

Abstract
The exact etiology of Parkinson’s disease (PD) remains obscure, lacking effective diagnostic and prognostic biomarkers. In search of novel molecular factors that may contribute to PD pathogenesis, emerging evidence highlights the multifunctional role of the calcium-binding protein S100B that is widely expressed in the brain and predominantly in astrocytes. Preclinical evidence points towards the possible time-specific contributing role of S100B in the pathogenesis of neurodegenerative disorders including PD, mainly by regulating neuroinflammation and dopamine metabolism. Although existing clinical evidence presents some contradictions, estimation of S100B in the serum and cerebrospinal fluid seems to hold a great promise as a potential PD biomarker, particularly regarding the severity of motor and non-motor PD symptoms. Furthermore, given the recent development of S100B inhibitors that are able to cross the blood brain barrier, novel opportunities are arising in the research field of PD therapeutics. In this review, we provide an update on recent advances in the implication of S100B protein in the pathogenesis of PD and discuss relevant studies investigating the biomarker potential of S100B in PD, aiming to shed more light on clinical targeting approaches related to this incurable disorder.
Keywords: S100, α-Synuclein, Cytokines, Apoptosis, RAGE
Introduction
The exact etiology of Parkinson’s disease (PD), the most common neurodegenerative movement disorder, remains obscure, with limited diagnostic biomarkers and only symptomatic therapy till date [1]. PD is a clinically heterogeneous disorder, and prognostic biomarkers reflecting disease severity are lacking.
The pathological hallmarks of PD involve the progressive death of dopaminergic neurons in the substantia nigra pars compacta (SNpc), as well as the accumulation of Lewy bodies and Lewy neurites containing α-synuclein in the degenerative neurons [2, 3]. Emerging evidence highlights the multifunctional role of astrocytes in PD pathogenesis [4]. Following various brain insults, such as intoxication with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or 6-hydroxydopamine (6-OHDA) (the main toxins used for mimicking PD in vivo), astrocytes become functionally activated [5]. Reactive astrocytes can phagocytose dead neurons and endocytose α-synuclein in synapses [6]. Τhey can also release neurotrophic factors, such as glial-derived neurotrophic factor (GDNF) which significantly contributes to the survival of dopaminergic neurons [7], but also potentially neurotoxic factors, such as glutamate, cytokines and S100B, thereby mediating neuroinflammatory responses [8–11]. Astrocytes play also a fundamental role in blood–brain barrier (BBB) formation that is often disrupted in PD [12].
S100B is a calcium-binding protein, widely expressed in the brain predominantly by astrocytes, but also by maturing oligodendrocytes, neural progenitor cells, dendritic cells and specific lymphocyte subpopulations [13, 14]. S100B is an acidic 21 kDa protein, and probably the most well studied member of S100 proteins [15]. It is a homodimer, and each subunit contains two EF-hand calcium-binding domains [15]. Intracellularly, S100B can interact with more than 20 different proteins in a Ca2+ sensitive manner, and is implicated in calcium homeostasis, energy metabolism, cell proliferation, mobility and cytoskeletal regulation [13]. S100B can be released extracellularly and is considered as a marker of brain damage, acting as a damage-associated molecular pattern (DAMP) protein through its interaction with the receptor for advanced glycation end products (RAGE) [13, 16]. RAGE is a multi-ligand receptor of the immunoglobulin superfamily mainly expressed by neurons and microglia that mediates inflammatory responses by activating multiple signaling pathways [17, 18]. Apart from S100B, AGEs, high mobility group box 1 (HMGB1), amyloid fibrils and other S100 proteins can interact with RAGE [19]. High extracellular Ca2+ levels have been associated with increased formation of S100B multimers, which induce stabilization of RAGE oligomers or RAGE dimerization, an important process in RAGE-mediated signal transduction [14]. S100B/RAGE interaction can activate small GTPases, including Ras, Rac1, and Cdc42, leading to the activation of the transcription factors NF-κB and activating protein-1 (AP-1), resulting in the increased expression of cyclooxygenase-2 (COX-2), interleukin (IL)-1β, and tumor necrosis factor (TNF)-α [20, 21]. Additionally, RAGE activation via S100B has been shown to induce microglia activation and migration via the activation of Ras/Rac1-Cdc42/NF-κB, Ras/MEK/extracellular signal-regulated kinase 1/2 (ERK1/2)/NF-κB, Ras/Rac1-Cdc42/c-Jun NH2 terminal protein kinase (JNK)/AP-1, and Src/Ras/PI3K/RhoA/ROCK pathway. ROCK phosphorylates LIM kinase (LIMK) that is involved in F-actin stabilization and cell motility [20]. At pro-inflammatory concentrations, S100B can stimulate the RAGE-dependent activation and release of CCL3, CCL5 and CXCL12 chemokines by microglia (Fig. 1) [20]. At nanomolar concentrations, S100B can upregulate the anti-apoptotic factor Bcl-2, whereas at micromolar concentrations, S100B triggers apoptosis, in a RAGE-dependent manner in neurons [22]. On the other hand, trophic and detrimental effects of low and high S100B levels, respectively, may be independent of RAGE in other cell types, such as myoblasts [23–25].
Fig. 1.
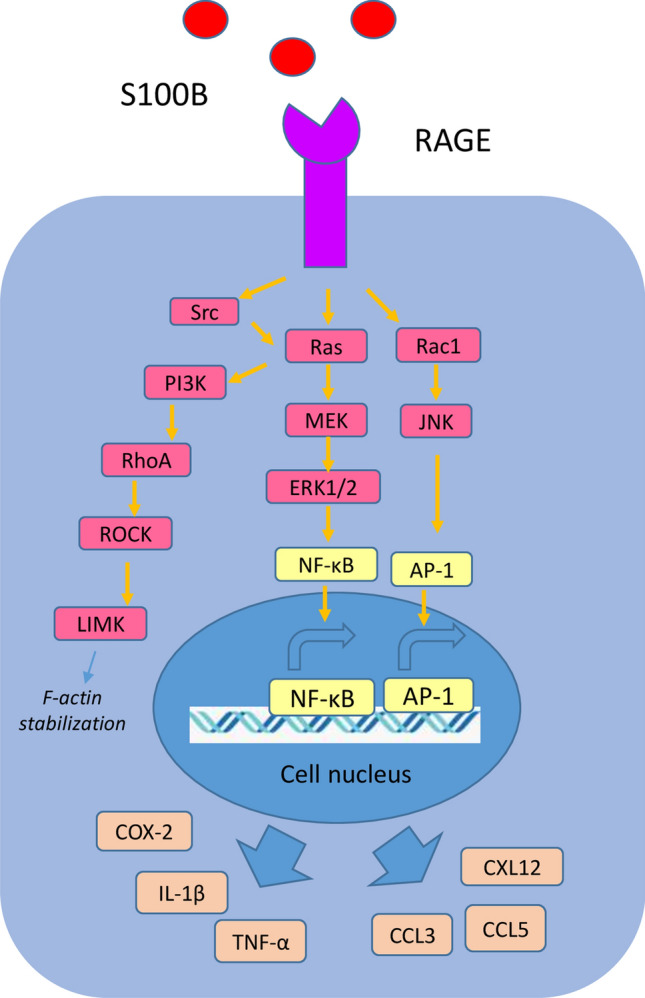
S100B/RAGE signaling. S100B/RAGE interaction activates small GTPases, such as Ras and Rac1, thereby activating the transcription factors NF-κB and activating protein-1 (AP-1), leading to the increased expression of cyclooxygenase-2 (COX-2), IL-1β, and TNF-α. S100B/RAGE interaction induces microglia activation and migration via the activation of Ras/Rac1-Cdc42/NF-κB, Ras/MEK/extracellular signal-regulated kinase 1/2 (ERK1/2)/NF-κB, Ras/Rac1-Cdc42/c-Jun NH2 terminal protein kinase (JNK)/AP-1, and Src/Ras/PI3K/RhoA/ROCK pathway. ROCK phosphorylates LIMK implicated in F-actin stabilization and cell motility. At pro-inflammatory concentrations, S100B has been also reported to stimulate the RAGE-dependent activation and release of CCL3, CCL5 and CXCL12 chemokines [14, 20]
At nanomolar concentrations, S100B has been shown to exert gliotrophic and neurotrophic roles by enhancing glial cell proliferation, neuronal survival, neurite outgrowth, synaptogenesis and neurogenesis, as well as by protecting neurons against toxic stimuli, delayed cellular damage after trauma and glutamate excitotoxicity [14, 26, 27]; however, at micromolar concentrations, it has been demonstrated to promote intracellular calcium overload, apoptosis, oxidative damage and excessive neuroinflammation by triggering the production of reactive oxygen species (ROS) and release of pro-inflammatory cytokines [14, 26, 27]. The specific role of S100B in the central nervous system (CNS) is still largely unknown. S100B expression has been detected elevated in the aging brain [14], and S100B dysregulation has been increasingly associated with various neurodegenerative diseases including Alzheimer’s disease (AD) and Parkinson’s disease (PD) [16]. For instance, at low doses, S100B protects neuroblastoma cells against Aβ-amyloid-induced neurotoxicity via RAGE, whereas at high doses, it enhances Aβ-induced neurotoxicity [27].
Although the role of S100B in neurological disorders has been already discussed [28], there is no recent review focusing on the role of S100B in PD. Given the controversial results of relevant studies and the increasing interest in the role of S100B in neurodegeneration, we critically discuss the accumulating preclinical and clinical evidence on the role of S100B in PD pathogenesis, its biomarker potential, as well as the therapeutic opportunities arising from the recent advances in the field.
The role of S100B in PD pathogenesis
Post-mortem human studies have revealed that S100B levels are increased in the substantia nigra of PD patients [8], as well as in the dorsomedial prefrontal cortex of patients with PD and multiple system atrophy (MSA, another synucleinopathy) at a transcriptional level, in comparison to controls [29], implying that S100B could represent a novel mediator of neurodegeneration in PD and paving the way for further exploration of its role in PD pathogenesis.
In vitro evidence has shown that MPTP enhanced the expression and release of S100B in C6 rat glioma cells co-cultured with neuronal PC12 cells, in a concentration- and time-dependent manner [30]. Elevation of S100B was associated with increased glial cell proliferation and reduced neuronal survival, while the pre-treatment of PC12 cells with S100B-antibodies prevented their death [30]. Interestingly, the administration of arachidonyl-2-chloroethylamide, a selective cannabinoid CB1 receptor agonist, inhibited the MPTP-induced glial cell proliferation, increased S100B expression and intracellular Ca2+ levels, as well as protected PC12 cells against apoptosis [30]. S100B and CB1 seem to exert opposing cellular effects. S100B results in Gs activation, as well as increased intracellular cyclic adenosine monophosphate (cAMP) and Ca2+ levels in neurons and glial cells [31], whereas CB1 receptor activation leads to Gi protein activation, reduced intracellular cAMP and decreased entry of Ca2+ into the cells [32]. CB1 receptor is increasingly gaining attention in PD research via yet unclear molecular mechanisms [33]. Another study has indicated that S100B is able to interact with the dopamine receptor D2 [15]. In addition, the co-expression of S100B and the D2 receptor increased D2 receptor-mediated activation of extracellular signal-regulated kinases (ERKs) and inhibition of adenylate cyclase in HEK293 cells [15]. S100B expression was detected inside the cultured neostriatal neurons expressing the D2 receptor, and their interaction (S100B/D2) was mediated via the third intracellular receptor loop (D2-IC3) [15], implying that intracellular S100B was responsible for these effects. However, in vivo evidence revealed that S100B is absent in dopaminergic neurons [8]. These discrepancies could be attributed to the fact that the results from the firstly mentioned study may pertain to cell lines, since many cell types in culture express S100B. Interestingly, however, forced expression of S100B in PC12 neuronal cells (a surrogate of noradrenergic neurons) has been demonstrated to result in enhanced proliferation and reduced responsiveness to nerve growth factor (NGF) via intracellular activation of Akt [34]. ERK1/2 signaling is highly implicated in various cellular processes underlying PD pathogenesis, including mitochondrial dysfunction, oxidative stress, neuronal apoptosis and inflammation [35]. Therefore, S100B seems to be a key player in endocannabinoid system and D2-mediated intracellular pathways in PD, although further research is needed to elucidate the exact molecular mechanisms involved.
In vivo evidence has shown that S100B is primarily localized in astrocytes and to a lesser extent in microglia, while it is absent in dopaminergic neurons [8]. Increased S100B secretion levels have been observed in the striatum of systematically MPTP and probenecid-treated mice [36], in the substantia nigra (SN) of MPTP-treated mice at both mRNA and protein levels [8], as well as in the striatum and SN of 6-OHDA-treated rats [37]. However, decreased striatal S100B levels have been also reported in intracerebroventricularly MPP+-treated mice [38], without evidence of significant alterations in the intracellular S100B levels in the striatum and substantia nigra of 6-OHDA-treated rats [39]. However, it is not completely clear whether the described effects of overexpressed S100B are due to the intracellular or extracellular S100B in the last two mentioned studies. Although S100B levels were initially increased in the striatum and SN upon MPTP treatment, their concentration tends to decrease the following days after the insult [8, 40, 41]. In accordance, an initial transitory increase of CSF S100B levels has been observed in 6-OHDA-treated rats, with 6-OHDA-treated astrocytes shown to exhibit increased S100B secretion in vitro [39]. Also, a significant reduction of S100B was demonstrated to follow the initial increase in the SN of MPTP-treated mice [42]. After 6-OHDA injection, striatal dopaminergic denervation has been accompanied by astrogliosis and S100B upregulation [10]. Inhibitory RAGE isoforms have been also observed in S100B-overexpressing astrocytes at an early stage after MPTP intoxication in mice [19], while striatal S100B levels were found elevated in pre-symptomatic MPTP-treated mice [43]. It has been proposed that the mechanical insult in the basal ganglia accompanying the intrastriatal intoxication of 6-OHDA could possibly explain the initial increase of S100B in this case [39]; however, the intraperitoneal MPTP injection has been associated with a transient S100B elevation in some of the abovementioned studies, thus weakening this hypothesis. The observed S100B reduction following the initial S100B increase could be potentially attributed to a compensatory process involving a self-inhibitory feedback loop [42], to protect the surviving neurons. Indeed, S100B gene expression can be suppressed by various negative regulatory elements in all cell types [14], suggesting that S100B expression is triggered by specific factors that counteract the function of the negative regulatory elements. When the neurotoxic effects of inducing factors such as MPTP or 6-OHDA are no longer active, S100B may, therefore, return to its baseline levels. S100B might also induce astrocytic apoptosis in an autocrine manner, thus leading to a decreased number of astrocytes as neuroinflammation progresses, lowering S100B levels [14, 44].
S100B has been demonstrated to significantly contribute to neurodegeneration in S100B-overexpressed or S100B-ablated animal models of PD. In particular, MPTP-induced dopaminergic degeneration has been shown to be partially inhibited in S100B-ablated mice [8]. S100B-mediated MPTP neurotoxicity involved RAGE upregulation, TNF-α release and enhanced microgliosis [8]. In accordance, S100B overexpression in transgenic mice has been shown to induce motor deficits similar to PD phenotype [45, 46] by suppressing the expression of G protein-coupled receptor kinase 2 (GRK2) and dopamine D2 receptor, thus affecting dopamine metabolism and promoting oxidative stress [45, 46]. S100B transgenic mice have been indicated to display impaired metabolism of several PD-related neurotransmitters, including Glu, GABA, Phe, His and Trp [47]. Hence, these findings suggest that constantly increased S100B levels may exert detrimental effects on neurons, whereas baseline S100B levels may be essential for normal neuronal function.
The loss of beneficial physiological functions such as release of neurotrophic and anti-inflammatory factors, and the gain of detrimental pro-inflammatory properties accompanying aberrant microglia activation contribute to PD pathogenesis [48]. RAGE activation via S100B can induce microglia activation and migration via the activation of Ras/Rac1-Cdc42/NF-κB, Ras/MEK/extracellular signal-regulated kinase 1/2 (ERK1/2)/NF-κB, Ras/Rac1-Cdc42/c-Jun NH2 terminal protein kinase (JNK)/AP-1, and Src/Ras/PI3K/RhoA/ROCK pathways [20, 21, 49, 50]. S100B has been shown to promote M1 and inhibit M2 polarization of microglia, respectively, via NF-κB, thus promoting cerebral ischemia [51]. However, the exact effects of extracellular S100B in microglia activation in PD still remains to be elucidated.
Collectively, the immediate astrocytic S100B upregulation after acute damage could reflect the initial reactive astrocytosis, which may trigger the activation of downstream factors, leading to the amplification of neuroinflammation, oxidative damage and dysregulation of neurotransmitter metabolism underlying PD pathogenesis (Fig. 2). Therefore, the role of S100B in PD pathogenesis seems to crucially depend not only on its concentration and specific animal model used, but also on the exact time of the disease course, highlighting the need for further studies to clarify the time-specific effects of S100B on PD.
Fig. 2.

Role of S100B in Parkinson’s disease (PD). S100B is mainly expressed by the astrocytes in the CNS. S100B-mediated MPTP neurotoxicity has been demonstrated to involve RAGE upregulation and secretion of pro-inflammatory cytokines, thereby enhancing neuroinflammation and oxidative damage. S100B has been shown to reduce the expression of dopamine D2 receptor, thereby affecting dopamine metabolism. S100B transgenic mice have been indicated to display impaired metabolism of several PD-related neurotransmitters, including Glu, GABA, Phe, His and Trp. Increased co-localization of S100B and A30P α-synuclein in astrocytes has been also observed in vivo, further supporting the implication of S100B in PD pathophysiology [8, 42, 43, 77]
S100B as a potential PD biomarker
Serum S100B levels have been elevated in patients with melanoma, acutely after brain damage, heart ischemia and intense physical exercise, potentially reflecting its release from damaged cells and entering into the peripheral circulation [14].
Clinical evidence points to the potential role of serum and CSF S100B as a PD biomarker. Although some studies have reported no differences in serum S100B levels between PD patients and controls [8, 52], serum S100B levels have been positively correlated with the severity of motor symptoms of PD patients, as assessed by Hoehn and Yahr (H&Y) and Activities of Daily Living (ADL) scales [52]. Overnight elevation of serum S100B levels has been further associated with increased disease severity and worse subjective sleep symptoms of patients with moderately advanced PD [53], implying that PD severity and non-restorative sleep may be related to sleep-related exaggerated brain inflammation or inadequate removal of potentially neurotoxic waste products. Deep brain stimulation (DBS) that is currently used for the treatment of advanced PD has been shown not to alter S100B levels in the serum of PD patients, suggesting that this surgical procedure may not affect serum S100B concentration [54]. Therefore, serum S100B might be a useful biomarker for disease severity or rate of progression of PD, potentially reflecting an “active”, pro-inflammatory molecular background of the disorder. However, given the elevation of S100B levels in other acute inflammatory conditions, caution is required for its specificity.
Additionally, serum autoantibodies against S100B (S100B-Abs) have been found four times higher in PD patients, 5 years after the diagnosis of the disease compared to controls, while they reached baseline levels at 10 years after PD diagnosis [55]. This biphasic pattern agrees with some of the abovementioned preclinical studies, reporting an initial transient increase of S100B in PD animal models and highlights the potential utility of S100B-Abs as a diagnostic tool at the initial stages of the disorder, which are diagnostically challenging. In accordance, increased CSF S100B levels have been detected in the early but not in late stages of Alzheimer’s disease [56]. However, the factor of age should be also carefully considered, given the fact that S100B levels are increased during normal aging [14]. Another study has demonstrated that serum S100B-Abs were higher in PD patients compared to controls [57], as well as in patients with PD dementia (PDD) and dementia with Lewy bodies (DLB) compared to AD, FTD, vascular dementia patients and controls [58], pointing towards a potential specificity of S100B for α-synucleinopathies, including PD. On the contrary, no significant differences in these autoantibodies have been detected between PD patients and controls, as well as in PD patients with and without dementia in another study [59].
In a large study of PD patients, S100B levels in the CSF were elevated compared to controls [8]. However, although increased CSF S100B levels have been demonstrated in patients with early PD (up to 36 months after clinical diagnosis) compared to controls, these results were not confirmed in an independent additional cohort [60]. In accordance, no significant differences have been reported in S100B levels of ventricular CSF derived from PD patients in a postmortem study [61]. Moreover, CSF S100B levels were higher in patients with PDD and DLB compared to controls [62]. While a gradual reduction of CSF S100B levels was also identified from DLB to PD, with PDD patients displaying intermediate values, no correlation was observed between S100B levels and the degree of cognitive impairment [62].
In summary, although existing evidence on the role of S100B as a PD biomarker are rather conflicting and the relevant results are characterized by relatively low reproducibility, S100B levels seem to be possibly associated with the severity of motor and non-motor PD symptoms, suggesting its potential efficacy as a marker of disease progression. Furthermore, S100B could facilitate the detection of PD endophenotypes, given the altered S100B levels in PD patients with dementia or sleep disorders. Since S100B levels are shown to differ significantly between PDD or DLB and other types of dementia, further studies are needed to validate this observed S100B specificity. In addition, given the fact that striatal S100B levels were found elevated in pre-symptomatic MPTP-treated mice [43], the potential serum or CSF S100B elevation in pre-symptomatic patients at risk for PD should be also further investigated. Finally, these controversial findings highlight the importance of the exact stage and severity of the disease that should be taken into consideration in further clinical studies investigating the role of S100B as a potential PD biomarker, since these factors could remarkably contribute to the observed differences.
S100B gene polymorphisms and PD risk
S100B gene is located on chromosome 21q22.2-q22.3 and consists of two exons [63]. Since the S100B rs3788266 variant has been associated with schizophrenia and psychosis in bipolar disorder [64, 65], it has been proposed that S100B gene variants may be implicated in the dopaminergic dysfunction accompanying PD.
A study screening a Chinese PD population for S100B gene polymorphisms associated with PD risk, detected two S100B variants, rs187503470 and rs1051169 [63] which, however, did not affect the amino acid sequence or splice site, suggesting that they may not be pathogenic mutations [63]. Moreover, no association was reported between the single-nucleotide polymorphism (SNP) rs3788266 of the S100B gene and PD occurrence [8]. Another recent study of a Swedish population has shown that the SNPs rs9722, rs881827, rs2239574, rs1051169 and rs9984765 of the S100B gene were not associated with PD development [66]. However, variants of rs9722, rs9984765, rs881827 and rs1051169 were indicated to be significantly more frequent in early-onset PD compared to late-onset, suggesting their potential role in determining the age of onset of the disease [66]. In particular, the higher frequency of the T allele of rs9722 in early-onset PD was confirmed in two independent Swedish cohorts in this study [66]. Importantly, healthy individuals carrying the T allele of rs9722, located in the 3′ untranslated region, displayed higher serum and frontal cortex S100B levels [67], implying a functional role of this variant, further supporting the detrimental role of S100B overexpression in PD development. These results suggest that S100B dysfunction may rather not be the initial cause of PD, but rather affect the rate of neurodegeneration and the “threshold” for the development of PD clinical symptoms. Early-onset and late-onset PD subtypes are characterized by clinical differences, potentially reflecting distinct pathophysiological backgrounds. Environmental factors, such as tobacco smoking and coffee consumption, have been shown to diversely affect the risk of PD development between these subgroups [68]. In this context, although existing evidence on the effects of smoking on S100B is limited [69], further studies could also examine these associations.
S100B as a potential therapeutic target against PD
Given the implication of S100B in PD pathogenesis, it has been proposed that it may represent a novel target for the development of treatment approaches. S100B neutralizing antibodies may represent a potentially relevant strategy, although there are no data regarding their use in clinical settings yet. At the transcriptional level, duloxetine, an antidepressant acting as a serotonin-norepinephrine reuptake inhibitor, has been identified as a transcriptional inhibitor of S100B [70]. Interestingly, duloxetine, which is able to cross BBB, has been demonstrated to exert antitumor properties in gliomas in vivo by downregulating S100B [70]. However, the selective serotonin reuptake inhibitor, fluoxetine, has been shown to stimulate S100B secretion from serotoninergic neurons, thus downregulating miR-16 in noradrenergic neurons, which consequently acquired properties of serotoninergic neurons [71]. Due to existing uncertainty of these results [72], the use of selective serotonin reuptake inhibitors to repress S100B expression should be viewed with caution. Intravenous administration of arundic acid, an agent indicated to inhibit astrocytic S100B biosynthesis at a transcriptional level, has shown promising results in cerebral ischemic brain damage in vivo [22, 73]. Oral administration of arundic acid to Tg2576 mouse models of AD has been demonstrated to ameliorate cerebral amyloidosis, reduce β-amyloid deposits and gliosis [74]. Importantly, in MPTP-treated mice, intraperitoneal administration of S100B has been indicated to protect dopaminergic neurons and ameliorate behavioral deficits [75]. Three distinct sites of S100B have been shown to bind to specific small molecule inhibitors, including SEN205A, compounds that covalently modify Cys84, and chlorpromazine that binds to site 1, 2 and 3 of S100B, respectively [76]. Currently, research efforts aim to improve the selectivity and bioavailability of these S100B inhibitors [76].
Pentamidine, an anti-protozoal agent that has been investigated in patients with melanoma in clinical trials, is an effective S100B inhibitor but with limited ability to cross BBB [77]. In a recent study of MPTP-treated mice, the intranasal delivery of chitosan-coated niosomes with entrapped pentamidine (inPentasomes) showed BBB penetration and ameliorated motor deficits as well as inhibited dopaminergic neuronal loss, RAGE/NF-κB pathway activation and subsequent neuroinflammation in the striatum and SN [77], paving the way for future studies investigating the potential S100B-targeted therapies against PD.
Given the fact that arachidonyl-2-chloroethylamide, a selective cannabinoid CB1 receptor agonist, inhibited the MPTP-induced increased S100B expression and neuronal apoptosis [30], non-psychotropic inhibitors of endocannabinoid metabolism, including AA-5-HT and VDM11 [30], could represent, novel tools against PD-related neurotoxicity.
Endurance exercise training has been associated with lower S100B expression in the striatum of PD mouse models [52]. Treatment with palmitoylethanolamide, a compound with anti-inflammatory effects mediated by PPAR-α receptor, has been shown to protect against dopaminergic loss and microglia activation in MPTP-treated mice, as well as inhibit S100B overexpression [78]. Oral treatment with chrysin, a natural flavonoid, has been indicated to protect against 6-OHDA-induced neurotoxicity and increase of S100B in the striatum of rats [79]. However, intraperitoneal treatment for 14 days with zonisamide, an antiepileptic agent that was shown to improve PD symptoms in clinical trials [80], has been demonstrated to be accompanied by increased astrocytic S100B levels in the basal ganglia of 6-OHDA-treated mice [81]. The combined treatment of the antioxidant N-acetylcysteine and the anti-inflammatory agent HA-1077 has been associated with enhanced dopaminergic death and astrocytic activation accompanied by elevated S100B expression in MPTP-treated mice [82]. Therefore, treatment approaches against PD may diversely affect S100B levels, probably depending on the specific molecular processes underlying each therapeutic strategy.
Final comments
Although the exact role of S100B in neurodegeneration still remains obscure, increasing evidence highlights the concentration-, time- and context-specific contribution of S100B to PD pathogenesis, possibly by regulating neuroinflammation, neurotransmitter metabolism and oxidative damage. S100B may not be a pathogenic factor specific to PD, since several studies have demonstrated its role in other neurodegenerative disorders such as AD, possibly suggesting that elevated levels of extracellular S100B in the brain might simply reflect ongoing inflammation and astrocyte activation. Although existing clinical evidence is conflicting, S100B seems to hold a promising potential as a PD biomarker, especially in terms of disease progression, severity and subtyping (early-onset versus late-onset PD, PD endophenotypes), as well as diagnosis at initial stages. However, concerns are raised in regard to its specificity and its altered levels during the course of the disease among others. Future studies with stratification of patients into subgroups (early-onset versus late-onset, early stages versus late stages of the disease, sporadic versus familial) may elucidate these unclear issues. Given the recent development of S100B inhibitors able to cross BBB, novel opportunities are also arising in the research field of PD therapeutics.
Another important aspect to be considered is the effects of levodopa or dopaminergic agonists administration on S100B levels, as PD patients at later disease stages probably use higher doses of these pharmaceutical agents [52] and levodopa usage has been indicated to enhance oxidative damage and inflammation [83].
Apart from RAGE, S100B can also bind to Toll-like receptors (TLRs) in the gut [84] and inflamed bronchi [85]. TLR-2 and TLR-4 have been shown upregulated in animal models of PD, and α-synuclein can act as a ligand for TLRs [86]. Although in general S100B/TLRs interaction has not been described in the brain, the potential role of the S100B/RAGE/TLRs axis in PD could be also explored.
Although MPTP and 6-OHDA have been widely used for mimicking PD in animal models, these neurotoxins induce rapid dopaminergic cell death, compared to the slower and progressive neuronal loss in humans [8]. Therefore, preclinical evidence obtaining from these experiments should be interpreted with caution. Increased co-localization of S100B and A30P α-synuclein in astrocytes has been also reported in transgenic mice expressing mutant A30P α-synuclein [87], further supporting the implication of S100B in PD pathogenesis. Nevertheless, the role of S100B should be further examined in genetic models of PD (SNCA, LRRK2), to better clarify its effects in PD pathogenesis.
Interestingly, S100B has been demonstrated to activate enteric glial cells and S100B overexpression has been associated with the initiation and maintenance of inflammatory response in the gut too [88]. Given the increasing evidence of the crucial involvement of the enteric glial cells in PD pathogenesis [89], the role of S100B as one of the molecules linking brain-gut communication should be further investigated.
Acknowledgements
YNP would like to acknowledge Monash University Malaysia for supporting with HDR Scholarship.
Abbreviations
- PD
Parkinson’s disease
- SNpc
Substantia nigra pars compacta
- MPTP
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- 6-OHDA
6-Hydroxydopamine
- GDNF
Glial-derived neurotrophic factor
- BBB
Blood–brain barrier
- DAMP
Damage-associated molecular pattern
- RAGE
Receptor for advanced glycation end products
- HMGB1
High mobility group box 1
- LIMK
LIM kinase
- AP-1
Activating protein-1
- COX-2
Cyclooxygenase-2
- TNF-α
Tumor necrosis factor- α
- CNS
Central nervous system
- ROS
Reactive oxygen species
- cAMP
Cyclic adenosine monophosphate
- ERKs
Extracellular signal-regulated kinases
- GRK2
G protein-coupled receptor kinase 2
- DBS
Deep brain stimulation
- SNP
Single-nucleotide polymorphism
Author contributions
EA carried out the literature review, conceptualized, and prepared the initial draft. YNP edited and contributed in the final manuscript. CP provided critical inputs, edited and contributed to the final version of the manuscript. All authors read and approved the final manuscript.
Funding
This research has not received any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Compliance with ethical standards
Conflict of interest
The authors have no conflict of interest to declare.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Radhakrishnan DM, Goyal V. Parkinson's disease: a review. Neurol India. 2018;66(Supplement):S26–S35. doi: 10.4103/0028-3886.226451. [DOI] [PubMed] [Google Scholar]
- 2.Angelopoulou E, Paudel YN, Piperi C. miR-124 and Parkinson's disease: a biomarker with therapeutic potential. Pharmacol Res. 2019;150:104515. doi: 10.1016/j.phrs.2019.104515. [DOI] [PubMed] [Google Scholar]
- 3.Angelopoulou E, Pyrgelis ES, Piperi C. Neuroprotective potential of chrysin in Parkinson's disease: molecular mechanisms and clinical implications. Neurochem Int. 2020;132:104612. doi: 10.1016/j.neuint.2019.104612. [DOI] [PubMed] [Google Scholar]
- 4.Booth HDE, Hirst WD, Wade-Martins R. The role of astrocyte dysfunction in Parkinson's disease pathogenesis. Trends Neurosci. 2017;40(6):358–370. doi: 10.1016/j.tins.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bahat-Stroomza M, et al. Induction of adult human bone marrow mesenchymal stromal cells into functional astrocyte-like cells: potential for restorative treatment in Parkinson's disease. J Mol Neurosci. 2009;39(1–2):199–210. doi: 10.1007/s12031-008-9166-3. [DOI] [PubMed] [Google Scholar]
- 6.Chung WS, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504(7480):394–400. doi: 10.1038/nature12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin LF, et al. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260(5111):1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- 8.Sathe K, et al. S100B is increased in Parkinson's disease and ablation protects against MPTP-induced toxicity through the RAGE and TNF-alpha pathway. Brain. 2012;135(Pt 11):3336–3347. doi: 10.1093/brain/aws250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morales I, et al. Striatal astrocytes engulf dopaminergic debris in Parkinson's disease: a study in an animal model. PLoS ONE. 2017;12(10):e0185989. doi: 10.1371/journal.pone.0185989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morales I, et al. The astrocytic response to the dopaminergic denervation of the striatum. J Neurochem. 2016;139(1):81–95. doi: 10.1111/jnc.13684. [DOI] [PubMed] [Google Scholar]
- 11.Niranjan R. The role of inflammatory and oxidative stress mechanisms in the pathogenesis of Parkinson's disease: focus on astrocytes. Mol Neurobiol. 2014;49(1):28–38. doi: 10.1007/s12035-013-8483-x. [DOI] [PubMed] [Google Scholar]
- 12.Gray MT, Woulfe JM. Striatal blood-brain barrier permeability in Parkinson's disease. J Cereb Blood Flow Metab. 2015;35(5):747–750. doi: 10.1038/jcbfm.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sorci G, et al. S100B protein, a damage-associated molecular pattern protein in the brain and heart, and beyond. Cardiovasc Psychiatry Neurol. 2010;2010:656481. doi: 10.1155/2010/656481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donato R, et al. S100B's double life: intracellular regulator and extracellular signal. Biochim Biophys Acta. 2009;1793(6):1008–1022. doi: 10.1016/j.bbamcr.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 15.Liu Y, Buck DC, Neve KA. Novel interaction of the dopamine D2 receptor and the Ca2+ binding protein S100B: role in D2 receptor function. Mol Pharmacol. 2008;74(2):371–378. doi: 10.1124/mol.108.044925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cristovao JS, Gomes CM. S100 proteins in Alzheimer's disease. Front Neurosci. 2019;13:463. doi: 10.3389/fnins.2019.00463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang X, et al. RAGE and its emerging role in the pathogenesis of Parkinson's disease. Neurosci Lett. 2018;672:65–69. doi: 10.1016/j.neulet.2018.02.049. [DOI] [PubMed] [Google Scholar]
- 18.Angelopoulou E, Piperi C, Papavassiliou AG. High-mobility group box 1 in Parkinson's disease: from pathogenesis to therapeutic approaches. J Neurochem. 2018;146(3):211–218. doi: 10.1111/jnc.14450. [DOI] [PubMed] [Google Scholar]
- 19.Viana SD, et al. Regulation of striatal astrocytic receptor for advanced glycation end-products variants in an early stage of experimental Parkinson's disease. J Neurochem. 2016;138(4):598–609. doi: 10.1111/jnc.13682. [DOI] [PubMed] [Google Scholar]
- 20.Bianchi R, et al. S100B protein stimulates microglia migration via RAGE-dependent up-regulation of chemokine expression and release. J Biol Chem. 2011;286(9):7214–7226. doi: 10.1074/jbc.M110.169342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bianchi R, et al. S100B binding to RAGE in microglia stimulates COX-2 expression. J Leukoc Biol. 2007;81(1):108–118. doi: 10.1189/jlb.0306198. [DOI] [PubMed] [Google Scholar]
- 22.Mori T, Asano T, Town T. Targeting S100B in cerebral ischemia and in Alzheimer's disease. Cardiovasc Psychiatry Neurol. 2010;2010:687067. doi: 10.1155/2010/687067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sorci G, et al. S100B causes apoptosis in a myoblast cell line in a RAGE-independent manner. J Cell Physiol. 2004;199(2):274–283. doi: 10.1002/jcp.10462. [DOI] [PubMed] [Google Scholar]
- 24.Riuzzi F, Sorci G, Donato R. S100B stimulates myoblast proliferation and inhibits myoblast differentiation by independently stimulating ERK1/2 and inhibiting p38 MAPK. J Cell Physiol. 2006;207(2):461–470. doi: 10.1002/jcp.20580. [DOI] [PubMed] [Google Scholar]
- 25.Riuzzi F, et al. S100B engages RAGE or bFGF/FGFR1 in myoblasts depending on its own concentration and myoblast density. Implications for muscle regeneration. PLoS ONE. 2012;7(1):e28700. doi: 10.1371/journal.pone.0028700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huttunen HJ, et al. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J Biol Chem. 2000;275(51):40096–40105. doi: 10.1074/jbc.M006993200. [DOI] [PubMed] [Google Scholar]
- 27.Businaro R, et al. S100B protects LAN-5 neuroblastoma cells against Abeta amyloid-induced neurotoxicity via RAGE engagement at low doses but increases Abeta amyloid neurotoxicity at high doses. J Neurosci Res. 2006;83(5):897–906. doi: 10.1002/jnr.20785. [DOI] [PubMed] [Google Scholar]
- 28.Michetti F, et al. The S100B story: from biomarker to active factor in neural injury. J Neurochem. 2019;148(2):168–187. doi: 10.1111/jnc.14574. [DOI] [PubMed] [Google Scholar]
- 29.Rydbirk R, et al. Cytokine profiling in the prefrontal cortex of Parkinson's disease and multiple system atrophy patients. Neurobiol Dis. 2017;106:269–278. doi: 10.1016/j.nbd.2017.07.014. [DOI] [PubMed] [Google Scholar]
- 30.Iuvone T, et al. Cannabinoid CB1 receptor stimulation affords neuroprotection in MPTP-induced neurotoxicity by attenuating S100B up-regulation in vitro. J Mol Med (Berl) 2007;85(12):1379–1392. doi: 10.1007/s00109-007-0233-y. [DOI] [PubMed] [Google Scholar]
- 31.Barger SW, Van Eldik LJ. S100 beta stimulates calcium fluxes in glial and neuronal cells. J Biol Chem. 1992;267(14):9689–9694. [PubMed] [Google Scholar]
- 32.Twitchell W, Brown S, Mackie K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol. 1997;78(1):43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- 33.Stampanoni Bassi M, et al. Cannabinoids in Parkinson's disease. Cannabis Cannabinoid Res. 2017;2(1):21–29. doi: 10.1089/can.2017.0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arcuri C, et al. S100B increases proliferation in PC12 neuronal cells and reduces their responsiveness to nerve growth factor via Akt activation. J Biol Chem. 2005;280(6):4402–4414. doi: 10.1074/jbc.M406440200. [DOI] [PubMed] [Google Scholar]
- 35.Bohush A, Niewiadomska G, Filipek A. Role of mitogen activated protein kinase signaling in Parkinson's disease. Int J Mol Sci. 2018;19(10):2973. doi: 10.3390/ijms19102973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Jarrah MD, Jamous M. Effect of endurance exercise training on the expression of GFAP, S100B, and NSE in the striatum of chronic/progressive mouse model of Parkinson's disease. NeuroRehabilitation. 2011;28(4):359–363. doi: 10.3233/NRE-2011-0664. [DOI] [PubMed] [Google Scholar]
- 37.Gomide V, Chadi G. Glial bFGF and S100 immunoreactivities increase in ascending dopamine pathways following striatal 6-OHDA-induced partial lesion of the nigrostriatal system: a sterological analysis. Int J Neurosci. 2005;115(4):537–555. doi: 10.1080/00207450590521064. [DOI] [PubMed] [Google Scholar]
- 38.Cunha MP, et al. MPP(+)-lesioned mice: an experimental model of motor, emotional, memory/learning, and striatal neurochemical dysfunctions. Mol Neurobiol. 2017;54(8):6356–6377. doi: 10.1007/s12035-016-0147-1. [DOI] [PubMed] [Google Scholar]
- 39.Batassini C, et al. Striatal injury with 6-OHDA transiently increases cerebrospinal GFAP and S100B. Neural Plast. 2015;2015:387028. doi: 10.1155/2015/387028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muramatsu Y, et al. Cerebral alterations in a MPTP-mouse model of Parkinson's disease—an immunocytochemical study. J Neural Transm (Vienna) 2003;110(10):1129–1144. doi: 10.1007/s00702-003-0021-y. [DOI] [PubMed] [Google Scholar]
- 41.Muramatsu Y, et al. Expression of S-100 protein is related to neuronal damage in MPTP-treated mice. Glia. 2003;42(3):307–313. doi: 10.1002/glia.10225. [DOI] [PubMed] [Google Scholar]
- 42.Teismann P, et al. Receptor for advanced glycation endproducts (RAGE) deficiency protects against MPTP toxicity. Neurobiol Aging. 2012;33(10):2478–2490. doi: 10.1016/j.neurobiolaging.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Viana SD, et al. Presymptomatic MPTP mice show neurotrophic S100B/mRAGE striatal levels. CNS Neurosci Ther. 2016;22(5):396–403. doi: 10.1111/cns.12508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu J, Van Eldik LJ. S100 beta induces apoptotic cell death in cultured astrocytes via a nitric oxide-dependent pathway. Biochim Biophys Acta. 1996;1313(3):239–245. doi: 10.1016/0167-4889(96)00095-x. [DOI] [PubMed] [Google Scholar]
- 45.Liu J, et al. S100B transgenic mice develop features of Parkinson's disease. Arch Med Res. 2011;42(1):1–7. doi: 10.1016/j.arcmed.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 46.Liu J, et al. Preliminary analysis of parkinson-like motor coordination abnormityin brain-specific hS100B transgenic mice. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2017;39(2):240–246. doi: 10.3881/j.issn.1000-503X.2017.02.013. [DOI] [PubMed] [Google Scholar]
- 47.Liu JL, et al. Metabonomics study of brain-specific human S100B transgenic mice by using high-performance liquid chromatography coupled with quadrupole time of flight mass spectrometry. Biol Pharm Bull. 2011;34(6):871–876. doi: 10.1248/bpb.34.871. [DOI] [PubMed] [Google Scholar]
- 48.Lecours C, et al. Microglial implication in Parkinson's disease: loss of beneficial physiological roles or gain of inflammatory functions? Front Cell Neurosci. 2018;12:282. doi: 10.3389/fncel.2018.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bianchi R, Giambanco I, Donato R. S100B/RAGE-dependent activation of microglia via NF-kappaB and AP-1 Co-regulation of COX-2 expression by S100B, IL-1beta and TNF-alpha. Neurobiol Aging. 2010;31(4):665–677. doi: 10.1016/j.neurobiolaging.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 50.Adami C, et al. S100B expression in and effects on microglia. Glia. 2001;33(2):131–142. [PubMed] [Google Scholar]
- 51.Zhou S, et al. S100B promotes microglia M1 polarization and migration to aggravate cerebral ischemia. Inflamm Res. 2018;67(11–12):937–949. doi: 10.1007/s00011-018-1187-y. [DOI] [PubMed] [Google Scholar]
- 52.Schaf DV, et al. S100B and NSE serum levels in patients with Parkinson's disease. Parkinsonism Relat Disord. 2005;11(1):39–43. doi: 10.1016/j.parkreldis.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 53.Carvalho DZ, et al. Overnight S100B in Parkinson's disease: a glimpse into sleep-related neuroinflammation. Neurosci Lett. 2015;608:57–63. doi: 10.1016/j.neulet.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 54.Themistocleous MS, et al. The insertion of electrodes in the brain for electrophysiological recording or chronic stimulation is not associated with any biochemically detectable neuronal injury. Neuromodulation. 2017;20(5):424–428. doi: 10.1111/ner.12598. [DOI] [PubMed] [Google Scholar]
- 55.Gruden MA, et al. Immunoprotection against toxic biomarkers is retained during Parkinson's disease progression. J Neuroimmunol. 2011;233(1–2):221–227. doi: 10.1016/j.jneuroim.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 56.Peskind ER, et al. Cerebrospinal fluid S100B is elevated in the earlier stages of Alzheimer's disease. Neurochem Int. 2001;39(5–6):409–413. doi: 10.1016/s0197-0186(01)00048-1. [DOI] [PubMed] [Google Scholar]
- 57.Wilhelm KR, et al. Immune reactivity towards insulin, its amyloid and protein S100B in blood sera of Parkinson's disease patients. Eur J Neurol. 2007;14(3):327–334. doi: 10.1111/j.1468-1331.2006.01667.x. [DOI] [PubMed] [Google Scholar]
- 58.Maetzler W, et al. Autoantibodies against amyloid and glial-derived antigens are increased in serum and cerebrospinal fluid of Lewy body-associated dementias. J Alzheimers Dis. 2011;26(1):171–179. doi: 10.3233/JAD-2011-110221. [DOI] [PubMed] [Google Scholar]
- 59.Maetzler W, et al. Comparable autoantibody serum levels against amyloid- and inflammation-associated proteins in Parkinson's disease patients and controls. PLoS ONE. 2014;9(2):e88604. doi: 10.1371/journal.pone.0088604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dos Santos MCT, et al. Evaluation of cerebrospinal fluid proteins as potential biomarkers for early stage Parkinson's disease diagnosis. PLoS ONE. 2018;13(11):e0206536. doi: 10.1371/journal.pone.0206536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maarouf CL, et al. Quantitative appraisal of ventricular cerebrospinal fluid biomarkers in neuropathologically diagnosed Parkinson's disease cases lacking Alzheimer's disease pathology. Biomark Insights. 2013;8:19–28. doi: 10.4137/BMI.S11422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gmitterova K, et al. Cerebrospinal fluid markers analysis in the differential diagnosis of dementia with Lewy bodies and Parkinson's disease dementia. Eur Arch Psychiatry Clin Neurosci. 2018;270(4):461–470. doi: 10.1007/s00406-018-0928-9. [DOI] [PubMed] [Google Scholar]
- 63.Guo Y, et al. Genetic analysis of the S100B gene in Chinese patients with Parkinson disease. Neurosci Lett. 2013;555:134–136. doi: 10.1016/j.neulet.2013.09.037. [DOI] [PubMed] [Google Scholar]
- 64.Liu J, et al. SNPs and haplotypes in the S100B gene reveal association with schizophrenia. Biochem Biophys Res Commun. 2005;328(1):335–341. doi: 10.1016/j.bbrc.2004.12.175. [DOI] [PubMed] [Google Scholar]
- 65.Roche S, et al. Candidate gene analysis of 21q22: support for S100B as a susceptibility gene for bipolar affective disorder with psychosis. Am J Med Genet B Neuropsychiatr Genet. 2007;144B(8):1094–1096. doi: 10.1002/ajmg.b.30556. [DOI] [PubMed] [Google Scholar]
- 66.Fardell C, et al. S100B polymorphisms are associated with age of onset of Parkinson's disease. BMC Med Genet. 2018;19(1):42. doi: 10.1186/s12881-018-0547-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hohoff C, et al. Risk variants in the S100B gene predict elevated S100B serum concentrations in healthy individuals. Am J Med Genet B Neuropsychiatr Genet. 2010;153B(1):291–297. doi: 10.1002/ajmg.b.30950. [DOI] [PubMed] [Google Scholar]
- 68.Angelopoulou E, et al. The relationship between environmental factors and different Parkinson's disease subtypes in Greece: data analysis of the Hellenic Biobank of Parkinson's disease. Parkinsonism Relat Disord. 2019;67:105–112. doi: 10.1016/j.parkreldis.2019.08.013. [DOI] [PubMed] [Google Scholar]
- 69.Kahyaoglu I, et al. Umbilical CORD S100B levels in active and passive smoker women. Eur Rev Med Pharmacol Sci. 2014;18(5):723–727. [PubMed] [Google Scholar]
- 70.Gao H, et al. S100B suppression alters polarization of infiltrating myeloid-derived cells in gliomas and inhibits tumor growth. Cancer Lett. 2018;439:91–100. doi: 10.1016/j.canlet.2018.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baudry A, et al. miR-16 targets the serotonin transporter: a new facet for adaptive responses to antidepressants. Science. 2010;329(5998):1537–1541. doi: 10.1126/science.1193692. [DOI] [PubMed] [Google Scholar]
- 72.Donato R, et al. Functions of S100 proteins. Curr Mol Med. 2013;13(1):24–57. [PMC free article] [PubMed] [Google Scholar]
- 73.Matsui T, et al. Astrocytic activation and delayed infarct expansion after permanent focal ischemia in rats. Part I: enhanced astrocytic synthesis of s-100beta in the periinfarct area precedes delayed infarct expansion. J Cereb Blood Flow Metab. 2002;22(6):711–722. doi: 10.1097/00004647-200206000-00010. [DOI] [PubMed] [Google Scholar]
- 74.Hsiao K, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274(5284):99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 75.Kato H, et al. Arundic acid, an astrocyte-modulating agent, protects dopaminergic neurons against MPTP neurotoxicity in mice. Brain Res. 2004;1030(1):66–73. doi: 10.1016/j.brainres.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 76.Bresnick AR. S100 proteins as therapeutic targets. Biophys Rev. 2018;10(6):1617–1629. doi: 10.1007/s12551-018-0471-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rinaldi F, et al. inPentasomes: An innovative nose-to-brain pentamidine delivery blunts MPTP parkinsonism in mice. J Control Release. 2019;294:17–26. doi: 10.1016/j.jconrel.2018.12.007. [DOI] [PubMed] [Google Scholar]
- 78.Esposito E, et al. Neuroprotective activities of palmitoylethanolamide in an animal model of Parkinson's disease. PLoS ONE. 2012;7(8):e41880. doi: 10.1371/journal.pone.0041880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goes ATR, et al. Protective role of chrysin on 6-hydroxydopamine-induced neurodegeneration a mouse model of Parkinson's disease: involvement of neuroinflammation and neurotrophins. Chem Biol Interact. 2018;279:111–120. doi: 10.1016/j.cbi.2017.10.019. [DOI] [PubMed] [Google Scholar]
- 80.Bermejo PE, Anciones B. A review of the use of zonisamide in Parkinson's disease. Ther Adv Neurol Disord. 2009;2(5):313–317. doi: 10.1177/1756285609338501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Asanuma M, et al. Neuroprotective effects of zonisamide target astrocyte. Ann Neurol. 2010;67(2):239–249. doi: 10.1002/ana.21885. [DOI] [PubMed] [Google Scholar]
- 82.Gil-Martinez AL, et al. Unexpected exacerbation of neuroinflammatory response after a combined therapy in old parkinsonian mice. Front Cell Neurosci. 2018;12:451. doi: 10.3389/fncel.2018.00451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dorszewska J, et al. Molecular effects of l-dopa therapy in Parkinson's disease. Curr Genom. 2014;15(1):11–17. doi: 10.2174/1389202914666131210213042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Esposito G, et al. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-alpha activation. Gut. 2014;63(8):1300–1312. doi: 10.1136/gutjnl-2013-305005. [DOI] [PubMed] [Google Scholar]
- 85.Sorci G, et al. The danger signal S100B integrates pathogen- and danger-sensing pathways to restrain inflammation. PLoS Pathog. 2011;7(3):e1001315. doi: 10.1371/journal.ppat.1001315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kouli A, Horne CB, Williams-Gray CH. Toll-like receptors and their therapeutic potential in Parkinson's disease and alpha-synucleinopathies. Brain Behav Immun. 2019;81:41–51. doi: 10.1016/j.bbi.2019.06.042. [DOI] [PubMed] [Google Scholar]
- 87.Marxreiter F, et al. Glial A30P alpha-synuclein pathology segregates neurogenesis from anxiety-related behavior in conditional transgenic mice. Neurobiol Dis. 2013;59:38–51. doi: 10.1016/j.nbd.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cirillo C, et al. S100B protein in the gut: the evidence for enteroglial-sustained intestinal inflammation. World J Gastroenterol. 2011;17(10):1261–1266. doi: 10.3748/wjg.v17.i10.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Santos SF, et al. The gut and Parkinson's disease—a bidirectional pathway. Front Neurol. 2019;10:574. doi: 10.3389/fneur.2019.00574. [DOI] [PMC free article] [PubMed] [Google Scholar]