

Abstract
Induction of bone formation by Wnt ligands is inhibited when sclerostin (Scl), an osteocyte-produced antagonist, binds to its receptors, the low-density lipoprotein receptor-related proteins 5 or 6 (LRP5/6). Recently, it was shown that enhanced inhibition is achieved by Scl binding to the co-receptor LRP4. However, it is not clear if the binding of Scl to LRP4 facilitates Scl binding to LRP5/6 or inhibits the Wnt pathway in an LRP5/6-independent manner. Here, using the yeast display system, we demonstrate that Scl exhibits a stronger binding affinity for LRP4 than for LRP6. Moreover, we found stronger Scl binding to LRP6 in the presence of LRP4. We further show that a Scl mutant (SclN93A), which tightly binds LRP4 but not LRP6, does not inhibit the Wnt pathway on its own. We demonstrate that SclN93A competes with Scl for a common binding site on LRP4 and antagonizes Scl inhibition of the Wnt signaling pathway in osteoblasts in vitro. Finally, we demonstrate that 2 weeks of bi-weekly subcutaneous injections of SclN93A fused to the fragment crystallizable (Fc) domain of immunoglobulin (SclN93AFc), which retains the antagonistic activity of the mutant, significantly increases bone formation rate and enhances trabecular volumetric bone fraction, trabecular number, and bone length in developing mice. Our data show that LRP4 serves as an anchor that facilitates Scl–LRP6 binding and that inhibition of the Wnt pathway by Scl depends on its prior binding to LRP4. We further provide evidence that compounds that inhibit Scl–LRP4 interactions offer a potential strategy to promote anabolic bone functions.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-022-04127-2.
Keywords: Sclerostin, LRP4, LRP6, Wnt pathway, Bone formation, Drug targets
Introduction
Bone tissue constantly undergoes modeling and remodeling, mediated by the coordinated activity of bone-resorbing osteoclasts, bone-building osteoblasts, and regulatory osteocytes. In healthy tissue, bone resorption is coupled with bone formation to maintain bone homeostasis [1, 2]. Unbalanced performance of bone cells leads to diverse bone pathologies, including osteoporosis, a common skeletal disorder characterized by a decrease in bone mass and bone strength, which significantly reduces quality of life and life expectancy in a large affected population [3]. A major cellular pathway responsible for sustaining physiological bone mass is the canonical Wnt pathway.
Signaling in the Wnt pathway is initiated when cell-secreted Wnt ligands form complexes with low-density lipoprotein receptor-related protein (LRP) 5 or 6 (LRP5/6) and Frizzled co-receptors. Binding of Wnt ligands to LRP5/6 triggers a signaling cascade that results in stabilization and nuclear translocation of β-catenin protein. In the nucleus, β-catenin associates with the DNA-bound T-cell factor/lymphoid enhancer factor (TCF/LEF) family of transcription factors, leading to expression of genes responsible for osteoblast differentiation and osteoclast inhibition [4–6].
Reduced signaling activity in the canonical Wnt pathway due to mutations affecting its signaling components is associated with various alterations in skeletal phenotype. Loss of LRP6 function is linked to osteoporosis in humans [7] and in mouse models [8, 9], whereas mutations in Wnt1 [10–12] and Wnt16 [13–15] are associated with decreased bone mineral density (BMD), shortening of long bones, bone fragility, and increased fracture risk. Similarly, Wnt3a+/− mice have reduced BMD as well as lower trabecular bone volume and number [16], and Wnt10a KO mice show trabecular bone loss with impaired bone mineralization [17, 18]. Patients with loss-of-function mutations in LRP5 suffer from osteoporosis pseudoglioma syndrome [19, 20] and idiopathic juvenile osteoporosis [21, 22], both of which cause early-onset severe osteoporosis, reduced BMD, and a tendency to fracture. On the other hand, missense gain-of-function mutations in the extracellular domain (ECD) of LRP5 were found in individuals with a high bone mass (HBM) phenotype [23, 24]. Similarly, patients with gain-of-function mutations in LRP6 that are homologous to LRP5 HBM variations exhibit generalized osteosclerosis, hyperostosis, and resistance to fracture [25].
The Wnt pathway is negatively regulated by sclerostin (Scl), a glycoprotein secreted mainly by osteocytes [26]. Loss of Scl expression leads to an exceptionally HBM phenotype, which is observed in two rare monogenic bone disorders, sclerosteosis and van Buchem disease [27–29]. The similarity between the phenotypes seen in sclerosteosis, van Buchem, and LRP5 HBM establishes that Scl antagonizes the Wnt pathway by binding to LRP5/6 [30, 31].
Recently, it was demonstrated that Scl inhibition of the Wnt pathway is facilitated by a co-receptor, LRP4, as Scl inhibition of bone mineralization is blocked by silencing LRP4 in osteoblastic cells and LRP4 overexpression enhances the inhibitory effect of Scl in the Wnt signaling assay [32]. Additionally, three mutations in LRP4 described in patients with the sclerosteosis bone phenotype result in impaired binding of Scl to LRP4, reduce Scl inhibitory activity in the Wnt signaling pathway, and increase serum Scl levels [32, 33]. The sclerosteosis phenotype was replicated in LRP4 knock-in mice bearing mutations that disrupt Scl binding, emphasizing the importance of Scl–LRP4 interactions for bone homeostasis [34, 35].
LRP5/6 and LRP4 are members of the same protein family with similar domain organization. They are single pass transmembrane proteins with large ECDs, which consist of four repeating YWTD β-propeller domains (E1, E2, E3, and E4) flanked by epidermal growth factor-like domains [36]. The interaction between Scl and LRP6 E1 is mediated through the conserved NXI motif located in Scl loop 2 [37], whereas the C-terminus of Scl has recently been shown to bind LRP6 E2 [38]. Furthermore, some mutations of the residues Asn93 or Ile95 in the NXI motif significantly impair Scl inhibition potency toward Wnt signaling, as shown using luciferase reporter assay [37, 39]. The binding interface between LRP4 and Scl has not yet been described, although some mutations in the E3 of LRP4 cause impaired Scl binding [32]. Furthermore, it was suggested that the interaction of Scl with LRP6 and LRP4 involves different binding sites, as Scl mutated at the Asn93 position to Ala (i.e., SclN93A), interacted with LRP4 but not with LRP6 in a co-immunoprecipitation assay. In the same assay, an Scl antibody with a loop 2-specific recognition site blocked Scl binding to LRP6 but not to LRP4 [40]. However, it is not clear if simultaneous binding to both LRP4 and LRP6 is required for stronger inhibition of the Wnt pathway.
Results
Scl exhibits superior binding to LRP4 compared with LRP6
To understand the mechanism responsible for enhanced inhibition of the Wnt pathway by Scl in presence of LRP4, we compared Scl binding to the ECDs of LRP4 and LRP6 using yeast surface display (YSD). Yeast surface-displayed Scl (YSD Scl) bound soluble LRP4 with an apparent KD value of 0.73 nM (Fig. 1a). Under the same experimental conditions, YSD Scl showed a remarkably lower apparent affinity for soluble LRP6 (by more than two orders of magnitude). This also prevented us from obtaining an apparent KD value for YSD Scl-LRP6 due to the high concentration of soluble LRP6 required to reach a binding equilibrium (Fig. 1a).
Fig. 1.

LRP4 binds Scl with higher affinity than LRP6 and enhances Scl binding to LRP6. a Yeast surface display (YSD) affinity titration curve. Recombinant yeast cells expressing Scl were incubated with soluble LRP6 (0.01–650 nM) or LRP4 (0.1–100 nM). b Geo MFI, presented as fold change. Recombinant yeast cells expressing Scl were pre-incubated with the indicated amounts of soluble LRP4, followed by incubation with soluble LRP6-Fc (200 nM). Points represent individual Geo MFI values of three independent experiments. Significance was assessed using unpaired, two-tailed Student's t test
We then asked whether sclerostin binding to LRP4 affects its binding to LRP6. For this purpose, YSD Scl was pre-incubated with different concentrations of soluble LRP4, followed by incubation with soluble LRP6-Fc. Soluble recombinant human Fc (rhFc) was used as a control for the effect of Fc fusion on binding. We observed an increase in the geometric mean fluorescence intensity (Geo MFI) signal of LRP6-Fc in YSD Scl cells that were pre-incubated with a saturation concentration of LRP4 (Fig. 1b). This indicates that the binding of LRP6 to Scl is stronger in the presence of LRP4. Additionally, these results suggest that Scl can bind both receptors simultaneously, since no decrease in Geo MFI signal was observed after pre-incubation with different concentrations of LRP4.
N93A mutation in the NXI motif of Scl profoundly lowers its affinity for LRP6 but not for LRP4
In earlier reports, cell binding assay and biolayer interferometry showed that mutating Asn to Ala in position 93 of the mature Scl sequence greatly reduces its binding affinity for LRP6 [37, 41]. To elucidate whether this mutation also affects Scl affinity for LRP4, yeast surface-displayed single mutant Scl (YSD SclN93A) was titrated either with soluble LRP4 or LRP6. YSD SclN93A bound to LRP4 with high affinity (an apparent KD = 0.48 nM; Fig. 2a), similar to YSD Scl (Fig. 1a). Accurate Kd values for SclN93A and Scl could not be obtained due to limited availability of the soluble ligands. To overcome this limitation for reactions that do not reach saturation within the tested concentration range, it is a common practice to compare affinity titration plots in the specific ligand concentration range. Such comparison of the binding plots allows a qualitative, but still reliable, comparison between binding curves without establishing an actual precise Kd value [42–45]. A qualitative assessment of the binding plots clearly showed that the binding of SclN93A to LRP6 (Fig. 2a) was significantly reduced relative to Scl (Fig. 1a) in agreement with former studies.
Fig. 2.

SclN93A binds with high affinity to LRP4, but with greatly reduced affinity to LRP6 and competes with Scl for the LRP4 binding site. a Yeast surface display (YSD) affinity titration curve. Recombinant yeast cells expressing SclN93A were assayed as described in Fig. 1a. b Binding competition. Recombinant yeast cells expressing SclN93A were incubated with increasing concentrations of soluble purified Scl (50–1000 nM) and a fixed concentration (5 nM) of soluble LRP4
SclN93A and Scl compete to bind LRP4, and SclN93A promotes Wnt signaling in osteoblasts
As both Scl and SclN93A were shown to interact with LRP4, we suggested they share a common binding site on LRP4. To test this, YSD SclN93A was titrated with increasing concentrations of soluble recombinant Scl, purified from P. Pastoris yeast (Suppl. Fig. 1). We observed a dose-dependent reduction in binding of YSD SclN93A to soluble LRP4 (Fig. 2b). This finding indicates there is a competition between Scl and SclN93A for the same binding site on LRP4.
Next, we endeavored to determine whether this competition would stimulate canonical Wnt signaling in a physiological system. Scl binding to LRP6 inhibits canonical signaling activated by the Wnt1 and Wnt3a ligand classes. Regarding in vitro studies related to the Wnt pathway in particular, overexpression of signaling components may be misleading [46]. Experimentally, SclN93A showed contradicted effects on Wnt pathway activation. One study showed that SclN93A moderately enhanced Wnt1 signaling in HEK293T cells [40], while another group showed it exhibited a reduced ability to inhibit Wnt1- and Wnt3a signaling in a similar experiment [41]. To examine the effects of SclN93A in a more physiological related context, we conducted a competition assay in MC3T3-E1 pre-osteoblastic cells, which endogenously express canonical Wnt pathway-relevant receptors without the need for their overexpression. As expected, addition of Wnt3a CM to the cell culture increased TCF/LEF activity while addition of Scl to the culture media reduced activation of Wnt signaling (Fig. 3a). Addition of SclN93A to culture media did not inhibit Wnt3a-induced TCF/LEF activity (Fig. 3b). When cells were treated with both Scl and SclN93A, we observed a dose-dependent increase in Wnt3a-activated luciferase expression. At a molar Scl:SclN93A ratio of 1:100, the signal of activation was similar to base-line activation with Wnt3a CM (Fig. 3a). To verify that the effects of SclN93A on the Wnt signaling activation is downstream of Wnt3a, we cultured MC3T3 cells with Scl and/or SclN93A, in the absence of Wnt3a CM (Suppl. Fig. 2a). No activation of luciferase in the absence of Wnt3a was detected in cultures of cells treated with either Scl (or SclN93A) or with a combination of both. To confirm that SclN93A ability to relieve inhibition of the Wnt pathway by Scl is LRP4 dependent, competition assays were reproduced in cells treated with small interfering RNA (siRNA) that target mouse LRP4 sequence. Immunoblot with LRP4 specific antibodies confirmed efficient knockdown of LRP4 by two different siRNAs (Suppl. Fig. 2b). When LRP4 was knocked down, addition of SclN93A to cells treated with Scl did not increase luciferase activity (Fig. 3c and Suppl. Fig. 2c). These results indicate that SclN93A stimulates Wnt3a signaling by competing with Scl for binding to LRP4.
Fig. 3.
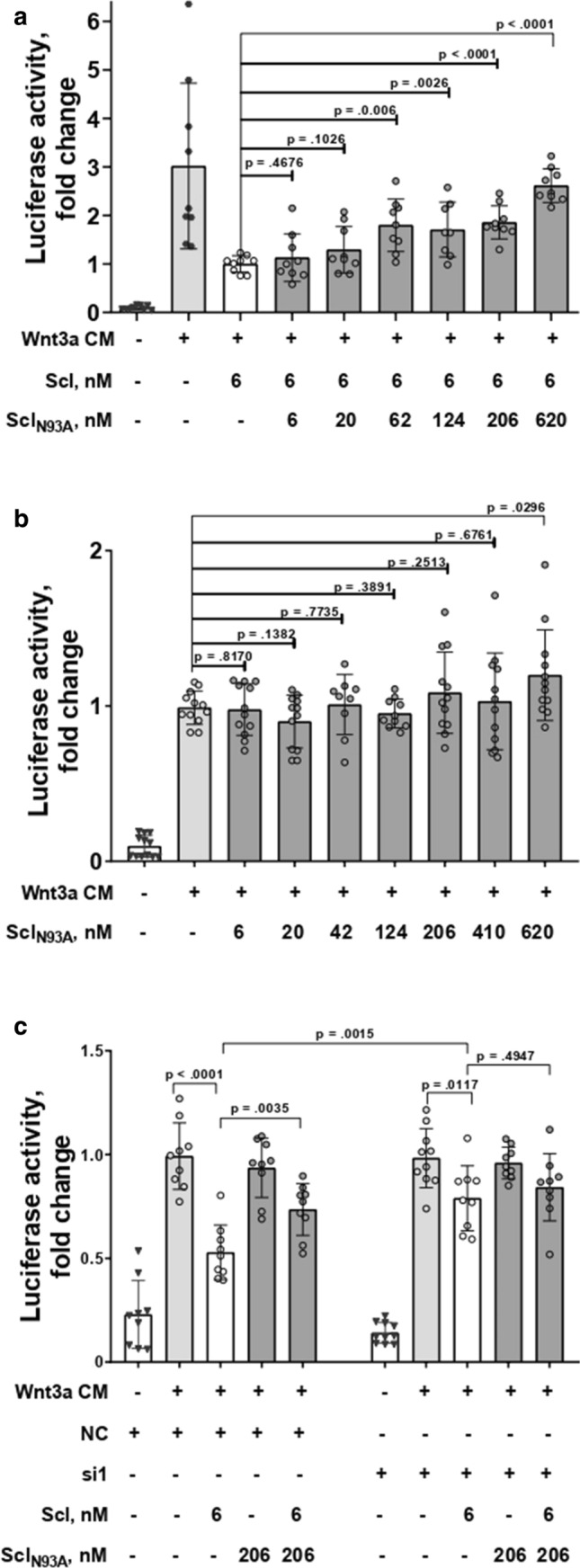
SclN93A relieves Wnt3a-activated signaling inhibition by Scl in MC3T3-E1 cells and this activity is LRP4 dependent. Wnt pathway activation in MC3T3 demonstrated by luciferase activity (measured as fold change) in a super-TOPFlash assay (a–c). Cells were treated with Wnt3a conditioned medium (CM) and a SclN93A (6–620 nM) or b Scl (6 nM) in the absence or presence of SclN93A (6–620 nM). c Cells were transiently transfected with LRP4 specific siRNA (si1), or nontargeting siRNA (NC) and different combinations of Scl and SclN93A. Points represent individual measurements of biological replicates (A–C, n = 9) collected in three separate experiments. Significance was assessed using unpaired, two-tailed Student's t test. An outlier identified using Grubbs' test was removed in two cases (treatment with Wnt3a CM and 124 nM SclN93A, and treatment with Wnt3a CM, 6 nM Scl, and 124 nM SclN93A)
Treatment with purified SclN93AFc increases the volumetric fraction of trabecular bone and bone length in developing mice
Next, we determined whether competition on Scl binding to LRP4 can promote bone anabolic functions. To the best of our knowledge, the retention time for Scl in circulating blood has not been reported. However, due to its relatively low molecular weight (around 28 kDa for mature glycosylated protein) and positive charge, we reasoned that it would be rather rapidly cleared by renal filtration [47]. Therefore, we conjugated the fragment crystallizable domain of immunoglobulin to the C-terminal end of SclN93A (designated SclN93AFc), which is an established strategy to prolong protein retention in circulating blood [48]. Similarly to unmodified SclN93A, treatment with SclN93AFc increased TCF/LEF activity in cells co-cultured with Wnt3a CM and SclWT (Suppl. Fig. 3), confirming that modification with Fc did not affect SclN93A activity.
Thereafter, we administered subcutaneous injections of SclN93AFc (0.5 mg/kg) to healthy 8-week-old female C57BL/6 mice twice a week for 2 weeks, a length of one bone remodeling cycle in mice. This concentration is equivalent to an approximately 150-fold molar excess of SclN93AFc on endogenous serum-circulating Scl, calculated based on average mouse weight, blood volume, and previously reported serum Scl levels (about 50 pg/mL for female mice) [49]. After 2 weeks, mice were killed and femurs were extracted and scanned using microcomputed tomography (µCT). We observed an increase in trabecular volume (Fig. 4a) and a 45% increase in volumetric fractions (BV/TV) in the femurs of SclN93AFc-injected mice, compared to the vehicle controls (Fig. 4b). Trabecular number was 35% higher in the SclN93AFc-injected group (Fig. 4c). Mean trabecular thickness and separation values were different in the treated group compared with the control group but did not reach statistical significance for the cohort size used in this study (Fig. 4d–e). We did not observe any differences in femoral cortical bone parameters in the midshaft between the treated and control groups (Suppl. Fig. 4). Nevertheless, femur length in SclN93AFc-treated mice was significantly greater compared with the control group (Fig. 4f). To corroborate the increase in bone volume observed in µCT, proximal trabecular region of mice tibiae was analyzed by static and dynamic histomorphometry. We found a 57% increase in the mineralizing surface to bone surface ratio (Fig. 4g) along with no change in mineral apposition rate (Fig. 4h), resulting in a net increase of 58% in the bone formation rate (Fig. 4i). Furthermore, osteoclast surface was significantly decreased in SclN93AFc-treated mice compared with the control group (Fig. 4j), by TRAP enzymatic activity staining. These data indicate that disrupting LRP4-Scl binding interface can promote bone formation in vivo.
Fig. 4.

SclN93AFc antagonizes Scl inhibition of the Wnt pathway, promotes bone formation, and increases femoral bone mass and length. Eight-week-old female C57BL/6 mice treated with purified SclN93AFc (0.5 mg/kg) for 2 weeks by bi-weekly subcutaneous injections. a Three-dimensional reconstructions of distal femurs from X-ray computed µCT scans. b–e Femoral µCT analysis of trabecular bone architecture in terms of b bone volume as a percentage of total trabecular volume (BV/TV) and c–e trabecular number (Tb.N), thickness (Tb.Th), and separation (Tb.Sp). f Femoral length measurement. Group size, n = 7 per vehicle group, n = 8 per SclN93AFc group. g–i Dynamic histomorphometry analysis of tibial bone formation parameters: mineralizing surface to bone surface ratio (MS/BS), mineral apposition rate (MAR), and bone formation rate (BFR). Group size, n = 6 per vehicle group, n = 5 per SclN93AFc group, randomly selected. j Quantitative analysis of osteoclast surface per bone surface (Oc.S/BS). Group size, n = 5 per vehicle group, n = 6 per SclN93AFc group, randomly selected. Significance was assessed using unpaired, two-tailed Student's t test
Discussion
Our data show, for the first time, that Scl binds more strongly to LRP4 compared with LRP6. Furthermore, the binding of Scl to LRP4 increases Scl–LRP6 binding, consistently both with the previously suggested role of LRP4 as an anchor to increase local Scl concentrations in bone [33] and with studies showing increased Scl serum concentrations in mice lacking LRP4 or in which Scl–LRP4 interactions were blocked by a specific antibody [50]. In contrast to our binding affinity results, it was previously shown using surface plasmon resonance (SPR) that Scl bound immobilized LRP4 and LRP6 with similar affinities in the low nanomolar range [32]. Nevertheless, in a different SPR study, Scl exhibited profoundly lower affinity for immobilized LRP6 (KD = 8.6 µM) [51].
This controversy may be explained by the fact that immobilization of a ligand via non-specific residues may affect the results and reproducibility of binding assays by causing conformational changes, or by blocking or reducing the number of available binding sites, as opposed to measuring free interactions between binding partners in solution. Although, in our binding assay, Scl was displayed on the yeast cell surface, which also implies a certain degree of conformational strain, our YSD platform was shown previously to enable the determination of KD values with an accuracy comparable to those obtained for soluble protein complexes [52, 53]. Moreover, our results show, for the first time, that Scl binds more strongly to LRP6 when LRP4 is present. Given the superior affinity of Scl for LRP4 compared with LRP6, we propose that Scl binds first to LRP4, which is followed by a conformational change in either Scl or LRP4 that promotes an optimal orientation for subsequent Scl binding to LRP6 and thereby achieves efficacious inhibition of Wnt3a-activated canonical Wnt signaling. This mechanism of activation may not apply to the Wnt1 class of ligands, as they have a different binding site on LRP6.
It was previously shown that deleting LRP4 or blocking its interaction with Scl activates the Wnt1 pathway in osteoblasts [32, 54]. However, it remained unclear whether the inhibitory effect of the Scl–LRP4 interaction on the Wnt pathway is LRP6 dependent. For the first time, we show here that LRP4 promotes Scl inhibitory action on Wnt3a-activated signaling, in addition to previously described facilitator role for inhibition of Wnt1. Using the YSD system, we show that SclN93A binding to soluble LRP6 is negligible compared with that of Scl, but the affinities of SclN93A and Scl toward soluble LRP4 are identical. We also show that, although SclN93A binds strongly to LRP4, it fails, unlike Scl, to inhibit Wnt3a signaling in osteoblasts. This suggests that Scl binding to LRP4 alone is insufficient for Wnt3a pathway inhibition, possibly because it does not affect the availability of binding sites for Wnt ligands on LRP6. When SclN93A is added in combination with Scl, a dose-dependent relief of Scl inhibition is observed. Low doses of SclN93A alone do not activate the Wnt pathway, suggesting that SclN93A competes with Scl on binding to LRP4, which was also demonstrated in our YSD binding competition experiment. As a result, fewer LRP4 receptors are available for Scl binding, thus greatly reducing the instances of Scl–LRP6 interaction. Consequently, this allows free interaction of Wnt ligands with LRP6 and promotes Wnt pathway activation. Taken together, these results provide evidence that the inhibitory effect of Scl–LRP4 interactions on the Wnt3a pathway is LRP6 dependent. Of note, our study is focused on Scl interaction with LRP4 and LRP6. Nevertheless, it is known that LRP5 and LRP6 share high structure similarity [55], most residues implicated in binding to Scl are conserved in these receptors [38] and they exhibit alike behavior in binding to both Scl and SclN93A [39]. Given this, we predict that replication of our experiments with LRP5 would yield similar results.
Mice treated with SclN93AFc exhibited a significant increase in trabecular BV/TV ratio and number as a result of higher bone formation parameters than in the vehicle group.
Moreover, osteoclast surfaces were significantly lower in SclN93AFc treated mice. This phenotype is consistent with observations made in skeletally mature mice with LRP4-deficient osteoblasts and osteocytes [54], in Scl-deficient mice [56, 57], and in mouse models bearing LRP4 mutations that compromise LRP4–Scl binding [34, 35]. In addition, the femurs of SclN93AFc-treated mice were significantly longer, consistently with results obtained previously in Scl-deficient mice [56]. The similarities between the phenotypes indicate a common mechanism in which the binding of sclerostin to LRP4 is essential for its binding to LRP6 in order to antagonize the Wnt pathway. Worthy of mention, besides being a bone formation inhibitor, LRP4 is a key component in the formation, stabilization and maintenance of the neuromuscular junction through complex formation with agrin and muscle-specific kinase [58–60]. Thus, a major concern in targeting LRP4 with a large molecule would be disruption of these interactions. Agrin or LRP4 deficient mice die at birth or unable to breathe independently [50, 58, 61]. Adult mice with conditional muscle LRP4 deletion quickly lose weight and exhibit muscle weakness within three weeks [60]. For this reason, we closely monitored SclN93AFc-injected mice and did not observe any weight loss, general weakness or other signs of distress. This suggests that no apparent damage to neuromuscular junction was caused by SclN93AFc treatment, although a longer experiment may be required to ensure safety.
Our results suggest that it may be possible to modulate Wnt signaling in bone by targeting LRP4, without the need to neutralize endogenous Scl. Such an approach could be advantageous in the treatment of bone disorders, given the complexity of the Wnt pathway and its involvement in various pathologies. For example, the anabolic response of bone lessens after prolonged treatment with Scl–Ab, accompanied by significant upregulation in the expression of other Wnt antagonists, possibly by inducing a negative feedback mechanism [62]. Complete loss of endogenous Scl was shown to promote osteoarthritis, rheumatoid arthritis, and posttraumatic osteoarthritis in mice [63–66], with another study demonstrating a protective role for endogenous Scl in prostate cancer invasions [67]. In addition, reports regarding the role of circulating serum Scl are controversial. Some studies linked increased Scl levels with inflammation and vascular lesions in chronic kidney disease patients [68] or with cardiovascular events [69, 70], whereas others reported an association between higher Scl levels and improved survival and lower short-term cardiovascular mortality in dialysis patients and patients with vascular calcification [71–73]. To the best of our knowledge, an association between Scl–LRP4 interactions and any of these pathologies was not reported and remains to be elucidated.
Experimental procedures
Mice and ethics statement
Six-week-old C57BL/6J female mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA). Upon arrival, mice were randomly assigned to the vehicle group or the sclerostinN93AFc treatment group. Animals were maintained in a 12-h light/dark cycle and provided ad libitum access to food and water at all times. Upon reaching 8 weeks of age, both groups were injected subcutaneously twice a week for 2 weeks. In this manner, treatment group mice received recombinant SclN93AFc (0.5 mg/kg; calculated based on an average mouse weight of 20 g) dissolved in PBS (Biological Industries, Beit Haemek, Israel) at a SclN93AFc:PBS ratio of 1:6.5, whereas the vehicle group mice received PBS diluted with HEPES‐buffered saline (HBS150; 20 mM HEPES, 150 mM NaCl, pH 7.4) at a HBS150:PBS ratio of 1:6.5. On the 10th and 13th days of subcutaneous treatment, mice were subcutaneously injected with the fluorescent dye calcein (10 mg/kg; Sigma Aldrich, St. Louis, MO, USA) and then killed on the 14th day, at age 10 weeks.
All mouse studies were carried out according to protocols approved by the Ben- Gurion University Committee for the Ethical Care and Use of Animals in Experiments (permit number: IL-04-01-2019(D)).
Cell culture
MC3T3-E1 cells (ATCC® CRL-2593™; an osteoblastic mouse calvaria cell line) were purchased from ATCC (Manassas, VA, USA). The cells were cultured in alpha-minimal essential medium (alpha-MEM) supplemented with 10% FBS, l-glutamine (2 mM), penicillin (100 U/mL), and streptomycin (0.1 mg/mL) in a humidified incubator at 37 °C under a 5% CO2 atmosphere. L-Wnt3a-transfected cells (a gift from Michal Hershfinkel, Ben Gurion University of the Negev (BGU), Beer-Sheva, Israel) were cultured in DMEM supplemented with G-418 (0.4 mg/mL). Conditioned media were prepared from these cells according to the ATCC protocol.
Purification of recombinant SclWT and SclN93A
To express SclWT and SclN93A variants in P. pastoris strain GS115, the sequences were amplified from pCTCON template vector, while adding recognition sites for AvrII and ECORI restriction enzymes (NEB) using 5’-ACA AAG AAT TCC GTC AAG GGT GGC AAG CGT-3’ as the forward primer and 5’-AAA ACC TAG GGT ACG CGT TCT CTA ATT CGG-3’ as the reverse primer. The amplified DNA and pPICK9K plasmid (Invitrogen) were digested with AvrII and ECORI (NEB), ligated using Quick Ligase (NEB) and transformed into competent E. coli cells by heat shock. Next, the plasmids were transformed into electrocompetent P. Pastoris GS115 strain cells, as previously described [74]. The highest expressing clones were chosen based on Western blot analysis of cell cultures using a 1:2000 dilution of primary human SOST/Sclerostin Antibody Polyclonal Goat IgG (R&D Systems), followed by 1:5000 dilution of donkey anti-goat IgG H&L (HRP) antibody (Abcam) and signal detection using the ECL substrate kit (Biological Industries). All antibodies were diluted in TBST with 5% bovine serum albumin. Protein purification was scaled up as described previously [74], with small modifications, namely, protein elution buffer was exchanged to HBS150 buffer (10 mM HEPES, 150 mM NaCl, pH 7.5) using a 5,000 MWCO Vivaspin centrifugal concentrator (GE Healthcare). The proteins were further purified using an AKTA pure 150 FPLC Superdex 75/10/300 size-exclusion column (GE Healthcare) pre-equilibrated with HBS150. Protein-containing size-exclusion fractions were pooled together after relevant fractions were determined by SDS-PAGE and staining with InstantBlue (Expedeon). Protein concentration was measured using a NanoDrop spectrophotometer (Thermo Scientific), based on protein absorbance at 280 nm (percent extinction coefficient = 11.36). Protein yields were 1.5 mg/L of yeast culture for SclWT and 1.1 mg/L for SclN93A.
Purification of recombinant SclN93AFc
pFUSE-hIgG1e3-Fc1 plasmid with IL2 signal peptide (a gift from Amir Aharoni, BGU) was used as a template for Gibson assembly using a set of overlapping primers. The forward and reverse primers for amplification were 5’-GTG CTA GCT GGC CAG ACA TG-3’ and 5’-CGA ATT CGT GAC AAG TGC-3’, respectively, for pFUSE-hIgG1e3-Fc1. For SclN93A the forward and reverse primers for amplification were 5’-CAC TTG TCA CGA ATT CGC AAG GGT GGC AAG CGT TTA AAA ATG-3’ and 5’-CTG GCC AGC TAG CAC TCA GTG ATG GTG ATG GTG ATG G-3’, respectively, while adding 6 × His tag for further affinity purification, followed by assembly reaction using Gibson assembly master mix (NEB). All constructs were confirmed by sequencing (Hylabs, Rehovot, Israel).
For protein expression, FreeStyle 293-F cells (a gift from Angel Porgador, BGU) were cultivated at 37 ℃ in an 8% CO2 atmosphere in a humidified orbital shaker incubator at 135 rpm. The cells were grown in serum-free FreeStyle™ 293 Expression medium (Gibco) and maintained at 1.5–2 × 106 cells/mL in a volume that did not exceed 30% of the total volume of the culture flask. The cells were transiently transfected using GeneTranIII transfection reagent (Biomiga, San Diego, CA) according to the manufacturer’s instructions. On day 4 after transient transfection, the cells were lysed in a lysis buffer (50 mM Tris, 50 mM NaCl, 1% Triton, pH 7.4) and centrifuged at 15,000 × g for 10 min at 4 ℃ to separate all cell debris to the sediment. The lysate supernatant was loaded into a Ni–NTA agarose (Invitrogen) gravity column, washed with binding buffer (50 mM Tris, 10 mM imidazole, 300 mM NaCl, pH 7.5) and eluted with an elution buffer (50 mM Tris, 300 mM imidazole, 300 mM NaCl, pH 7.5). The buffer was exchanged to HBS150 using a 30 kDa Amicon Ultra centrifugal filter unit (Merck KGaA, Darmstadt, Germany). The proteins were further purified using an AKTA pure 150 FPLC Superdex 200/16/600 size-exclusion column (GE Healthcare) pre-equilibrated with HBS150. Protein-containing size-exclusion fractions were pooled together after relevant fractions were determined by SDS-PAGE and staining with InstantBlue (Expedeon). Protein concentration was measured using a NanoDrop spectrophotometer (Thermo Scientific), based on protein absorbance at 280 nm (percent extinction coefficient = 12.41). Protein yield was 0.65 mg/L of the starting cell culture.
Protein expression on yeast cell surface and binding analysis by flow cytometry
Full length human sclerostin cDNA encoding for mature sclerostin protein (residues Gln24–Tyr213) was synthesized by Integrated DNA Technologies. A mutation changing asparagine 93 to alanine was introduced to the SclWT sequence by site-directed mutagenesis using back-to-back 5’ phosphorylated primers carrying the mutation (forward, 5’-/Phos/TGC TGC CAG CTG CGA TTG GT-3’; reverse, 5’-/Phos/AAC GTG CAG GAC CAC ACT GAC C-3’). The sequences were subcloned to a pCTCON vector (a gift from Dane Wittrup, MIT) by restriction cloning using BamHI [New England Biolabs (NEB)] and NheI (NEB) restriction enzymes and T4 DNA ligase (NEB). The plasmids were transformed into a competent EBY100 Saccharomyces cerevisiae yeast strain (a gift from Amir Aharoni, BGU) by electroporation (MicroPulser electroporator, Bio-Rad). To confirm the DNA sequences of the transformed yeast cells by Sanger sequencing (Hylabs, Rehovot, Israel), the plasmids were extracted from the yeast using Zymoprep™ yeast plasmid miniprep I kit (Zymo Research, USA) and transformed to competent Escherichia coli cells by electroporation, followed by extraction using HiYield plasmid mini kit (RBC Bioscience, Taiwan).
For flow cytometry analysis, yeast cells transformed with wildtype or mutated Scl (Scl or SclN93A) were grown in expression-inducing SGCAA medium (2% galactose, 0.67% yeast nitrogen base, 0.5% bacto casamino acids, 1.47% sodium citrate, 0.429% citric acid monohydrate) overnight at 30 °C, until the culture achieved an optical density at 600 nm of 5.0 (i.e., OD600 = 5.0). Cells were washed with PBS with 1% bovine serum albumin (BSA; Caisson Laboratories, Utah, USA). The expression of displayed proteins was detected with 1:50 mouse anti-c-Myc antibody, 9E10 (Abcam, Cambridge, MA, USA) followed by 1:50 anti-mouse IgG (whole molecule)—R-phycoerythrin antibody produced in goat (Sigma-Aldrich).
For binding analysis of displayed SclWT or SclN93A, cells were incubated with different concentrations of either soluble His-tagged mouse LRP6 (R&D Systems) or with soluble His-tagged human LRP4 (R&D Systems) for 2 h at room temperature, following detection with a 1:50 FITC-conjugated anti-6xHis-tag monoclonal antibody (Invitrogen). For competition binding between LRP4 and LRP6, cells displaying Scl were pre-incubated with soluble LRP4 (1, 5 or 200 nM) for 2 h, followed by incubation with 200 nM soluble human LRP6-Fc (R&D Systems) for 2 h and detection with a 1:50 monoclonal anti-human IgG (Fc specific)—FITC antibody produced in mouse (Sigma Aldrich). All Geo MFI values were normalized to the Geo MFI of control cells incubated with only the secondary fluorescent antibody. For competition binding between YSD Scl and YSD SclN93A, cells displaying SclN93A were incubated with soluble His-tagged human LRP4 (5 nM; R&D Systems) and various concentrations of soluble SclWT for 2 h at room temperature, followed by detection with a 1:50 FITC-conjugated anti-6xHis-tag monoclonal antibody (Invitrogen). All antibodies were diluted in PBS with 1% bovine serum albumin. Labeled cells (30,000 per experimental condition) were analyzed on an Accuri C6 flow cytometer (BD Biosciences). Further FACS analysis was performed using FlowJo software (BD, Ashland, OR, USA). All values were normalized to the highest binding signal of the respective yeast-displayed protein combination. Apparent KD values were calculated by non-linear regression.
Dual luciferase reporter assay
MC3T3-E1 cells (7 × 103 cell per well) were seeded in a 96-well plate in triplicate, so that the cells were 70–80% confluent on the following day. After 24 h, the cells were transiently transfected with M50 Super 8 × TOPFlash reporter plasmid (50 ng/well; a gift from Randall Moon, Addgene plasmid # 12456; http://n2t.net/addgene:12456; RRID:Addgene 12456) and Renilla control plasmid (5 ng/well; a gift from Ramon Birnbaum, BGU, Beer-Sheva, Israel), using Lipfectamine2000 (Invitrogen) according to the manufacturer’s instructions. Twenty-four hours after transfection, cells were treated with Wnt3a conditioned media (CM) from L-Wnt3a-transfected cells and various concentrations of purified recombinant SclWT, SclN93A, and/or SclN93AFc for an additional 19 h. The cells were lysed with passive lysis buffer (Promega, Madison, WI) and luciferase activity was measured on a SPARK microplate reader (Tecan Austria GmbH, Grödig, Austria) using the dual-luciferase reporter assay kit (Promega). Each transfection was performed in triplicate and repeated in at least two separate experiments. All luciferase results were normalized to the Renilla control.
LRP4 knock down assay
Dicer-substrate short interfering RNAs (DsiRNAs) targeting mouse LRP4 (mm.Ri.Lrp4.13.1 and mm.Ri.Lrp4.13.3) and nontargeting negative control (51-01-14-03) were purchased from Integrated DNA Technologies (IDT, Coralville, IA). 80–90% confluent MC3T3-E1 cells were transfected with DsiRNA, M50 Super 8 × TOPFlash reporter and Renilla control mix using using Lipfectamine2000 (Invitrogen) according to the manufacturer’s instructions. After 48 h, cells were treated with Wnt3a CM and purified recombinant SclWT or SclN93A, alone or in combination, for an additional 19 h. The cells were assayed by Dual Luciferase Reporter Assay, as described above. Knockdown efficiency was detected at 48 h by western blot analysis using mouse monoclonal anti-LRP4 antibody (Abcam), mouse anti-Actin monoclonal antibody (MP Biomedicals) and Peroxidase AffiniPure Goat Anti-Mouse IgG (H + L) (Jackson ImmunoResearch, West Grove, PA, USA).
Microcomputed tomography and bone length measurement
Following euthanasia, femurs were extracted and stored in PBS-soaked gauze prior to imaging. Images of the left distal femurs were acquired using a microcomputed tomography Bruker SkyScan 1174 scanner (Skyscan, Aartselaar, Belgium). The femurs were scanned at 50 kV and 800 µA using a 0.25 mm aluminum filter with an isotropic voxel size of 10 µm. The mineralized tissues were segmented by a global thresholding procedure. Trabecular bone parameters were assessed in a region starting 2.1 mm below the growth plate (reference level) and extending down for 1.5 mm. Femur length was measured from the fovea capitis to the medial condyle with a caliper.
Dynamic histomorphometry
Left tibiae were fixed in 4% PFA with 0.1 M phosphate buffer for 24 h at room temperature and then embedded in methyl methacrylate. Sections (4 µm) were prepared with a Leica RM2255 Rotary Microtome (Leica Biosystems). Images of the trabecular region were captured at 20 × magnification using a CoolSNAP HQ2 Monochrome camera (Photometrics, Tucson, AZ, USA), Olympus IX81 microscope and CellSens Dimension imaging software (Olympus America, Inc., Center Valley, PA, USA). Three sections per mouse were analyzed using ImageJ2 software [75].
Tartrate-resistant acid phosphatase (TRAP) enzymatic activity staining
Right femurs were fixed in 4% PFA with 0.1 M phosphate buffer for 24 h at 4 °C, then transferred to 30% sucrose in 0.1 M phosphate buffer for additional 24 h at 4 °C. Femurs were embedded and frozen in SCEM-(L1) cryoembedding medium (SECTION-LAB Co. Ltd., Japan) and cut into 8 µm sections with Leica CM1860 Cryostat Microtome (Leica Biosystems). For detecting TRAP enzymatic activity, sections were first stained with Calcein blue (Sigma) for accumulated mineral, followed by TRAP enzymatic activity staining using ELF™ 97 Phosphatase Substrate (Invitrogen) [76]. Sections were imaged at 20 × magnification using a CoolSNAP HQ2 Monochrome camera, Olympus IX81 microscope and CellSens Dimension imaging software. Sections were analyzed using ImageJ2 software.
Statistical analysis
Statistical analyses were performed with GraphPad Prism 8 (GraphPad Software, La Jolla, CA, USA). Unpaired, two-tailed Student's t tests were used to analyze between-group differences following treatment with SclWT, SclN93A, and/or SclN93AFc in the dual luciferase experiments and in the YSD binding assays, and to evaluate differences between the mouse groups treated with SclN93AFc or vehicle. Differences at p < 0.05 were considered significant.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by grants from the ISRAEL SCIENCE FOUNDATION no. 357/18 (NL) and no. 1615/19 (NP).
Author contributions
SK, conceptualization, data curation, investigation, methodology, validation, visualization, writing—original draft, writing—review and editing. BC, methodology. CA-D, methodology. NP, conceptualization, formal analysis, funding acquisition, investigation, methodology, resources, supervision, validation, visualization, writing—original draft, writing—review and editing. NL, conceptualization, formal analysis, funding acquisition, investigation, methodology, resources, supervision, validation, visualization, writing—original draft, writing—review and editing.
Funding
This work was supported by grants from the ISRAEL SCIENCE FOUNDATION no. 357/18 (NL) and no. 1615/19 (NP).
Code availability
Not applicable.
Data availability
Data will be made available on reasonable request.
Declarations
Conflict of interest
The authors declare no conflict of interest.
Ethics approval
All mouse studies were carried out according to protocols approved by the Ben-Gurion University Committee for the Ethical Care and Use of Animals in Experiments (permit number: IL-04-01-2019(D)).
Consent to participate
Not applicable.
Consent for publication
All authors agree with the submission, and the work has not been published or submitted for publication elsewhere either completely or in part, or in another form or language.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Niv Papo, Email: papo@bgu.ac.il.
Noam Levaot, Email: levaot@bgu.ac.il.
References
- 1.Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem. 2010 doi: 10.1074/jbc.R109.041087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robling AG, Castillo AB, Turner CH. Biomechanical and molecular regulation of bone remodeling. Annu Rev Biomed Eng. 2006 doi: 10.1146/annurev.bioeng.8.061505.095721. [DOI] [PubMed] [Google Scholar]
- 3.National Institutes of Health (NIH) (2001) National Institutes of Health (NIH) consensus development panel on osteoporosis prevention, diagnosis, and therapy. JAMA
- 4.Zeng X, et al. Initiation of Wnt signaling: control of Wnt coreceptor Lrp6 phosphorylation/activation via frizzled, dishevelled and axin functions. Development. 2008 doi: 10.1242/dev.013540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khosla S, Westendorf JJ, Oursler MJ. Building bone to reverse osteoporosis and repair fractures. J Clin Investig. 2008 doi: 10.1172/JCI33612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weivoda MM, et al. Wnt signaling inhibits osteoclast differentiation by activating canonical and noncanonical cAMP/PKA pathways. J Bone Miner Res. 2016 doi: 10.1002/jbmr.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mani A, et al. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science. 2007 doi: 10.1126/science.1136370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kokubu C, et al. Skeletal defects in ringelschwanz mutant mice reveal that Lrp6 is required for proper somitogenesis and osteogenesis. Development. 2004 doi: 10.1242/dev.01405. [DOI] [PubMed] [Google Scholar]
- 9.Holmen SL, et al. Decreased BMD and limb deformities in mice carrying mutations in both Lrp5 and Lrp6. J Bone Miner Res. 2004 doi: 10.1359/JBMR.040907. [DOI] [PubMed] [Google Scholar]
- 10.Keupp K, et al. Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet. 2013 doi: 10.1016/j.ajhg.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pyott SM, et al. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am J Hum Genet. 2013 doi: 10.1016/j.ajhg.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu Y, et al. Novel WNT1 mutations in children with osteogenesis imperfecta: clinical and functional characterization. Bone. 2018 doi: 10.1016/j.bone.2018.06.018. [DOI] [PubMed] [Google Scholar]
- 13.Zheng HF, et al. WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PLoS Genet. 2012 doi: 10.1371/journal.pgen.1002745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Medina-Gomez C, et al. Meta-analysis of genome-wide scans for total body BMD in children and adults reveals allelic heterogeneity and age-specific effects at the WNT16 locus. PLoS Genet. 2012 doi: 10.1371/journal.pgen.1002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.García-Ibarbia C, et al. Missense polymorphisms of the WNT16 gene are associated with bone mass, hip geometry and fractures. Osteoporos Int. 2013 doi: 10.1007/s00198-013-2302-0. [DOI] [PubMed] [Google Scholar]
- 16.Takada I, et al. A histone lysine methyltransferase activated by non-canonical Wnt signalling suppresses PPAR-γ transactivation. Nat Cell Biol. 2007 doi: 10.1038/ncb1647. [DOI] [PubMed] [Google Scholar]
- 17.Zhou H, et al. Glucocorticoid-dependent Wnt signaling by mature osteoblasts is a key regulator of cranial skeletal development in mice. Development. 2009 doi: 10.1242/dev.027706. [DOI] [PubMed] [Google Scholar]
- 18.Tsukamoto M, et al. Findings as a starting point to unravel the underlying mechanisms of in vivo interactions involving Wnt10a in bone, fat and muscle. Bone. 2019 doi: 10.1016/j.bone.2018.10.009. [DOI] [PubMed] [Google Scholar]
- 19.Laine CM, et al. Novel mutations affecting LRP5 splicing in patients with osteoporosis-pseudoglioma syndrome (OPPG) Eur J Hum Genet. 2011 doi: 10.1038/ejhg.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gong Y et al (2001) LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell [DOI] [PubMed]
- 21.Hartikka H, et al. Heterozygous mutations in the LDL receptor-related protein 5 (LRP5) gene are associated with primary osteoporosis in children. J Bone Miner Res. 2005 doi: 10.1359/JBMR.050101. [DOI] [PubMed] [Google Scholar]
- 22.Korvala J, et al. Mutations in LRP5 cause primary osteoporosis without features of OI by reducing Wnt signaling activity. BMC Med Genet. 2012 doi: 10.1186/1471-2350-13-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boyden LM, et al. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002 doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 24.Roetzer KM, et al. Novel familial mutation of LRP5 causing high bone mass: genetic analysis, clinical presentation, and characterization of bone matrix mineralization. Bone. 2018 doi: 10.1016/j.bone.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 25.Whyte MP, et al. New explanation for autosomal dominant high bone mass: mutation of low-density lipoprotein receptor-related protein 6. Bone. 2019 doi: 10.1016/j.bone.2019.05.003. [DOI] [PubMed] [Google Scholar]
- 26.Winkler DG, et al. Osteocyte control of bone formation via sclerostin, a novel BMP antagonist. EMBO J. 2003 doi: 10.1093/emboj/cdg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brunkow ME, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001 doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balemans W, et al. Increased bone density in sclerosteosis is due to the deficiency of a novel secreted protein (SOST) Hum Mol Genet. 2001 doi: 10.1093/hmg/10.5.537. [DOI] [PubMed] [Google Scholar]
- 29.Balemans W, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002 doi: 10.1136/jmg.39.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Semënov M, Tamai K, He X. SOST is a ligand for LRP5/LRP6 and a Wnt signaling inhibitor. J Biol Chem. 2005 doi: 10.1074/jbc.M504308200. [DOI] [PubMed] [Google Scholar]
- 31.Li X, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005 doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 32.Leupin O, et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011 doi: 10.1074/jbc.M110.190330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fijalkowski I, et al. A novel domain-specific mutation in a sclerosteosis patient suggests a role of LRP4 as an anchor for sclerostin in human bone. J Bone Miner Res. 2016 doi: 10.1002/jbmr.2782. [DOI] [PubMed] [Google Scholar]
- 34.Boudin E, et al. The Lrp4R1170Q homozygous knock-in mouse recapitulates the bone phenotype of sclerosteosis in humans. J Bone Miner Res. 2017 doi: 10.1002/jbmr.3160. [DOI] [PubMed] [Google Scholar]
- 35.Bullock WA, et al. Lrp4 mediates bone homeostasis and mechanotransduction through interaction with sclerostin in vivo. iScience. 2019 doi: 10.1016/j.isci.2019.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strickland DK, Gonias SL, Argraves WS. Diverse roles for the LDL receptor family. Trends Endocrinol Metab. 2002 doi: 10.1016/S1043-2760(01)00526-4. [DOI] [PubMed] [Google Scholar]
- 37.Bourhis E, et al. Wnt antagonists bind through a short peptide to the first β-propeller domain of LRP5/6. Structure. 2011 doi: 10.1016/j.str.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 38.Kim J, et al. Sclerostin inhibits Wnt signaling through tandem interaction with two LRP6 ectodomains. Nat Commun. 2020 doi: 10.1038/s41467-020-19155-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Veverka V, et al. Characterization of the structural features and interactions of sclerostin. Molecular insight into a key regulator of Wnt-mediated bone formation. J Biol Chem. 2009 doi: 10.1074/jbc.M807994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holdsworth G, et al. Characterization of the interaction of sclerostin with the low density lipoprotein receptor-related protein (LRP) family of wnt co-receptors. J Biol Chem. 2012 doi: 10.1074/jbc.M112.350108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boschert V, et al. Mutational analysis of sclerostin shows importance of the flexible loop and the cystine-knot for Wnt-signaling inhibition. PLoS ONE. 2013 doi: 10.1371/journal.pone.0081710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jarmoskaite I, Alsadhan I, Vaidyanathan PP, Herschlag D. How to measure and evaluate binding affinities. Elife. 2020;9:1–34. doi: 10.7554/ELIFE.57264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Deventer JA, Kelly RL, Rajan S, Wittrup KD, Sidhu SS. A switchable yeast display/secretion system. Protein Eng Des Sel. 2015;28(10):317–325. doi: 10.1093/PROTEIN/GZV043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zahradník J, et al. SARS-CoV-2 RBD in vitro evolution follows contagious mutation spread, yet generates an able infection inhibitor. BioRxiv. 2021 doi: 10.1101/2021.01.06.425392. [DOI] [Google Scholar]
- 45.Bowen J, Schneible J, Bacon K, Labar C, Menegatti S, Rao BM. Screening of yeast display libraries of enzymatically treated peptides to discover macrocyclic peptide ligands. Int J Mol Sci. 2021;22:1634. doi: 10.3390/IJMS22041634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goel S, Chin EN, Fakhraldeen SA, Berry SM, Beebe DJ, Alexander CM. Both LRP5 and LRP6 receptors are required to respond to physiological Wnt ligands in mammary epithelial cells and fibroblasts. J Biol Chem. 2012 doi: 10.1074/jbc.M112.362137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cejka D, et al. Renal elimination of sclerostin increases with declining kidney function. J Clin Endocrinol Metab. 2014 doi: 10.1210/jc.2013-2786. [DOI] [PubMed] [Google Scholar]
- 48.Strohl WR. Fusion proteins for half-life extension of biologics as a strategy to make biobetters. BioDrugs. 2015 doi: 10.1007/s40259-015-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tu X, et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone. 2012 doi: 10.1016/j.bone.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang MK, et al. Disruption of Lrp4 function by genetic deletion or pharmacological blockade increases bone mass and serum sclerostin levels. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1413828111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krause C, et al. Distinct modes of inhibition by sclerostin on bone morphogenetic protein and Wnt signaling pathways. J Biol Chem. 2010 doi: 10.1074/jbc.M110.153890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chao G, Lau WL, Hackel BJ, Sazinsky SL, Lippow SM, Wittrup KD. Isolating and engineering human antibodies using yeast surface display. Nat Protoc. 2006 doi: 10.1038/nprot.2006.94. [DOI] [PubMed] [Google Scholar]
- 53.Lipovšek D, et al. Evolution of an interloop disulfide bond in high-affinity antibody mimics based on fibronectin type III domain and selected by yeast surface display: molecular convergence with single-domain camelid and shark antibodies. J Mol Biol. 2007 doi: 10.1016/j.jmb.2007.02.029. [DOI] [PubMed] [Google Scholar]
- 54.Xiong L, et al. Lrp4 in osteoblasts suppresses bone formation and promotes osteoclastogenesis and bone resorption. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1419714112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wehrli M, et al. Arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature. 2000;407(6803):527–530. doi: 10.1038/35035110. [DOI] [PubMed] [Google Scholar]
- 56.Li X, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008 doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 57.Lin C, et al. Sclerostin mediates bone response to mechanical unloading through antagonizing Wnt/β-catenin signaling. J Bone Miner Res. 2009 doi: 10.1359/jbmr.090411. [DOI] [PubMed] [Google Scholar]
- 58.Weatherbee SD, Anderson KV, Niswander LA. LDL-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development. 2006;133(24):4993–5000. doi: 10.1242/dev.02696. [DOI] [PubMed] [Google Scholar]
- 59.Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60(2):285–297. doi: 10.1016/j.neuron.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barik A, et al. LRP4 is critical for neuromuscular junction maintenance. J Neurosci. 2014;34(42):13892–13905. doi: 10.1523/JNEUROSCI.1733-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gautam M, Noakes P, Moscoso L (2021) F. R.-cell, and undefined 1996, Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Elsevier, New York. https://www.sciencedirect.com/science/article/pii/S0092867400812532. Accessed 27 Jun 2021 [DOI] [PubMed]
- 62.Holdsworth G, et al. Dampening of the bone formation response following repeat dosing with sclerostin antibody in mice is associated with up-regulation of Wnt antagonists. Bone. 2018 doi: 10.1016/j.bone.2017.11.003. [DOI] [PubMed] [Google Scholar]
- 63.Chan BY, et al. Increased chondrocyte sclerostin may protect against cartilage degradation in osteoarthritis. Osteoarthr Cartil. 2011 doi: 10.1016/j.joca.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 64.Bouaziz W, Funck-Brentano T, Lin H, Marty C, Hay E, Cohen-Solal M. Lack of sclerostin promotes osteoarthritis by activating canonical and non-canonical WNT pathways. Osteoarthr Cartil. 2014 doi: 10.1016/j.joca.2014.02.629. [DOI] [Google Scholar]
- 65.Chang JC, et al. SOST/sclerostin improves posttraumatic osteoarthritis and inhibits MMP2/3 expression after injury. J Bone Miner Res. 2018 doi: 10.1002/jbmr.3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li J, et al. SOST deficiency aggravates osteoarthritis in mice by promoting sclerosis of subchondral bone. Biomed Res Int. 2019 doi: 10.1155/2019/7623562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hudson BD, et al. SOST inhibits prostate cancer invasion. PLoS ONE. 2015 doi: 10.1371/journal.pone.0142058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Desjardins L, et al. Uremic toxicity and sclerostin in chronic kidney disease patients. Nephrol Ther. 2014 doi: 10.1016/j.nephro.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 69.Novo-Rodríguez C, et al. Circulating levels of sclerostin are associated with cardiovascular mortality. PLoS ONE. 2018 doi: 10.1371/journal.pone.0199504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kanbay M, et al. Serum sclerostin and adverse outcomes in nondialyzed chronic kidney disease patients. J Clin Endocrinol Metab. 2014 doi: 10.1210/jc.2014-2042. [DOI] [PubMed] [Google Scholar]
- 71.Viaene L, et al. Sclerostin: another bone-related protein related to all-cause mortality in haemodialysis? Nephrol Dial Transplant. 2013 doi: 10.1093/ndt/gft039. [DOI] [PubMed] [Google Scholar]
- 72.Brandenburg VM, et al. Relationship between sclerostin and cardiovascular calcification in hemodialysis patients: a cross-sectional study. BMC Nephrol. 2013 doi: 10.1186/1471-2369-14-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Drechsler C, et al. High levels of circulating sclerostin are associated with better cardiovascular survival in incident dialysis patients: results from the NECOSAD study. Nephrol Dial Transplant. 2015 doi: 10.1093/ndt/gfu301. [DOI] [PubMed] [Google Scholar]
- 74.Rosenfeld L, Shirian J, Zur Y, Levaot N, Shifman JM, Papo N. Combinatorial and computational approaches to identify interactions of macrophage colony-stimulating factor (M-CSF) and its receptor c-FMS. J Biol Chem. 2015 doi: 10.1074/jbc.M115.671271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rueden CT, et al. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017 doi: 10.1186/s12859-017-1934-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dyment NA, et al. High-throughput, multi-image cryohistology of mineralized tissues. J Vis Exp. 2016;2016(115):54468. doi: 10.3791/54468. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.
Data will be made available on reasonable request.