer cells. Furthermore, these results provide a basis for future analysis of EVs derived from archived devil serum samples to investigate their potential use as liquid biopsies in DFTD.
The functional pathways enriched in the DFTD EV proteome suggest that EVs may be involved in DFTD metastatic processes in the progression of both cancers. The focal adhesion pathway is formed by large protein complexes which are upregulated in cancer cells to colonise other organs during metastasis [50]. Several of the cell adhesion-related proteins that were highly enriched in DFTD EVs have been implicated in EV-associated metastasis, specifically in preparing the pre-metastatic niche in the lungs. In particular, the ras-related proteins RalA and RalB, shown to promote lung metastasis in breast cancer [51], were upregulated in both DFT1 and DFT2 EVs relative to fibroblast EVs. We also found that the integrin subunit ITGA6, which is involved in tumour EV organotropism to the lung [27], was also significantly upregulated. Considering the role of these proteins in lung-specific metastasis together with the finding that nearly 50% of tumour metastasis in DFTD involves the lungs [10], our findings raise the possibility that DFTD EVs play a role in preparing the pre-metastatic niche in the lung tissue of infected devils.
The differential expression of epithelial–mesenchymal transition (EMT) hallmark proteins in EVs derived from DFT2 cells relative to DFT1 cells identified potential markers for discrimination between these transmissible cancers. The EMT hallmark features could be only detected in EVs and not in the whole-cell proteome signatures, demonstrating the capacity of EVs to enrich molecules related to the pathogenesis of distinct disease subtypes that would otherwise go undetected. The EMT hallmark contains mesenchymal proteins known to increase cell motility and migration [52], which are linked to cancer phenotypes of increased aggression and metastatic behaviour [53–56]. The mesenchymal characteristics of the DFT2 EV proteome revealed by the enrichment of the EMT hallmark may suggest a ‘repair’ Schwann cell phenotype, in which de-differentiation via EMT pathways aid repair of peripheral nerve damage [57]. These results are consistent with transcriptomic analyses of DFT2 tumour biopsies which demonstrated enrichment of the repair Schwann cell phenotype relative to peripheral nerves, while DFT1 biopsies did not show significant enrichment of this phenotype [46]. Elements of the EMT mesenchymal phenotype in DFT2 EVs including serum of infected devils provide an ingress to investigate mechanisms of tumorigenesis and development of a potential DFT2-specific biomarker.
The extracellular matrix glycoprotein tenascin-C (TNC), one of the EMT markers for DFT2 EVs in vitro, also displayed a high predictive power to classify devils infected with DFT2 in serum EVs. TNC was not detected in cell culture lysates, suggesting specific packaging of this protein into EVs. TNC is produced by stroma and cancer cells, and it is highly expressed during embryogenesis, being barely detected in adult tissues [58]. TNC has been demonstrated to promote mesenchymal properties in several cancer cells such as glioblastoma, colorectal, and breast cancer [59–61]. High levels of TNC are associated with poor prognosis of patients of several types of cancer. Furthermore, TNC has been shown to participate in cancer proliferation, migration, invasion, and metastasis [62], and has been proposed as a pan-cancer EV biomarker, as it can differentiate a variety of cancer tissues from healthy controls with high sensitivity and specificity [63]. These lines of evidence suggest that TNC is a promising serum biomarker for DFTD differential diagnosis; therefore, we suggest further validation studies using a larger cohort of animals. At the time this study was initiated, only 25 DFT2 infected devils had been reported in the literature [6], as this recently emergent cancer is still confined to the geographically isolated D’Entrecasteaux Channel region. Thus, while only five devil samples were employed in this study, they represented 20% of all known DFT2 infected devils and were repurposed from prior DFT2 research [4]. However, with ongoing devil trapping in this region (Rodrigo Hamede, personal communication 2021), availability of DFT2-infected devils is increasing and future work with a larger cohort of samples will be warranted.
An interesting finding of this study was the significantly greater EV production of DFT2 cells relative to DFT1 or devil fibroblast cells. As EVs are key players in cell signalling mechanisms [64–66], this suggests the potential of increased rates of cell signalling of DFT2 cells in vivo relative to DFT1 cells. It has been demonstrated that cancer cells secrete more EVs than normal cells [67–69], and higher grade cancer cells secrete more EVs than lower grade cancer cells [69–71]. These lines of evidence and the enrichment of mesenchymal proteins in the EVs derived from DFT2 cells suggest that DFT2 may be a more aggressive cancer than DFT1. The greater EV production by DFT2 was consistent with the significantly greater nanoparticle abundance derived from serum of DFT2-infected devils relative to those infected with DFT1. These results further support the hypothesis that the DFT2 diagnosis biomarker TNC is a tumour-derived EV protein. This study demonstrates that the potential of EV analyses for biomarker discovery, previously demonstrated for human cancers (Melo et al., 2015, Lane et al. 2018, Hoshino et al., 2020), extends to non-model and wildlife species.
This is the first investigation, to our knowledge, of EVs in the context of a disease that affects wild animals. We have demonstrated the potential of EVs to shed light on mechanisms of DFTD, such as metastasis and cell phenotype, which was not identified in their parental whole-cell proteome. Additionally, we identified novel candidate proteins with potential value as diagnostic biomarkers in devil serum samples. Metastatic cancers have been increasingly reported in wildlife in the past few decades [72]. EV approaches offer a promising avenue to the development of sensitive and less-invasive clinical tools needed for wildlife cancer monitoring and management. One of the limitations of this study is the difficulties of targeted protein quantitation due to the current lack of species-specific antibodies for devil studies. The future development of devil specific antibodies, specifically against Tasmanian devil TNC, is necessary to further investigate and validate the protein’s biomarker value with other techniques, such as ELISA, flow cytometry, western blot, or immunohistochemistry. In addition, we suggest investigating other types of molecular cargo of devil derived EVs, such as miRNA, which would likely complement the EV protein investigation carried out in this study. The EV-based investigation of cancer in the wild will likely provide useful information for human cancers, as EVs are well-conserved structures through the tree of life [73], and wild animals have more similarities to humans than laboratory animals in terms of environmental exposures and life-span [46].
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We would like to acknowledge all the members of the devil and wild immunology group for their advice and guidance. We would also like to thank Ginny Ralph for providing care of captive devils, the Bonorong Wildlife Sanctuary for providing access to Tasmanian devils and Dr Alexandre Kreiss for collecting the blood and to the Save the Tasmanian Devil Program for provision of samples. The authors would also like to acknowledge La Trobe University Bioimaging Platform for their support with TEM analysis.
Funding
This work was supported by the National Geographic explorer early career grant, Holsworth Wildlife Research Endowment grants, the University of Tasmania Foundation through funds raised by the Save the Tasmanian Devil Appeal. Proteomics infrastructure was funded by ARC LE180100059. Sample collection from wild devils was funded by US National Institutes of Health (NIH) grant R01-GM126563-01 and US National Science Foundation (NSF) grant DEB1316549 as part of the joint NIH-NSF-USDA Ecology and Evolution of Infectious Diseases program.
Data availability
The mass spectrometry raw proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [74] partner repository with the dataset identifier PXD020766 (cell lysate and EV samples); and PXD025579 (serum EV samples).
Code availability
Not applicable.
Declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All animal procedures were performed under a Standard Operating Procedure approved by the General Manager, Natural and Cultural Heritage Division, Tasmanian Government Department of Primary Industries, Parks, Water and the Environment and under the auspices of the University of Tasmania Animal Ethics Committee (permit numbers: A0017550, A0013326, A0015835).
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Cunningham CX, Comte S, McCallum H, Hamilton DG, Hamede R, Storfer A, Hollings T, Ruiz-Aravena M, Kerlin DH, Brook BW, Hocking G, Jones ME. Quantifying 25 years of disease-caused declines in tasmanian devil populations: host density drives spatial pathogen spread. Ecol Lett. 2021;24:958–969. doi: 10.1111/ele.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murchison EP, Tovar C, Hsu A, Bender HS, Kheradpour P, Rebbeck CA, Obendorf D, Conlan C, Bahlo M, Blizzard CA, Pyecroft S, Kreiss A, Kellis M, Stark A, Harkins TT, Marshall GJA, Woods GM, Hannon GJ, Papenfuss AT. The Tasmanian devil transcriptome reveals Schwann cell origins of a clonally transmissible cancer. Science. 2010;327:84–87. doi: 10.1126/science.1180616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamede RK, McCallum H, Jones M. Biting injuries and transmission of Tasmanian devil facial tumour disease. J Anim Ecol. 2013;82:182–190. doi: 10.1111/j.1365-2656.2012.02025.x. [DOI] [PubMed] [Google Scholar]
- 4.Pye RJ, Pemberton D, Tovar C, Tubio JM, Dun KA, Fox S, Darby J, Hayes D, Knowles GW, Kreiss A, Siddle HV, Swift K, Lyons AB, Murchison EP, Woods GM. A second transmissible cancer in tasmanian devils. Proc Natl Acad Sci USA. 2016;113:374–379. doi: 10.1073/pnas.1519691113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murchison EP, Schulz-Trieglaff OB, Ning Z, Alexandrov LB, Bauer MJ, Fu B, Hims M, Ding Z, Ivakhno S, Stewart C, Ng BL, Wong W, Aken B, White S, Alsop A, Becq J, Bignell GR, Cheetham RK, Cheng W, Connor TR, Cox AJ, Feng ZP, Gu Y, Grocock RJ, Harris SR, Khrebtukova I, Kingsbury Z, Kowarsky M, Kreiss A, Luo S, Marshall J, McBride DJ, Murray L, Pearse AM, Raine K, Rasolonjatovo I, Shaw R, Tedder P, Tregidgo C, Vilella AJ, Wedge DC, Woods GM, Gormley N, Humphray S, Schroth G, Smith G, Hall K, Searle SM, Carter NP, Papenfuss AT, Futreal PA, Campbell PJ, Yang F, Bentley DR, Evers DJ, Stratton MR. Genome sequencing and analysis of the Tasmanian devil and its transmissible cancer. Cell. 2012;148:780–791. doi: 10.1016/j.cell.2011.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.James S, Jennings G, Kwon YM, Stammnitz M, Fraik A, Storfer A, Comte S, Pemberton D, Fox S, Brown B, Pye R, Woods G, Lyons B, Hohenlohe PA, McCallum H, Siddle H, Thomas F, Ujvari B, Murchison EP, Jones M, Hamede R. Tracing the rise of malignant cell lines: distribution, epidemiology and evolutionary interactions of two transmissible cancers in tasmanian devils. Evol Appl. 2019;12:1772–1780. doi: 10.1111/eva.12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stammnitz MR, Coorens TH, Gori KC, Hayes D, Fu B, Wang J, Martin-Herranz DE, Alexandrov LB, Baez-Ortega A, Barthorpe S. The origins and vulnerabilities of two transmissible cancers in tasmanian devils. Cancer Cell. 2018;33(607–619):e615. doi: 10.1016/j.ccell.2018.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruiz-Aravena M, Jones ME, Carver S, Estay S, Espejo C, Storfer A, Hamede RK. Sex bias in ability to cope with cancer: tasmanian devils and facial tumour disease. Proc Royal Soc. 2018;285:20182239. doi: 10.1098/rspb.2018.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCallum H, Jones M, Hawkins C, Hamede R, Lachish S, Sinn DL, Beeton N, Lazenby B. Transmission dynamics of Tasmanian devil facial tumor disease may lead to disease-induced extinction. Ecology. 2009;90:3379–3392. doi: 10.1890/08-1763.1. [DOI] [PubMed] [Google Scholar]
- 10.Loh R, Bergfeld J, Hayes D, O'Hara A, Pyecroft S, Raidal S, Sharpe R. The pathology of devil facial tumor disease (DFTD) in Tasmanian Devils (Sarcophilus harrisii) Vet Pathol. 2006;43:890–895. doi: 10.1354/vp.43-6-890. [DOI] [PubMed] [Google Scholar]
- 11.Siddle HV, Kreiss A, Tovar C, Yuen CK, Cheng Y, Belov K, Swift K, Pearse AM, Hamede R, Jones ME, Skjodt K, Woods GM, Kaufman J. Reversible epigenetic down-regulation of MHC molecules by devil facial tumour disease illustrates immune escape by a contagious cancer. Proc Natl Acad Sci USA. 2013;110:5103–5108. doi: 10.1073/pnas.1219920110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caldwell A, Coleby R, Tovar C, Stammnitz MR, Kwon YM, Owen RS, Tringides M, Murchison EP, Skjodt K, Thomas GJ, Kaufman J, Elliott T, Woods GM, Siddle HV. The newly-arisen devil facial tumour disease 2 (DFT2) reveals a mechanism for the emergence of a contagious cancer. Elife. 2018;7:e35314. doi: 10.7554/eLife.35314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hamede R, Lachish S, Belov K, Woods G, Kreiss A, Pearse AM, Lazenby B, Jones M, Mccallum H. Reduced effect of tasmanian devil facial tumor disease at the disease front. Conserv Biol. 2012;26:124–134. doi: 10.1111/j.1523-1739.2011.01747.x. [DOI] [PubMed] [Google Scholar]
- 14.Pyecroft SB, Pearse A-M, Loh R, Swift K, Belov K, Fox N, Noonan E, Hayes D, Hyatt A, Wang L. Towards a case definition for devil facial tumour disease: what is it? EcoHealth. 2007;4:346. doi: 10.1007/s10393-007-0126-0. [DOI] [Google Scholar]
- 15.Tovar C, Obendorf D, Murchison EP, Papenfuss AT, Kreiss A, Woods GM. Tumor-specific diagnostic marker for transmissible facial tumors of Tasmanian devils: immunohistochemistry studies. Vet Pathol. 2011;48:1195–1203. doi: 10.1177/0300985811400447. [DOI] [PubMed] [Google Scholar]
- 16.Kwon YM, Stammnitz MR, Wang J, Swift K, Knowles GW, Pye RJ, Kreiss A, Peck S, Fox S, Pemberton D, Jones ME, Hamede R, Murchison EP. Tasman-PCR: a genetic diagnostic assay for Tasmanian devil facial tumour diseases. R Soc Open Sci. 2018;5:180870. doi: 10.1098/rsos.180870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hogg C, Fox S, Pemberton D, Belov K. Chapter 3: Pathology and diagnostics of DFTD and other devil diseases. In: saving the Tasmanian devil: recovery through science-based management. CSIRO Publishing; 2019. [Google Scholar]
- 18.Zaborowski MP, Balaj L, Breakefield XO, Lai CP. Extracellular vesicles: composition, biological relevance, and methods of study. Bioscience. 2015;65:783–797. doi: 10.1093/biosci/biv084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stahl PD, Raposo G. Extracellular vesicles: exosomes and microvesicles, integrators of homeostasis. Physiology (Bethesda) 2019;34:169–177. doi: 10.1152/physiol.00045.2018. [DOI] [PubMed] [Google Scholar]
- 20.Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 21.Vlassov AV, Magdaleno S, Setterquist R, Conrad R. Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Bba-Gen Subjects. 2012;1820:940–948. doi: 10.1016/j.bbagen.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 22.Yang M, Wu SY. The advances and challenges in utilizing exosomes for delivering cancer therapeutics. Front Pharmacol. 2018;9:735. doi: 10.3389/fphar.2018.00735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng L, Sharples RA, Scicluna BJ, Hill AF. Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J Extracell Vesicles. 2014;3:23743. doi: 10.3402/jev.v3.23743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mashouri L, Yousefi H, Aref AR, Ahadi AM, Molaei F, Alahari SK. Exosomes: composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol Cancer. 2019;18:75. doi: 10.1186/s12943-019-0991-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peinado H, Aleckovic M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, Garcia-Santos G, Ghajar CM, Nitadori-Hoshino A, Hoffman C, Badal K, Garcia BA, Callahan MK, Yuan JD, Martins VR, Skog J, Kaplan RN, Brady MS, Wolchok JD, Chapman PB, Kang YB, Bromberg J, Lyden D. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883. doi: 10.1038/nm.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell. 2016;30:836–848. doi: 10.1016/j.ccell.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic MM, Molina H, Kohsaka S, Di Giannatale A, Ceder S, Singh S, Williams C, Soplop N, Uryu K, Pharmer L, King T, Bojmar L, Davies AE, Ararso Y, Zhang T, Zhang H, Hernandez J, Weiss JM, Dumont-Cole VD, Kramer K, Wexler LH, Narendran A, Schwartz GK, Healey JH, Sandstrom P, Labori KJ, Kure EH, Grandgenett PM, Hollingsworth MA, de Sousa M, Kaur S, Jain M, Mallya K, Batra SK, Jarnagin WR, Brady MS, Fodstad O, Muller V, Pantel K, Minn AJ, Bissell MJ, Garcia BA, Kang Y, Rajasekhar VK, Ghajar CM, Matei I, Peinado H, Bromberg J, Lyden D. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329–335. doi: 10.1038/nature15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adem B, Vieira PF, Melo SA. Decoding the biology of exosomes in metastasis. Trends Cancer. 2020;6:20–30. doi: 10.1016/j.trecan.2019.11.007. [DOI] [PubMed] [Google Scholar]
- 29.Willms E, Cabanas C, Mager I, Wood MJA, Vader P. Extracellular vesicle heterogeneity: subpopulations, isolation techniques, and diverse functions in cancer progression. Front Immunol. 2018;9:738. doi: 10.3389/fimmu.2018.00738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 31.LeBleu VS, Kalluri R. Exosomes as a multicomponent biomarker platform in cancer. Trends Cancer. 2020;6:767–774. doi: 10.1016/j.trecan.2020.03.007. [DOI] [PubMed] [Google Scholar]
- 32.Théry C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006;30:332231–332229. doi: 10.1002/0471143030.cb0322s30. [DOI] [PubMed] [Google Scholar]
- 33.Abramowicz A, Marczak L, Wojakowska A, Zapotoczny S, Whiteside TL, Widlak P, Pietrowska M. Harmonization of exosome isolation from culture supernatants for optimized proteomics analysis. PLoS ONE. 2018;13:e0205496. doi: 10.1371/journal.pone.0205496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hughes CS, Moggridge S, Muller T, Sorensen PH, Morin GB, Krijgsveld J. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat Protoc. 2019;14:68–85. doi: 10.1038/s41596-018-0082-x. [DOI] [PubMed] [Google Scholar]
- 35.R Core Team . R: a language and environment for statistical computing. R Foundation for Statistical Computing; 2020. [Google Scholar]
- 36.Pathan M, Fonseka P, Chitti SV, Kang T, Sanwlani R, Van Deun J, Hendrix A, Mathivanan S. Vesiclepedia 2019: a compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 2019;47:D516–D519. doi: 10.1093/nar/gky1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keerthikumar S, Chisanga D, Ariyaratne D, Al SH, Anand S, Zhao K, Samuel M, Pathan M, Jois M, Chilamkurti N, Gangoda L, Mathivanan S. ExoCarta: a web-based compendium of exosomal cargo. J Mol Biol. 2016;428:688–692. doi: 10.1016/j.jmb.2015.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 39.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Glaab E, Baudot A, Krasnogor N, Schneider R, Valencia A. EnrichNet: network-based gene set enrichment analysis. Bioinformatics. 2012;28:i451–i457. doi: 10.1093/bioinformatics/bts389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 43.Reimand J, Isserlin R, Voisin V, Kucera M, Tannus-Lopes C, Rostamianfar A, Wadi L, Meyer M, Wong J, Xu CJ, Merico D, Bader GD. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, cytoscape and enrichmentmap. Nat Protoc. 2019;14:482–517. doi: 10.1038/s41596-018-0103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 45.Thery C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin-Smith GK, Ayre DC, Bach JM, Bachurski D, Baharvand H, Balaj L, Baldacchino S, Bauer NN, Baxter AA, Bebawy M, Beckham C, Zavec AB, Benmoussa A, Berardi AC, Bergese P, Bielska E, Blenkiron C, Bobis-Wozowicz S, Boilard E, Boireau W, Bongiovanni A, Borras FE, Bosch S, Boulanger CM, Breakefield X, Breglio AM, Brennan MA, Brigstock DR, Brisson A, Broekman MLD, Bromberg JF, Bryl-Gorecka P, Buch S, Buck AH, Burger D, Busatto S, Buschmann D, Bussolati B, Buzas EI, Byrd JB, Camussi G, Carter DRF, Caruso S, Chamley LW, Chang YT, Chen CC, Chen S, Cheng L, Chin AR, Clayton A, Clerici SP, Cocks A, Cocucci E, Coffey RJ, Cordeiro-da-Silva A, Couch Y, Coumans FAW, Coyle B, Crescitelli R, Criado MF, D'Souza-Schorey C, Das S, Chaudhuri AD, Candia P, De Santana EF, De Wever O, del Portillo HA, Demaret T, Deville S, Devitt A, Dhondt B, Di Vizio D, Dieterich LC, Dolo V, Rubio APD, Dominici M, Dourado MR, Driedonks TAP, Duarte FV, Duncan HM, Eichenberger RM, Ekstrom K, Andaloussi SEL, Elie-Caille C, Erdbrugger U, Falcon-Perez JM, Fatima F, Fish JE, Flores-Bellver M, Forsonits A, Frelet-Barrand A, Fricke F, Fuhrmann G, Gabrielsson S, Gamez-Valero A, Gardiner C, Gartner K, Gaudin R, Gho YS, Giebel B, Gilbert C, Gimona M, Giusti I, Goberdhan DCI, Gorgens A, Gorski SM, Greening DW, Gross JC, Gualerzi A, Gupta GN, Gustafson D, Handberg A, Haraszti RA, Harrison P, Hegyesi H, Hendrix A, Hill AF, Hochberg FH, Hoffmann KF, Holder B, Holthofer H, Hosseinkhani B, Hu GK, Huang YY, Huber V, Hunt S, Ibrahim AGE, Ikezu T, Inal JM, Isin M, Ivanova A, Jackson HK, Jacobsen S, Jay SM, Jayachandran M, Jenster G, Jiang LZ, Johnson SM, Jones JC, Jong A, Jovanovic-Talisman T, Jung S, Kalluri R, Kano S, Kaur S, Kawamura Y, Keller ET, Khamari D, Khomyakova E, Khvorova A, Kierulf P, Kim KP, Kislinger T, Klingeborn M, Klinke DJ, Kornek M, Kosanovic MM, Kovacs AF, Kramer-Albers EM, Krasemann S, Krause M, Kurochkin IV, Kusuma GD, Kuypers S, Laitinen S, Langevin SM, Languino LR, Lannigan J, Lasser C, Laurent LC, Lavieu G, Lazaro-Ibanez E, Le Lay S, Lee MS, Lee YXF, Lemos DS, Lenassi M, Leszczynska A, Li ITS., Liao K, Libregts SF, Ligeti E, Lim R, Lim SK, Line A, Linnemannstons K, Llorente A, Lombard CA, Lorenowicz MJ, Lorincz AM, Lotvall J, Lovett J, Lowry MC, Loyer X, Lu Q, Lukomska B, Lunavat TR, Maas SLN, Malhi H, Marcilla A, Mariani J, Mariscal J, Martens-Uzunova ES, Martin-Jaular L, Martinez MC, Martins VR, Mathieu M, Mathivanan S, Maugeri M, McGinnis LK, McVey MJ, Meckes DG, Meehan KL, Mertens I, Minciacchi VR, Moller A, Jorgensen MM, Morales-Kastresana A, Morhayim J, Mullier F, Muraca M, Musante L, Mussack V, Muth DC, Myburgh KH, Najrana T, Nawaz M, Nazarenko I, Nejsum P, Neri C, Neri T, Nieuwland R, Nimrichter L, Nolan JP, Nolte-'t Hoen ENM, Noren Hooten N, O'Driscoll L, O'Grady T, O'Loghlen A, Ochiya T, Olivier M, Ortiz A, Ortiz LA, Osteikoetxea X, Ostegaard O, Ostrowski M, Park J, Pegtel DM, Peinado H, Perut F, Pfaffl MW, Phinney DG, Pieters BCH, Pink RC, Pisetsky DS, von Strandmann EP, Polakovicova I, Poon IKH, Powell BH, Prada I, Pulliam L, Quesenberry P, Radeghieri A, Raffai RL, Raimondo S, Rak J, Ramirez MI, Raposo G, Rayyan MS, Regev-Rudzki N, Ricklefs FL, Robbins PD, Roberts DD, Rodrigues SC, Rohde E, Rome S, Rouschop KMA, Rughetti A, Russell AE, Saa P, Sahoo S, Salas-Huenuleo E, Sanchez C, Saugstad JA, Saul MJ, Schiffelers RM, Schneider R, Schoyen TH, Scott A, Shahaj E, Sharma S, Shatnyeva O, Shekari F, Shelke GV, Shetty AK, Shiba K, Siljander PRM, Silva AM, Skowronek A, Snyder OL, Soares RP, Sodar BW, Soekmadji C, Sotillo J, Stahl PD, Stoorvogel W, Stott SL, Strasser EF, Swift S, Tahara H, Tewari M, Timms K, Tiwari S, Tixeira R, Tkach M, Toh WS, Tomasini R, Torrecilhas AC, Tosar JP, Toxavidis V, Urbanelli L, Vader P, van Balkom BWM, van der Grein SG, Van Deun J, van Herwijnen MJC, Van Keuren-Jensen K, van Niel G, van Royen ME, van Wijnen AJ, Vasconcelos MH, Vechetti IJ, Veit TD., Vella LJ, Velot E, Verweij FJ, Vestad B, Vinas JL, Visnovitz T, Vukman KV, Wahlgren J, Watson DC, Wauben MHM, Weaver A, Webber JP, Weber V., Wehman AM, Weiss DJ, Welsh JA, Wendt S, Wheelock AM, Wiener Z, Witte L, Wolfram J, Xagorari A, Xander P, Xu J, Yan XM., Yanez-Mo M, Yin H, Yuana Y, Zappulli V, Zarubova J, Zekas V, Zhang JY, Zhao ZZ, Zheng L, Zheutlin AR, Zickler AM, Zimmermann P, Zivkovic AM, Zocco D, Zuba-Surma EK (2018) Minimal information for studies of extracellular vesicles (MISEV): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 7:1535750. 10.1080/20013078.2018.1535750 [DOI] [PMC free article] [PubMed]
- 46.Patchett AL, Coorens THH, Darby J, Wilson R, McKay MJ, Kamath KS, Rubin A, Wakefield M, McIntosh L, Mangiola S, Pye RJ, Flies AS, Corcoran LM, Lyons AB, Woods GM, Murchison EP, Papenfuss AT, Tovar C. Two of a kind: transmissible Schwann cell cancers in the endangered Tasmanian devil (Sarcophilus harrisii) Cell Mol Life Sci. 2020;77:1847–1858. doi: 10.1007/s00018-019-03259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, Dingli F, Loew D, Tkach M, Thery C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci U S A. 2016;113:E968–977. doi: 10.1073/pnas.1521230113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.D'Alessio S, Thorgeirsdottir S, Kraev I, Skirnisson K, Lange S. Post-translational protein deimination signatures in plasma and plasma evs of reindeer (Rangifer tarandus) Biology. 2021;10:222. doi: 10.3390/biology10030222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Magnadottir B, Uysal-Onganer P, Kraev I, Svansson V, Hayes P, Lange S. Deiminated proteins and extracellular vesicles - Novel serum biomarkers in whales and orca. Comp Biochem Physiol Part D Genomics Proteomics. 2020;34:100676. doi: 10.1016/j.cbd.2020.100676. [DOI] [PubMed] [Google Scholar]
- 50.Phillips RA, Kraev I, Lange S. Protein deimination and extracellular vesicle profiles in antarctic seabirds. Biology. 2020;9:15. doi: 10.3390/biology9010015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maziveyi M, Alahari SK. Cell matrix adhesions in cancer. The proteins that form the glue. Oncotarget. 2017;8:48471–48487. doi: 10.18632/oncotarget.17265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghoroghi S, Mary B, Larnicol A, Asokan N, Klein A, Osmani N, Busnelli I, Delalande F, Paul N, Halary S, Gros F, Fouillen L, Haeberle AM, Royer C, Spiegelhalter C, Andre-Gregoire G, Mittelheisser V, Detappe A, Murphy K, Timpson P, Carapito R, Blot-Chabaud M, Gavard J, Carapito C, Vitale N, Lefebvre O, Goetz JG, Hyenne V. Ral GTPases promote breast cancer metastasis by controlling biogenesis and organ targeting of exosomes. Elife. 2021;10:e61539. doi: 10.7554/eLife.61539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 55.Ksiazkiewicz M, Markiewicz A, Zaczek AJ. Epithelial-mesenchymal transition: a hallmark in metastasis formation linking circulating tumor cells and cancer stem cells. Pathobiology. 2012;79:195–208. doi: 10.1159/000337106. [DOI] [PubMed] [Google Scholar]
- 56.Wang Y, Zhou BP. Epithelial-mesenchymal transition—a hallmark of breast cancer metastasis. Cancer Hall. 2013;1:38–49. doi: 10.1166/ch.2013.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Syn N, Wang L, Sethi G, Thiery J-P, Goh B-C. Exosome-mediated metastasis: from epithelial–mesenchymal transition to escape from immunosurveillance. Trends Pharmacol Sci Trends Pharmacol Sci. 2016;37:606–617. doi: 10.1016/j.tips.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 58.Jessen KR, Mirsky R. The success and failure of the schwann cell response to nerve injury. Front Cell Neurosci. 2019;13:33. doi: 10.3389/fncel.2019.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Midwood KS, Chiquet M, Tucker RP, Orend G. Tenascin-C at a glance. J Cell Sci. 2016;129:4321–4327. doi: 10.1242/jcs.190546. [DOI] [PubMed] [Google Scholar]
- 60.Angel I, Kerman OP, Rousso-Noori L, Friedmann-Morvinski D. Tenascin C promotes cancer cell plasticity in mesenchymal glioblastoma. Oncogene. 2020;39:6990–7004. doi: 10.1038/s41388-020-01506-6. [DOI] [PubMed] [Google Scholar]
- 61.Takahashi Y, Sawada G, Kurashige J, Matsumura T, Uchi R, Ueo H, Ishibashi M, Takano Y, Akiyoshi S, Iwaya T. Tumor-derived tenascin-C promotes the epithelial-mesenchymal transition in colorectal cancer cells. Anticancer Res. 2013;33:1927–1934. doi: 10.3892/ol.2021.12831. [DOI] [PubMed] [Google Scholar]
- 62.Nagaharu K, Zhang X, Yoshida T, Katoh D, Hanamura N, Kozuka Y, Ogawa T, Shiraishi T, Imanaka-Yoshida K. Tenascin C induces epithelial-mesenchymal transition–like change accompanied by SRC activation and focal adhesion kinase phosphorylation in human breast cancer cells. Am J Pathol. 2011;178:754–763. doi: 10.1016/j.ajpath.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoshida T, Akatsuka T, Imanaka-Yoshida K. Tenascin-C and integrins in cancer. Cell Adh Migr. 2015;9:96–104. doi: 10.1080/19336918.2015.1008332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hoshino A, Kim HS, Bojmar L, Gyan KE, Cioffi M, Hernandez J, Zambirinis CP, Rodrigues G, Molina H, Heissel S, Mark MT, Steiner L, Benito-Martin A, Lucotti S, Di Giannatale A, Offer K, Nakajima M, Williams C, Nogues L, Pelissier Vatter FA, Hashimoto A, Davies AE, Freitas D, Kenific CM, Ararso Y, Buehring W, Lauritzen P, Ogitani Y, Sugiura K, Takahashi N, Aleckovic M, Bailey KA, Jolissant JS, Wang H, Harris A, Schaeffer LM, Garcia-Santos G, Posner Z, Balachandran VP, Khakoo Y, Raju GP, Scherz A, Sagi I, Scherz-Shouval R, Yarden Y, Oren M, Malladi M, Petriccione M, De Braganca KC, Donzelli M, Fischer C, Vitolano S, Wright GP, Ganshaw L, Marrano M, Ahmed A, DeStefano J, Danzer E, Roehrl M HA, Lacayo NJ, Vincent TC, Weiser MR, Brady MS, Meyers PA, Wexler LH, Ambati SR, Chou AJ, Slotkin EK, Modak S, Roberts SS, Basu EM, Diolaiti D, Krantz BA, Cardoso F, Simpson AL, Berger M, Rudin CM, Simeone DM, Jain M, Ghajar CM, Batra SK, Stanger BZ, Bui J, Brown KA, Rajasekhar VK, Healey JH, de Sousa M, Kramer K, Sheth S, Baisch J, Pascual V, Heaton TE, La Quaglia MP, Pisapia DJ, Schwartz R, Zhang H, Liu Y, Shukla A, Blavier L, DeClerck YA, LaBarge M, Bissell MJ, Caffrey TC, Grandgenett PM, Hollingsworth MA, Bromberg J, Costa-Silva B, Peinado H, Kang Y, Garcia BA, O'Reilly EM, Kelsen D, Trippett TM, Jones DR, Matei IR, Jarnagin WR, Lyden D (2020) Extracellular vesicle and particle biomarkers define multiple human cancers. Cell 182:1044-1061e1018. 10.1016/j.cell.2020.07.009 [DOI] [PMC free article] [PubMed]
- 65.Wan Z, Gao X, Dong Y, Zhao Y, Chen X, Yang G, Liu L. Exosome-mediated cell-cell communication in tumor progression. Am J Cancer Res. 2018;8:1661–1673. doi: 10.1002/jev2.12125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Samuelson I, Vidal-Puig AJ. Fed-EXosome: Extracellular vesicles and cell–cell communication in metabolic regulation. Essays Biochem. 2018;62:165–175. doi: 10.1042/EBC20170087. [DOI] [PubMed] [Google Scholar]
- 67.Maia J, Caja S, Strano MMC, Couto N, Costa-Silva B. Exosome-based cell-cell communication in the tumor microenvironment. Front Cell Dev Biol. 2018;6:18. doi: 10.3389/fcell.2018.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Redzic JS, Kendrick AA, Bahmed K, Dahl KD, Pearson CG, Robinson WA, Robinson SE, Graner MW, Eisenmesser EZ. Extracellular vesicles secreted from cancer cell lines stimulate secretion of MMP-9, IL-6, TGF-β1 and EMMPRIN. PLoS ONE. 2013;8:e71225. doi: 10.1371/journal.pone.0071225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu Y, Zhang Y, Wang L, Zhao R, Qiao Y, Han D, Sun Q, Dong N, Liu Y, Wu D, Zhang X, Huang N, Ma N, Zhao W, Liu Y, Gao X. miR-200a targets Gelsolin: a novel mechanism regulating secretion of microvesicles in hepatocellular carcinoma cells. Oncol Rep. 2017;37:2711–2719. doi: 10.3892/or.2017.5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu KR, Xing F, Wu SY, Watabe K. Extracellular vesicles as emerging targets in cancer: recent development from bench to bedside. Bba-Rev Cancer. 2017;1868:538–563. doi: 10.1016/j.bbcan.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hasselmann DO, Rappl G, Tilgen W, Reinhold U. Extracellular tyrosinase mRNA within apoptotic bodies is protected from degradation in human serum. Clin Chem. 2001;47:1488–1489. doi: 10.1093/clinchem/47.8.1488. [DOI] [PubMed] [Google Scholar]
- 72.Zernecke A, Bidzhekov K, Noels H, Shagdarsuren E, Gan L, Denecke B, Hristov M, Koppel T, Jahantigh MN, Lutgens E, Wang S, Olson EN, Schober A, Weber C. Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection. Sci Signal. 2009;2:81. doi: 10.1126/scisignal.2000610. [DOI] [PubMed] [Google Scholar]
- 73.Abu-Helil B, van der Weyden L. Metastasis in the wild: investigating metastasis in non-laboratory animals. Clin Exp Metastasis. 2019;36:15–28. doi: 10.1007/s10585-019-09956-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Askenase PW. Ancient evolutionary origin and properties of universally produced natural exosomes contribute to their therapeutic superiority compared to artificial nanoparticles. Int J Mol Sci. 2021;22:1429. doi: 10.3390/ijms22031429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, Inuganti A, Griss J, Mayer G, Eisenacher M, Perez E, Uszkoreit J, Pfeuffer J, Sachsenberg T, Yilmaz S, Tiwary S, Cox J, Audain E, Walzer M, Jarnuczak AF, Ternent T, Brazma A, Vizcaino JA. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry raw proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [74] partner repository with the dataset identifier PXD020766 (cell lysate and EV samples); and PXD025579 (serum EV samples).
Not applicable.