

Abstract
Ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) is a unique component of the ubiquitin–proteasome system (UPS), which has multiple activities in maintaining intracellular ubiquitin levels. We previously reported the aberrant low expression of UCHL1 in podocytes of non-immune complex-mediated glomerulonephritis, and recent studies indicate that anti-UCHL1 antibody was responsible for the refractory minimal change disease (MCD), but the specific effect of UCHL1 to the podocytopathy has not been determined. Therefore, we generated podocyte-specific UCHL1 gene knockout (UCHL1cre/cre) rats model. Podocyte-specific UCHL1 knockout rats exhibited severe kidney damage, including segmental/global glomerulosclerosis, kidney function damage and severe proteinuria, compared with littermate control. Subsequently, by carrying out mass spectrometry analysis of isolated glomeruli of rats, abnormal protein accumulation of ECM-receptor Interaction was found in UCHL1cre/cre rats. Mechanistic studies in vivo and in vitro revealed that aberrant protein accumulation after UCHL1 deficiency induced endoplasmic reticulum (ER) stress, unfolded protein reaction (UPR) to reduce the protein level of podocyte skeleton proteins, and CHOP mediated apoptosis as well, which related to the dysfunction of the ubiquitin–proteasome system with decreased free monomeric ubiquitin level, thereby affecting protein ubiquitination and degradation. In addition, inhibition of ER stress by 4-PBA could attenuate the degree of ER stress and podocyte dysfunction. Our study indicates that UCHL1 is a potential target for preventing podocytes injury in some non-immune complex-mediated glomerulopathy.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-023-04747-2.
Keywords: Ubiquitin carboxyl-terminal hydrolase L1 (UCHL1), Endoplasmic reticulum stress, Podocyte apoptosis, Ubiquitin–proteasome system (UPS), Podocytopathy
Introduction
Podocyte is the terminally differentiated epithelial cell within the glomerulus, with an important function in maintaining a filtration barrier, which is composed together of podocytes, endothelial cells and the basement membrane. However, podocyte is hard to repair when it is injured, which may cause glomerulopathy with heavy proteinuria as the main manifestation [1, 2].
Ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) is a very specific regulator with multi-function in the ubiquitin–proteasome system, involving cell proliferation, differentiation and apoptosis [3, 4]. Although it has been originally characterized as a deubiquitinating enzyme which shows a preference for specific targets [5, 6], recent studies indicate that it also exhibits dimerization-dependent ubiquitin ligase activity and a mono-ubiquitin stabilizer [7, 8]. Compared to the weak hydrolase activity of UCHL1, the ubiquitin ligase activity of UCHL1 is relatively strong, linking the poly-ubiquitin chain at the site of K63 but not K46, thus affecting the signaling transduction but not the proteasome-mediated protein degradation [6, 9, 10]. Of note, UCHL1 has a very strong ability in stabilizing the pool of mono-ubiquitin through its combination with ubiquitin [7]. In the gad mouse, a significantly reduced level of mono-ubiquitin in neurons was caused by loss of UCHL1 function, which led to the consequent inadequate ubiquitylation of proteins and accumulation of amyloid β protein and synuclein in the inclusions of AD brains [11, 12].
Our previous studies have shown increased expression of UCHL1 in the immune complex-mediated glomerulonephritis and very low expression in non-immune complex-mediated glomerulonephritis, such as minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS), with podocyte foot effacement and proteinuria [13]. However, the detailed role of UCHL1 in podocytopathy remains unclear. Radon et al. reported that UCHL1-deficient C57BL/6 mice exhibited no significant morphologic changes in the glomerular filtration barrier and slightly increased proteinuria with no significance [14]. Of interest, Jamin et al. demonstrated that the plasma level of anti-UCHL1 IgG was significantly increased in relapsing idiopathic steroid-sensitive nephrotic syndrome patients, and injection of purified patient anti-UCHL1 antibodies into BALB/c mice could induce proteinuria and podocyte foot effacement which similar to human MCD [15]. The differences in morphology and biofunction between the two mice model in the above studies suggested the sensitivity to injury stimuli in different species (C57BL/6 mice vs. BALB/c mice) for the study of UCHL1 in podocytes.
Therefore, in this study, we generated podocyte-specific UCHL1 gene knockout rats (the species susceptible to stimuli causing glomerulopathy) to explore the role of podocyte UCHL1 in renal morphology, proteinuria and renal function. Mechanistic studies were performed by both in vivo and in vitro studies to investigate the expression of differential proteins from the glomerulus and examined the change of endoplasmic reticulum stress indicators and ubiquitin–proteasome pathway, thus uncovering the effect of UCHL1 deficiency on podocyte function and its detailed mechanism.
Materials and methods
Antibodies
Goat anti-rabbit secondary antibody (111-035-003, WB at 1:5000) and anti-mouse secondary antibody (115-035-003, WB at 1:5000) were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). Antibodies against BiP (3177, WB at 1:1000, IHC/IF at 1:100), PERK (5683, WB at 1:1000), Phospho-PERK (Thr980) (3179, WB at 1:1000), IRE1 (3294, WB at 1:1000), ATF6 (65800, WB at 1:1000), TRAF2 (4724, WB at 1:1000), SAPK/JNK (9252, WB at 1:1000), Phospho-SAPK/JNK (Thr183/Tyr185) (4671, WB at 1:1000), cleaved caspase 3(9664, WB at 1:1000), integrin β3 (13166, WB at 1:1000), ubiquitin (3936, WB at 1:1000) and normal rabbit IgG (2729, WB at 1:1000) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against β-actin (66009-1-Ig, WB at 1:5000), ATF4 (10835-1-AP, WB at 1:1000), podocin (20384-1-AP, WB at 1:1000, IF at 1:100) and synaptopodin (21064–1-AP, WB at 1:1000, IF at 1:100) were purchased from Proteintech (Rosemont, IL, USA). Antibodies against fibronectin (ab45688, WB at 1:1000), laminin (ab11575, WB at 1:1000), WT1 (ab89901, WB at 1:1000, IHC/IF at 1:100), CHOP (ab11419, WB at 1:1000), IRE1 (phospho S724) (ab124945, WB at 1:1000), Alexa Fluor 594 anti-rabbit (150080, IF at 1:100) and anti-goat IgG (ab150132, IF at 1:100) were purchased from Abcam (Cambridge, MA, USA). UCHL1 (AB1761-I, WB at 1:1000, IF at 1:100) antibody was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Generation of podocyte-specific UCHL1 gene-knockout rats
The rats carrying the loxP UCHL1 construct (UCHL1flox/wt) and podocyte-specific Cre recombinase (Podcre+/−) were purchased from Biocytogen Company, Beijing, China. A line of floxed rats in UCHL1 exons 4 is flanked by loxP sites. Transgenic rats expressing Cre recombinase under the control of the rat podocin promoter were generated. Rats that were homozygous for the UCHL1 floxed allele were generated by mating heterozygous UCHL1 floxed rats. When bred to podocin-Cre transgenic rats, the podocin-Cre gene in one allele rats was generated. And then these rats were crossbred to knock out both UCHL1 alleles by Cre-mediated excision in both alleles, thereby creating conditional knockout rats in which the UCHL1 gene was specifically knockout in podocytes. UCHL1flox/flox littermates lacking the Podocin-Cre transgene (UCHL1flox/flox; Podcre−/−) were used as control (UCHL1Ctrl) rats. Rats were housed at the Animal Care Facility, Shanghai Medical School, Fudan University.
PCR primers and genotyping
Genomic DNA isolated from rat ear or tail was subjected to PCR genotyping using DNA polymerase (Takara, Japan). PCR conditions were 94 °C for 3 min followed by a round of 35 cycles: 94 °C for 30 s, 62 °C for 30 s, 72 °C for 60 s, with an additional 10 min extension at 72 °C. Two pairs of primers at the 5’end-floxed and 3′end-floxed alleles were used to detect the UCHL1flox/wt and UCHL1flox/flox. PCR primers used for 5’end-floxed alleles were 5′-GAATAGCGCAACAAACACCAGCTG-3′ (forward) and 5′-GGTTCTAGAGGCTGTTGCATGC-3′ (reverse). While 5′-GCCTGTGGTCTCAAAGAGTTCG-3′ (forward) and 5′-ATCCGGGTTAAGTGGCAGATGTG-3′ (reverse) were used to detect 3′end-floxed allele. For podocyte-specific Cre recombinase, 5′-GCACAAGGAAGCATCAACTACCCAA-3′ (forward), 5′-CTCATGCGTGTGTCCATCTTGTGA-3′ (wildtype reverse), and 5′-CGCGAACATCTTCAGGTTCTGC-3′ (mutant reverse)were used to detect wildtype, Podcre+/− and Podcre+/+, which revealed Cre recombinase gene was wildtype, heterozygous, or homozygous.
Quantitative real-time PCR
Total RNA was isolated with TRIzol reagent and reverse transcribed into cDNA with reverse transcriptase (Invitrogen). The cDNA products were subjected to quantitative real-time PCR analysis using a universal PCR master mix (Invitrogen). The PCR primers were used for amplification of rat genes UCHL1. The RT-PCR program consisted of 1 cycle at 95 °C for 5 min followed by 40 cycles at 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 30 s. The expression level of the gene was normalized to the level of GAPDH. The primer sequences were GAPDH, 5′-GGTGGACCTCATGGCCTACA-3′ and 5′-CTCTCTTGCTCTCAGTATCCTTGCT-3′; UCHL1, 5′-GCTTGTTCTTCCCTGTGC-3′ and 5′-GGCTAACTTCTTGTCCCTTC-3′.
Cell culture
Rat glomerular podocytes were kindly provided by Dr Guangping Chen (Department of Molecular Pathology, Tokyo Metropolitan Institute of Gerontology)[16]. Podocytes were cultured in RPMI-1640 containing 10% fetal bovine serum (Thermo Scientific, NY, USA), 100 U/ml penicillin, 100 μg/ml streptomycin (Sigma-Aldrich) at 37 °C in 95% air and 5% CO2.
Transfection with UCHL1 siRNA or UCHL1 plasmid and treatment with 4-PBA
UCHL1 siRNA (Genomeditech, Shanghai, China) or UCHL1 plasmid was incubated in transfection buffer with GenMute transfection reagent (SignaGen, MD, USA) for 15 min at room temperature to let transfection complex form, according to the manufacturer’s instructions. Add the transfection mix to the podocytes dropwise followed by incubation for 60 h at which time podocytes were lysed. For 4-PBA, podocytes were transfected with UCHL1 siRNA for 48 h at which time administered by 2.5 mM, 4-PBA(Sigma-Aldrich), then cultured for an additional 12 h.
UCHL1 forward primer: 5′-GGAUGGAUCCGUCCUGAAA-3′ and reverse primer: 5′-UUUCAGGACGGAUCCAUCC-3′.
Histologic examination, immunohistochemistry and immunofluorescence staining
For light microscopic analysis, rat kidneys were fixed with a 10% formalin solution and then processed for paraffin embedded and sectioned at 5 μm thickness, followed by HE staining.
For immunohistochemistry, kidney sections were deparaffinized and rehydrated. After heating sections in citrate buffer for antigen retrieval, applying 3% H2O2 for 30 min at room temperature, and blocking with 5% goat serum for 1 h at 37 °C, kidney sections were incubated with primary antibody diluted in blocking buffer overnight at 4 °C. The sections were subsequently incubated with a secondary antibody for 1 h at room temperature. DAB (DAKO, Glostrup, Denmark) was added to each kidney section. The sections were counterstained with hematoxylin and images were captured by a light microscope (Nikon, Tokyo, Japan).
For immunofluorescence, after heat-induced antigen retrieval, blocking with 5% goat serum for 1 h at 37 °C and incubating with the primary antibodies, the sections were incubated with Alexa Fluor® 594 secondary antibodies for 1 h at room temperature. The images were captured by a fluorescence microscope (Zeiss, Germany) with the ZEN software.
Western blot analysis and immunoprecipitation (IP)
Tissue or cells were homogenized in ice-cold RIPA lysis buffer (Bimake, Shanghai, China). After high-speed centrifugation, supernatants were collected and 30 μg of total protein was resolved by 8–10% SDS-PAGE and transferred onto a PVDF membrane (Millipore, MA, USA). Membranes were blocked with 5% nonfat milk and probed with appropriate primary antibody at 4 °C overnight, followed by incubation with secondary antibody conjugated with HRP for 1 h at room temperature. The blots were visualized by enhanced chemiluminescence (Bio-Rad, CA, USA). The intensity of the bands was quantitated by Image J software.
For immunoprecipitation, cells were lysed in NP-40 lysis buffer and the protein samples were collected after high-speed centrifugation. The magnetic beads (Thermo Scientific) were mixed with antibody or IgG and incubated with rotation for 10 min at room temperature. Beads were washed twice with lysis/wash buffer and once with purified water. The antigen/antibody complex was eluted. Then, samples were subjected to SDS-PAGE for separation and detection.
TUNEL stain
Rat kidneys were sectioned at 5 μm thickness and stained according to the manufacturer's instructions of In Situ Cell Death Detection Kit, TMR red (Roche, Basel, Switzerland). Staining was visualized using fluorescence microscopy (Zeiss, Germany) with the ZEN software.
Isolation of glomeruli
The rats were anesthetized with 3% pentobarbital sodium and perfused with ice-cold phosphate-buffered saline (PBS). The rats were euthanized. The removed kidneys were chopped up, collected and digested in 1.5 mg/ml collagenase A (Roche) for 30 min at 37 °C. The renal tissue was passed through a 150 μm cell strainer (Corning, NY, USA) with gentle pressure and washed the cell strainer with phosphate-buffered saline. All the cell suspension was collected followed by passing through a 100 μm cell strainer. The collected cell suspension was passed through a 40 μm cell strainer. The glomeruli were retained above the strainer followed by being washed off from the strainer with PBS and centrifuged.
Mass spectrometry analysis
The isolated glomeruli were ground by liquid nitrogen and then lysed in four volumes of lysis buffer (8 M urea, 1% Protease Inhibitor Cocktail), followed by sonication on ice using an ultrasonic processor (Scientz, China). The debris was removed by centrifugation. The supernatant was collected and the protein concentration was determined by a BCA kit (Thermo Scientific). Then, the protein samples were sent to PTM Biolabs (Hangzhou, China) and analyzed by mass spectrometry.
Measurement of kidney function
Blood samples were centrifuged at 2500 rpm for 10 min and the supernatant was plasma. Serum creatinine and blood urea nitrogen (BUN) were analyzed by commercially available rat assay kit (Abcam). For urinary albumin excretion, urine samples were measured by a commercially available ELISA kit (Abcam).
Quantification of podocyte loss
Section Method for Estimation of Podocyte Number. Briefly, the rat renal block was cut into two thickness slices (3 μm and 6 μm), which were then stained for the podocyte marker WT1. The double-blind method was used to count WT1-positive cells. Ten rat kidney sections were selected from each group, and all the positive podocytes which the brownish-yellow granules appeared in the nuclear were counted on each section under HPF (× 400). The number of podocytes (P) is WT1-positive cells, and the glomerular tuft area (GA) is measured according to the circle formula. P/GA means the mean podocyte number per glomerular tuft area. The P/GA difference in thick and thin sections could be obtained (P/GA△ = P/GAthick − P/GAthin) by dividing the known difference in section thickness by the P/GA△to get GV/P. According to the formula, the mean glomerular tuft volume (GV) was calculated based on the maximum sphere diameter. Finally, the podocyte number per glomerular tuft was calculated [17].
Flow cytometry analysis of podocyte apoptosis
The Annexin V-PE and 7-AAD double staining kit (BD Biosciences, CA, USA) was used to detect the apoptosis rate of podocytes, according to the manufacturer's instructions. Briefly, podocytes were washed twice with cold PBS and resuspended in 1 × binding buffer at a concentration of 1 × 106 cells/ml. After transferring 100 μl (5 × 105 cells) of the solution to a new tube, the Annexin V-PE and 7-AAD were added and incubated for 15 min at room temperature in the dark. Then, 400 μl 1 × binding buffer was added to each group. Finally, flow cytometry analysis was performed with a FACScan flow cytometer using the CellQuest software (BD Biosciences) within 1 h. Early apoptotic cells (Annexin V+/7-AAD−) and late apoptotic/necrotic cells (Annexin V+/7-AAD+) were considered to evaluate the apoptosis of podocytes.
Statistics analysis
Statistics analyses were performed using GraphPad Prism 8.0. Statistical comparisons between groups were carried out by Student’s t test and ANOVA. Statistical difference was defined as *P < 0.05.
Results
UCHL1 deletion in podocytes caused segmental/global glomerulosclerosis and proteinuria
By crossing UCHL1-floxed mice (UCHL1flox/flox) with transgenic mice expressing Cre recombinase under the control of the podocin (NPHS2) promoter, we generated lines of podocyte-specific UCHL1 gene knockout in one allele (UCHL1cre/w) or in both alleles (UCHL1cre/cre), as illustrated in Supplementary Fig. 1a. Gender-matched UCHL1flox/flox; PodCre−/− (UCHL1ctrl in short hereafter) littermates were used as control rats. PCR genotyping identified UCHL1cre/cre, UCHL1cre/w and UCHL1ctrl carrying only 5′end-floxed and 3′end-floxed UCHL1 allele, while the heterozygous floxed rat (UCHL1flox/w; PodCre+/−) carrying the 5′end-floxed, 3′end-floxed UCHL1 allele, and the WT allele as well; and the podocin-Cre gene was detected in UCHL1cre/cre, UCHL1cre/w or UCHL1flox/w; PodCre+/− (Supplementary Fig. 1b). Immunoblotting of glomerulus homogenates confirmed effective deletion of UCHL1 protein (Supplementary Fig. 1c). Double immunofluorescence staining of UCHL1 and WT1, a nucleus marker expressed both in PECs and podocytes, showed successful partial deletion of UCHL1 in podocytes from UCHL1cre/w rats and almost complete deletion of UCHL1 in podocytes from UCHL1cre/cre rats with an unchanged UCHL1 expression in PECs in both rats (Supplementary Fig. 2a). UCHL1cre/cre rats showed a remarkable decrease in survival rate, volume and body weight (Fig. 1a–b, d), compared to UCHL1cre/w and UCHL1ctrl rats. The kidneys from UCHL1cre/cre rats showed pale color with red miliary dots on the surface (Fig. 1c). H&E staining showed glomeruli with mesangial proliferation (mesangial cell proliferation and mesangial matrix accumulation) and occlusion of some capillary lumen in UCHL1cre/cre rats by 1 week of age. By 3 weeks of age, UCHL1cre/cre rats showed segmental or global glomerulosclerosis, with more tubular atrophy or dilation, and formation of proteinaceous casts. Increased interstitial fibrosis was confirmed by Masson’s trichrome staining in UCHL1cre/cre rats. However, no significant differences by 3 weeks of age between UCHL1cre/w and UCHL1ctrl rats were observed (Fig. 1e). Electron microscopy showed extensive foot process effacement (arrows) and shrink of some glomerular basement membrane (GBM) in UCHL1cre/cre rats, whereas the foot process was well remained in UCHL1cre/w and UCHL1ctrl rats (Fig. 1f). As shown in Fig. 1g–j, serum creatinine, BUN, urine albumin levels and urine albumin/creatinine ratios (ACR) were significantly elevated in UCHL1cre/cre rats. Coomassie-Brilliant Blue staining of urine samples processed by SDS-PAGE confirmed that the urine albumin levels were elevated in UCHL1cre/cre rats by 1 week of age, which were increased obviously by 3 weeks of age, while no significant changes were observed between UCHL1cre/w and UCHL1ctrl littermates by 1st and 3rd week of age. (Fig. 1k).
Fig. 1.

Characterization of UCHL1cre/cre and UCHL1cre/w rats. a Graph depicting the survival rate of UCHL1cre/cre rats, UCHL1cre/w rats and UCHL1w/w rats. n = 10–12 rats per group. Representative rats (b) and kidney (c) images at 3 weeks of age. (c) Pale kidneys and red miliary dots (arrows) in kidneys were observed in UCHL1cre/cre rats. d Body weight of all three genotypes of rats at the indicated age. Values were expressed as means ± SEM, n = 8–10 rats per group, **P < 0.01, ***P < 0.001, ****P < 0.0001. e H&E staining and Masson trichome staining showed mesangial proliferation by 1 week of age, and segmental or global glomerulosclerosis with tubular atrophy and interstitial fibrosis by 3 weeks of age in UCHL1cre/cre rats, compared with UCHL1cre/w and UCHL1ctrl rats. Scale bar: 50 μm. f Electron microscopy showed extensive foot process effacement (arrows) in UCHL1cre/cre rats at 1 and 3 weeks of age, compared with UCHL1cre/w and UCHL1ctrl littermates. Scale bar:1 μm. Serum creatinine (g), BUN (h), urine albumin levels (i) and urine albumin/creatinine ratios (ACR) (j) were significantly elevated in UCHL1cre/cre rats by 1 and 3 weeks of age. Values were expressed as means ± SEM, n = 8–10 rats per group, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (k) Coomassie-Brilliant Blue staining of urine samples processed by SDS-PAGE indicated that the urine albumin levels (~ 67 kDa bands) were elevated in UCHL1cre/cre rats at 1 week, which were increased obviously by 3 weeks. Standard BSA concentrations are shown in lanes 2 and 3. n = 6 rats per group. Shown are representative immunoblots from at least three independent experiments with similar results
UCHL1 deletion in podocytes caused abnormal accumulation of protein acting in ECM-receptor Interaction
To investigate the protein changes induced by UCHL1 deletion, glomeruli protein was isolated from UCHL1cre/cre, UCHL1cre/w and UCHL1ctrl rats by 3 weeks of age, and analyzed by mass spectrometry analysis and functional enrichment (fold change > 2). Subcellular location identified the significantly changed proteins in isolated glomeruli of UCHL1cre/cre rats mostly distributed in the extracellular region (Supplementary Fig. 3a). GO analysis found significantly up-regulated proteins for cellular component, biological process and molecular function, and KEGG pathway analysis found complement and coagulation cascades, focal adhesion and ECM-receptor interaction pathway in UCHL1cre/cre rats, compared with UCHL1ctrl rats (data not shown). Most ECM-receptor interaction proteins were significantly up-regulated in UCHL1cre/cre rats by KEGG pathway schematic (KEGG pathway map04512) and heat map, compared with UCHL1ctrl rats. There were no significant protein changes in UCHL1cre/w rats, compared with UCHL1ctrl rats (Supplementary Fig. 3b, c).
We examined three selected ECM-receptor interaction proteins including fibronectin, laminin and integrin β3 by western blot, and found the protein expression of all three proteins increased mildly in UCHL1cre/w rats but significantly in UCHL1cre/cre rats (Fig. 2a). Podocytes transfected with UCHL1 siRNA also showed a significantly increased protein level of fibronectin, laminin and integrin β3, whereas transfected with UCHL1 plasmids showed a decrease in these three proteins (Fig. 2b, c).
Fig. 2.
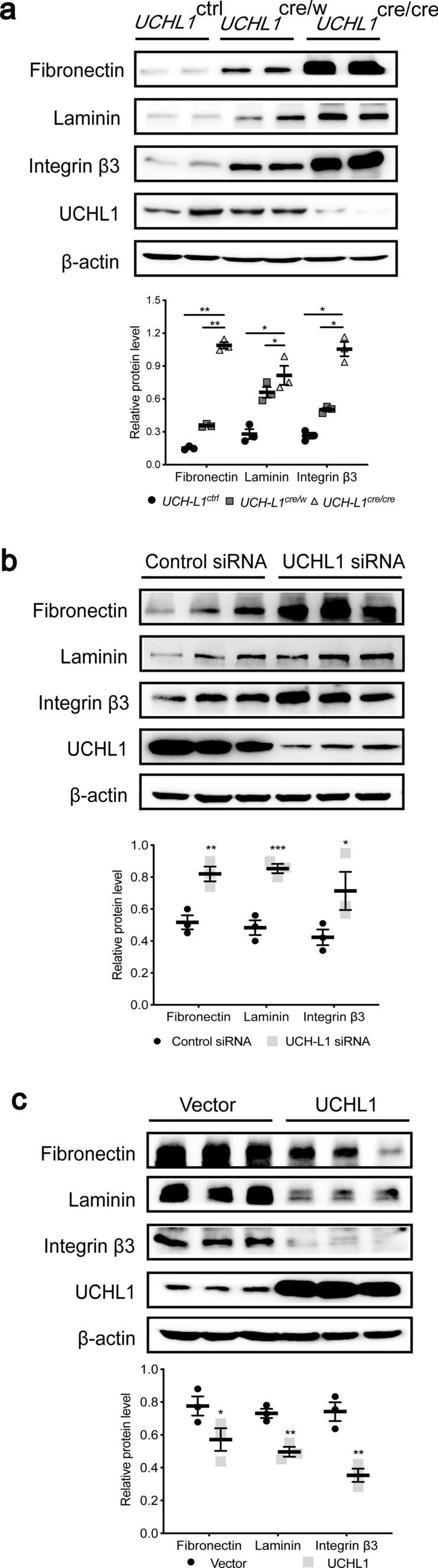
UCHL1 deletion induced abnormal protein accumulation including fibronectin, laminin and integrin β3 in podocytes. a Western blot examined the protein expression of fibronectin, laminin and integrin β3 in isolated glomeruli of UCHL1cre/cre, UCHL1cre/w and UCHL1ctrl rats. n = 6 rats per group. b, c Western blot examined the protein level of fibronectin, laminin and integrin β3 in podocytes transfected with control siRNA or UCHL1 siRNA (b), or in podocytes expressing vector control (Vector) or UCHL1 plasmids (c) in vitro. Values were expressed as means ± SEM, *P < 0.05, **P < 0.01, ***P < 0.001. β-Actin was as a loading control for all western blots. Shown are representative immunoblots from at least three independent experiments with similar results
UCHL1 deletion in podocytes induced endoplasmic reticulum (ER) stress
Because those ECM-receptor interaction proteins are folded and packaged in the endoplasmic reticulum before they are secreted to the extracellular area, so we wonder whether an accumulation of ECM-receptor interaction proteins causes endoplasmic reticulum stress. Double immunofluorescence staining showed obviously an increased expression of BiP (the marker of ER stress) in podocytes of UCHL1cre/cre rats. (Fig. 3a and Supplementary Fig. 2b). Immunoblotting of glomerulus proteins showed a high level of BiP and ATF4, two ER stress-related proteins (Fig. 3b). Compared to control siRNA, transfection of UCHL1 siRNA into podocytes in vitro also induced increased expression of BiP and ATF4 (Fig. 3c), whereas control siRNA had no effect on those proteins compared to blank control (Supplementary Fig. 4).
Fig. 3.

Endoplasmic reticulum (ER) stress of podocytes was activated by UCHL1 deletion. a Double immunofluorescence staining of BiP, an ER stress marker, and nephrin in podocytes of UCHL1cre/cre, UCHL1cre/w and UCHL1ctrl rats. Scale bar: 50 μm. n = 3 rats per group. b, c Western blot examined the protein level of BiP and ATF4 in isolated glomeruli of UCHL1cre/cre, UCHL1cre/w and UCHL1ctrl rats (b), or podocytes transfected with control siRNA or UCHL1 siRNA (c) in vitro. n = 6 rats per group. d Western blot examined the protein level of UPR containing PERK, IRE1 and ATF6 in isolated glomeruli of UCHL1cre/cre, UCHL1cre/w and UCHL1ctrl rats. n = 6 rats per group. e, f Western blot examined the three downstream of UPR (PERK, IRE1 and ATF6) in podocytes transfected with control siRNA or UCHL1 siRNA (e), or in podocytes expressing vector control (Vector) or UCHL1 plasmids (f) in vitro. Values were expressed as means ± SEM, *P < 0.05, **P < 0.01, n.s. (no significance). β-Actin was a loading control for all western blots. Shown are representative immunoblots from at least three independent experiments with similar results
Since ER stress may induce unfolded protein response (UPR) including a decrease of protein synthesis and acceleration of protein folding to alleviate the degree of stress [18], we examined the downstream of ER stress, and found, compared to the UCHL1ctrl rats, that the protein levels of p-IRE1, t-IRE1, p-PERK and t-PERK had no significant change in UCHL1cre/w rats, but remarkably increased in UCHL1cre/cre rats (Fig. 3d). We then knocked down UCHL1 in podocytes in vitro and found that the protein levels of p-IRE1, p-PERK and TRAF2 were increased with no change of t-IRE1, t-PERK and ATF6 (Fig. 3e). Additionally, BiP, p-PERK and p-IRE1 were decreased by overexpression of UCHL1 (Fig. 3f). The results above indicated that protein accumulation caused by deficiency of UCHL1 induced ER stress and stimulated its downstream IRE1 and PERK rather than ATF6 which related to UPR.
4-PBA inhibited abnormal protein accumulation and ER Stress induced by UCHL1 deficiency in podocytes
When the podocyte with knocking down of UCHL1 was treated with 4-PBA, an inhibitor of ER stress, the protein level of fibronectin and laminin was decreased, and surprisingly UCHL1 was increased (Fig. 4a). Furthermore, we found that ER stress markers, BiP and ATF4 were decreased (Fig. 4b), and UPR-related proteins including p-IRE1, p-PERK and TRAF2 were decreased as well (Fig. 4c). Additionally, IRE1 phosphorylation and BIP level, which were upregulated in podocyte with UCHL1 knockdown, decreased after TUDCA, another well-established inhibitor of ER stress, treatment (Supplementary Fig. 5d). The results demonstrated that inhibition of ER stress could attenuate the degree of stress as well as the accumulation of ECM-receptor interaction proteins. On the contrary, tunicamycin, an activator of ER stress, not only stimulated the protein expression of p-IRE1, t-IRE1, p-PERK, t-PERK, ATF6, TRAF2, BiP and ATF4 in podocytes, but also reverse the inhibition effect on ER stress and UPR caused by UCHL1 overexpression in podocyte showing markedly increased protein expression of BiP, p-IRE1 and p-PERK (Supplementary Fig. 5).
Fig. 4.

4-PBA inhibited abnormal protein accumulation and ER stress induced by UCHL1 deficiency in podocytes. a Western blot showed that the increase of fibronectin and laminin after UCHL1 siRNA transfection in podocytes was significantly reduced with treatment of 2.5 mM 4-PBA for 12 h. b, c Western blot examined the protein level of BiP, ATF4 and UPR in podocytes transfected with UCHL1 siRNA and treated with or without 2.5 mM 4-PBA for 12 h. Values were expressed as means ± SEM, *P < 0.05, **P < 0.01, n.s. (no significance). β-Actin was a loading control for all western blots. Shown are representative immunoblots from at least three independent experiments with similar results
UCHL1 deficiency in podocytes reduced the expression of podocyte-specific marker and induced apoptosis
UPR could induce a decrease in protein synthesis and persistent ER stress could cause cellular dysfunction and apoptosis; we further investigated whether the UCHL1 deficiency in podocytes affects protein expression and arrangement of podocyte skeleton proteins and apoptosis in rats. Double Immunofluorescence staining of the podocyte skeleton proteins (synaptopodin, podocin and nephrin) with UCHL1 showed that podocyte-specific markers distributed evenly in podocytes of UCHL1ctrl rats and UCHL1cre/w rats, but decreased and discontinuously distributed with fragmented pattern in podocytes of UCHL1cre/cre rats (Fig. 5a). Transfection with UCHL1 siRNA in podocytes in vitro also reduced the expression of podocin and nephrin (Fig. 5b). To confirm the changes of podocyte skeleton proteins, phalloidin and α-actinin4 in podocytes were stained. Phalloidin staining showed disappear of long filaments and decrease in the number of actin filaments after UCHL1 siRNA transfection, suggesting F-actin arrangement was disrupted. What is more, podocytes with UCHL1 knockdown lose the fibrous α-Actinin 4 structures which were prominent and ordered in control cells, indicating that the downregulation of UCHL1 induced an intense disruption of the cytoskeleton in podocyte (Fig. 5c).
Fig. 5.

UCHL1 deficiency reduced the expression of podocyte-specific marker and induced apoptosis in podocytes. a Representative double immunofluorescence images showed the expression and distribution of synaptopodin, podocin and nephrin in glomeruli of UCHL1cre/cre, UCHL1cre/w or UCHL1ctrl rats. Scale bar: 50 μm. n = 3 rats per group. b Western blot examined the protein level of nephrin and podocin in podocytes transfected with control siRNA or UCHL1 siRNA in vitro. c Phalloidin and α-actinin 4 staining of podocyte after UCHL1 siRNA or control siRNA transfection. d Quantification of podocytes per glomerular. Double immunofluorescence staining with WT1 to label podocyte nuclei and nephrin to label podocyte cytoplasm, and Immunohistochemistry staining of WT1 in a thick and thin section method were used to count the podocytes per glomerular. The mean number of WT1/nephrin co-positive cells per glomerulus of each rat was calculated by counting 20 randomly selected glomeruli of each rat (n = 6 rats per group) and is shown as the upper right graphic. The mean number of WT1 positive cells per glomerulus was calculated from 20 randomly selected glomeruli of each rat (n = 10 rats per group) in two thickness slices and is shown as the lower right graphic. Number of podocytes per glomerular tuft was obviously decreased in UCHL1cre/cre rats, compared with UCHL1cre/w rats and UCHL1ctrl rats. Scale bar: 50 μm. e Representative double immunofluorescence images of WT1 and TUNEL of all three genotypes of rats showing the TUNEL-positive nuclei in podocytes (yellow arrows are podocytes). Scale bar: 50 μm. n = 3 rats per group. f Flow cytometry analysis showing the apoptosis rate of podocytes with control siRNA or UCHL1 siRNA transfection. The cells in the upper right and lower right were considered as apoptotic cells. The level of apoptosis rate was higher in podocytes with UCHL1 siRNA transfection. g, h Western blot examined the protein level of cleaved caspase3 in podocytes transfected with control siRNA or UCHL1 siRNA (g), or in podocytes expressing vector control (Vector) or UCHL1 plasmids (h) in vitro. Values were expressed as means ± SEM, *P < 0.05, ***P < 0.001, ***P < 0.0001. β-Actin was a loading control for all western blots. Shown are representative immunoblots from at least three independent experiments with similar results
Counting the podocyte number by double immunofluorescence and cutting different thickness of sections that staining with WT1 as described in the Methods, we found that compared to UCHL1ctrl rats, the mean podocytes number per glomerulus was significantly decreased in UCHL1cre/cre rats, but without change in UCHL1cre/w rats (Fig. 5d). Double Immunofluorescence staining of TUNEL and WT1 showed that there were many apoptotic podocytes in a peripheral area of the glomerulus in UCHL1cre/cre rats (Yellow arrow in Fig. 5e).
In vitro study showed that knocking down of UCHL1 induced an increase of apoptotic cells and elevated expression of cleaved caspase3 (Fig. 5f, g), whereas overexpressing of UCHL1 decreased the protein level of cleaved caspase3 (Fig. 5h). The above data showed an important role of UCHL1 in maintaining the skeleton proteins and preventing podocyte from apoptosis.
UCHL1 deficiency induced ER stress-associated apoptosis with CHOP activation
Once the UPR could not compensate for the continuous ER stress, the apoptotic pathway related to ER stress will be activated[19]. To investigate whether the podocyte apoptosis caused by UCHL1 deficiency (as shown above) was mediated by ER stress-associated apoptotic pathway, CHOP and JNK were examined. In vitro knocking down of UCHL1 induced activation of CHOP rather than JNK (Fig. 6a). Increased CHOP accompanied by increased cleaved caspase3 was also observed in UCHL1cre/cre rats (Fig. 6b). Treating podocytes with 4-PBA not only markedly reversed the decrease of nephrin and podocin caused by UCHL1 knocking down (Fig. 6c), but also reduced CHOP and cleaved caspase3 (Fig. 6d), suggesting the podocyte injury induced by UCHL1 deficiency largely mediated via ER stress.
Fig. 6.

UCHL1 deficiency induced ER stress-associated apoptosis and injury of podocytes, which was ameliorated by 4-PBA. a Western blot examined the protein level of CHOP and JNK signaling in podocytes with control siRNA or UCHL1 siRNA transfection, showing UCHL1 siRNA transfection upregulated CHOP rather than p-JNK and t-JNK. b Western blot examined the protein level of CHOP and cleaved caspase3 in isolated glomeruli of all three genotypes of the rat. n = 6 rats per group. c Western blot showed that the decreased protein level of nephrin and podocin after UCHL1 siRNA transfection in podocytes was increased with treatment of 2.5 mM 4-PBA for 12 h. d Western blot showed the increased protein level of CHOP and cleaved caspase 3 after UCHL1 siRNA transfection in podocytes were significantly reduced with treatment of 2.5 mM 4-PBA for 12 h. Values were expressed as means ± SEM, *P < 0.05, **P < 0.01, ****P < 0.0001. β-Actin was a loading control for all western blots. Shown are representative immunoblots from at least three independent experiments with similar results
UCHL1 deficiency reduced the ubiquitinated fibronectin, laminin or integrin β3 and the free monomeric ubiquitin level
The unfolded or misfolded proteins in ER are degraded through the ubiquitin–proteasome pathway, in which UCHL1 is of importance, so we investigated the influence of UCHL1 deficiency on it. Firstly, we confirmed fibronectin, laminin and integrin β3 were ubiquitinated and degraded by the proteasome (Fig. 7a–c). However, they were not the specific targets of UCHL1 as the deubiquitinase because they could not combine with UCHL1 directly (Supplementary Fig. 6a–c). Then, we transfected podocytes with UCHL1 siRNA together with MG132, a proteasome inhibitor, and observed a significantly decreased ubiquitination level of these three proteins (Fig. 7d–f). Because UCHL1 has a strong ability to stabilize the ubiquitin pool, we further examined the level of ubiquitin and found that the free monomeric ubiquitin was markedly decreased and the polyubiquitin at site of around 20 kDa as well by interference of UCHL1 (Fig. 7g), but increased by overexpression of UCHL1 (Fig. 7h), which consistent with the change in ubiquitination level of fibronectin, laminin and integrin β3, indicating that UCHL1 influences the ubiquitination of such ECM-receptor interaction proteins by regulating the homeostasis of ubiquitin pool rather than its deubiquitinase activity, thus reducing their degradation and causing ER stress.
Fig. 7.

UCHL1 deficiency reduced the ubiquitinated fibronectin, laminin or integrin β3 and the free monomeric ubiquitin level. a–c Podocytes were cultured with MG132 for 12 h before being collected. Immunoprecipitation and western blot analysis showed the ubiquitination level of fibronectin, laminin and integrin β3 were significantly elevated, which indicated all of them were degraded by the ubiquitin–proteasome pathway in podocytes. d–f Immunoprecipitation and western blot analysis demonstrated that compared to the control group, the ubiquitination level of fibronectin, laminin and integrin β3 were significantly decreased in podocytes transfected with UCHL1 siRNA. g, h Immunoprecipitation and western blot analysis examined the levels of poly-ubiquitin and free monomeric ubiquitin in podocytes transfected with control siRNA or UCHL1 siRNA (g), or podocytes expressing vector or exogenous UCHL1 (h) in vitro. β-Actin was a loading control for all western blots. Shown are representative immunoblots from at least three independent experiments with similar results
Discussion
Literatures have shown the abnormality in the expression and function of UCHL1 plays important role in the development of neurodegenerative diseases, type 2 diabetes and several types of cancer [20–22]. Although our group has demonstrated for the first time that UCHL1 is expressed in the normal human podocyte and exhibited a very low level in podocytopathy such as MCD and FSGS [13], its detailed role in podocyte injury remains unclear. Radon et al. reported that UCHL1-deficient C57BL/6 mice resulted in proteinuria from 9 to 11 weeks of age and increased with age without significant morphologic changes of the glomerular filtration barrier. Furthermore, their study showed that podocyte-specific UCHL1-deficient mice developed very mild albuminuria with no significance [14], which indicates that the proteinuria in the UCHL1-deficient mice was most likely caused by the tubulointerstitial injury with UCHL1 knockout. However, Jamin et al. showed that injection of purified patient anti-UCHL1 antibodies into BALB/c mice induced proteinuria and extensive podocyte foot process effacement [15]. As we know, rat and BALB/c mouse are more susceptible to various stimuli causing podocyte injury than C57BL/6 mouse [23], especially a number of rat models exhibit kidney disease phenotype obviously [24], so we generated a podocyte-specific UCHL1 gene knockout rat model to investigate the phenotypes and mechanisms underlying podocyte injury due to UCHL1 deficiency, which was further confirmed by in vitro studies.
Our podocyte-specific UCHL1 gene knockout (UCHL1cre/cre) rats showed a notable phenotype of decreased renal function, obvious albuminuria, segmental or global glomerulosclerosis and extensive foot process effacement by biochemical and morphological analysis. Of note, UCHL1cre/cre rats showed a premature death within 3 weeks of age. Shirato et al. demonstrated that UCHL1 expressed with an abundant amount in the early stages of the nephron precursor, and continuously decreased thereafter with the differentiation of epithelial cells and growth of glomeruli [25], indicating its important functions for nephrogenesis, which could explain the premature death in our study.
In gracile axonal dystrophy (gad) mice with a null mutant of UCHL1, there was an abnormal accumulation of amyloid β protein [26], synuclein [11], ubiquitin-positive deposits [27] and GAPDH in nerves, causing dysfunction of the neurons and a wide range of neurodegenerative diseases characterized by the aberrant accumulation of intracellular and extracellular proteins [28]. Consistent with the previous observation in neurodegenerative diseases, we observed remarkably up-regulated ECM-receptor interaction proteins, including fibronectin, laminin and integrin β3, in the glomerulus of UCHL1cre/cre rats. However, all those three proteins did not bind to UCHL1 directly, which excluded the possibility of being the specific targets of UCHL1 as its deubiquitinated enzyme activity. Intriguingly, the free monomeric ubiquitin was significantly decreased after UCHL1 knocking down, while it increased after UCHL1 overexpression, suggesting a strong role of UCHL1 in maintaining available ubiquitin pool via its activity to bind to monomeric ubiquitin for inhibiting mono-ubiquitin degradation [7]. The ability was independent of its hydrolase or ligase activity, as it was found that the C90S mutant of UCHL1, which remained ubiquitin binding ability and lacked hydrolase activity, still retained the ability to bind to free monomeric ubiquitin and maintain ubiquitin stability. It suggested that the binding of UCHL1 and monomeric ubiquitin was a physical association rather than through the enzymatic activity of UCHL1 [7, 8]. Since the loss of mono-ubiquitin in UCHL1 deficiency will interfere with the UPS process of the cell, it reflected that the ECM-receptor interaction proteins accumulation after UCHL1 deficiency was caused by homeostasis dysregulation of the whole ubiquitin pool.
UPS is crucial for the proper operation of ER as it is involved in ER-associated protein degradation (ERAD) to degrade those accumulated unfolded or misfolded proteins. When ERAD is blocked partially or completely, abundant proteins accumulate in the ER, thus causing ER stress [29]. One compensatory way to ER stress is unfolded protein response (UPR) to reduce the protein synthesis by stimulating the signaling sensor PERK, IRE1 or ATF6 through a chaperone BiP [18, 30, 31]. We observed that UCHL1 deficiency not only enhanced the expression of BiP, but also activated two downstream of UPR, PERK and IRE1, and decreased the protein expression of nephrin and podocin as well, indicating protein synthesis was weakened by UPR via PERK and IRE1 rather than ATF6. The reason behind this selective activation in UPR to ER stress is not clear. Park et al. also observed activation of PERK and IRE1 pathways, but not ATF6 pathway was induced by ER stress in Lamb2−/− mice with C321R mutation, further causing extensive podocyte foot process effacement and heavy proteinuria [32, 33]. Furthermore, ER-associated apoptosis will be induced by long-term ER stress, and CHOP is a critical initiator [19]. Further study showed that CHOP was up-regulated by UCHL1 deficiency, for its transcription could be induced by all three downstream pathways of UPR[34–37], accompanied by an increase of podocyte apoptosis. Strikingly, we found that treatment of podocytes knocking down UCHL1 with the ER stress inhibitor 4-PBA could rescue the podocyte injury significantly. The results were consistent with the reported in the brain of diabetic rats, accompanied by declined UCHL1 expression, increased ubiquitinated proteins and apoptosis, which could be intervened by 4-PBA [38]. Together with our results, it suggested that targeting ER could antagonize the ERAD dysfunction caused by UCHL1 deficiency.
In conclusion, our study describes a previously unreported renal phenotype of UCHL1 deficiency specifically in podocytes. Mechanistically, we show that UCHL1 deficiency resulted in a decrease of free monomeric ubiquitin, leading to ECM-receptor interaction proteins accumulation, ER stress and UPR, and finally podocyte skeleton protein dysregulation and apoptosis. This study has important implications to explain the newly defined anti-UCHL1 antibody-related minimal change disease clinically. Our study reveals that inhibition of ER stress could be a potential way for preventing podocyte injury.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We would like to thank Dr. Zhihuang Zheng and Chuanlei Li for their help throughout the project.
Author contributions
HW, ZZ and YX designed the study. YH, CQ, JS and ZZ carried out experiments. WT and AA analyzed data. HW, YH and CQ drafted the paper. HW, YX and ZZ revised the paper. All the authors approved the submitted and published versions of the paper.
Funding
This work was supported by the National Natural Science Foundation of China, Grant/Award (no. 82170719).
Availability of data and material
The datasets generated during the current study are available from the corresponding author upon reasonable request.
Declarations
Conflict of interests
The authors have no relevant financial or non-financial interests to disclose.
Ethics approval
The ethics governing the use and conduct of experiments on animals were strictly observed. All animal experiments were approved by the Animal Research Ethics Committee of the School of Basic Medical Sciences of Fudan University (no. 20100223).
Consent for publication
Not applicable.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Yuan Hu and Chenyang Qi have contributed equally to this work and share the first authorship.
Contributor Information
Yanyong Xu, Email: yanyong_xu@fudan.edu.cn.
Huijuan Wu, Email: hjwu@shmu.edu.cn.
Zhigang Zhang, Email: zzg@shmu.edu.cn.
References
- 1.Shankland SJ. The podocyte’s response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69(12):2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- 2.Nagata M. Podocyte injury and its consequences. Kidney Int. 2016;89(6):1221–1230. doi: 10.1016/j.kint.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 3.Bishop P, Rocca D, Henley JM. Ubiquitin C-terminal hydrolase L1 (UCH-L1): structure, distribution and roles in brain function and dysfunction. Biochem J. 2016;473(16):2453–2462. doi: 10.1042/BCJ20160082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Day IN, Thompson RJ. UCHL1 (PGP 9.5): neuronal biomarker and ubiquitin system protein. Prog Neurobiol. 2010;90(3):327–362. doi: 10.1016/j.pneurobio.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 5.Bett JS, Ritorto MS, Ewan R, et al. Ubiquitin C-terminal hydrolases cleave isopeptide- and peptide-linked ubiquitin from structured proteins but do not edit ubiquitin homopolymers. Biochem J. 2015;466(3):489–498. doi: 10.1042/BJ20141349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Y, Fallon L, Lashuel HA, et al. The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson’s disease susceptibility. Cell. 2002;111(2):209–218. doi: 10.1016/s0092-8674(02)01012-7. [DOI] [PubMed] [Google Scholar]
- 7.Osaka H, Wang YL, Takada K, et al. Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum Mol Genet. 2003;12(16):1945–1958. doi: 10.1093/hmg/ddg211. [DOI] [PubMed] [Google Scholar]
- 8.Cartier AE, Djakovic SN, Salehi A, et al. Regulation of synaptic structure by ubiquitin C-terminal hydrolase L1. J Neurosci. 2009;29(24):7857–7868. doi: 10.1523/JNEUROSCI.1817-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erpapazoglou Z, Walker O, Haguenauer-Tsapis R. Versatile roles of k63-linked ubiquitin chains in trafficking. Cells. 2014;3(4):1027–1088. doi: 10.3390/cells3041027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Susor A, Liskova L, Toralova T, et al. Role of ubiquitin C-terminal hydrolase-L1 in antipolyspermy defense of mammalian oocytes. Biol Reprod. 2010;82(6):1151–1161. doi: 10.1095/biolreprod.109.081547. [DOI] [PubMed] [Google Scholar]
- 11.Wang YL, Takeda A, Osaka H, et al. Accumulation of beta- and gamma-synucleins in the ubiquitin carboxyl-terminal hydrolase L1-deficient gad mouse. Brain Res. 2004;1019(1–2):1–9. doi: 10.1016/j.brainres.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 12.Lohmann F, Sachs M, Meyer TN, et al. UCH-L1 induces podocyte hypertrophy in membranous nephropathy by protein accumulation. Biochim Biophys Acta. 1842;7:945–958. doi: 10.1016/j.bbadis.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y, Wu J, Wu H, et al. UCH-L1 expression of podocytes in diseased glomeruli and in vitro. J Pathol. 2009;217(5):642–653. doi: 10.1002/path.2511. [DOI] [PubMed] [Google Scholar]
- 14.Radon V, Czesla M, Reichelt J, et al. Ubiquitin C-Terminal Hydrolase L1 is required for regulated protein degradation through the ubiquitin proteasome system in kidney. Kidney Int. 2018;93(1):110–127. doi: 10.1016/j.kint.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 15.Jamin A, Berthelot L, Couderc A, et al. Autoantibodies against podocytic UCHL1 are associated with idiopathic nephrotic syndrome relapses and induce proteinuria in mice. J Autoimmun. 2018;89:149–161. doi: 10.1016/j.jaut.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 16.Chen G, Nagasawa R, Imasawa T, et al. Identification of soluble interleukin-4 receptor in rat glomerular epithelial cells(1) Biochim Biophys Acta. 1999;1452(1):79–88. doi: 10.1016/s0167-4889(99)00117-2. [DOI] [PubMed] [Google Scholar]
- 17.Sanden SK, Wiggins JE, Goyal M, et al. Evaluation of a thick and thin section method for estimation of podocyte number, glomerular volume, and glomerular volume per podocyte in rat kidney with Wilms’ tumor-1 protein used as a podocyte nuclear marker. J Am Soc Nephrol. 2003;14(10):2484–2493. doi: 10.1097/01.asn.0000089829.45296.7c. [DOI] [PubMed] [Google Scholar]
- 18.Frakes AE, Dillin A. The UPR(ER): sensor and coordinator of organismal homeostasis. Mol Cell. 2017;66(6):761–771. doi: 10.1016/j.molcel.2017.05.031. [DOI] [PubMed] [Google Scholar]
- 19.Iurlaro R, Munoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016;283(14):2640–2652. doi: 10.1111/febs.13598. [DOI] [PubMed] [Google Scholar]
- 20.Reinicke AT, Laban K, Sachs M, et al. Ubiquitin C-terminal hydrolase L1 (UCH-L1) loss causes neurodegeneration by altering protein turnover in the first postnatal weeks. Proc Natl Acad Sci USA. 2019;116(16):7963–7972. doi: 10.1073/pnas.1812413116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu S, Gonzalez-Prieto R, Zhang M, et al. Deubiquitinase activity profiling identifies UCHL1 as a candidate oncoprotein that promotes TGFbeta-induced breast cancer metastasis. Clin Cancer Res. 2020;26(6):1460–1473. doi: 10.1158/1078-0432.CCR-19-1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costes S, Huang CJ, Gurlo T, et al. beta-cell dysfunctional ERAD/ubiquitin/proteasome system in type 2 diabetes mediated by islet amyloid polypeptide-induced UCH-L1 deficiency. Diabetes. 2011;60(1):227–238. doi: 10.2337/db10-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pippin JW, Brinkkoetter PT, Cormack-Aboud FC, et al. Inducible rodent models of acquired podocyte diseases. Am J Physiol Renal Physiol. 2009;296(2):F213–229. doi: 10.1152/ajprenal.90421.2008. [DOI] [PubMed] [Google Scholar]
- 24.Garrett MR, Korstanje R. Using genetic and species diversity to tackle kidney disease. Trends Genet. 2020;36(7):499–509. doi: 10.1016/j.tig.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shirato I, Asanuma K, Takeda Y, et al. Protein gene product 9.5 is selectively localized in parietal epithelial cells of Bowman’s capsule in the rat kidney. J Am Soc Nephrol. 2000;11(12):2381–2386. doi: 10.1681/ASN.V11122381. [DOI] [PubMed] [Google Scholar]
- 26.Ichihara N, Wu J, Chui DH, et al. Axonal degeneration promotes abnormal accumulation of amyloid beta-protein in ascending gracile tract of gracile axonal dystrophy (GAD) mouse. Brain Res. 1995;695(2):173–178. doi: 10.1016/0006-8993(95)00729-a. [DOI] [PubMed] [Google Scholar]
- 27.Saigoh K, Wang YL, Suh JG, et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat Genet. 1999;23(1):47–51. doi: 10.1038/12647. [DOI] [PubMed] [Google Scholar]
- 28.Goto A, Wang YL, Kabuta T, et al. Proteomic and histochemical analysis of proteins involved in the dying-back-type of axonal degeneration in the gracile axonal dystrophy (gad) mouse. Neurochem Int. 2009;54(5–6):330–338. doi: 10.1016/j.neuint.2008.12.012. [DOI] [PubMed] [Google Scholar]
- 29.Fujimoto D, Kuwabara T, Hata Y, et al. Suppressed ER-associated degradation by intraglomerular cross talk between mesangial cells and podocytes causes podocyte injury in diabetic kidney disease. FASEB J. 2020;34(11):15577–15590. doi: 10.1096/fj.202000078RR. [DOI] [PubMed] [Google Scholar]
- 30.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 31.Kopp MC, Larburu N, Durairaj V, et al. UPR proteins IRE1 and PERK switch BiP from chaperone to ER stress sensor. Nat Struct Mol Biol. 2019;26(11):1053–1062. doi: 10.1038/s41594-019-0324-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park SJ, Kim Y, Yang SM, et al. Discovery of endoplasmic reticulum calcium stabilizers to rescue ER-stressed podocytes in nephrotic syndrome. Proc Natl Acad Sci USA. 2019;116(28):14154–14163. doi: 10.1073/pnas.1813580116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen YM, Zhou Y, Go G, et al. Laminin beta2 gene missense mutation produces endoplasmic reticulum stress in podocytes. J Am Soc Nephrol. 2013;24(8):1223–1233. doi: 10.1681/ASN.2012121149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han J, Back SH, Hur J, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 2013;15(5):481–490. doi: 10.1038/ncb2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Y, Gui D, Chen J, et al. Down-regulation of PERK-ATF4-CHOP pathway by Astragaloside IV is associated with the inhibition of endoplasmic reticulum stress-induced podocyte apoptosis in diabetic rats. Cell Physiol Biochem. 2014;33(6):1975–1987. doi: 10.1159/000362974. [DOI] [PubMed] [Google Scholar]
- 36.Yoshida H, Okada T, Haze K, et al. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol. 2000;20(18):6755–6767. doi: 10.1128/MCB.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madhusudhan T, Wang H, Dong W, et al. Defective podocyte insulin signalling through p85-XBP1 promotes ATF6-dependent maladaptive ER-stress response in diabetic nephropathy. Nat Commun. 2015;6:6496. doi: 10.1038/ncomms7496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shruthi K, Reddy SS, Chitra PS, et al. Ubiquitin-proteasome system and ER stress in the brain of diabetic rats. J Cell Biochem. 2019;120(4):5962–5973. doi: 10.1002/jcb.27884. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study are available from the corresponding author upon reasonable request.