

Abstract
Oxidative stress impairs functional recovery after intracerebral hemorrhage (ICH). Histone deacetylase 6 (HDAC6) plays an important role in the initiation of oxidative stress. However, the function of HDAC6 in ICH and the underlying mechanism of action remain elusive. We demonstrated here that HDAC6 knockout mice were resistant to oxidative stress following ICH, as assessed by the MDA and NADPH/NADP+ assays and ROS detection. HDAC6 deficiency also resulted in reduced neuronal apoptosis and lower expression levels of apoptosis-related proteins. Further mechanistic studies showed that HDAC6 bound to malate dehydrogenase 1 (MDH1) and mediated-MDH1 deacetylation on the lysine residues at position 121 and 298. MDH1 acetylation was inhibited in HT22 cells that were challenged with ICH-related damaging agents (Hemin, Hemoglobin, and Thrombin), but increased when HDAC6 was inhibited, suggesting an interplay between HDAC6 and MDH1. The acetylation-mimetic mutant, but not the acetylation-resistant mutant, of MDH1 protected neurons from oxidative injury. Furthermore, HDAC6 inhibition failed to alleviate brain damage after ICH when MDH1 was knockdown. Taken together, our study showed that HDAC6 inhibition protects against brain damage during ICH through MDH1 acetylation.
Keywords: Hemin, Collagenase VII, Acetyl-mimetic mutants, Non-acetylatable mutants, AAV9 administration, Lentiviral administration
Introduction
Intracerebral hemorrhage (ICH), an acute cerebrovascular disease caused by ruptured blood vessels within the brain, is associated with high morbidity and mortality [1–3]. Hitherto, there is no effective treatment for ICH. Accumulating evidence suggests that oxidative stress is important in brain injury following ICH [4–7]. Neurons are susceptible to oxidative stress and are predisposed to apoptosis upon oxidative challenge [8, 9]. Therefore, targeting the oxidative stress response is an important therapeutic strategy for ICH.
Histone deacetylase 6 (HDAC6), a member of the histone deacetylase family, is found to play an important role in the response toward oxidative stress [10, 11]. Tubastatin A (TubA), a selective inhibitor of HDAC6, inhibits oxidative stress and apoptosis in acute kidney injury (AKI) mice [12]. Tubacin, another HDAC6 inhibitor, ameliorates endothelial cell injury caused by low-density lipoprotein-induced oxidative stress [13]. The role of HDAC6 in the central nervous system (CNS) has been reported recently. Treatment with TubA or interference with HDAC6 expression by siRNA significantly alleviates oxidative stress, and reduces neuro-apoptosis and cerebral infarction volume in mice with experimental stroke [14, 15]. The current study aims to determine whether HDAC6 mediates oxidative stress and subsequent neuronal apoptosis during ICH.
Malate dehydrogenase 1 (MDH1) is one of the key enzymes in biological glucose metabolism and plays an important role in oxidative stress. MDH1 participates in the malate-aspartic acid shuttle, and coordinates glycolysis and mitochondrial respiration, thereby reducing coenzyme II (NADPH)/oxidized coenzyme (NADP+) ratio and cellular reactive oxygen species (ROS) activity [16, 17]. The oxidative stress is enhanced when MDH1 expression or activity is decreased but reduced when MDH1 activity is upregulated by post-translational acetylation. A study by Kim EY et al. demonstrate that the increased activity of MDH1 after acetylation promotes fatty acid synthesis via increased acetyl CoA and NADPH generation [16]. These studies suggest that MDH1 acetylation acts as a negative regulator of oxidative stress.
In the present study, we demonstrated that knockout of HDAC6 alleviated brain damage by protecting against oxidative stress-induced neuronal apoptosis following ICH. Further examination of the mechanism of action revealed that HDAC6 bound to and mediated-MDH1 deacetylation at lysine 121 and 298. MDH1 acetylation promoted NADPH generation and reduced ROS levels, hence protecting neurons from oxidative damage. TubA treatment failed to rescue brain damage following ICH in MDH1 knockdown mice. These results suggest that HDAC6 inhibition ameliorates neuronal injury after ICH through enhancing MDH1 acetylation.
Materials and methods
Animals
Dr. YAO (Duke University) provided the HDAC6 knockout mice (HDAC6−/−) on a C57 BL/6J background. The animals used in this experiment were male wild-type mice (C57 BL/6J) and HDAC6−/− mice at the age of 2–3 months, which were sex and age matched. The experiment procedures were approved by the Institutional Animal Use and Care Committee of Xuzhou Medical University. Mice had access to water and food ad libitum.
Cell culture
HT22 or HEK293T cells were obtained from the Chinses type culture collection. Both cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum, 120 U/ml penicillin, and 100 mg/l streptomycin in a 37 °C incubator containing 5% CO2.
ICH model
ICH model was established as previously described [18]. Briefly, mice were anesthetized intraperitoneally with pentobarbital sodium (50 mg/kg), and collagenase VII (0.045 U in 1 μl of sterile saline) (Sigma-Aldrich, Germany) was injected at a rate of 0.2 μl/min into the right striatum, which was located 0.2 mm anterior, 2.5 mm lateral, and 3.5 mm deep relative to the bregma. The sham mice received similar procedures with an injection of the sterile saline instead of collagenase VII.
In vitro ICH model
To mimic ICH-like condition in vitro, HT22 cells were stimulated with hemin at a concentration of 10 μM for 24 h, according to our previous study [18].
Expression vectors and plasmids’ transfection
The plasmids used in the study were purchased from Sangon Bioengineering (Shanghai, China), and the plasmids were as follows: Myc-tagged MDH1 and Flag-tagged HDAC6 vectors were generated using the corresponding full-length sequences of wild-type MDH1 and HDAC6, respectively. Myc-MDH1K118R, Myc-MDH1K121R, Myc-MDH1K298R, and MDH13KR mutant vectors were generated by replacing lysine with arginine at K118, K121, and K298 sites individually or simultaneously. Myc-MDH1K121Q and Myc-MDH1K298Q mutant vectors were generated by replacing lysine with glutamine at K121 and K298 sites individually. Non-acetylate MDH12KR (K to R) and acetyl-mimetic MDH12KQ (K to Q) constructs were generated via mutations at both the K121 and K298 sites. The plasmids were transiently transfected using the reagent lipofectamine 2000 according to the manufacturer’s instructions. The primers used for the indicated mutant plasmids are shown in Table 1 (the capital letters denote the mutation sites).
Table 1.
List of oligonucleotides used in this study
| Name | Sequence |
|---|---|
| Myc-MDH1K118R | F 5′-gggcacagccttggagAGAtacgccaagaaatcag-3′ |
| Myc-MDH1K118R | R 5′-ctgatttcttggcgtaTCTctccaaggctgtgccc-3′ |
| Myc-MDH1K121R | F 5′-ccttggagaaatacgccAGAaaatcagttaaggtcattgt-3′ |
| Myc-MDH1K121R | R 5′-acaatgaccttaactgatttTCTggcgtatttctccaagg-3′ |
| Myc-MDH1K298R | F 5′-gatcaagaataagacctggAGAtttgttgaaggcctccccat-3′ |
| Myc-MDH1K298R | R 5′-atggggaggccttcaacaaaTCTccaggtcttattcttgatc-3′ |
| Myc-MDH1K121Q | F 5′-ggcacagccttggagaaatacgccCAGaaatcagttaagg-3′ |
| Myc-MDH1K121Q | R 5′-ccttaactgatttCTGggcgtatttctccaaggctgtgcc-3′ |
| Myc-MDH1K298Q | F 5′-caagaataagacctggCAGtttgttgaaggcc-3′ |
| Myc-MDH1K298Q | R 5′-ggccttcaacaaaCTGccaggtcttattcttg-3′ |
| Myc-MDH13KR | F1 5′-gcacagccttggagAGAtacgccAGAaaatcagttaaggtcattgttgtggg-3′ |
| Myc-MDH13KR | R1 5′-acaacaatgaccttaactgatttTCTggcgtaTCTctccaaggctgtgccc-3′ |
| Myc-MDH13KR | F2 5′-gatcaagaataagacctggAGAtttgttgaaggcctccccat-3′ |
| Myc-MDH13KR | R2 5′-atggggaggccttcaacaaaTCTccaggtcttattcttgatc-3′ |
| Myc-MDH12KR | F1 5′-ccttggagaaatacgccAGAaaatcagttaaggtcattgt-3′ |
| Myc-MDH12KR | R1 5′-acaatgaccttaactgatttTCTggcgtatttctccaagg-3′ |
| Myc-MDH12KR | F2 5′-gatcaagaataagacctggAGAtttgttgaaggcctccccat-3′ |
| Myc-MDH12KR | R2 5′-atggggaggccttcaacaaaTCTccaggtcttattcttgatc-3′ |
| Myc-MDH12KQ | F1 5′-ggcacagccttggagaaatacgccCAGaaatcagttaagg-3′ |
| Myc-MDH12KQ | R1 5′-ccttaactgatttctgggcgtaTTTctccaaggctgtgcc-3′ |
| Myc-MDH12KQ | F2 5′-caagaataagacctggCAGtttgttgaaggcc-3′ |
| Myc-MDH12KQ | R2 5′-ggccttcaacaaaCTGccaggtcttattcttg-3′ |
| shMDH1 | F 5′-ccggggatcttactgcaaaggaactctcgagagttcctttgcagtaagatcctttttg-3′ |
| shMDH1 | R 5′-attcaaaaaggatcttactgcaaaggaactctcgagagttccttggcagtaagatcc-3′ |
Primers used for the mutant MDH1 vectors and shRNAs against MDH1 were listed
siRNA and transfection
The HDAC6 small interfering RNA (siRNA) used in this study was purchased from GenePharma (Suzhou, China) and transfected into cells with lipofectamine 2000 reagent according to the manufacturer’s protocols. The HDAC6 siRNA sense chain was 5′-GCUUCUAACUGGUCCACUATT-3′ and the antisense chain 5′-UAGUGGACCAGUUAGAAGCTT-3′.
AAV9, lentiviral, and administration
Flag-MDH1WT/Flag-MDH12KR Adeno-Associated virus 9 (AAV9) and MDH1 lentiviral were obtained from Sangon Bioengineering (Shanghai, China). The primer applied for shRNA against MDH1 is shown in Table 1.
AAV9 or lentiviral was administered into the brain through intracerebroventricular injection. The administration site was located at 0.1 mm anterior, 1.0 mm lateral, and 3.0 mm deep relative to the bregma.
Behavior analyses
Modified Neurological Severity Score (mNSS) [19], which includes motor, sensory, balance, and reflex tests, was employed to evaluate neurological deficits before and 3 days post ICH. mNSS scores range from 0 to 18, with 1 point indicating an inability to perform one task. The higher the score, the more severe the mice's loss of ability. The corner-turning test is also a key index of neurological dysfunction, which was performed under a condition with an angle of 30 °C. The right or left turn made by the mouse was recorded when the mouse walked deep into the angle. The percentage of right turns was then calculated to indicate neurological dysfunction [20].
Determination of brain water content
Brain water content was analyzed as previously described [18]. Briefly, brains of experimental mice were removed and divided into three parts as follows: the ipsilateral, contralateral hemisphere, and the cerebellum. These different parts were first weighed to determine the wet weight, and then, the dry weight was determined after drying the samples at 100 °C for 24 h. Brain water content was determined using the formula: (wet weight-dry weight)/wet weight*100%.
Nissl and TUNEL staining
After successively transcardial perfused with PBS and 4% paraformaldehyde (PFA), brains were harvested and post-fixed with 4% PFA at 4 °C overnight. Furthermore, coronal mouse brain specimens were cut serially into 40–50-μm-thick slices from the frontal lobe to the visual cortex for Nissl and TUNEL staining.
Nissl staining was conducted as described previously [18]. Briefly, the sections were first incubated in 100, 95, and 80% ethanol for 30 s and then treated with FD Cresyl Violet SolutionTM (FD Neoro Technologies, Columbia, MD, USA) for 2 min, followed by dehydration through a series of alcohol concentrations (70, 80, 90, and 100%) for 30 s, and finally sealed with neutral resin.
Apoptotic neuron cells were detected using a TUNEL Apoptosis Detection Kit according to its manufacturer’s instructions (Roche, Germany). Briefly, slices were permeabilized with 0.5% Triton X-100 in PBS (0.5% PBST) and blocked with 10% goat serum for 1 h before being incubated in a TUNEL Reaction Mixture for 1 h at 37 °C in a light-resistant container. Thereafter, the slices were co-incubated with a rabbit anti-NeuN antibody (ab177487, 1:500, Abcam, UK) overnight at 4 °C and followed by incubation with an Alexa Fluor 488 secondary antibody (1:500, abcam, USA) at 37 °C for 2 h. Finally, the images of NeuN and TUNEL positive immunostaining from three different peri-hematoma fields in each slice were taken with an Olympus Inverted fluorescence microscope (Olympus America, Center Valley, PA, USA) or Zeiss LSM880 confocal microscope (Zeiss, Germany). All images were analyzed and quantified in ImageJ software (NIH) by 2 independent observers in a blind manner. Positively stained cells and NeuN+/TUNEL+ co-labeling cells were counted automatically with ImageJ software. The mean number counts of positively stained cells were calculated from three separate microscopic fields of perilesional areas of each section on high-power images (×200 magnification). Three section per sample (n = 6 samples/group) were used for quantitative analysis. The numbers of positive cells and the ratio of NeuN+/TUNEL+ positive cells among all NeuN+ cells and among all TUNEL+ cells per 0.1 mm2 were calculated.
Hemorrhagic injury volume analysis
A series of adjacent 40 μm-thick coronal sections were subjected to Nissl staining for injury volume assessment as previously described [19, 21]. Seven brain sections were traced by the Global Laboratory Image analysis system (Data Translation). Injury volume was determined by subtraction of the ipsilateral nondamaged regional volume from the contralateral regional volume. The regional injury volume was presented as a percent of the injury volume that with respect to the contralateral region.
Western blot analysis
Proteins isolated from peri-hematoma tissues and hemin-induced HT22 cells were employed for western blot analysis. Briefly, these tissues were lysed with RIPA (Beyotime Biotechnology, China) buffer for 10 min and centrifuged at 4 °C at 13,000g for 30 min, and the proteins in the supernatant were then collected for western blot analysis. Equal amounts of protein were separated by electrophoresis and transferred to nitrocellulose membranes (Merck Millipore, Germany) and blocked with 5% non-fat milk. The target membranes were then incubated with the primary antibodies that listed in Table 2 and exposed to enhanced chemiluminescence kit (ECL) (ThermoFisher Scientific, USA) for image development after incubation with the corresponding second antibodies at room temperature. Protein band densities from three independent biological replicates were analyzed using ImageJ software. Quantitation of protein expression was performed by densitometry of the representative bands of the immunoblots and normalized with respect to interior reference band (such as GAPDH, Tubulin) and then normalized against the sham control group for all quantitative comparisons.
Table 2.
List of primary antibodies used in this study
| Antibody | Company | Lot number | Dilution ratio |
|---|---|---|---|
| Rabbit anti-BAX | Cell signaling technology | #14796 | 1:1000 |
| Rabbit anti-cleaved-caspase-3 | Cell signaling technology | #9661 | 1:1000 |
| Rabbit anti-HDAC6 antibody | Cell signaling technology | #7558 | 1:1000 |
| Rabbit anti-MDH1 antibody | Abcam | ab180152 | 1:20000 |
| Mouse anti-Myc | Abcam | ab32 | 1:20000 |
| Rabbit anti-Flag | Abcam | ab205606 | 1:20000 |
| Mouse anti-GAPDH antibody | Cell signaling technology | #51332 | 1:5000 |
| Rabbit anti-tubulin antibody | Cell signaling technology | #2128 | 1:10000 |
Primary antibodies used for WB assay were listed
Co-immunoprecipitation and immunoblotting assays
Brain tissues or HT22 cells or 293T cells were lysed with Nonidet P-40 lysis buffer for 10 min and then centrifuged at 13,000g at 4 °C for 30 min to obtain the soluble proteins. A total of 1 mg protein was precipitated using 1 μg of indicated primary antibodies at 4 °C overnight, and followed by incubated with 30 μl of protein G agarose beads (Sigma Technology, Germany) at 4 °C for 4 h. Rabbit IgG was used as a control. Subsequently, the mixture was centrifuged at 13,000g at 4 °C to obtain immunoprecipitants, which were then eluted and boiled in 30 μl of loading buffer following thrice washes with ice-cold PBS buffer. The immunocomplexes were finally subjected to WB assay. The primary antibodies used are listed in Table 2. A total of 100 μg of the lysates without precipitation with antibodies were analyzed directly by WB assay following normalization of total protein content.
Cell immunofluorescence staining
Cells were plated on coverslips and fixed with 4% paraformaldehyde for 20 min at room temperature. Furthermore, cells were permeabilized and blocked with a mixture of 0.5% Triton X-100 and 10% goat serum for 1 h at room temperature. Cells were then incubated with a rabbit anti-Myc antibody (#2272, 1:500, Cell signaling technology) or a mouse anti-Flag antibody (ab18230, 1:500, Abcam) at 4 °C overnight. Cells were washed and incubated with a secondary antibody conjugated with anti-rabbit Alexa 488 (ab150077, 1:500, Abcam) or anti-mouse Alexa 594 (ab150108, 1:500, Abcam) at room temperature for 1 h. Images of the cells were acquired using Zeiss LSM880 confocal microscope (Zeiss, Germany).
Malondialdehyde (MDA) and NADPH/NADP+ assay
Perihematomal tissues were isolated and washed with cold PBS on day 3 post-ICH, followed by homogenization with 1 ml/0.1 g pre-cooled lysis buffer. Homogenized samples were centrifuged at 13,000g at 4 °C for 10 min. The supernatant was obtained for determination of the concentration of MDA and NAPDH/NADP+ using the corresponding MDA and NADPH/NADP+ assay kits (Beyotime Biotechnology, China) following the manufacturer’s instructions.
ROS analysis
HT22 cells were cultured in 96-well plates, and HDAC6 siRNA or the indicated MDH1 plasmids were transfected into the cells when cell density reached 70–80%. The cells were then subjected to hemin 12 h later and followed by oxidant-sensitive probe DCFH-DA labeled ROS detection, using a relative assay kit (Beyotime Biotechnology, China) according to the manufacturer’s instructions.
Statistical analysis
All data were presented in the form of mean ± standard deviation (mean ± SD). Graphpad Prism 8.0 was performed to statistical analysis of the data in our study. Comparisons among multiple groups were analyzed by one-way ANOVA with Tukey’s post hoc test. The T test was used to compare difference between the two groups. Statistical significance was determined as P < 0.05.
Results
HDAC6 knockout alleviates brain damage following ICH
To investigate the role of HDAC6 in ICH-induced brain damage, we employed HDAC6 knockout (HDAC6−/−) mice in comparison to WT control mice. It was revealed that the HDAC6−/− mice had no functional HDAC6 gene as indicated by gene identification assay (Fig. 1A) and no HDAC6 protein as detected by western blot (WB) analysis (Fig. 1B), indicating that the HDAC6 gene had been entirely knocked out. Using Nissl’s staining, the injury volume was compared between WT and HDAC6−/− mice at 3 days following ICH. As shown in Fig. 1C, D, when HDAC6 was knocked out, the injury volume caused by ICH was significantly decreased compared with WT mice. In addition, our findings indicated that HDAC6 knockout greatly ameliorated neurological dysfunction at 3 days following ICH, as seen by a reduction in the mNSS score and right turn percentage compared with WT mice (Fig. 1E, F). Similarly, when HDAC6 was knocked out, the resulting cerebral edema caused by ICH was significantly reduced compared to WT mice at 3 days following ICH (Fig. 1G). Taken together, our findings indicated that HDAC6 knockout protected against ICH-induced brain damage.
Fig. 1.
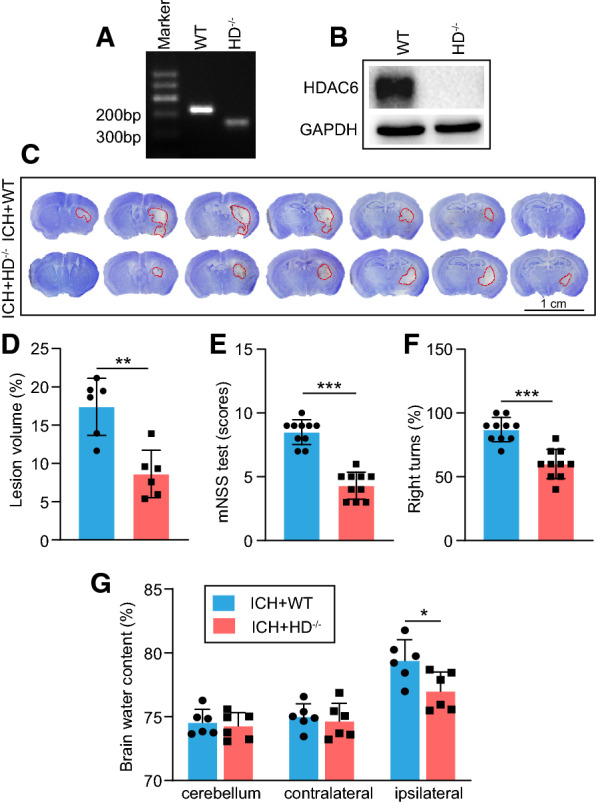
Knockout of HDAC6 alleviates brain damage following ICH. A–B HDAC6−/− mice had defects in HDAC6 gene amplification and HDAC6 protein expression compared with WT mice. At day 3 following ICH, C–D Nissl staining and analysis were employed to evaluate hematoma volume (n = 6 mice/group), E–F mNSS score and corner turn test were applied to detect neurological dysfunction (n = 10 mice/group), and G brain water content analysis was used to assess cerebral edema (n = 6 mice/group). *P < 0.05, **P < 0.01, ***P < 0.001
HDAC6 knockout alleviates oxidative stress and neuro-apoptosis following ICH
We investigated the involvement of HDAC6 in oxidative stress. At 3 days after collagenase VII injection, WT and HDAC6−/− mice were subjected to oxidative stress tests (Fig. 2A). Our findings confirmed that when WT mice were exposed to ICH, their malondialdehyde (MDA) levels increased significantly, but markedly decreased in HDAC6−/− mice (Fig. 2B). In comparison with WT mice, the ratio of NADPH/NADP+ was markedly elevated in HDAC6−/− mice following ICH (Fig. 2C). We further assessed intracellular ROS generation in HT22 cells using the oxidant-sensitive probe 2′,7′-Dichlorodihydrofluorescein diacetate (DCFH-DA). We first transfected HDAC6 siRNA into HT22 cells for 12 h to knockdown HDAC6, which was confirmed by WB analysis (Fig. 2D), and then exposed the cells to hemin for 24 h before collecting the samples for ROS tests. The results showed that when HT22 cells were challenged with hemin, the level of ROS increased considerably, but decreased when HDAC6 was knocked down using HDAC6 siRNA (Fig. 2E, F). Taken together, these findings indicated that HDAC6 knockdown could ameliorate the oxidative stress response during ICH.
Fig. 2.

Knockout of HDAC6 alleviates oxidative stress following ICH. A In vivo experimental design for results presented in Fig. 2B–C. B–C Malondialdehyde (MDA) level and NADPH/NADP+ ration in HDAC6−/− and control WT mice at day 3 following ICH (n = 6/group). D The effect of HDAC6 siRNA was verified by WB assay (n = 3/group). E Representative fluorescence images showing DCFH-DA labeled ROS levels at 24 h following hemin exposure. F Quantitation of ROS content in the group of E (n = 6/group). G Representative images of neuron death shown by double TUNEL and NeuN staining in the peri-hematoma zone of HDAC6−/− and control WT mice at day 3 following ICH (neuron: green and TUNEL: red) and magnified images of NeuN+/TUNEL+ staining were indicated with white dashed line squares. Scale bars: 50 μm. H–L Quantitative analysis of H total NeuN+ cells, I total TUNEL+ cells, J double NeuN+/TUNEL+ cells, K the ration of NeuN+/TUNEL+ cells among total NeuN+ cells, and L the ratio of NeuN+/TUNEL+ cells among total TUNEL+ cells in all groups of G (n = 6/group). M Representative western blot (WB) images and quantitative analysis of cleaved caspase-3 and Bax proteins at day 3 following ICH in vivo (n = 3/group). N Representative WB images and quantitative analysis of cleaved caspase-3 and Bax protein at 24 h following hemin exposure (n = 3/group). *P < 0.05, ***P < 0.001
Given that oxidative stress contributes to neuronal apoptosis, we further investigated the role of HDAC6 in neuronal apoptosis at 3 days following ICH. In vivo, TUNEL staining showed that the number of dead/dying neurons was considerably lower in HDAC6−/− mice compared with WT mice, and the total number of neuron between WT and HDAC6−/− mice showed no significant difference (Fig. 2G–L). Moreover, the apoptosis-associated proteins, including cleaved-caspase-3 and Bax, were significantly increased in WT mice 3 days after ICH; however, there was a marked reduction in the above proteins when HDAC6 was knockout (Fig. 2M). In vitro, immunoblotting analysis showed a significant increase in Bax and cleaved-caspase-3 expression in response to hemin, but a significant decrease after HDAC6 knockdown by HDAD6 siRNA (Fig. 2N). Taken together, our study demonstrated that knocking out HDAC6 can alleviate neuro-apoptosis induced by ICH.
Interaction between HDAC6 and MDH1
Since HDAC6 is a deacetylase, we determined potential substrates of HDAC6 by mass spectrometry to investigate the underlying mechanism of oxidative stress. Among the 11 substrates identified by mass spectrometry, MDH1 was chosen for this study due to its reported role in oxidative stress. The co-immunoprecipitation (coIP) technology in vitro showed that Myc-MDH1 protein can be precipitated by anti-Flag antibody in immunoprecipitation assays of 293T cells with both Flag-HDAC6 and Myc-MDH1 transfection. Flag-HDAC6 can similarly be precipitated by anti-Myc antibody (Fig. 3A). Additionally, we observed an in vivo interaction between endogenous HDAC6 and MDH1. Figure 3B shows that the MDH1 protein existed in the anti-HDAC6 antibody specific immunoprecipitates from WT mice brain extracts, but was absent in the control IgG immunoprecipitates. Similarly, we found that HDAC6 protein was also present in the anti-MDH1 antibody-specific immunoprecipitates. In addition, we co-transfected Flag-HDAC6 and Myc-MDH1 plasmids into both 293T and HT22 cells, and immunofluorescence confocal assay found that HDAC6 co-localized with MDH1 (Fig. 3C). Therefore, our findings suggested that HDAC6 could interact with MDH1.
Fig. 3.

Interaction between HDAC6 and MDH1. A 293T cells were single-transfected with Flag-HDAC6 plasmid or con-transfected with Myc-MDH1 plasmid. A total of 1 mg cell samples were immunoprecipitated by 1 μg of anti-Flag or anti-Myc antibody 24 h after transfection. Immunoprecipitants were then subjected to WB analysis with the indicated antibodies. B Brain samples of WT mice were subjected to IP assays with control anti-IgG or anti-HDAC6 or anti-MDH1 antibody, followed by WB analysis with the indicated antibodies. C Typical confocal images for HDAC6 (red) and MDH1 (green) in 293T and HT22 cells that co-transfected with Flag-HDAC6 and Myc-MDH1 plasmids. D Cell lysates of 293T cells transfected with Myc-MDH1 plasmid alone or combined with Flag-HDAC6 plasmid were precipitated by anti-acetyl-lysine antibody. WB analysis of acetylated MDH1 level was then conducted. E 293T cells transfected with Flag-HDAC6 were treated with 10 μM TubA. Cells were further subjected to IP assay to determine ac-MDH1 level. F Immunoprecipitation of brain tissue lysates of WT and HDAC6−/− mice using anti-acetyl-lysine antibody, followed by WB analysis of ac-MDH1 level
Next, we investigated whether HDAC6 deacetylated MDH1. We found that when 293T cells were co-treated with MDH1 and HDAC6, MDH1 acetylation was significantly decreased compared to when 293T cells were treated with MDH1 alone (Fig. 3D). Second, as shown in Fig. 3E, when 293T cells were treated with TubA rather than DMSO, there was a considerable increase in MDH1 acetylation. We further conducted validation experiments in vivo. Immunoprecipitation with anti-acetyl-lysine antibody showed that the level of MDH1 acetylation was significantly upregulated in HDAC6 knockout mice compared to WT mice (Fig. 3F). Therefore, we concluded that HDAC6 was capable of deacetylating MDH1.
HDAC6 negatively regulates MDH1 acetylation in response to ICH
Since the enzymatic activity of MDH1 can be enhanced by acetylation, we investigated whether the ICH-related damaging agents affected MDH1 acetylation. As shown in Fig. 4A–C, the damaging agents (Hemin, Hemoglobin, Thrombin) significantly decreased MDH1 acetylation in HT22 cells. We further examined whether HDAC6 altered the degree of MDH1 acetylation during ICH. Immunoprecipitation analysis showed that the level of acetylated MDH1 was significantly decreased following hemin induction compared to the control group, but significantly increased following HDAC6 siRNA knockdown (Fig. 4D). Additionally, we examined the effect of TubA on the level of acetylated MDH1 following ICH in mice. As shown in Fig. 4E, TubA treatment can significantly increase the level of acetylated MDH1 during ICH when compared to the DMSO-treated group. Consistently, the level of MDH1 acetylation was significantly increased in HDAC6−/− mice compared to WT mice in the presence of ICH (Fig. 4F). Collectively, the vivo and in vitro findings revealed that HDAC6 inhibition significantly increased the level of acetylated MDH1 following ICH.
Fig. 4.

HDAC6 negatively regulates MDH1 acetylation in response to ICH. A–C IP and WB analysis of the ac-MDH1 in HT22 cells at 24 h following treated with A Hemin, B Hemoglobin, and C Thrombin. D IP assay performed to determine ac-MDH1 level in the indicated group (n = 3/group). E IP assay determined to examine acetylation level of MDH1 in the indicated group (n = 3/group). F IP and WB analysis of the acetylation level of MDH1 in HDAC6−/− and WT mice at day 3 after subjection to ICH surgery (n = 3/group). G coIP assay of the interaction between HDAC6 and MDH1 at day 3 after ICH surgery or under 30 mg/kg TubA treatment in WT mice. *P < 0.05, ***P < 0.001
To obtain additional insight into the role of HDAC6 in the ICH-induced decrease in MDH1 acetylation, we explored whether the ICH state affected the interaction between HDAC6 and MDH1. CoIP assay revealed that when mice were subjected to ICH surgery (Fig. 4G), the interaction between HDAC6 and MDH1 was enhanced, whereas decreased upon TubA treatment. Therefore, this study suggested that the decreased interaction between HDAC6 and MDH1 resulted in an increase in MDH1 acetylation when HDAC6 was inhibited.
HDAC6 deacetylates MDH1 at the K121 and K298 residues
To identify the specific lysine residues in MDH1 that may be acetylated by HDAC6, we first used the lysine residues prediction site (http://www.uniprot.org/) and a relevant published article, which showed the potential acetylation sites of MDH1 may be in Lys 118 (K), Lys 121 (K), and Lys 298 (K). To examine if HDAC6 acts on these MDH1 residues, we generated Lys-to-arginine mutants in these sites either separately or simultaneously. Our findings indicated that mutations in K121, K298 rather than K118 could significantly reduce MDH1 acetylation, and that acetylation was completely lost due to mutations in the three lysine residues (3KR) (Fig. 5A). Additionally, the coIP assay showed that the interaction between HDAC6 and MDH1 was significantly impaired or hardly detectable when lysine residues 121 or 298 were mutated alone or co-mutated at the three 118, 121, 298 lysine residues compared with wild-type MDH1, but not when lysine 118 was mutated alone (Fig. 5B). Therefore, we concluded that HDAC6 may deacetylate lysine residues 121 and 298 of MDH1. Levels of acetylated MDH1 were indeed markedly decreased in 293T cells transfected with the acetyl-mimetic mutants (Myc-MDH1K121Q, Myc-MDH1K298Q) and non-acetylatable mutants (Myc-MDH1K121R, Myc-MDH1K298R), further confirming the acetylation sites of MDH1 (Fig. 5C). We next generated Myc-tagged MDH1 acetyl-mimetic mutants (Myc-MDH1 2KQ) and non-acetylatable mutants (Myc-MDH1 2KR), in which the mutations in the two lysine residues K121 and K298 were substituted with glutamine (Q) or arginine (R), respectively. The IP assay showed that the acetylation of MDH1 was lost when the lysines in 121 and 298 sites were replaced with arginine or glutamine (Fig. 5D, E). Our findings collectively revealed that HDAC6 deacetylated MDH1 at the K121 and K298 residues.
Fig. 5.

HDAC6 deacetylates lysine residues of MDH1 at position 121 and 298. A 293T cells were transfected with wild-type and mutated plasmids of Myc-MDH1 (K118R, K121R, K298R, 3KR) for 24 h, and then subjected to IP and immunoblotting assay with the indicated antibodies. B 293T cells were co-transfected with Flag-HDAC6 and Myc-MDH1 (WT, K118R, K121R, K298R, 3KR) plasmids for 24 h, and then subjected to IP and immunoblotting assay with the indicated antibodies. C 293T cells were transfected with Myc-MDH1 (WT, K121R, K121Q, K298R, K298Q) plasmids for 24 h, and then subjected to IP and immunoblotting assay with the indicated antibodies. D 293T or E HT22 cells were transfected with Myc-MDH1 plasmids (WT, 2KR, 2KQ) for 24 h and then subjected to IP and immunoblotting assay with the indicated antibodies
Acetylated MDH1 reduces oxidative stress and neuro-apoptosis
We examined whether acetylated MDH1 affected oxidative stress during ICH. First, we conducted a study in vitro. Our results showed that ROS was overproduced when HT22 cells were stimulated with hemin, but was significantly reduced following wild-type MDH1 or MDH12KQ treatment. In contrast, the MDH12KR treatment failed to exhibit a defense effect against ROS production by hemin (Fig. 6A). This finding revealed that acetylation of MDH1 played a critical role in decreasing ROS generation in vitro. We further established a model with MDH1 protein overexpression in the WT mouse by injecting MDH1 AAV9 virus (Flag-MDH1WT, Flag-MDH12KR) with or without the corresponding green-fluorescent protein (GFP) into brain; autofluorescence without primary antibody labeling suggested that MDH1 AAV9 could be successfully injected into brain (Fig. 6B), and western blot analysis suggested that WT-MDH1WT and MDH12KR overexpression could be detected in the intracranial cavity (Fig. 6C, D). As shown in Fig. 6E, when WT mice were subjected to ICH surgery, the level of MDA was significantly increased, but markedly decreased when Flag-MDH1WT was overexpressed rather than Flag-MDH12KR. The ratio of NADPH/NADP+, that significantly decreased following ICH, was found to be markedly increased when treated with wild-type MDH1, but not with MDH12KR (Fig. 6F). Overall, our findings revealed that acetylation of MDH1 played an important role in reducing oxidative stress following ICH.
Fig. 6.

Acetylated MDH1 alleviated oxidative stress and neuro-apoptosis following ICH. A Representative fluorescence images showing DCFH-DA labeled ROS levels in the indicated groups. B–C Results for green-fluorescent protein (GFP)-MDH1 AAV9 virus (Flag-MDH1, Flag-MDH12KR) in the brain as identified by B autofluorescence without primary antibody staining and C WB assay. D WB results of MDH1 AAV9 virus without GFP labeling in the brain. E–F MDA level and NADPH/NADP+ ratio were assayed using relative assay kit (n = 6/group). G HT22 cells were transfected with WT-MDH1 and its mutant MDH1 (2KR, 2KQ), respectively, for 12 h and then subjected to hemin for 24 h. Expression of cleaved caspase-3 and Bax protein was assayed by WB (n = 4/group). H WT mice were transfected with Flag-MDH1-AAV9 and Flag-MDH12KR-AAV9 for 21d, and then subjected to ICH surgery. Expression of cleaved caspase-3 and Bax level was assayed by WB at day 3 following ICH (n = 4/group). I Representative images and quantitative analysis of neuron death in the peri-hematoma zone of indicated groups (neuron: green and TUNEL: red). Magnified images of NeuN+/TUNEL+ staining were indicated by white dashed line squares. Scale bar: 50 μm. J–N Quantitative analysis of J total NeuN+ cells, K total TUNEL+ cells, L double NeuN+/TUNEL+ cells, M the ration of NeuN+/TUNEL+ cells among total NeuN+ cells, and N the ratio of NeuN+/TUNEL+ cells among total TUNEL+ cells in all groups of I (n = 6/group). *P < 0.05, **P < 0.01, ***P < 0.001
We further investigated the role of acetylated MDH1 in neuro-apoptosis during ICH in vivo and vitro. Immunoblotting of samples from the indicated groups in vitro revealed that the hemin-induced group had significantly higher Bax and cleaved-caspase-3 levels than the control group. However, the MDH1WT or MDH12KQ treated group, but not the MDH12KR, displayed significant decrease in Bax and cleaved-caspase-3 protein levels (Fig. 6G). In vivo, intracranial targeted overexpression of Flag-MDH1WT (wild-type MDH1-AAV9 vector) significantly reduced the increased expression of Bax and cleaved-caspase-3 induced by ICH in mice, while Flag-MDH12KR-AAV9 treatment failed to exert a protective effect (Fig. 6H). In addition, the neuronal death (NeuN+/TUNEL+) induced by ICH was significantly reduced when mice treated with Flag-MDH1WT, but not Flag-MDH12KR (Fig. 6I–N). Taken together, our findings indicated that acetylation MDH1 may help protect against neuro-apoptosis.
Protective function of HDAC6 inhibition is acetylated MDH1 dependent
To elucidate the underlying mechanism of the protective role of HDAC6 inhibition, we established MDH1 knockdown mice by injecting MDH1 lentivirus for 2 weeks and then subjected them to ICH surgery (Fig. 7A). Autofluorescence and WB analysis revealed that 2 weeks after intraventricular injection, intracranial MDH1 protein could significantly be reduced by the MDH1 lentivirus (Fig. 7B, C). Following that, we used TubA to determine if TubA's effect on brain protection was acetylated MDH1 dependent. Nissl’s staining revealed that TubA treatment significantly reduced hematoma volume following ICH in WT mice. However, TubA exhibited a weakened protective effect on hematoma volume reduction when MDH1 protein was knocked down using lentivirus (Fig. 7D, E). In addition, administration of TubA significantly alleviated brain edema and improved neurological dysfunction, but failed to exhibit this protective effect when MDH1 knockdown (Fig. 7F–H). Therefore, our findings indicated that HDAC6 inhibition exerted its protective effect in an acetylated MDH1-dependent manner.
Fig. 7.

Protective effect of HDAC6 inhibition depends upon acetylation level of MDH1. A Experimental design for results presented in Fig. 7B–H. B–C Results of green-fluorescent protein (GFP)-MDH1 lentivirus in the brain were identified by B autofluorescence without primary antibody staining and C WB assay (n = 3/group). At day 3 following ICH, D–E brain injury was assayed by Nissl staining (n = 6/group), F cerebral edema was determined by the brain water content (n = 6/group), and G–H neurological function was assayed by mNSS score and corner-turning test (n = 10/group). I Model illustration of mechanism by which HDAC6/MDH1 signaling mediates oxidative stress-induced neuron apoptosis following ICH. Upon ICH injury, the interaction between HDAC6 and MDH1 was enhanced, which promoted HDAC6 mediated-MDH1 deacetylation at K121 and K298, thereby inhibiting NADP+ shift to NADPH; consequently, ROS was overproduced, which contributed to neuron apoptosis. However, administration of TubA (a specific HDAC6 inhibitor) alleviated the oxidative stress response by disrupting HDAC6 and MDH1 association, thus recovered neuron from apoptosis. **P < 0.01, ***P < 0.001
Discussion
The identification of HDAC6 as a regulator of oxidative stress sheds light not only on the possible mechanism underlying ICH-induced brain damage but also on the development of novel ICH therapies. In this study, we found that HDAC6 depletion protected against ICH-induced brain damage via oxidative stress and neuronal apoptosis defense. Mechanically, HDAC6 was found to interact with and deacetylate MDH1 at lysine residues 121 and 298. In the present study, we also demonstrated that the depletion of HDAC6 increased MDH1 acetylation, and protected neurons against oxidative stress damage. Moreover, the protective effect of the HDAC6 inhibitor TubA in ICH was weakened with MDH1 knockdown (Fig. 7I). Collectively, targeting HDAC6/MDH1 signaling may provide a potential therapeutic approach for ICH treatment.
Oxidative stress characterized by excessive production of reactive oxygen species (ROS) is a vital pathophysiological event after intracerebral hemorrhage [5, 6]. Excessive ROS production results in DNA damage and protein and lipid oxygenation, which alters cell signaling pathways and eventually results in neuronal death. Thus, reducing the oxidative stress response is an important target for intracerebral hemorrhage treatment. Numerous studies have established the critical role of HDAC6 in oxidative stress. For example, in diabetes and Alzheimer's disease (AD), inhibiting HDAC6 expression can reduce elevated ROS and Ca2+ levels [22, 23]. Zhang et al. have demonstrated that AC-1215, an HDAC6 inhibitor, can protect liver cells from necrosis in mice with acute liver failure (ALF) by reducing the production of granulose-dependent ROS [24]. Additionally, Yuan et al. found that tubacin, a specific HDAC6 inhibitor, could reduce neuronal necrosis and increase neuronal survival by regulating the level of ROS in a rat cortical neuron model of oxygen–glucose deprivation [25]. Butler et al. also found that TubA administration preserved neuronal survival in an oxidative stress-induced model [26]. In line with these studies, our present study demonstrated that HDAC6 knockout reduced oxidative stress response and the neuron-apoptosis following ICH mouse model.
HDAC6 has been shown to regulate a variety of critical biological processes, such as ciliary disassembly [27], DNA repair [28], and axon growth [29] by deacetylating of its specific substrates. We hypothesized that HDAC6 may exert its oxidative stress effect by mediating the deacetylation of some certain substrates associated with oxidative stress. Our current study determined an interaction between HDAC6 and Malate dehydrogenase 1 (MDH1), and found that HDAC6 can modify the level of MDH1 acetylation during ICH. ICH-associated damaging agents (hemin, hemoglobin, and thrombin) reduced MDH1 acetylation in an HDAC6-dependent manner. MDH1, a dehydrogenase subtype, predominantly expressed in tissues with high aerobic metabolic demands, including brain [30–32]. Previous studies have showed that MDH1 involved in oxidative stress by regulating the NAPDH/NADP+ ratio and cellular ROS activity [33]. Additionally, a reduction in MDH1 activity may induce pancreatic ductal adenocarcinoma (PDAC) cells death via enhanced oxidative stress and the corresponding damaged mitochondria [34]. More importantly, it is accepted that post-translational can be used to modulate MDH1 function [16]. Cumulative studies have indicated that MDH1 deeply involved in the central nervous system diseases, such as aging [35], somatic retardation, epilepsy, and progressive microcephaly [17], and rather than being confined to tumors [33, 36]. However, the involvement of MDH1, particularly its acetylation in the development of oxidative stress during intracerebral hemorrhage remains unknown. In this study, we determined that acetylation of lysine residues in MDH1 were K121 and K298 (2K), and that mutants altered MDH1 acetylation and the association between HDAC6 and MDH1. We further demonstrated the wild-type or the acetylation-mimetic 2KQ mutant, but not the acetylation-resistant 2KR mutant treatment significantly decreased the ROS levels and apoptosis proteins (Cleaved-caspase-3 and Bax) expression in HT22 cells challenged with hemin. Additionally, we demonstrated in vivo that AAV-mediated overexpression of non-acetylation mutant MDH1 rather than acetylation-resistant MDH1 can significantly reduce oxidative stress response and neuronal apoptosis following intracerebral hemorrhage. Yi-Ping Wang et al. have reported that the arginine methylation of MDH1 by CARM1 (another post-translational modification of MDH1) was negatively regulated by MDH1-dependent antioxidant stress activities [34]. Thus, our findings showed that MDH1 acetylation plays a critical role in cellular ROS defense and neuronal protection during ICH. We speculate that the manipulation between HDAC6 and MDH1 will facilitate the investigation of novel anti-oxidative mechanisms during ICH.
We further demonstrated that the inhibition of HDAC6 protected against ICH-induced brain damage through elevating the acetylation of MDH1. This conclusion is based on the following observation. TubA treatment significantly alleviated brain damage during ICH, as measured by decreased hematoma volume, cerebral edema, and neurological dysfunction score. However, the protective effect of TubA against brain damage was significantly weakened in the presence of MDH1 lentiviral knockdown. TubA's inability to execute its protective role was hypothesized to be due to its failure to enhance the MDH1 acetylation level in ICH when MDH1 was knocked down. However, a specific knockdown of the MDH1 acetylation site in mice to investigate the effect of MDH1 acetylation on oxidative stress levels will be a more effective approach.
Although the current study revealed a protective effect of HDAC6 on neuro-apoptosis in ICH, potential effects on other cells cannot be excluded. For example, HDAC6 has been shown to regulate LPS tolerance in astrocyte [37]. Additionally, HDAC6 inhibition reverses long-term doxorubicin-induced cognitive dysfunction by restoring microglia homeostasis and synaptic integrity [38]. Indeed, as shown in Fig. 2 in our study, quite a few TUNEL+ cells are not neurons. Therefore, HDAC6 is likely to play a role on the death of other types of cells in response to ICH. Further studies are warranted to explore the additional cellular targets of HDAC6. In addition, the DNA fragmentation identified by the TUNEL staining occurs characteristically, but not exclusively, in apoptotic cells. Other forms of cell death, such as pyroptosis [39, 40] and necrosis [41, 42], also happen after ICH and can trigger inflammatory responses. Some studies have demonstrated that targeting HDAC6 alleviates macrophage pyroptosis [39] and inhibits inflammatory response in astrocyte [43]. It is thus possible that HDAC6 inhibition reduces the non-apoptotic death of microglia or astrocyte, thereby alleviating brain damage. Further studies are necessary to elucidate the modalities of cell death in astrocyte, microglia, or other types of cells mediated by HDAC6 during ICH.
In summary, this study established here support an important role for HDAC6 inhibition in preventing neurons from oxidative stress damage and promoting neuronal survival after an acute intracerebral hemorrhage insult, and the protective effects of HDAC6 inhibition were related to MDH1 acetylation. This study may open a new perspective for interventions of ICH.
Acknowledgements
We are grateful to the members of Dr Cui’s laboratory for their kind suggestions.
Abbreviations
- Bax
Bcl-2-associated X protein
- Caspase-3
Cysteiny aspartate specific proteinase-3
- Co-IP
Co-immunoprecipitation
- IP
Immunoprecipitation
- DMSO
Dimethyl sulfoxide
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- HDACs
Histone deacetylases
- HDAC6
Histone deacetylases 6
- ICH
Intracerebral hemorrhage
- MDH1
Malate dehydrogenase 1
- MDA
Malondialdehyde
- mNSS
Modified neurological severity score
- NAD
Nicotinamide adenine Dinucleotide
- NADPH
Nicotinamide adenine dinucleotide phosphate
- ROS
Reactive oxygen species
- siRNA
Small interfering RNA
- TubA
Tubastatin A
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
Author contributions
GYC and MW: contributed to research design and manuscript writing. MW: executed most experiments. CZ: contributed to data acquirement and analyzation. LY: assisted experiments accomplishment. WJM and BCL: helped with most of the mouse experiments. YW: assisted protein immunoprecipitation. WFW and MYZ: assisted protein extraction and animal experiments. All authors read and approved the final manuscript.
Funding
This work was supported by grants from National Natural Science Foundation of China (81571210, 81771282, and 82171305) and Xuzhou Innovation Capacity Building Program (KC19239) to Dr. Guiyun Cui, by grants from the National Natural Science Foundation of China (81971134), the Natural Science Foundation of Jiangsu Province (BK20191152), Medical Scientific Research Project of Jiangsu Provincial Health Commission (ZDB2020017), and Xuzhou Key Research and Development Program (KC19131) to Dr. Xinchun Ye, by grants from the National Natural Science Foundation of China (82001276) to Dr. Hu, and by grants from Development Fund of Affiliated Hospital of Xuzhou Medical University (XYFM2020033) to Miao Wang.
Data availability
The datasets used and/or analyzed during the present study are available from the corresponding author upon reasonable request.
Declarations
Conflict of interest
Authors declare no competing financial interests.
Study approval
All animal experiments carried out were approved by the Jiangsu Provincial Animal Care, and all mice procedures were approved by the institutional Animal Use and Care Committee of XuZhou Medical University.
Consent for publication
All authors have read and approved the manuscript.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Miao Wang and Chao Zhou have contributed equally to this work.
Contributor Information
Miao Wang, Email: WANGMIAO125430@163.com.
Guiyun Cui, Email: GYC171722014@163.com, Email: guiyuncui@foxmail.com.
References
- 1.Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, et al. Heart disease and stroke statistics-2020 update: a report from the American heart association. Circulation. 2020;141(9):e139–e596. doi: 10.1161/CIR.0000000000000757. [DOI] [PubMed] [Google Scholar]
- 2.Galyfos G, Sianou A, Filis K. Cerebral hyperperfusion syndrome and intracranial hemorrhage after carotid endarterectomy or carotid stenting: a meta-analysis. J Neurol Sci. 2017;381:74–82. doi: 10.1016/j.jns.2017.08.020. [DOI] [PubMed] [Google Scholar]
- 3.Kang M, Yao Y. Oligodendrocytes in intracerebral hemorrhage. CNS Neurosci Ther. 2019;25(10):1075–1084. doi: 10.1111/cns.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duan X, Wen Z, Shen H, Shen M, Chen G. Intracerebral hemorrhage, oxidative stress, and antioxidant therapy. Oxid Med Cell Longev. 2016;2016:1203285. doi: 10.1155/2016/1203285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu X, Tao C, Gan Q, Zheng J, Li H, You C. Oxidative stress in intracerebral hemorrhage: sources, mechanisms, and therapeutic targets. Oxid Med Cell Longev. 2016;2016:3215391. doi: 10.1155/2016/3215391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yao Z, Bai Q, Wang G. Mechanisms of oxidative stress and therapeutic targets following intracerebral hemorrhage. Oxid Med Cell Longev. 2021;2021:8815441. doi: 10.1155/2021/8815441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imai T, Matsubara H, Hara H. Potential therapeutic effects of Nrf2 activators on intracranial hemorrhage. J Cereb Blood Flow Metab: Official J Int Soc Cereb Blood Flow and Metab. 2021;41(7):1483–1500. doi: 10.1177/0271678X20984565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kang R, Li R, Dai P, Li Z, Li Y, Li C. Deoxynivalenol induced apoptosis and inflammation of IPEC-J2 cells by promoting ROS production. Environ Pollut (Barking, Essex: 1987) 2019;251:689–698. doi: 10.1016/j.envpol.2019.05.026. [DOI] [PubMed] [Google Scholar]
- 9.Zheng J, Shi L, Liang F, Xu W, Li T, Gao L, Sun Z, Yu J, Zhang J. Sirt3 Ameliorates oxidative stress and mitochondrial dysfunction after intracerebral hemorrhage in diabetic rats. Front Neurosci. 2018;12:414. doi: 10.3389/fnins.2018.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leyk J, Daly C, Janssen-Bienhold U, Kennedy BN, Richter-Landsberg C. HDAC6 inhibition by tubastatin A is protective against oxidative stress in a photoreceptor cell line and restores visual function in a zebrafish model of inherited blindness. Cell Death Dis. 2017;8(8):e3028. doi: 10.1038/cddis.2017.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Q, Li S, Zhou Z, Fu M, Yang X, Hao K, Liu Y. HDAC6 inhibitor Cay10603 inhibits high glucose-induced oxidative stress, inflammation and apoptosis in retinal pigment epithelial cells via regulating NF-κB and NLRP3 inflammasome pathway. Gen Physiol Biophys. 2020;39(2):169–177. doi: 10.4149/gpb_2019058. [DOI] [PubMed] [Google Scholar]
- 12.Shi Y, Xu L, Tang J, Fang L, Ma S, Ma X, Nie J, Pi X, Qiu A, Zhuang S, et al. Inhibition of HDAC6 protects against rhabdomyolysis-induced acute kidney injury. Am J Physiol Renal Physiol. 2017;312(3):F502–F515. doi: 10.1152/ajprenal.00546.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leucker TM, Nomura Y, Kim JH, Bhatta A, Wang V, Wecker A, Jandu S, Santhanam L, Berkowitz D, Romer L, et al. Cystathionine γ-lyase protects vascular endothelium: a role for inhibition of histone deacetylase 6. Am J Physiol Heart Circ Physiol. 2017;312(4):H711–H720. doi: 10.1152/ajpheart.00724.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Z, Leng Y, Wang J, Liao HM, Bergman J, Leeds P, Kozikowski A, Chuang DM. Tubastatin A, an HDAC6 inhibitor, alleviates stroke-induced brain infarction and functional deficits: potential roles of α-tubulin acetylation and FGF-21 up-regulation. Sci Rep. 2016;6:19626. doi: 10.1038/srep19626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Zhao Y, Shi J, Ren Z, Chen F, Tang W. Histone deacetylase 6 interference protects mice against experimental stroke-induced brain injury via activating Nrf2/HO-1 pathway. Animal Cells Syst. 2019;23(3):192–199. doi: 10.1080/19768354.2019.1601132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim EY, Kim WK, Kang HJ, Kim JH, Chung SJ, Seo YS, Park SG, Lee SC, Bae KH. Acetylation of malate dehydrogenase 1 promotes adipogenic differentiation via activating its enzymatic activity. J Lipid Res. 2012;53(9):1864–1876. doi: 10.1194/jlr.M026567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Broeks MH, Shamseldin HE, Alhashem A, Hashem M, Abdulwahab F, Alshedi T, Alobaid I, Zwartkruis F, Westland D, Fuchs S, et al. MDH1 deficiency is a metabolic disorder of the malate-aspartate shuttle associated with early onset severe encephalopathy. Hum Genet. 2019;138(11–12):1247–1257. doi: 10.1007/s00439-019-02063-z. [DOI] [PubMed] [Google Scholar]
- 18.Wang M, Ye X, Hu J, Zhao Q, Lv B, Ma W, Wang W, Yin H, Hao Q, Zhou C, et al. NOD1/RIP2 signalling enhances the microglia-driven inflammatory response and undergoes crosstalk with inflammatory cytokines to exacerbate brain damage following intracerebral haemorrhage in mice. J Neuroinflammation. 2020;17(1):364. doi: 10.1186/s12974-020-02015-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J, Li Y, Wang L, Zhang Z, Lu D, Lu M, Chopp M. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke. 2001;32(4):1005–1011. doi: 10.1161/01.STR.32.4.1005. [DOI] [PubMed] [Google Scholar]
- 20.Yang H, Ni W, Jiang H, Lei Y, Su J, Gu Y, Zhou L. Histone deacetylase inhibitor scriptaid alleviated neurological dysfunction after experimental intracerebral hemorrhage in mice. Behav Neurol. 2018;2018:6583267. doi: 10.1155/2018/6583267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ye XC, Hu JX, Li L, Li Q, Tang FL, Lin S, Sun D, Sun XD, Cui GY, Mei L, et al. Astrocytic Lrp4 (low-density lipoprotein receptor-related protein 4) contributes to ischemia-induced brain injury by regulating ATP release and adenosine-A(2A)R (adenosine A2A receptor) signaling. Stroke. 2018;49(1):165–174. doi: 10.1161/STROKEAHA.117.018115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leng Y, Wu Y, Lei S, Zhou B, Qiu Z, Wang K, Xia Z. Inhibition of HDAC6 activity alleviates myocardial ischemia/reperfusion injury in diabetic rats: potential role of peroxiredoxin 1 acetylation and redox regulation. Oxid Med Cell Longev. 2018;2018:9494052. doi: 10.1155/2018/9494052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi H, Kim HJ, Kim J, Kim S, Yang J, Lee W, Park Y, Hyeon SJ, Lee DS, Ryu H, et al. Increased acetylation of peroxiredoxin1 by HDAC6 inhibition leads to recovery of Aβ-induced impaired axonal transport. Mol Neurodegener. 2017;12(1):23. doi: 10.1186/s13024-017-0164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang WB, Zhang HY, Wang Y, Jiao FZ, Wang LW, Gong ZJ. Quantitative proteomic analysis reveals the sites related to acetylation and mechanism of ACY-1215 in acute liver failure mice. Front Pharmacol. 2019;10:653. doi: 10.3389/fphar.2019.00653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yuan L, Wang Z, Liu L, Jian X. Inhibiting histone deacetylase 6 partly protects cultured rat cortical neurons from oxygen-glucose deprivation-induced necroptosis. Mol Med Rep. 2015;12(2):2661–2667. doi: 10.3892/mmr.2015.3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am Chem Soc. 2010;132(31):10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ran J, Yang Y, Li D, Liu M, Zhou J. Deacetylation of α-tubulin and cortactin is required for HDAC6 to trigger ciliary disassembly. Sci Rep. 2015;5:12917. doi: 10.1038/srep12917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang M, Xiang S, Joo HY, Wang L, Williams KA, Liu W, Hu C, Tong D, Haakenson J, Wang C, et al. HDAC6 deacetylates and ubiquitinates MSH2 to maintain proper levels of MutSα. Mol Cell. 2014;55(1):31–46. doi: 10.1016/j.molcel.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalinski AL, Kar AN, Craver J, Tosolini AP, Sleigh JN, Lee SJ, Hawthorne A, Brito-Vargas P, Miller-Randolph S, Passino R, et al. Deacetylation of Miro1 by HDAC6 blocks mitochondrial transport and mediates axon growth inhibition. J Cell Biol. 2019;218(6):1871–1890. doi: 10.1083/jcb.201702187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joh T, Takeshima H, Tsuzuki T, Setoyama C, Shimada K, Tanase S, Kuramitsu S, Kagamiyama H, Morino Y. Cloning and sequence analysis of cDNAs encoding mammalian cytosolic malate dehydrogenase. Comparison of the amino acid sequences of mammalian and bacterial malate dehydrogenase. J Biol Chem. 1987;262(31):15127–15131. doi: 10.1016/S0021-9258(18)48147-1. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka T, Inazawa J, Nakamura Y. Molecular cloning and mapping of a human cDNA for cytosolic malate dehydrogenase (MDH1) Genomics. 1996;32(1):128–130. doi: 10.1006/geno.1996.0087. [DOI] [PubMed] [Google Scholar]
- 32.Lo AS, Liew CT, Ngai SM, Tsui SK, Fung KP, Lee CY, Waye MM. Developmental regulation and cellular distribution of human cytosolic malate dehydrogenase (MDH1) J Cell Biochem. 2005;94(4):763–773. doi: 10.1002/jcb.20343. [DOI] [PubMed] [Google Scholar]
- 33.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496(7443):101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang YP, Zhou W, Wang J, Huang X, Zuo Y, Wang TS, Gao X, Xu YY, Zou SW, Liu YB, et al. Arginine methylation of MDH1 by CARM1 inhibits glutamine metabolism and suppresses pancreatic cancer. Mol Cell. 2016;64(4):673–687. doi: 10.1016/j.molcel.2016.09.028. [DOI] [PubMed] [Google Scholar]
- 35.Lee SM, Dho SH, Ju SK, Maeng JS, Kim JY, Kwon KS. Cytosolic malate dehydrogenase regulates senescence in human fibroblasts. Biogerontology. 2012;13(5):525–536. doi: 10.1007/s10522-012-9397-0. [DOI] [PubMed] [Google Scholar]
- 36.Kim BS, Lee K, Jung HJ, Bhattarai D, Kwon HJ. HIF-1α suppressing small molecule, LW6, inhibits cancer cell growth by binding to calcineurin b homologous protein 1. Biochem Biophys Res Commun. 2015;458(1):14–20. doi: 10.1016/j.bbrc.2015.01.031. [DOI] [PubMed] [Google Scholar]
- 37.Beurel E. HDAC6 regulates LPS-tolerance in astrocytes. PLoS One. 2011;6(10):e25804. doi: 10.1371/journal.pone.0025804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McAlpin BR, Mahalingam R, Singh AK, Dharmaraj S, Chrisikos TT, Boukelmoune N, Kavelaars A, Heijnen CJ. HDAC6 inhibition reverses long-term doxorubicin-induced cognitive dysfunction by restoring microglia homeostasis and synaptic integrity. Theranostics. 2022;12(2):603–619. doi: 10.7150/thno.67410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu S, Chen H, Ni H, Dai Q. Targeting HDAC6 attenuates nicotine-induced macrophage pyroptosis via NF-κB/NLRP3 pathway. Atherosclerosis. 2021;317:1–9. doi: 10.1016/j.atherosclerosis.2020.11.021. [DOI] [PubMed] [Google Scholar]
- 40.Xu P, Hong Y, Xie Y, Yuan K, Li J, Sun R, Zhang X, Shi X, Li R, Wu J, et al. TREM-1 exacerbates neuroinflammatory injury via NLRP3 inflammasome-mediated pyroptosis in experimental subarachnoid hemorrhage. Transl Stroke Res. 2021;12(4):643–659. doi: 10.1007/s12975-020-00840-x. [DOI] [PubMed] [Google Scholar]
- 41.Collins MK, Marvel J, Malde P, Lopez-Rivas A. Interleukin 3 protects murine bone marrow cells from apoptosis induced by DNA damaging agents. J Exp Med. 1992;176(4):1043–1051. doi: 10.1084/jem.176.4.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fukuda K, Kojiro M, Chiu JF. Demonstration of extensive chromatin cleavage in transplanted Morris hepatoma 7777 tissue: apoptosis or necrosis? Am J Pathol. 1993;142(3):935–946. [PMC free article] [PubMed] [Google Scholar]
- 43.Youn GS, Ju SM, Choi SY, Park J. HDAC6 mediates HIV-1 tat-induced proinflammatory responses by regulating MAPK-NF-kappaB/AP-1 pathways in astrocytes. Glia. 2015;63(11):1953–1965. doi: 10.1002/glia.22865. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author upon reasonable request.