

Abstract
Bile acid biosynthesis is subjected to feedback regulation whereby bile acids down-regulate their own synthesis. The major point of this regulation is at the level of cholesterol 7α-hydroxylase (7α-hydroxylase), which controls bile acid output from the classic pathway. This regulation is at the level of transcription of the gene. Two bile acid response elements have been localized within the 5′-flanking region of the rat gene and these elements overlap three nuclear receptor binding sites for hepatocyte nuclear factor (HNF-4), liver X receptor (LXR) and α1-fetoprotein transcription factor (FTF). Recently it has been shown that bile acids are physiological ligands for the farnesyl X receptor (FXR), which suggested that FXR could function by binding to one of the three nuclear receptor sites to mediate regulation of 7α-hydroxylase transcription by bile acids. In this study we show that FXR is indeed a crucial factor for bile acid-mediated regulation, but that it functions without binding to DNA. Furthermore, we also demonstrate that neither the LXR nor the HNF-4 sites are involved in bile acid-mediated regulation of 7α-hydroxylase transcription. Most importantly, we show that the FTF site is essential for regulation of 7α-hydroxylase by bile acids, similar to what we have recently demonstrated for another gene of the bile acid biosynthetic pathway, the sterol 12α-hydroxylase gene. These studies demonstrate the crucial role of FTF in the expression and regulation of a critical gene in the bile acid biosynthetic pathways.
INTRODUCTION
Cholesterol conversion to bile acids occurs via the ‘classic’ (neutral) or the ‘alternative’ (acidic) bile acid biosynthesis pathways (1). Cholic acid and chenodeoxycholic acid are the end products of these pathways and the major primary bile acids found in most vertebrates. Cholic acid is hydroxylated at position 12α whereas chenodeoxycholic acid is not. There are three enzymes that play major regulatory roles in these two pathways. Cholesterol 7α-hydroxylase/CYP7a1 (7α-hydroxylase1) is the rate limiting enzyme in the classic pathway. Sterol 27-hydroxylase/CYP27 (27-hydroxylase) is the first enzyme in the alternative pathway. Sterol 12α-hydroxylase/CYP8b1 (12α-hydroxylase) is the specific enzyme for cholic acid synthesis and determines the ratio of cholic acid to chenodeoxycholic acid and thus the hydrophobicity of the circulating pool.
Bile acids exert negative feedback regulation on their own synthesis (2). Interruption of the enterohepatic circulation, by biliary diversion or by feeding bile acid-binding resins (cholestyramine), enhances cholesterol and bile acid synthesis (3). A major point of this regulation is at the level of 7α-hydroxylase. Our laboratory has demonstrated that bile acids negatively regulate transcription of the 7α-hydroxylase gene, which controls output from the classic pathway (4). Two bile acid response elements (BARE) have been localized within the 5′-flanking region of the rat gene (5,6). Bile acids also down-regulate transcription of the 27-hydroxylase gene (7) and it has been reported that hepatocyte nuclear factor 1 (HNF-1) is involved in this regulation (8).
Recently it has been shown that bile acids are physiological ligands for the farnesyl X receptor (FXR), an orphan nuclear receptor (9–11). FXR binds to an inverted repeat of 6 nt separated by 1 nt (IR-1) (12) and it was originally found to bind farnesol, a precursor of cholesterol and hence of bile acids. Transcriptional activation of the intestinal bile acid-binding protein (I-BABP) was shown to require binding of bile acid-activated FXR to an IR-1 sequence (9). In contrast, a modest repression (∼2-fold) of the 7α-hydroxylase promoter by bile acids occurred only when FXR was overexpressed, using a hepatocyte-derived cell line (HepG-2) as host. For 7α-hydroxylase, however, no DNA sequence has been identified that binds FXR and mediates this effect and the molecular mechanisms involved in bile acid-mediated repression of gene transcription remain unknown.
Several nuclear receptors have been reported to bind to the rat 7α-hydroxylase promoter. Liver enriched receptor (LXR), a nuclear receptor that binds oxysterols, has been shown to mediate the cholesterol-mediated induction of rat and mouse 7α-hydroxylase transcription upon binding to a direct repeat sequence separated by 4 nt (DR-4) (13–15). This LXR element overlaps BARE I (6). It has been suggested that FXR might mediate bile acid-mediated suppression of 7α-hydroxylase transcription, either by binding to the same element or by interfering with binding of LXR (11). However, the human 7α-hydroxylase promoter does not have a LXR element (16), but bile acids still down-regulate 7α-hydroxylase transcription. In addition, HNF-4 and COUP-TFII bind sequences overlapping BARE II (5,17) and COUP-TFII has also been described to bind sequences overlapping the LXR site (18,19), but the biological role of binding of these receptors to the rat 7α-hydroxylase promoter is unclear.
More recently it was shown that a transcriptional factor, named CYP7A promoter-binding factor (CPF), is required for expression of the 7α-hydroxylase gene (20). CPF was previously isolated as α1-fetoprotein transcription factor (FTF) (21) and throughout this study we use both names, CPF and FTF, interchangeably. This factor is a member of the Ftz-F1 family of the class IV orphan nuclear receptor superfamily (22). CPF binds to the region of the 7α-hydroxylase promoter previously characterized as BARE II (6). Recently our laboratory has also characterized two DNA elements that bind FTF and are required for both expression and bile acid-mediated regulation of the 12α-hydroxylase gene (23).
In spite of localization of these two BARE and nuclear receptor elements, the mechanism for bile acid-mediated regulation of 7α-hydroxylase transcription remains unknown. In this study we have characterized, at the nucleotide level, the three nuclear receptor elements located in the rat 7α-hydroxylase promoter. We show that FXR enhances bile acid-mediated suppression of the 7α-hydroxylase promoter without binding to the DNA and that binding of FTF/CPF is crucial not only to render a fully active 7α-hydroxylase promoter but, most importantly, for bile acid-mediated regulation of its transcription.
MATERIALS AND METHODS
Materials
Reagents used in DNA cloning and sequencing were from New England Biolabs, Boehringer Mannheim, US Biochemical Corp. or Gibco BRL. Common laboratory chemicals were from Fisher, Sigma or Bio-Rad. The luciferase promoter-less vector pGL3-Basic was purchased from Promega. Oligonucleotides were prepared in the Medical College of Virginia DNA Synthesis Facility by the phosphoramidite method on an automated DNA synthesizer. pRSV-FXR, an expression plasmid containing the rat FXR cDNA in the expression vector pRSV, was a gift from Dr Cary Weinberger. pRSV-GHR contains the rabbit growth hormone receptor (GHR) cDNA. pCI-FTF, an expression plasmid containing the human FTF cDNA in the expression vector pCI (Promega), was a generous gift from Dr Luc Bélanger (21). pCMX, a plasmid for expression in mammalian cells and in vitro, contains the CMV and T7 promoters. This plasmid, as well as pCMX-rFXR and pCMX-hRXR, were a gift from Dr Ronald M. Evans.
Preparation of chimeric 7α-hydroxylase promoter–luciferase reporter constructs
Standard recombinant DNA procedures were carried out essentially as described previously (4). pGL3R7α-342 was prepared by PCR and contains nucleotides –342 to +59 ligated into the SmaI site of pGL3-Basic (Promega). Mutation constructs were generated by oligonucleotide-directed mutagenesis in M13 (24) or using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). All constructs were confirmed by DNA sequencing.
Transient transfection and luciferase assays
Rat primary hepatocytes were prepared as previously described (24). Six hours after plating, hepatocytes were transfected with Lipofectin (Gibco) using 1.5 µg total DNA on 35 mm plates. An aliquot of 40 ng test plasmid, 5 ng pCMV-Gal [a plasmid containing the human cytomegalovirus (CMV) promoter in front of the bacterial β-galactosidase gene], to normalize for transfection efficiencies, and 5 ng pRSV-FXR were used unless indicated otherwise. After 16 h the DNA was removed and, where indicated, 50 µM taurocholic acid (TCA) was added. Cells were harvested 24 h later and luciferase and β-galactosidase assays were performed with a kit from Tropix (Bedford, MA), according to the manufacturer’s protocol. All transfections were performed in duplicate. Background activity (pGL3-Basic) was subtracted in each case. Average values are for the number of experiments indicated.
In vitro transcription/translation and electrophoretic mobility shift analysis
Transcription/translation of cDNAs encoding FXR, RXR, LXR, FTF or GHR, as a control, was performed using the TNT T7-coupled rabbit reticulocyte lysate system according to the manufacturer’s protocol (Promega).
DNA binding reactions were set up in 50 mM KCl, 20 mM Tris–HCl, pH 8.0, 0.2 mM EDTA, 4% Ficoll, 1.0 µg poly(dI-dC), 4 µl of translated protein and a 1500-fold molar excess of an irrelevant single-stranded DNA in a final volume of 20 µl on ice. After 15 min incubation, 320 fmol of the indicated 32P-labeled DNA probes (∼2 × 105 c.p.m.) were added. All probes were adjusted to the same specific radioactivity. After incubation for 20 min on ice, samples were loaded onto a 4.5% polyacrylamide gel and subjected to electrophoresis at 4°C. Gels were dried and exposed to XAR-5 film (Kodak).
RESULTS
In order to study the molecular mechanisms involved in bile acid-mediated regulation of 7α-hydroxylase transcription, we first needed to optimize the extent of regulation. We co-transfected a chimeric gene containing 342 nt from the rat 7α-hydroxylase promoter in front of the luciferase gene as a reporter (pGL3R7α-342), with or without an expression plasmid containing rat FXR cDNA. We used rat primary hepatocytes, which have been shown to regulate expression of the endogenous 7α-hydroxylase gene by TCA, as the cell host (25).
When FXR was not co-transfected, TCA-dependent suppression of 7α-hydroxylase promoter activity was only 2-fold (Fig. 1A). Co-transfection of FXR increased the suppression by TCA up to 5-fold in a concentration-dependent manner. This result suggested that FXR is involved in the regulation of 7α-hydroxylase transcription by bile acids and that in primary hepatocytes FXR is in limiting amounts. To further prove this, we performed a test plasmid concentration curve experiment in the absence and presence of the FXR expression plasmid and quantified the extent of regulation by TCA. Figure 1B shows that in the absence of exogenous FXR, 7α-hydroxylase promoter activity is regulated up to 4-fold when low quantities (≤50 ng) of reporter construct where transfected. Increasing the reporter construct level to 1.5 µg virtually eliminated the observed regulation. However, when a FXR expression plasmid was co-transfected, bile acid-dependent down-regulation was >5-fold, even when high amounts of the reporter construct were used.
Figure 1.


FXR overexpression enhances bile acid-mediated regulation of the CYP7A1 promoter. (A) Male rat primary hepatocytes were transfected with 0.5 µg of pGL3/R7α-342 in the presence of pRSV/FXR or pRSV/GHR and treated with the indicated amounts of TCA as described in Materials and Methods. Relative transcription was determined by normalizing luciferase activity to β-galactosidase activity. The data were normalized to the activity produced by the construct in the absence of TCA. Results of a typical experiment are shown. (B) Male rat primary hepatocytes were co-transfected with the indicated amounts of pGL3/R7α-342 (20, 50, 500 or 1500 ng) with or without pRSV/FXR as indicated. Transfected cells were incubated with and without TCA (50 µM final concentration) and harvested after 24 h. Each value represents the percentage of promoter activity in the presence of TCA as compared to promoter activity in the absence of TCA. Values represent the averages of two experiments ± SD.
Figure 2 shows the nucleotide sequence of the 7α-hydroxylase proximal promoter and highlights the positions of the previously characterized nuclear receptor sites (DR-1, CPF and LXR) and the two BARE sites that overlap the receptor elements. Previously reported studies have shown that only these regions of the 7α-hydroxylase promoter are important for bile acid-mediated regulation (5,6). To study the role of these DNA elements, we introduced specific mutations in these sites and studied their effects on promoter activity, regulation by bile acids and binding to the corresponding nuclear receptors.
Figure 2.

Sequence of the rat 7α-hydroxylase promoter. The first 160 nt of the rat 7α-hydroxylase are shown. The two previously proposed BARE elements are shown in bold and underlined. The LXR, HNF4 and CPF/FTF sites and the TATA box are shown in boxes.
First, we studied the LXR site (–49 to –72), which overlaps the previously described BARE I, using two mutations that we created (Fig. 3A). pGL3R7α-342/LXRm1,2 mutates the two repeats of the LXR site, whereas pGL3R7α-342/DR1 converted the DR-4 site into a DR-1 site by deleting 3 nt. Neither mutation decreased promoter activity. In fact, pGL3R7α-342/DR1 had significantly higher activity than the wild-type, perhaps by making it a target for another nuclear receptor with higher activation potential than LXR, such as HNF-4. Most importantly, regulation by bile acids of these two promoter mutants was essentially identical to the wild-type promoter. To characterize the altered binding of LXR to these two mutants, we performed gel retardation assays using the wild-type and mutant probes and in vitro made LXR/RXR. Figure 3B shows that, as expected, the wild-type 7α-hydroxylase probe containing the LXR element binds the dimer LXR/RXR (Fig. 3B, lane 2). LXR or RXR alone did not bind (data not shown). The two mutants did not bind LXR/RXR (Fig. 3B, lanes 4 and 6). As a negative control we used in vitro made GHR (Fig. 3B, lanes 1, 3 and 5), which, as expected, did not bind any of the probes.
Figure 3.

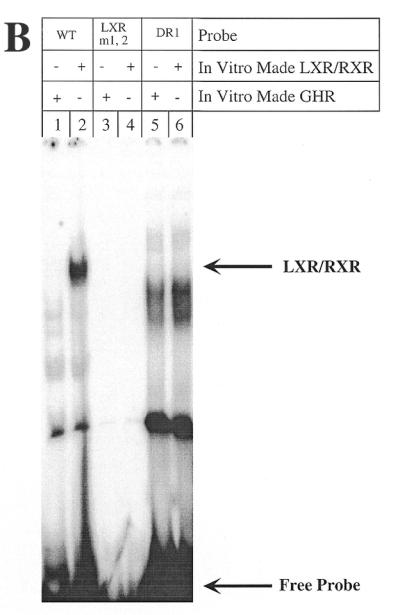
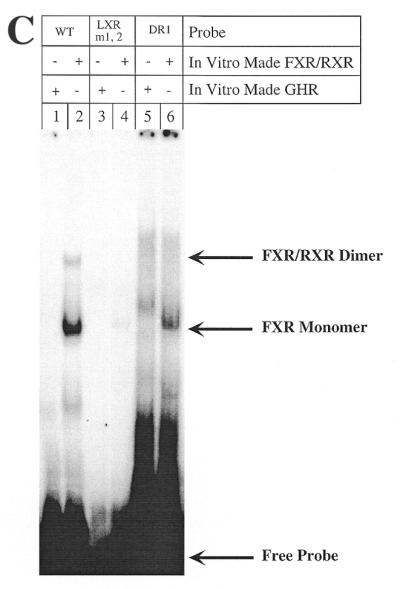

Mutations of the 7α-hydroxylase LXR site abolish binding of both LXR and FXR but do not alter promoter activity or bile acid-mediated regulation. (A) Male rat primary hepatocytes were transfected with wild-type 7α-hydroxylase promoter construct (pGL3R7α-342) or the indicated mutants and 5 ng of FXR expression plasmid. After transfection cells were treated with TCA as described in Materials and Methods. The wild-type sequence of DR-1 and CPF as well as the LXR sites are shown in boxes with the BARE sites in bold. The exact nucleotides mutated in each site are indicated for each construct. The data were normalized to the activity produced by the wild-type construct, pGL3R7α-342, in cells grown in the absence of TCA and represent the averages of n experiments ± SD. (B) Gel shift experiments were performed as described in Materials and Methods using in vitro made LXR/RXR and the indicated probes. The wild-type probe (WT) corresponds to nucleotides –72 to –49 (Fig. 2) and the mutant probes had the mutations indicated in (A). Arrows indicate the retarded complex and the free probe. (C) In vitro made FXR/RXR was used as indicated with the same wild-type and mutant probes. Arrows indicate the complex with the dimer FXR/RXR, with the monomer FXR and the free probe. (D) In vitro made FXR, FXR/RXR or GHR (as a control) was used as indicated with the wild-type 7α-hydroxylase probe. Anti-FXR antibody was used where indicated.
FXR has been described as functioning by binding as the heterodimer FXR/RXR to an inverted repeat separated by 1 nt (IR-1) (12). It can also function by binding to DR-1 or DR-4 elements in heterologous promoters (26). The 7α-hydroxylase LXR site, a direct repeat separated by 4 nt (DR-4), is capable of binding FXR monomers as well as FXR/RXR dimers (Fig. 3C, lane 2). Although this binding is much weaker than to an IR-1 site (Fig. 4B), it could perhaps mediate the bile acid-mediated regulation observed when FXR is overexpressed (Fig. 1). However, the two LXR mutants that we created lost the capability to bind FXR/RXR dimer and most of the FXR monomer (Fig. 3C, lanes 4 and 6), but these mutants were still fully regulated by bile acids (Fig. 3A). The identities of the FXR monomer and FXR/RXR dimer bands formed with the 7α-hydroxylase LXR probe were demonstrated by the antibody supershift experiment shown in Figure 3D.
Figure 4.

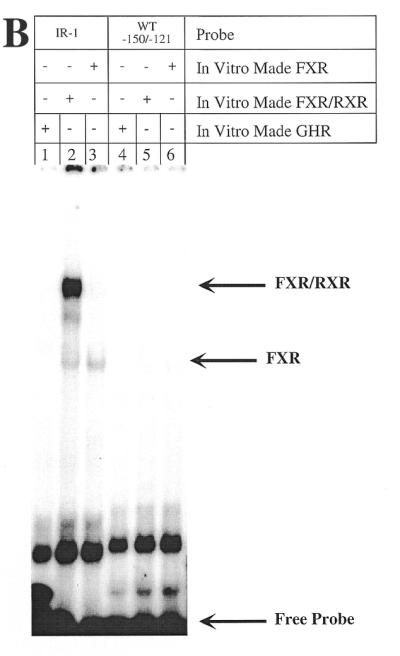
Mutations of the rat 7α-hydroxylase DR-1 site do not alter promoter activity or bile acid-mediated regulation. (A) Male rat primary hepatocytes were transfected with the indicated constructs and 5 ng of FXR expression plasmid and treated with TCA as described in Materials and Methods. The exact nucleotide mutated in each site is also indicated for each construct. The data were normalized as in Figure 3. (B) In vitro made FXR/RXR was used as indicated with a wild-type probe containing the 7α-hydroxylase DR-1 site (nucleotides –150 to –121) and a perfect IR-1 as a positive control.
To analyze the potential role of the DR-1/BARE II site (–121 to –150) on bile acid-mediated regulation of 7α-hydroxylase promoter activity, two mutations were created (Fig. 4). One mutation, pGL3R7α-342/DR1m1, mutated the first DR-1 repeat. The second mutation, pGL3R7α-342/DR1m1,2, mutated both DR-1 repeats. These two mutants left the CPF site virtually unmodified. Only the 5′ T within the CPF site in mutant pGL3R7α-342/DR1m1,2 was mutated to an A, and this mutation maintains an active CPF/FTF site (23). Both mutants were active (∼50% activity of wild-type) and, most importantly, they were fully regulated by bile acids (Fig. 4A). A double mutant was also created, pGL3R7α-342/LXR m1,2, DR1m1, which mutates both LXR repeats and one DR-1 repeat. This mutant should abolish both BAREs, including the LXR and DR-1 sites, but, as shown in Figure 4, this mutant is still fully regulated by bile acids.
The 7α-hydroxylase DR-1 (TGGACT t AGTTCA) actually resembles an IR-1, with only one nucleotide different. To analyze whether this site is actually capable of binding FXR/RXR as well as does a perfect IR-1, we performed gel retardation assays with in vitro made FXR/RXR and FXR alone. As a positive control we used a perfect IR-1 probe. Figure 4B shows that the 7α-hydroxylase DR-1 probe has no binding capability for either FXR/RXR or FXR (lanes 5 and 6), whereas our positive control probe, a perfect IR-1 element, strongly bound the heterodimer (lane 2).
To characterize the role of the CPF/FTF site, we created two mutants (Fig. 5A). pGL3R7α-342/CPFm1 mutates only two nucleotides within the CPF recognition element, leaving the DR-1 site unchanged. pGL3R7α-342/CPFm2 mutates the two DR-1 repeats with one nucleotide overlapping the CPF site. Both promoter mutants showed low activity. Most importantly, these were the only two mutants tested that showed no regulation by bile acids. Neither of these two mutants was capable of binding FTF in gel retardation assays (Fig. 5B).
Figure 5.

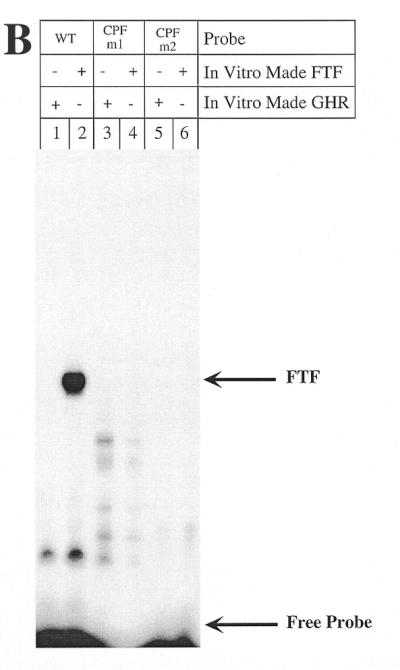
Mutations of the rat 7α-hydroxylase CPF site abolish promoter activity. (A) Male rat primary hepatocytes were transfected with the indicated constructs and 5 ng of FXR expression plasmid and treated with TCA as described in Materials and Methods. The exact nucleotide mutated in each site is also indicated for each construct. The data were normalized as in Figure 3. (B) In vitro made CPF was used as indicated with a wild-type probe containing the 7α-hydroxylase CPF site (–150 to –121) and the mutant probes as indicated.
DISCUSSION
This study provides important evidence on the role of two transcriptional factors, FXR and FTF/CPF, involved in expression of the 7α-hydroxylase gene and in its regulation by bile acids. We show that FXR is a factor involved in bile acid-mediated regulation of 7α-hydroxylase transcription that functions without binding to DNA. Most importantly, we also show that binding of FTF is required for bile acid-mediated regulation of the 7α-hydroxylase promoter.
Recently three laboratories independently reported that bile acids are ligands for the nuclear receptor FXR (9–11). It was shown that FXR-mediated induction of I-BABP promoter activity by bile acids requires an IR-1 site, a DNA element known to bind FXR (9). FXR also mediated activation by bile acids of a heterologous promoter containing IR-1 sites. This suggested that FXR may mediate suppression of bile acid biosynthetic genes, such as 7α-hydroxylase and 12α-hydroxylase, by binding of ligand-occupied FXR to a similar, but not identical, site within the promoters of these genes. The human 7α-hydroxylase promoter was regulated ∼2-fold only when FXR was overexpressed using HepG2 cells as host cells. However, the endogenous 7α-hydroxylase gene was more than 90% regulated without FXR overexpression (9). In this study we show that FXR is indeed involved in bile acid-mediated regulation of 7α-hydroxylase transcription, as shown by the increased regulation when FXR is overexpressed in primary hepatocytes (Fig. 1).
Wang et al. (11) suggested that FXR could function by displacing LXR from its binding site within the 7α-hydroxylase promoter. This LXR site overlaps the previously described BARE I (6). Here we show that FXR does in fact bind, albeit weakly, to the LXR site found in the rat 7α-hydroxylase promoter (Fig. 3C). However, two different mutants that disrupt the LXR site and abolish binding of both LXR and FXR (Fig. 3B and C) have no effect on regulation by bile acids (Fig. 3A). These results demonstrate that neither LXR nor its binding site within the rat 7α-hydroxylase promoter are involved in bile acid-mediated regulation of 7α-hydroxylase.
Another site within the rat 7α-hydroxylase promoter has also been described as mediating regulation by bile acids (5; Fig. 2). This site, BARE II, overlaps what has been characterized as a DR-1 site that binds HNF-4 (17). Examination of this site (TGGACT t AGTTCA) indicates that it is highly homologous to an IR-1 site, the binding site for FXR. However, this site has no binding affinity for FXR (Fig. 4B) nor does it mediate regulation by bile acids. Two different mutants that had been altered in either one or both repeats were still fully regulated by bile acids (Fig. 4A). In addition, both mutants lost the capability to bind HNF-4 (data not shown).
The BARE II site also overlaps the recently described CPF/FTF site which is important for activity of the human 7α-hydroxylase promoter in HepG2 cells (20). However, in rat primary hepatocytes constructs with the FTF mutated still showed promoter activity (Fig. 5A). We believe that this is because primary hepatocytes maintain expression of some other factor that is important for 7α-hydroxylase expression whose expression has been lost in HepG2 cells. Mutations within this FTF site were the only mutations that abolished bile acid regulation of rat 7α-hydroxylase promoter activity (Fig. 5A) and they also abolished FTF binding, as expected (Fig. 5B). Additionally, scanning mutants of the rest of the promoter did not alter its regulation by bile acids (5), pointing to FTF as a crucial factor involved in this regulation.
In a recent report we showed that FTF is also required for both transcription and regulation by bile acids of the 12α-hydroxylase gene (23). However, attempts in our laboratory to show regulation of FTF expression or activity by bile acids have failed. We conclude that FTF is a common factor involved in bile acid-mediated down-regulation of gene transcription of at least two genes, 7α-hydroxylase and 12α-hydroxylase. We propose that bile acids down-regulate gene transcription by altering the binding capability or activity of FTF by interacting with an as yet unidentified co-repressor.
The role of FXR in bile acid-mediated suppression of gene transcription is unclear at this point. Experiments in this study demonstrate that FXR enhances regulation by bile acids of the 7α-hydroxylase promoter without binding to DNA, suggesting an indirect effect. While this manuscript was in preparation, another study reported similar results with regard to the role of FXR in bile acid-mediated regulation of 7α-hydroxylase promoter activity (27). It should be pointed out that FXR does not seem to be required for regulation of 12α-hydroxylase (23). This leaves FTF as the only common factor involved in suppression of gene transcription by bile acids of two genes of the bile acid biosynthetic pathway, 7α-hydroxylase and 12α-hydroxylase.
Acknowledgments
ACKNOWLEDGEMENTS
Lesley D. Johnson provided invaluable technical help and we thank Pat Bohdan for preparation of rat hepatocyes. We thank Dr Luc Bélanger for plasmid pCI-FTF and Dr Cary Weinberger for pRSV-FXR. We are grateful to Drs Hylemon and Vlahcevic for critical comments and support. We also thank Dr Wells for discussions and critical review of the manuscript. This research was supported in part by National Institutes of Health Grant DK44218. A.d.C.-O. is a post-doctoral fellow of the North Atlantic Treaty Organization.
REFERENCES
- 1.Javitt N.B. (1994) FASEB J., 8, 1308–1311. [DOI] [PubMed] [Google Scholar]
- 2.Carey M.C. and Cahalene,M.J. (1988) In Arias,I.M., Jakoby,W.B., Popper,H., Schachter,D. and Shafritz,D.A. (eds), The Liver: Biology and Pathobiology. Raven Press, New York, NY, pp. 573–616.
- 3.Danielsson J., Einarsson,K. and Johansson,G. (1967) Eur. J. Biochem., 2, 44–49. [DOI] [PubMed] [Google Scholar]
- 4.Ramirez M.I., Karaoglu,D., Haro,D., Barillas,C., Bashirzadeh,R. and Gil,G. (1994) Mol. Cell. Biol., 14, 2809–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stroup D., Crestani,M. and Chiang,J.Y.L. (1997) Am. J. Physiol., 273, G508–G517. [DOI] [PubMed] [Google Scholar]
- 6.Chiang J.Y.L. and Stroup,D. (1994) J. Biol. Chem., 269, 17502–17507. [PubMed] [Google Scholar]
- 7.Vlahcevic Z.R., Jairath,S.K., Heuman,D.M., Stravitz,R.T., Hylemon,P.B., Avadhani,N.G. and Pandak,W.M. (1996) Am. J. Physiol., 270, G646–G652. [DOI] [PubMed] [Google Scholar]
- 8.Rao Y.P., Vlahcevic,Z.R., Stravitz,R.T., Mallonee,D.H., Mullick,J., Avadhani,N.G. and Hylemon,P.B. (1999) J. Steroid Biochem. Mol. Biol., 70, 1–14. [DOI] [PubMed] [Google Scholar]
- 9.Makishima M., Okamoto,A.Y., Repa,J.J., Tu,H., Learned,R.M., Luk,A., Hull,M.V., Lustig,K.D., Mangelsdorf,D.J. and Shan,B. (1999) Science, 284, 1362–1365. [DOI] [PubMed] [Google Scholar]
- 10.Parks D.J., Blanchard,S.G., Bledsoe,R.K., Chandra,G., Consler,T.G., Kliewer,S.A., Stimmel,J.B., Willson,T.M., Zavacki,A.M., Moore,D.D. and Lehmann,J.M. (1999) Science, 284, 1365–1368. [DOI] [PubMed] [Google Scholar]
- 11.Wang H., Chen,J., Hollister,K., Sowers,L.C. and Forman,B.M. (1999) Mol. Cell, 3, 543–553. [DOI] [PubMed] [Google Scholar]
- 12.Forman B.M., Goode,E., Chen,J., Oro,A.E., Bradley,D.J., Perlmann,T., Noonan,D.J., Burka,L.T., McMorris,T., Lamph,W.W., Evans,R.W. and Weinberger,C. (1995) Cell, 81, 687–693. [DOI] [PubMed] [Google Scholar]
- 13.Janowski B.A., Willy,P.J., Devi,T.R., Falck,J.R. and Mangelsdorf,D.J. (1996) Nature, 383, 728–731. [DOI] [PubMed] [Google Scholar]
- 14.Lehmann J.M., Kliewer,S.A., Moore,L.B., Smith-Oliver,T.A., Oliver,B.B., Su,J.L., Sundseth,S.S., Winegar,D.A., Blanchard,D.E., Spencer,T.A. and Willson,T.M. (1997) J. Biol. Chem., 272, 3137–3140. [DOI] [PubMed] [Google Scholar]
- 15.Peet D.J., Turley,S.D., Ma,W., Janowski,B.A., Lobaccaro,J.M., Hammer,R.E. and Mangelsdorf,D.J. (1998) Cell, 93, 693–704. [DOI] [PubMed] [Google Scholar]
- 16.Molowa D.T., Chen,W.S., Cimis,G.M. and Tan,C.P. (1992) Biochemistry, 31, 2539–2544. [DOI] [PubMed] [Google Scholar]
- 17.Stroup D. and Chiang,J.Y. (2000) J. Lipid Res., 41, 1–11. [PubMed] [Google Scholar]
- 18.Crestani M., Sadeghpour,A., Stroup,D., Galli,G. and Chiang,J.Y. (1998) J. Lipid Res., 39, 2192–2200. [PubMed] [Google Scholar]
- 19.Stroup D., Crestani,M. and Chiang,J.Y. (1997) J. Biol. Chem., 272, 9833–9839. [DOI] [PubMed] [Google Scholar]
- 20.Nitta M., Ku,S., Brown,C., Okamoto,A.Y. and Shan,B. (1999) Proc. Natl Acad. Sci. USA, 96, 6660–6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galarneau L., Pare,J.F., Allard,D., Hamel,D., Levesque,L., Tugwood,J.D., Green,S. and Belanger,L. (1996) Mol. Cell. Biol., 16, 3853–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mangelsdorf D.J., Thummel,C., Beato,M., Herrlich,P., Schutz,G., Umesono,K., Blumberg,B., Kastner,P., Mark,M., Chambon,P. and Evan,R.M. (1995) Cell, 83, 835–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.del Castillo-Olivares A. and Gil,G. (2000) J. Biol. Chem., 275, 17793–17799. [DOI] [PubMed] [Google Scholar]
- 24.Subramanian A., Teixeira,J., Wang,J. and Gil,G. (1995) Mol. Cell. Biol., 15, 4672–4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hylemon P.B., Gurley,E.C., Stravitz,R.T., Litz,J.S., Pandak,W.M., Chiang,J.Y.L. and Vlahcevic,Z.R. (1992) J. Biol. Chem., 267, 16866–16871. [PubMed] [Google Scholar]
- 26.Laffitte B.A., Kast,H.R., Nguyen,C.M., Zavacki,A.M., Moore,D.D. and Edwards,P.A. (2000) J. Biol. Chem., 275, 10638–10647. [DOI] [PubMed] [Google Scholar]
- 27.Chiang J.Y., Kimmel,R., Weinberger,C. and Stroup,D. (2000) J. Biol. Chem., 275, 10918–10924. [DOI] [PubMed] [Google Scholar]