

Abstract
The ɛ4 allele of the apolipoprotein E gene (APOE) is the strongest genetic risk factor for late-onset Alzheimer disease (AD). Within the CNS, APOE is produced by a variety of cell types under different conditions, posing a challenge for studying its roles in AD pathogenesis. However, through powerful advances in research tools and the use of novel cell culture and animal models, researchers have recently begun to study APOE4’s roles in AD in a cell type-specific manner and at a deeper and more mechanistic level than ever before. In particular, cutting-edge omics studies have enabled APOE4 to be studied at the single-cell level and have allowed for the identification of critical APOE4 effects in AD-vulnerable cellular subtypes. Through these studies, it has become evident that APOE4 produced in various types of CNS cells — including astrocytes, neurons, microglia, oligodendrocytes and vascular cells — has diverse roles in AD pathogenesis. Here, we review these scientific advances and propose a novel cell type-specific APOE4 cascade model of AD. In this model, neuronal APOE4 emerges as a crucial pathological initiator and driver of AD pathogenesis, instigating glial responses and, ultimately, neurodegeneration. In addition, we provide perspectives on future directions for APOE4 research and related therapeutic developments in the context of AD.
Introduction
Alzheimer disease (AD) is a neurodegenerative disorder that is clinically characterized by a progressive decline in cognitive functions. Pathologically, AD is characterized by loss of synapses and neurons, extracellular deposition of amyloid-β (Aβ) peptides in the form of amyloid plaques, neuritic dystrophy, intraneuronal accumulation of hyperphosphorylated tau (p-tau) in the form of neurofibrillary tangles (NFTs), vascular alterations and inflammatory responses mediated by microglia and astrocytes1,2. AD starts with a long preclinical stage, lasting 20–30 years, during which amyloid plaques form without clinical symptoms3–5. This is followed by a prodromal stage, usually lasting 3–5 years, characterized by mild cognitive impairment (MCI). Finally, some but not all cases progress to dementia3–5. Worldwide, the total number of people with preclinical AD, prodromal AD and AD dementia is estimated to be approximately 416 million6 (Fig. 1a).
Fig. 1: CNS APOE expression and links to Alzheimer disease.
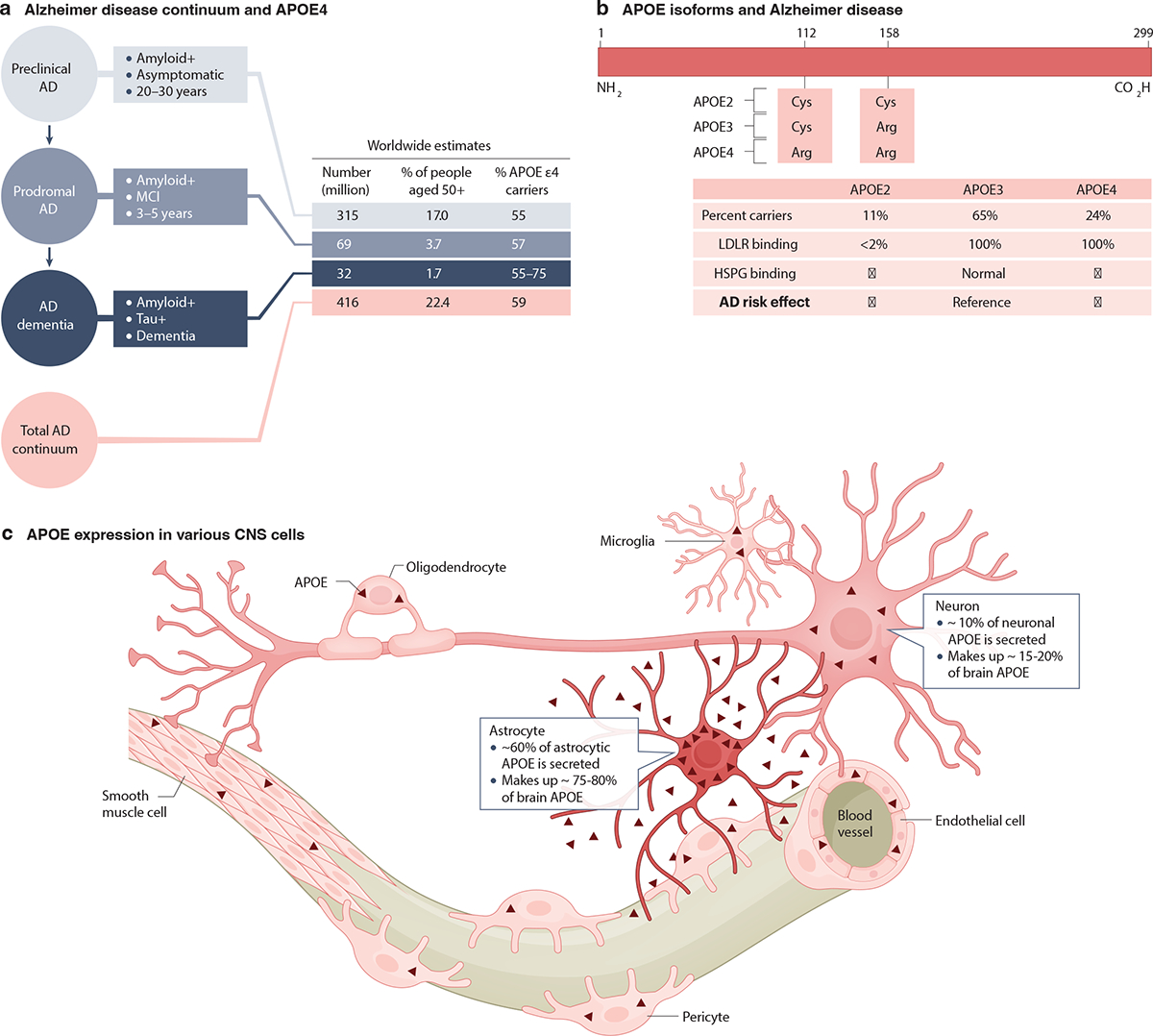
a, An overview of Alzheimer disease (AD) continuum3–5, worldwide estimates of numbers of individuals with preclinical AD, prodromal AD and AD dementia6, and their relationship with the E4 isoform of apolipoprotein E (APOE4)6. b, There are three common APOE isoforms: APOE2, APOE3, and APOE41,11. They exhibit sequence differences at amino acid residues 112 and 15812, differential receptor binding properties13 and varying impacts on AD risk6,10,14–17. Downward-pointing arrows indicate reduced heparan sulfate proteoglycan (HSPG) binding or AD risk; upward-pointing arrows indicate increased HSPG binding or AD risk. c, Multiple cell types in the CNS can produce APOE at different levels (indicated by the depth of red shading) under normal conditions1,20,39. These include astrocytes, neurons, microglia, oligodendrocytes and vascular cells (endothelial cells, pericytes and smooth muscle cells). In studies in which astrocytic APOE was selectively removed in mice, there was a loss of 75–80% of brain APOE levels42–44, while removing neuronal APOE led to a 15–20% reduction of brain APOE levels24,42. Thus, astrocytic and neuronal APOE seem to account for approximately 75–80%42–44 and 15–20%24,42 of total APOE in the brain, respectively. After production, approximately 60% of astrocytic APOE is secreted into the extracellular space47,48, whereas most APOE produced in neurons remains intracellular48. The proportions of total and secreted APOE in the brain that are contributed by microglia, oligodendrocytes and vascular cells have yet to be determined. LDLR, low-density lipoprotein receptor; MCI, mild cognitive impairment.
Despite over a century of AD research, we lack effective treatments or cures and those that have been proposed remain controversial. This is likely due to the complex etiology of AD pathogenesis, which involves interactions among diverse genetic, epigenetic and environmental factors. Causative genetic mutations have been identified for early-onset autosomal dominant AD (ADAD)7,8; however, ADAD accounts for fewer than 1% of all AD cases. For late-onset AD, which accounts for the vast majority of AD cases, researchers so far have identified only genetic loci that can alter disease risk, the strongest of which is the apolipoprotein E (APOE) locus9,10.
The human APOE gene is polymorphic, comprising three common alleles (APOE ε4, ε3 and ε2) that translate into three APOE isoforms: APOE4, APOE3 and APOE21,11. APOE’s canonical function is as a transporter of lipids between cells and organs. As a result of amino acid differences at residue 112 or 15812, the three APOE isoforms exhibit different binding properties with the APOE receptors through which they mediate this transport13 (Fig. 1b). Carriers of the APOE ε4 allele, the strongest genetic risk factor for late-onset AD10,14,15, account for approximately 24% of the healthy population16 (Fig. 1b) but make up 55–75% of AD dementia cases (as well as approximately 57% of those with prodromal AD and approximately 55% of those with preclinical AD) (Fig 1a)14,17.
Along with its unique structural and functional properties12,13,18, the presence of APOE4 has been linked to more severe AD pathologies1,2,19–21. Having a copy of the APOE ε4 allele increases the formation of amyloid plaques1,2,19,20 and NFTs in humans and animal models1,2,22–24. APOE4 is also associated with increased gliosis and elevated levels of proinflammatory cytokines23–26 This APOE4-enhanced gliosis is, in turn, associated with other pathological phenotypes such as lipid accumulation in glial cells27–29, neurodegeneration23,24,28,30 and hippocampal atrophy23,24,28,30. Moreover, humans with AD, especially those carrying the APOE ε4 allele, and AD mouse models carrying the same allele exhibit oligodendrocyte dysfunction and myelination defects24,31–33 as well as blood-brain barrier (BBB) impairments34–36. Given this complicated picture, it is critical to dissect the detrimental effects of APOE4 at a cell type-specific level and to identify the underlying molecular mechanisms that drive AD pathogenesis in the most relevant cell populations. Although numerous studies and recent reviews have examined the pathobiological effects of APOE4 in general20,25,37,38, a comprehensive review of APOE4’s roles in AD pathogenesis at a cell type-specific level is lacking: this is therefore the focus of this Review.
Several cell types in the mammalian CNS can produce APOE, including astrocytes, neurons, microglia, oligodendrocytes and vascular cells, although the production levels vary by cell type and condition1,20,39 (Fig. 1c). Studies leveraging new research tools and experimental models have revealed that the effects of APOE4 on AD pathologies depend on its cellular sources and expression levels. Here, we review the roles of APOE4 derived from various CNS cell types, summarize the potential mechanisms underlying the cell type-specific effects of APOE4, present a novel cell type-specific APOE4 cascade model of AD, and provide perspectives on future APOE4 research and related therapeutic developments.
Astrocytic APOE4 in AD pathogenesis
Expression and regulation of astrocytic APOE
Astrocytes produce most of the APOE in the CNS39–41 (Fig. 2a), contributing approximately 75–80% of the total APOE in the mouse brain42–44 (Fig. 1c). Although the regulation of astrocytic APOE production remains to be fully elucidated, the transcription factor C/EBPβ is known to bind the APOE promoter and regulate APOE expression in astrocytes45. Mitochondrial function may also modulate APOE levels, as treatment of human astrocytes with an inhibitor of mitochondrial respiration increased APOE mRNA and protein levels46. After production, approximately 60% of astrocytic APOE is secreted into the extracellular space47,48. This process may be regulated by 25-hydroxycholesterol (25-HC), an oxysterol secreted by microglia, as treatment of primary mouse astrocytes expressing human APOE with 25-HC led to a 2-fold increase in secreted APOE49. Since astrocytes are the major source of brain APOE, the roles that intracellular and extracellular astrocytic APOE may play in health and AD have been a major focus in APOE research (Fig. 2a–c).
Fig 2: Expression of APOE4 in astrocytes and its roles in AD pathogenesis.
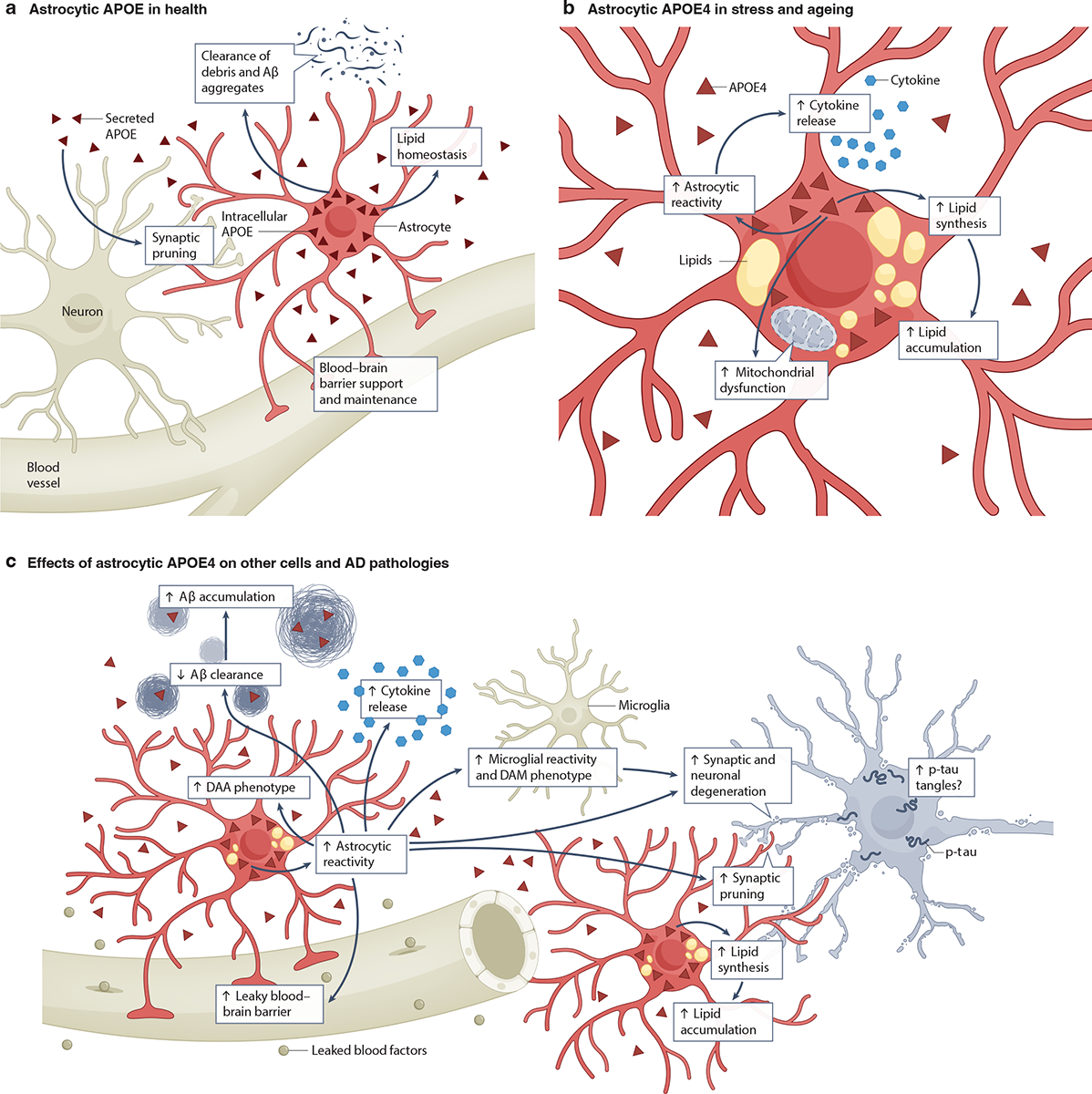
a, Healthy astrocytes produce and secrete most of the apolipoprotein E (APOE) found in the brain39–41. This APOE participates in many astrocyte functions, including synaptic pruning, lipid homeostasis, clearance of amyloid β (Aβ) aggregates and cell debris and blood-brain barrier (BBB) support and maintenance50–58. b, During stress and aging, astrocytic APOE4 contributes to astrocytic reactivity43,50,51,59, elevated proinflammatory cytokine release50,51,59, augmented mitochondrial dysfunction63–66, and increased lipid synthesis31,53–55,60 and intracellular lipid accumulation31,51,53–55,60. c, Astrocytic APOE4 also affects other CNS cells and can promote AD pathologies. For example, it drives an increase in microglial reactivity and adoption of a disease-associated microglia (DAM) phenotype43. In addition, it contributes to AD pathogenesis by increasing astrocytic reactivity43,50,51,59, the transition to a disease-associated astrocyte (DAA) phenotype78, cytokine release50,51,59, synaptic pruning43,52 and lipid accumulation31,51,53–55,60. In turn, these changes may lead to BBB leakiness57,58,70, Aβ accumulation51,54,71,74, tau accumulation43,77 and (in conjunction with neuronal APOE4) synaptic and neuronal degeneration43. The potential relationships among the effects of astrocytic APOE4 on other cells and AD pathologies are depicted using arrows. Note that non-astrocytic sources of APOE4 have been omitted from this figure for clarity. p-tau, hyperphosphorylated tau.
Astrocytic APOE4 in inflammation, metabolism and BBB function
Under normal conditions, astrocytic APOE helps astrocytes maintain homeostatic lipid metabolism, controls synaptic pruning, regulates clearance of extracellular Aβ aggregates and cell debris, and helps to preserve BBB integrity50–58 (Fig. 2a). However, APOE can also promote astrocytic inflammatory responses in an isoform-dependent manner43,50,51,59 (Fig. 2b,c). In primary mouse astrocytes and astrocytes derived from human induced pluripotent stem cells (hiPSCs), expression of APOE4 increased the secretion of inflammatory cytokines, including interleukin-1β (IL-1β), IL-6, and inducible NO synthase (NOS2), as compared to expression of APOE3 or APOE250,51,59. In a tauopathy model in which mice express mutant human tau (PS19 mice), additional expression of APOE4 (PS19-APOE4 mice) enhanced the reactive astrocytosis elicited by tau pathology, while its selective removal from astrocytes induced a more physiological state43, suggesting a role of astrocytic APOE4 in tauopathy-induced reactive astrocytosis.
APOE isoforms also differentially affect astrocytic lipid metabolism and accumulation (Fig. 2b,c). APOE4 expression increased cholesterol synthesis and accumulation in astrocytes of human APOE4 knock-in (APOE4-KI) mice and astrocytes derived from APOE4 hiPSCs31,53–55,60. Sterol regulatory element-binding protein 2, a key activator of cholesterol synthesis, was elevated in APOE4 hiPSC-derived astrocytes, suggesting a potential pathway through which astrocytic APOE4 may promote cholesterol production60. Primary astrocytes from APOE4-KI mice exhibited changes in lipid pathway proteins, including reduced levels of peroxisome proliferator-activated receptor-γ (PPARγ, a transcription factor involved in maintaining lipid homeostasis) and altered levels of receptors and transporters involved in lipid uptake and efflux55. Compared with APOE3 hiPSC-derived astrocytes, APOE4 hiPSC-derived astrocytes exhibited increased localization of cholesterol to lysosomes51. Furthermore, a recent preprint has reported that immortalized astrocytes from APOE4-KI mice developed larger lipid droplets and greatly impaired lipid turnover compared to their APOE3-KI counterparts61. In addition, selectively removing astrocytic APOE in mice markedly decreased the number of lipid-accumulating reactive astrocytes62. Beyond affecting lipid composition intracellularly, astrocytic APOE4 also reduces the lipidation of secreted lipoprotein particles, compared to APOE356.
The differential effects of the APOE isoforms also extend to astrocytic glucose metabolism. Primary astrocytes from APOE4-KI mice demonstrated reduced mitochondrial membrane potential and respiration compared to those from APOE3-expressing controls63. Similarly, immortalized astrocytes from APOE-KI mice, primary APOE-KI mouse astrocytes, and hiPSC-derived astrocytes displayed changes in glucose utilization, with APOE4-expressing astrocytes showing elevated levels of glycolytic activity relative to their APOE3 counterparts64–66. Interestingly, treatment with cholesterol-depleting agents restored mitochondrial respiration and activity in APOE4 hiPSC-derived astrocytes, suggesting that APOE4-induced cholesterol accumulation may contribute to glucose metabolism shifts in astrocytes66.
Astrocytic APOE4 also affects many other cell types, especially under stressed and AD-like conditions (Fig. 2c). For instance, selectively removing APOE4 from astrocytes prevented disease-associated gene signatures in neurons, oligodendrocytes and microglia in PS19-APOE4 mice: fewer oligodendrocytes expressed AD-associated immune pathway genes and microglia showed less phagocytosis of synapses and disease-associated microglial (DAM) transcriptional signatures43. These results suggest that astrocytic APOE4 enhances glial reactivity43. In addition, astrocytic APOE4 seems to affect lipid metabolism in neurons. For example, primary neurons from wildtype (WT) mice showed elevated levels of cholesterol biosynthesis when treated with APOE derived from primary astrocytes of APOE4-KI mice, as compared to those treated with APOE from APOE3-KI mouse astrocytes67. In a similar vein, in an organoid model containing hiPSC-derived neurons and astrocytes of various APOE genotype pairings, the presence of APOE4-producing human astrocytes elevated cholesterol levels and promoted lipid accumulation in human neurons68. Additionally, astrocytic APOE4 may also affect synaptic plasticity by impairing neuronal APOE receptor recycling via the endosomal pathway69.
Astrocytic APOE4 also influences BBB integrity and function (Fig. 2c). In an in vitro BBB model consisting of primary astrocytes derived from APOE-KI or ApoE knockout (ApoE-KO) mice together with WT primary brain endothelial cells and pericytes, astrocytic APOE4 impaired endothelial cell tight junctions by reducing the levels of threonine-phosphorylated occludin57. This effect was recapitulated in an in vivo model, in which selective removal of astrocytic APOE4 prevented the impairments of the BBB, endothelial tight junctions and astrocytic end-foot coverage of blood vessels observed in APOE4-KI mice70. In a mouse ApoE-KO model, selective astrocytic expression of APOE4, but not APOE3, led to BBB leakiness58. Taken together, these findings suggest that astrocytic APOE4 affects multiple CNS cell types that might contribute to AD pathogenesis.
Astrocytic APOE4 and hallmark AD pathologies
In mouse and cell culture models, astrocytic expression of APOE4 promotes amyloid plaque pathology and impairs astrocytic Aβ uptake, as compared to APOE351,54,71–73 (Fig. 2c). Furthermore, selective removal of astrocytic APOE4 reduced plaque loads in a model of amyloidosis in which mice co-express APOE4, mutant amyloid precursor protein (APP) and mutant presenilin 1 (PSEN1, a protein involved in Aβ production) (APP/PS1–21 mice)74. In another amyloidosis model (5xFAD mice) that co-expresses APOE4, selective ablation of astrocytic APOE4 reduced gliosis and BBB disruption and shifted Aβ deposition from the brain parenchyma to the vasculature44.
The connection between astrocytic APOE4 and tau pathology is less straightforward. Selective expression of APOE4 in astrocytes on an ApoE-KO background caused little or no accumulation of endogenous mouse p-tau75,76. However, selective removal of astrocytic APOE4 in PS19-APOE4 mice moderately lowered p-tau levels43. Glypican-4 (GPC4), a protein that is secreted by astrocytes and has been shown to preferentially bind APOE4 (relative to APOE2)77, may provide a link between astrocytic APOE4 and this effect. Notably, GPC4 treatment increased p-tau accumulation in primary neurons from PS19 mice77. Similarly, inducing greater GPC4 expression in astrocytes of young PS19 mice increased hippocampal p-tau levels77. Thus, the suggestion that astrocytic APOE4 might augment tau pathology warrants further studies.
APOE4 in disease-associated astrocytic subtypes
Single-nucleus RNA-sequencing (snRNA-seq) studies examining different types of cells in AD have identified a distinct group of disease-associated astrocytes (DAAs)78,79 (Fig. 2c). These DAAs generally have elevated APOE expression, suggesting an involvement of astrocytic APOE in their formation78. DAAs also exhibit increased expression of the genes encoding cathepsin and serine protease inhibitor A3N, which are thought to be involved in AD pathogenesis80,81, as well as genes involved in lipid and cholesterol pathways, the complement system, endocytosis and inflammatory responses78. APOE4 hiPSC-derived astrocytes also showed upregulation of lipid metabolism-related genes and downregulation of lipid transport-related genes54. Notably, selective ablation of neuronal APOE4 in PS19-APOE4 mice reduced the abundance of one DAA subtype, referred to as neuronal APOE4-promoted DAA (nE4-DAA), which showed increased expression of APOE and cathepsin genes24. Intriguingly, the reduction of nE4-DAAs corresponded with ameliorated AD pathologies24. Thus, the emergence of DAA subtypes may also be brought about indirectly by the effects of neuronal APOE4 expression (see below).
Taken together, these studies demonstrate that astrocytic APOE4 can enhance diverse aspects of AD pathogenesis directly or indirectly, through exacerbation of amyloid and tau pathology and the promotion of glial reactivity, adoption of a DAA phenotype, proinflammatory cytokine production and release, altered metabolism and BBB disruption (Fig. 2b,c).
Neuronal APOE4 in AD pathogenesis
Expression and regulation of neuronal APOE
Under physiological conditions, mammalian neurons express low levels of APOE (Fig. 3a), contributing approximately 15–20% of total brain APOE in mice24,42 (Fig. 1c). However, they upregulate APOE expression in response to stress and aging39,82–89 (Fig. 3b). snRNA-seq studies of WT mice, human APOE-KI mice and human brains from young subjects or from older individuals with AD have shown that APOE is expressed in both inhibitory and excitatory neurons, albeit at lower levels than in astrocytes24,90. Interestingly, unlike astrocytes, which secrete most of the APOE that they produce, neurons secrete only about 10% of the APOE that they generate48, likely setting the stage for its intracellular accumulation.
Fig. 3: Expression of APOE4 in neurons and its roles in AD pathogenesis.
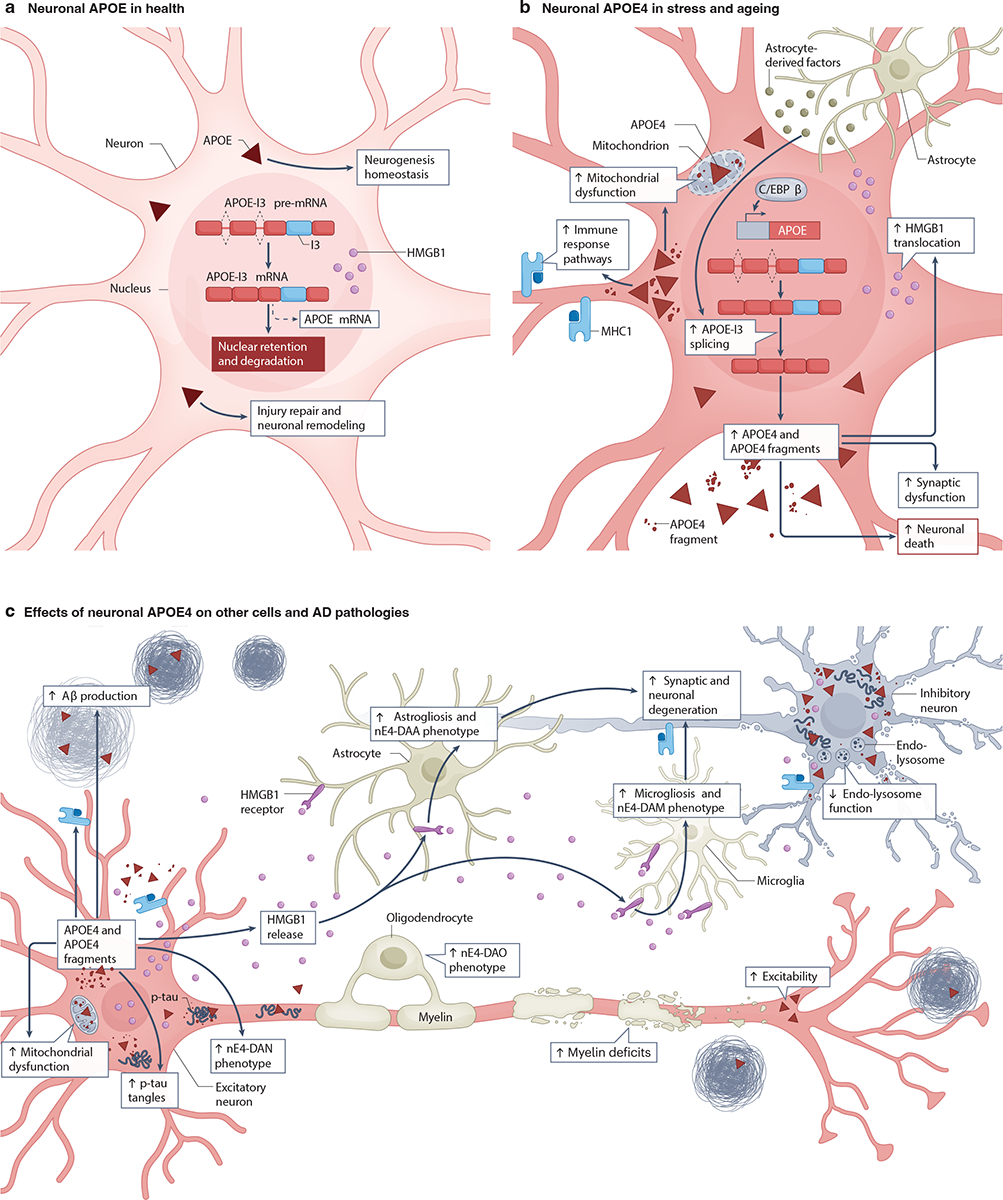
a, Healthy neurons express low levels of apolipoprotein E (APOE)39,82–89: the APOE mRNA they produce has an intron 3 retention (APOE-I3 mRNA), which ensures that it is largely retained in the nucleus, where it is degraded, rather than being processed into mature APOE mRNA91. Neurons also retain high mobility group box 1 (HMGB1), a proinflammatory molecule, within the nucleus129. Neuronal APOE is involved in the maintenance of neurogenesis94, injury repair92,93 and neuronal remodeling1,95. b, Under conditions of stress and aging, neurons increase APOE expression by active splicing of APOE-I3 into mature APOE mRNA91. The transcription factor C/EBPβ45,98 and astrocyte-derived factor(s)48 also upregulate APOE expression in neurons. Increased neuronal APOE4 production leads to increased APOE4 fragmentation22,75,85,126,128, expression of immune response pathway genes (such as major histocompatibility complex class 1 (MHC1) genes)90, nucleo-cytoplasmic translocation of HMGB1129, mitochondrial dysfunction95,109–113, synaptic dysfunction99,101, and neuronal death24,85,90,98,114. c, Neuronal APOE4 affects other CNS cells. For example, it increases astrogliosis and the adoption of a neuronal APOE4-promoted disease-associated astrocyte (nE4-DAA) phenotype24, microgliosis and the adoption of a neuronal APOE4-promoted disease-associated microglial (nE4-DAM) phenotype24, the adoption of neuronal APOE4-promoted disease-associated oligodendrocyte (nE4-DAO) phenotype24 and myelin deficits24. Neuronal APOE4 also increases intraneuronal tau tangles24,75,76,85,98, endo-lysosomal dysfunction115,116, neuronal hyperexcitability103–106, amyloid β (Aβ) production and accumulation85,98, the adoption of a neuronal APOE4-promoted disease-associated neuronal (nE4-DAN) phenotype24, HMGB1 release (resulting in binding with its receptors on glial cells and consequently inducing gliosis)129, neuronal MHC1-mediated microglia engagement90 and synaptic and neuronal degeneration24,85,90,98,114. The potential relationship among these neuronal APOE4 effects on other cells and AD pathologies is depicted using arrows. Note that non-neuronal sources of APOE4 have been omitted for clarity.
The cellular and molecular mechanisms that control neuronal APOE expression are not fully understood. One study identified a neuron-specific splicing variant of APOE mRNA with an intron-3 retention (APOE-I3) that may help regulate neuronal APOE expression91 (Fig. 3a). The authors suggested that APOE transcripts are continuously produced in neurons under normal conditions, and the subsequently generated APOE-I3 pre-mRNA is retained in the nucleus without being translated. However, in response to neuronal stress or injury, it is quickly processed into mature APOE mRNA for protein translation91 (Fig. 3b). Consistent with this model, neuronal APOE expression is increased in response to CNS injury, potentially enabling time-sensitive neuronal protection and repair92,93. Indeed, neuronal APOE may be important for neurogenesis94 and neuronal remodeling1,95 (Fig. 3a). RNA-seq analysis has shown that the abundance of APOE-I3 transcripts in the brains of individuals with AD is related to the severity of tau and amyloid pathologies96. Interestingly, the level of APOE-I3 transcripts increased with the number of APOE ɛ4 alleles that an individual possessed96, possibly reflecting increasing levels of neuronal stress and injury. A recent study identified a stress-inducible transcriptional mechanism involving R-loops and RNA abasic sites that regulates an enhancer RNA that activates APOE transcription in non-neuronal cells97. An interesting question is whether this enhancer RNA also regulates APOE expression in stressed neurons and, if so, whether it affects the production or splicing of APOE-I3 pre-mRNA.
Like astrocytic APOE expression, neuronal APOE expression is also regulated by C/EBPβ45,98 (Fig. 3b). Notably, C/EBPβ promotes the expression of APOE4 more than APOE345,98. Interestingly, neuronal APOE has also been found to stimulate C/EBPβ activation, resulting in a feed-forward loop in which downstream AD pathology can be further exacerbated98. In addition, astrocytes may secrete factor(s) that regulate neuronal APOE expression through an extracellular signal-regulated kinases (ERK)-dependent pathway48, possibly by promoting the splicing of APOE-I3 into mature APOE mRNA91. Clearly, further studies are needed to fully elucidate the mechanisms that regulate neuronal APOE expression.
Neuronal APOE4 causes cellular and network dysfunction
Neuronal APOE4 can have detrimental effects on synaptic and neuronal network functions (Fig. 3b,c) that occur early in life. Neuron-specific APOE4 expression in mice with a mouse ApoE-KO background resulted in synaptic loss as well as learning and memory deficits at 6–9 months of age99–101. This APOE4-induced synaptic loss may be related to an increase in phosphorylation of vasodilator-stimulated phosphoprotein (VASP) S235 that disrupts its interactions with numerous actin cytoskeletal and microtubular proteins102. A recent preprint reported that ex vivo hippocampal neurons taken from 7–9-month-old APOE4-KI mice, compared to APOE3-KI controls, displayed hyperexcitability that was prevented by removal of neuronal, but not astrocytic, APOE4103. This neuronal hyperexcitability persisted as the mice aged, probably due to an age- and APOE4-dependent loss of inhibitory tone103,104. Recordings of local field potentials in 6–8-month-old APOE4-KI mice revealed reductions in hippocampal sharp wave ripple (SWR) activity and in the power of associated slow gamma oscillations, which are critical for memory consolidation105,106. Strikingly, the presence of a SWR-associated slow gamma oscillation deficit at a younger age predicted the development of learning and memory impairments 10 months later in these mice105,106. Moreover, selective removal of neuronal, but not astrocytic, APOE4 — especially from GABAergic interneurons — prevented these abnormalities42,103,105, highlighting the pathogenic importance of neuronal APOE4. In fact, the extent of APOE4-induced GABAergic interneuron loss (especially somatostatin (SST)-expressing interneuron loss) correlated with the severity of learning and memory deficits in aged APOE4-KI mice42,107,108. Interestingly, in APOE-KI mice, APOE expression in SST interneurons was ~40% higher than it was in dentate gyrus granule cells and CA1 neurons, suggesting that higher APOE expression (especially APOE4) renders SST interneurons selectively vulnerable to degeneration and loss in AD90.
Although the mechanisms by which neuronal APOE4 impairs the functions and survival of neurons remain to be fully defined, several studies suggest that impairments of specific subcellular compartments may be involved. Neuronal APOE4 impairs the function and integrity of mitochondria95,109–111 (Fig. 3b,c), which could result in energy failure. Primary neurons from transgenic mice with neuron-specific expression of APOE4 had reduced levels of mitochondrial respiratory complexes, as compared to those expressing APOE3112. Proteomic analysis of mouse neuroblastoma cell lines expressing human APOE4 versus APOE3 yielded compatible results113. Notably, no significant differences in the levels of neuronal mitochondrial complexes were detected in transgenic mice expressing APOE4 versus APOE3 specifically in astrocytes, making it unlikely that these changes result from the indirect effects of astrocytic APOE4112. In a similar vein, neuronal (but not astrocytic) expression of APOE4 sensitized mice to excitotoxin-induced neurodegeneration114. In humans with early-stage AD115 and in cultured neuronal cells116, APOE4 is also associated with neuronal endo-lysosomal defects (Fig. 3c), which could contribute to its detrimental effect on receptor recycling and synaptic plasticity69.
Neuronal APOE4 and hallmark AD pathologies
Neuronal APOE4 also affects major AD pathologies, including amyloid and tau pathologies, gliosis, neurodegeneration and myelin deficits (Fig. 3c). APOE4 hiPSC-derived neurons produced more Aβ, had higher levels of APOE fragments and p-tau and more degeneration of GABAergic neurons than APOE3 controls85. These pathologies were rescued by gene-editing of APOE4 to APOE385. In another study, APOE4 hiPSC-derived neurons were more susceptible to cytotoxicity than APOE3 controls and had higher p-tau levels117. Although the mechanisms that link neuronal APOE4 and p-tau remain to be fully defined, several interesting leads have emerged. For example, heparan sulfate proteoglycans (HSPGs) are cell-surface receptors that interact more strongly with APOE4 than APOE313,118 and are involved in the inter-neuronal spread of tau119–121. Treatment of APOE4 neurons with an HSPG inhibitor reduced their p-tau levels117. Moreover, APOE4 hiPSC-derived neurons carrying the APOE-R136S mutation, which strongly protects against ADAD122,123, had lower p-tau levels and reduced tau uptake via HSPG than isogenic APOE4 neurons without this mutation28. Taken together, these results identify HSPGs as a critical mediator of copathogenic interactions between APOE4 and tau.
The effects of neuronal APOE4 on AD pathologies are also evident in vivo. In transgenic mice, expressing APOE4 in neurons increased neuronal p-tau levels, whereas expressing APOE4 in astrocytes did not75,76, indicating that neuronal APOE4 is more directly linked than astrocytic APOE4 to neuronal p-tau accumulation. In PS19-APOE4 mice, selective removal of APOE4 from neurons markedly reduced tau pathology24, whereas selective removal of astrocytic APOE4 in a similar model lowered p-tau levels only moderately43 (an effect that might have resulted secondarily from a reduction in astrocytosis and microgliosis124). Moreover, neuronal, but not astrocytic APOE4 promoted excitotoxic neuronal death114. A recent study further suggests that neuronal APOE4 activates C/EBPβ, which regulates the expression of APP, MAPT (the gene encoding tau) and beta-secretase 1 (BACE1, encoding an enzyme involved in APP processing), promoting Aβ deposition, tau aggregation, and neurodegeneration98. A recent preprint has reported that, in experiments in which APOE4 hiPSC-derived neurons were transplanted into the hippocampi of APOE4-KI mice in conjunction with microglial depletion, both human neuronal APOE4 and microglia were important for the formation of dense-core Aβ plaques and tau aggregates125.
Although the mechanisms linking neuronal APOE4 to the pathological hallmarks of AD are likely multifarious, diverse lines of evidence suggest several mechanisms that have a prominent role and may make interesting targets for therapeutic interventions. One of these mechanisms involves the intraneuronal proteolytic cleavage of APOE, to which APOE4 is much more susceptible than APOE3, and which results in the generation of C-terminally truncated, neurotoxic APOE fragments22. Such APOE4 fragments can elicit the formation of intraneuronal p-tau accumulation and neurodegeneration in neuronal cultures and transgenic mice22,126 and reduce Aβ clearance in mutant APP transgenic mice127. Transgenic mice expressing full-length APOE4 in neurons showed an age-dependent accumulation of APOE4 fragments in the hippocampus, whereas transgenic mice expressing APOE4 in astrocytes did not75. Furthermore, APOE4 hiPSC-derived neurons85 and AD brains from APOE4 carriers126,128 had higher levels of APOE fragments than those from non-carriers. Taken together, these findings indicate that APOE4 proteolysis in neurons promotes the intraneuronal accumulation of p-tau as well as other AD-related neuronal deficits (Fig. 3b,c).
Another mechanism linking APOE4 to such deficits is the interaction between neuronal APOE and major histocompatibility complex class 1 (MHC1) molecules (Fig. 3b,c). Neuronal expression of APOE and MHC1 molecules have been shown to be tightly and positively correlated, and reducing neuronal MHC1 molecule levels resulted in reduced intraneuronal accumulation and mislocalization of p-tau in mouse primary neurons overexpressing APOE490. It was proposed that the APOE4-induced increase in neuronal MHC1 molecule expression serves as an ‘eat me’ signal to recruit microglia (and potentially T cells) to engage with the ‘tagged’ neurons, causing synaptic and neuronal loss90. In line with this model, selective removal of APOE4 from neurons in PS19-APOE4 mice markedly reduced not only tau pathology, but also gliosis, neurodegeneration and myelin deficits in the hippocampus24. More specifically, PS19-APOE4 mice exhibited significantly higher levels of pathological tau accumulation, astrocytosis, microgliosis, hippocampal neuronal loss and myelin deficits compared to PS19-APOE4 mice in which neuronal APOE4 was selectively removed, suggesting that neuronal APOE4 is an upstream initiator and potent driver of these AD-related pathologies and that its removal is sufficient to attenuate these phenotypes24.
In addition to MHC1 molecules, another proinflammatory molecule that may promote pathogenic neuronal–glial interactions is high mobility group box 1 (HMGB1). Compared to APOE3, neuronal expression of APOE4, together with tau pathology, increased the nucleo-cytoplamic translocation and cellular release of HMGB1. This triggered gliosis, likely by binding to toll-like receptors (TLRs) on glial cells129 (Fig. 3c). Clinical studies of individuals with MCI have revealed an increase in HMGB1 levels in their cerebrospinal fluid (CSF)130 and an interactive association of APOE4 and plasma HMGB1 levels with widespread cortical thinning131. Notably, pharmacological inhibition of HMGB1 release from neurons ameliorated neuroinflammation, neurodegeneration and myelin deficits in PS19-APOE4 mice129. Interestingly, HMGB1 can upregulate MHC1 molecule expression in response to cellular stress132,133. Thus, in response to neuronal APOE4 expression, HMGB1 and MHC1 molecules may act in concert to trigger glia-mediated synaptic and neuronal degeneration (Fig. 3c).
APOE4 in Disease-associated neuronal subtypes
snRNA-seq studies have identified neuronal states that are enriched in disease, known as disease-associated neuron (DAN) phenotypes. A comparative snRNA-seq analysis identified two excitatory neuronal clusters with abnormally high expression of genes encoding major heat shock proteins, calmodulin, and ubiquitin B in the hippocampus of PS19-APOE4 mice that were not seen in PS19-APOE4 mice with neuronal APOE4 removal (these were referred to as neuronal APOE4-promoted DAN or nE4-DAN)24. nE4-DAN had high levels of APOE4 expression, as compared to other excitatory neuron subpopulations, and were associated with high levels of tau pathology and hippocampal atrophy24 (Fig. 3c).
Relative levels of APOE expression may also provide insight into the vulnerability of different subtypes of neurons in AD. In APOE4-KI mice, snRNA-seq revealed higher levels of APOE4 expression in inhibitory than excitatory neurons90, paralleling the earlier onset of the detrimental effects of APOE4 on inhibitory neurons42,107,108. In a similar vein, snRNA-seq analyses of human cortices from individuals with AD revealed that disease progression was related to the proportion of neuronal cells, especially inhibitory interneurons, expressing high levels of APOE90,134. Specifically, the proportion of APOE-expression-high neurons, especially inhibitory neurons, was relatively low in individuals with no cognitive impairment, was highest in individuals with MCI, and was relatively low in individuals with AD. This pattern suggests that neuronal (especially inhibitory neuronal) APOE expression is upregulated early in the clinical progression of AD and declines thereafter as neurodegeneration advances90, which is in line with the observation that vulnerable inhibitory neuron subtypes were selectively depleted in the brains of individuals with AD135.
In summary, in response to stress, aging or disease, some (but not all) neurons increase their APOE expression (Fig. 3b). In APOE4 carriers (to a greater extent than in APOE3 carriers), this process results in the intraneuronal generation of neurotoxic APOE fragments, neuronal dysfunction, the emergence of nE4-DAN and the overproduction or increased release of proinflammatory molecules, triggering glial responses that further promote synaptic and neuronal degeneration (Fig. 3b,c). Specific subpopulations of inhibitory neurons produce more APOE and may also be more vulnerable to detrimental APOE4 effects than other types of neurons. Their dysfunction and loss lead to excitation-inhibition imbalance and network hyperexcitability, which is associated with faster cognitive decline in AD136,137. Thus, neuronal APOE4 is likely to be a critical upstream pathogenic initiator and driver that directly or indirectly affects many AD-related cell types, genes and pathways (Fig. 3c).
Microglial APOE4 in AD pathogenesis
Expression and regulation of microglial APOE
Like neurons, microglia can express APOE in a stress and injury-dependent manner39,47,138–140 (Fig. 4a,b). While APOE expression is low in microglia under normal conditions (Fig. 4a), microglia in both aging and diseased brains have been shown to greatly upregulate their APOE expression39,138–142 (Fig. 4b). For instance, microglial APOE expression was increased in aged human APOE-KI mice141, PS19 mice33, and 5xFAD mice139. Notably, microglia surrounding Aβ plaques displayed the greatest increase in APOE expression124,140,143.
Fig. 4: Expression of APOE4 in microglia and its roles in AD pathogenesis.
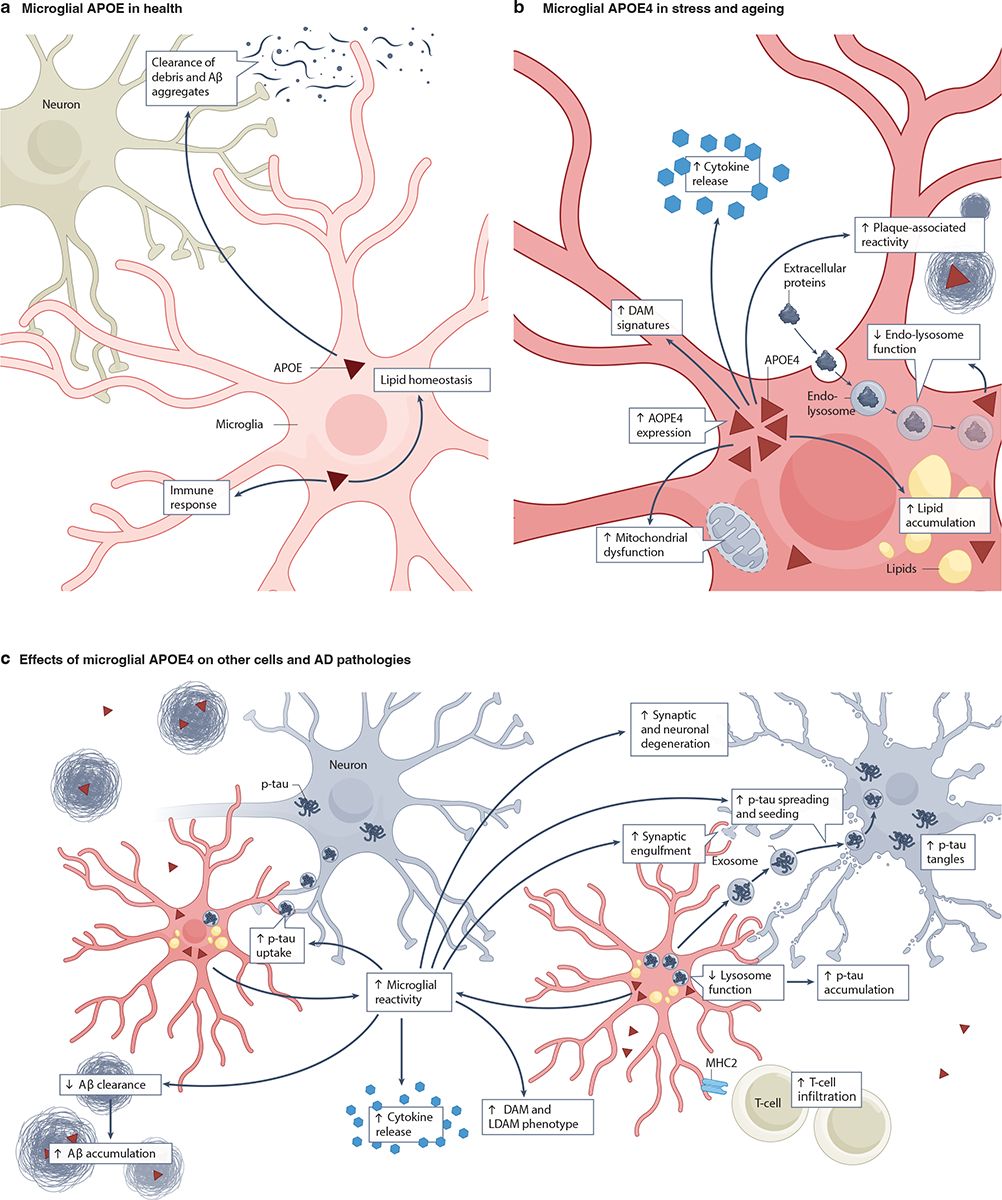
a, Healthy microglia express low levels of apolipoprotein E (APOE)39. This APOE is involved in immune responses, lipid homeostasis and in clearing amyloid β (Aβ) aggregates and cell debris23,53,143. b, During stress and aging, microglia increase their APOE expression138–141. Microglial APOE4 leads to an increase in amyloid plaque-associated reactivity54, lipid accumulation27,148, lysosomal defects151,152, mitochondrial dysfunction141, disease-associated microglial (DAM) signatures140,167 and proinflammatory cytokine release167. c, Microglial APOE4 affects other cells, including neurons and T cells166, and contributes to AD pathogenesis by reducing Aβ clearance157 and lysosome function151 and by increasing synaptic engulfment161, p-tau uptake and accumulation86,151,152, DAM signatures140,152, lipid-droplet-accumulating microglia LDAM29,147, and cytokine release167. Together, these APOE4-induced microglial dysfunctions facilitate tau spreading and seeding162–164 and, ultimately, synaptic and neuronal degeneration (in conjunction with neuronal APOE4)140,152. The potential relationships among these microglial APOE4 effects on other cells and AD pathologies is depicted using arrows. Note that non-microglial sources of APOE have been omitted for clarity.
The low-density lipoprotein receptor (LDLR) pathway may directly or indirectly contribute to the regulation of APOE expression in microglia, as LDLR overexpression in microglia in mice suppressed microglial activation and expression of APOE33. APOE produced by other CNS cells may also affect microglia, as selective removal of neuronal APOE4 in PS19-APOE4 mice decreased the sizes of populations of microglia exhibiting high levels of APOE expression24. This suggests that neuronal APOE4, or the pathological alterations that it causes, augment microglial APOE expression. Since microglia have been implicated in a variety of APOE4-related AD pathologies23,144, it is crucial to understand the effects of microglia-derived APOE4.
Microglial APOE4 in metabolism and lipid accumulation
APOE genotype influences microglial metabolism and lipid accumulation (Fig. 4b). Homeostatic microglia have been shown to rely on oxidative phosphorylation for energy production, while activated microglia with high APOE expression shift toward the glycolytic pathway and increase their expression of hypoxia inducible factor 1-alpha (HIF1α), a gene highly upregulated during glycolysis141,145. These findings are in line with observations indicating that aged APOE4-KI mice had increased aerobic glycolysis and higher HIF1α expression141.
Emerging evidence highlights the role of APOE in microglial lipid metabolism and accumulation (Fig. 4b). APOE is a major lipid transporter between neurons and glial cells11,19. APOE-containing lipid particles produced by APOE4-KI mouse microglia were smaller than those produced by astrocytes, implying cellular source-dependent differences in the lipidation state of APOE146. Furthermore, lipid droplet accumulation was a major phenotype observed in aged and diseased microglia27,29,147,148 (Fig. 4b,c). Similarly, APOE4 hiPSC-derived microglia (hiMGL) accumulated more lipid droplets than APOE3 microglia27,149. Additionally, when human APOE was selectively expressed in microglia in an amyloidosis model in which mice expressing APP harboring the Swedish FAD mutation are crossed with mice in which PSEN1 exon 9 is deleted (APPswe/PS1ΔE9 mice) on a mouse ApoE-KO background, APOE4 microglia showed increased lipid accumulation compared to APOE3 microglia149. APOE4 microglia also demonstrated excessive buildup of lipid droplets in APOE-KI mice fed with cuprizone, which induces demyelination, compared to APOE3 and APOE2 microglia, indicating an impairment in lipid metabolism in APOE4 microglia in response to demyelination150.
Notably, in hiMGL, human post mortem brains, and APOE-KI mouse brains, APOE4 increased de novo cholesterol synthesis in microglia and impaired cholesterol trafficking, possibly through lysosomal sequestration60. Several other studies also showed that microglial APOE4 expression caused lysosomal dysfunction through aberrant lysosomal trafficking151 and impaired endosomal-lysosomal activities86,151,152. Additionally, during de novo fat synthesis, APOE4 hiMGL exhibited lipogenesis that promoted proinflammatory signals and reduced the uptake of extracellular fatty acids and lipoproteins, thereby disrupting neuronal network activity147.
Microglial APOE4, functional deficits and AD pathologies
Microglial APOE appears to be a key regulator of microglial function and reactivity, which consequently affect global brain health and disease progression (Fig. 4b,c). Ingenuity pathway analysis (IPA), a pathway analysis application used for high throughput omics data analysis, on isolated microglia from APP-PS1 mice revealed that APOE affected the relative proportions of homeostatic microglia versus microglia associated with neurodegeneration143. In APOE-KI mice, APOE4 microglia displayed increased activation and exacerbated pro-inflammatory responses86,152 and, in postmortem human brains, APOE4 carriers had increased numbers of microglia compared to non-carriers153,154. In hiMGL, APOE4 promoted a more reactive morphology and enhanced inflammatory gene signatures compared to APOE354. Lastly, when stimulated with lipopolysaccharide (LPS), microglia from APOE4-KI mice exhibited higher levels of proinflammatory signals compared to microglia from APOE3-KI mice155.
APOE genotype also influences vital microglial functions such as phagocytosis and chemotaxis53,156 (Fig. 4a,b). For instance, in murine N9 microglia expressing human APOE isoforms, those expressing APOE4 showed impaired phagocytosis of Aβ42 (an aggregation prone form of Aβ peptides) and dysfunctional motility156. Moreover, in APPswe/PS1ΔE9 mice that received bone marrow transplants from human APOE4-KI mice (APOE4-KI-BMT), microglia exhibited reduced association with Aβ deposits and dystrophic changes and amyloid plaques were larger and more abundant than in APPswe/PS1ΔE9 mice that received APOE3-KI-BMT157. Meanwhile, selectively removing microglial APOE4 in APP/PS1 mice led to significant reduction in plaque loads compared to APOE4-APP/PS1 mice158, suggesting a ‘gain-of-toxic’ effect of APOE4 on microglia-mediated Aβ clearance. Similarly, when APOE was selectively expressed in microglia in APPswe/PS1ΔE9 mice on a mouse ApoE-KO background, the presence of APOE3 microglia sufficiently protected against the development of amyloid pathology, whereas APOE4 microglia did not149. This defective response of APOE4 microglia to Aβ pathology seemed to be mediated by the APOE4–ITGB8–TGFβ pathway, which acted as a negative regulator158. A similar defective response of APOE4 microglia to Aβ was also observed in a chimeric model in which hiPSC-derived neurons were transplanted into APOE-KI mouse hippocampus: here, APOE4 microglia tended to colocalize less with Aβ deposits than APOE3 microglia159. Mechanistically, IL-33 has been shown to mediate microglial migration towards APOE-associated Aβ plaques by inducing microglial VCAM1, and when this VCAM1–APOE interaction was disrupted, Aβ clearance was reduced in APP/PS1 mice160.
In contrast to these effects, APOE4-expressing microglia actually exhibited increased phagocytosis of apoptotic cells in culture (N9 murine microglia overexpressing APOE4)156 and increased synaptic engulfment in PS19-APOE4 mice161 (Fig. 4c). Some studies even suggest that microglia may actively participate in tau spreading and seeding162–164. According to these studies, reactive microglia may phagocytose tau or tau-containing synapses and/or neurons. Due to APOE4-dependent lysosomal defects and resulting inefficiencies in processing the internalized tau86,151,152, pathological tau aggregates then begin to accumulate within the microglia and are released via exosome transfer to spread and seed tau pathology162–164 (Fig. 4c). Consistent with this model, a human brain imaging study found greater levels of microglial activation and tau accumulation in medial temporal structures of APOE4 carriers than in non-carriers, possibly contributing to their increased neurodegeneration and clinical impairment165. Likewise, a recent preprint has reported that, in a chimeric AD model in which APOE4 hiPSC-derived neurons were transplanted into APOE4-KI mouse hippocampus, microglial depletion reduced human neuronal APOE4-driven tau pathology125. Interestingly, selectively removing microglial APOE4 in PS19-APOE4 mice restored microglia to a neuroprotective phenotype158 and microglial depletion in these mice reduced tau pathology and neurodegeneration, possibly by reducing cerebral infiltration by T cells responding to MHC2 molecule-expressing microglia166 (Fig. 4c).
APOE4 in disease-associated microglial subtypes
Microglia are an extremely heterogeneous cell type whose roles and functions in the CNS confer unique spatial-temporal gene signatures. The transcriptional profile of microglia is markedly altered in AD and in the presence of APOE4, with an increased proportion of microglia showing disease-associated rather than homeostatic profiles140,152,167. Disease-associated subtypes or states of microglia include disease-associated microglia (DAM)140,152, lipid-droplet-accumulating microglia (LDAM)29, interferon responsive microglia (IRM)168, and antigen-presenting microglia (MHC2-M)125,168. Among these, DAM and LDAM have been studied most extensively in the context of APOE4 (Fig. 4c).
DAM are characterized by increased expression of genes associated with proinflammatory immune responses, phagocytosis and lipid metabolism, and decreased expression of genes associated with surveillant microglia140,152. Interestingly, even in the absence of overt AD pathology, the combination of APOE4 and aging was sufficient to elicit changes in microglia of APOE4-KI mice that strongly resembled the DAM phenotype141. In AD-related mouse models with amyloid or tau pathology, DAM localized around Aβ plaques and NFT-bearing neurons169. Remarkably, selectively ablating endogenous microglial APOE using Cx3cr1-Cre:ApoE mice prevented the emergence of microglia with a neurodegenerative phenotype (MGnD, another type of DAM), compared to WT mice143. The same study showed that global ApoE-KO conferred no additional protection, suggesting that microglial APOE is particularly crucial in initiating the appearance of MGnD143. However, selectively removing microglial APOE4 in APP/PS1/APOE4 or PS19-APOE4 mice increased MGnD158 and, conversely, selectively expressing APOE4 in microglia in APPswe/PS1ΔE9 mice on a mouse ApoE-KO background decreased activated response microglia (ARM, another type of DAM), which highly expressed MHC genes149. Together, these findings suggest that mouse APOE and human APOE4 might affect MGnD and ARM differently, which warrants further in-depth study. It should be noted that the proportion of DAM was not reduced in PS19-APOE4 versus PS19-APOE3 mice in other studies24,28,43,129.
As may be expected, DAM (or MGnD) have increased APOE expression, which precedes the decrease in homeostatic markers (such as P2Y purinoceptor 12 (P2RY12) and C–X–C motif chemokine ligand 3 (CXCL3)) and increase in disease-associated signatures (including lipoprotein lipase (LPL), cystatin-F (CST7), and secreted phosphoprotein-1 (SPP1)140,143, suggesting that APOE influences microglial gene expression. Human APOE4 carriers and human APOE-KI mice crossed with APP/PS1 mice or stimulated with LPS demonstrate that APOE4 primes microglia for conversion into the phagocytic and proinflammatory state of DAM or promotes the proliferation of such cells167. In PS19-APOE4 mice with or without neuronal APOE4 removal, neuronal APOE4-promoted DAM (nE4-DAM) subtypes were identified and found to positively associate with tau pathology and hippocampal degeneration24. Furthermore, introduction of the APOE-R136S mutation, which protects against ADAD122,123, markedly reduced the number of DAM as well as tau pathology and neurodegeneration in PS19-APOE4 mice28. Interestingly, removing APOE4 selectively from astrocytes43 or neurons24 in PS19-APOE4 mice also decreased DAM and nE4-DAM, respectively, suggesting that both astrocytic and neuronal APOE4 promote the emergence of DAM subtypes.
LDAM are another emerging subtype of diseased microglia, defined by an increased expression of genes, such as ACSL1, that are involved in pathways related to lipid handling, transport and metabolism29. LDAM also exhibit an accumulation of lipid droplets in the cell soma, which may indicate increased endogenous lipid production, excessive uptake of extracellular lipids from degenerating neurons, or an inability to degrade lipids29. Notably, APOE4 hiMGL show more lipid droplet accumulation than APOE3 microglia27,147,149. Recent research has raised the possibility that dysfunctional lipid metabolism and lipid droplet formation in APOE4 microglia impair microglial surveillance of neuronal network activity147. Furthermore, a recent preprint has reported that this can promote neuronal p-tau accumulation and neurotoxicity27. Interestingly, introduction of the protective APOE-R136S mutation markedly reduced lipid droplet accumulation in microglia of PS19-APOE4 mice28.
Beyond DAM and LDAM, other disease-associated microglial subtypes continue to be identified. For example, in a chimeric AD model in which APOE4 hiPSC-derived neurons were transplanted into APOE4-KI mouse hippocampus, there was an enrichment in microglial subpopulations highly expressing MHC2 genes (MHC2-M) that contributed to neuronal APOE4-promoted Aβ and tau pathologies125. Comparing to the data from APPswe/PS1ΔE9 mice selectively expressing APOE4 in microglia on a mouse ApoE-KO background149, the data from the chimeric AD model suggest that APOE4 microglia might respond differently to human neuron-produced versus mouse neuron-produced AD pathologies, which warrants further in-depth study. MHC2-M have been found to be enriched in human AD patient brains, close to amyloid plaques and tau tangles170,171. MHC2-M were also enriched in the hippocampus of PS19-APOE4 mice, which was important for recruiting infiltrating T cells166.
Altogether, these findings have begun to highlight the heterogeneity of microglia and their differential contributions to AD pathogenesis, especially in the context of APOE4 (Fig. 4c). Clearly, microglial APOE4 promotes the emergence of DAM subtypes. Notably, APOE4 in DAM and LDAM may increase the phagocytosis of neuronal synapses containing pathological tau, leading to increased synaptic loss and neurodegeneration. APOE4-associated endo-lysosomal defects in microglia may impede the degradation of pathological tau and promote the accumulation of tau in microglia, a process that may contribute to tau spreading and seeding (Fig. 4c). APOE4 also seems to enhance MHC2-M-dependent T-cell infiltration, which may further exacerbate tau pathology and neurodegeneration.
Oligodendrocytic APOE4 in AD pathogenesis
Expression and regulation of oligodendrocytic APOE
Like the other glial cells of the CNS, oligodendrocytes can express APOE24,31,90 (Fig. 5a). In PS19-APOE4 mice, a subgroup of oligodendrocytes exhibited increased expression of APOE compared to PS19-APOE3 mice24. This suggests that increased APOE expression is part of their injury response, and that this is augmented in PS19-APOE4 mice because they have more tauopathy and neurodegeneration than PS19-APOE3 mice (Fig. 5b). Consistent with this interpretation, rat primary oligodendrocytes increased their expression and secretion of APOE when they were treated with conditioned medium from mechanically injured neurons172. Notably, oligodendrocytic APOE expression is also affected by neuronal APOE, as selective ablation of APOE4 from neurons reduced oligodendrocytic APOE expression in PS19-APOE4 mice24.
Fig. 5: Expression of APOE4 in oligodendrocytes and vascular cells and its roles in AD pathogenesis.
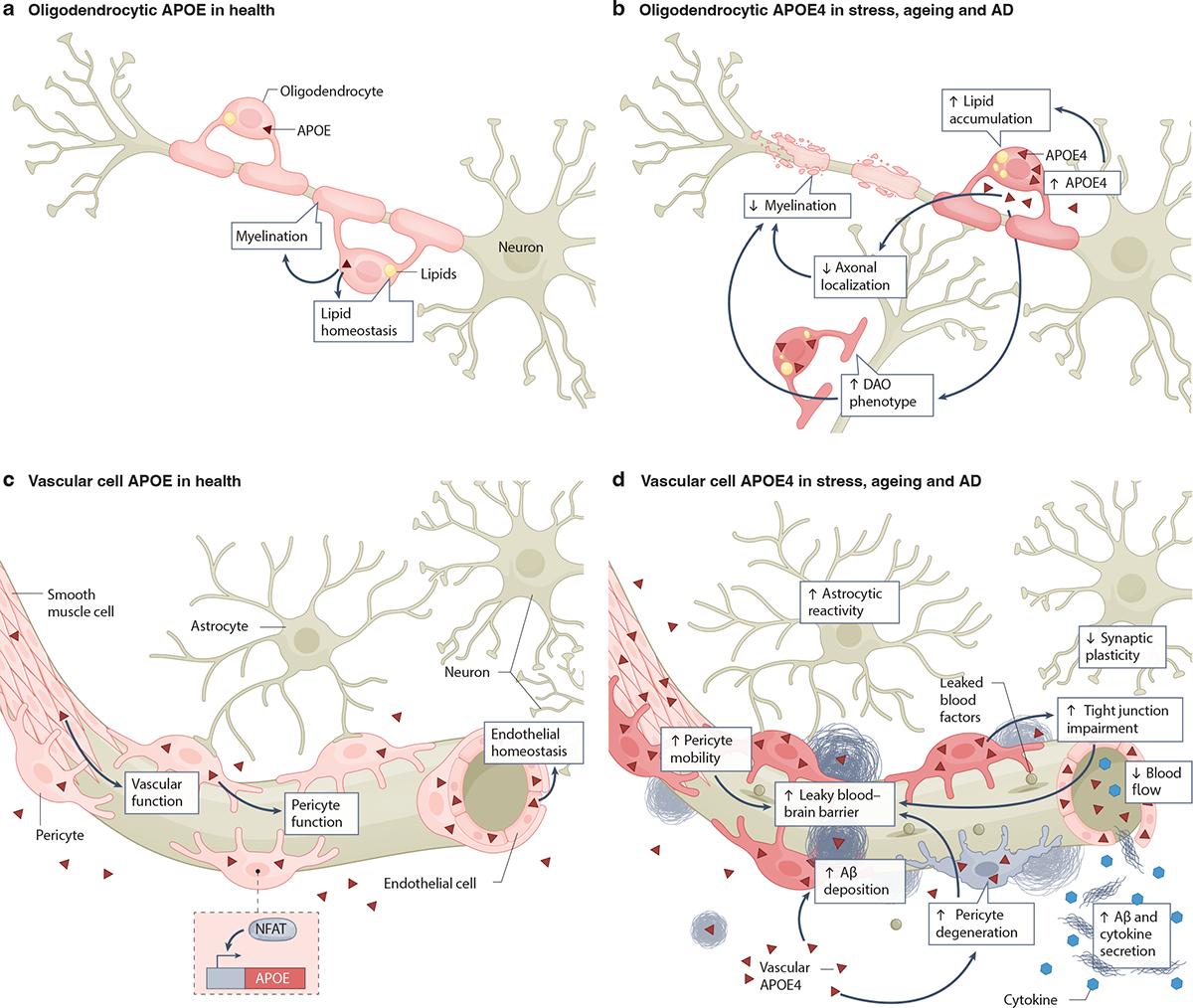
a, Healthy oligodendrocytes express low levels of apolipoprotein E (APOE)24,31,90. Oligodendrocytic APOE is involved in lipid homeostasis and myelination of neurons31. b, Under conditions of stress, aging, and AD, oligodendrocytes increase their APOE4 expression24,172. Oligodendrocytic APOE4 increases intracellular lipid accumulation and disease-associated oligodendrocyte (DAO) phenotypes24,31 and decreases oligodendrocytic axonal localization and myelination of neuronal axons31. The potential relationship among oligodendrocytic APOE4 effects on AD pathologies is depicted using arrows. c, Healthy cells of the brain vasculature, including smooth muscle cells, pericytes and endothelial cells, produce and secrete APOE39,176–182. The transcription factor NFAT regulates APOE expression in pericytes178. Vascular cell-produced APOE is involved in maintaining vascular function, pericyte function and endothelial homeostasis176–178,183. d, Under conditions of stress, aging, and AD, vascular cell APOE4 leads to tight junction impairments36,183, astrocyte reactivity185, increased pericyte migration and degeneration176,177, decreased synaptic plasticity185, reduced blood flow185, elevated secretion of amyloid β (Aβ) and cytokines180, increased Aβ deposition in the form of cerebral amyloid angiopathy (CAA)178, and blood-brain barrier (BBB) leakage35,36,176. The potential relationship among vascular APOE4 effects on other cells and AD pathologies is depicted using arrows. Note that non-oligodendrocytic and non-vascular sources of APOE4 have been omitted for clarity.
Oligodendrocytic APOE4 in lipid metabolism and myelination
APOE isoforms differentially affect oligodendrocytic lipid metabolism and myelination (Fig. 5a,b). Notably, oligodendrocytes of the prefrontal cortex of human APOE4 carriers exhibited elevation of cholesterol biosynthesis-associated gene expression compared to oligodendrocytes of non-carriers31. Interestingly, this APOE4 effect was gene dose-dependent31. Similarly, APOE4 hiPSC-derived oligodendrocytes displayed increased cholesterol synthesis and intracellular cholesterol storage compared to APOE3 oligodendrocytes31. The APOE4 oligodendrocytes also had reduced cholesterol localization in the plasma membrane, increased cholesteryl esters and lipid droplets, and elevated endoplasmic reticulum (ER) stress31. When co-cultured with hiPSC-derived neurons, APOE4 oligodendrocytes displayed less myelin basic protein (MBP) and axonal colocalization than their APOE3 counterparts31 (Fig. 5b). Furthermore, aged APOE4-KI mice showed fewer cells in the oligodendrocyte lineage and an abnormal accumulation of oligodendrocyte precursors, suggesting that APOE4 disrupts oligodendrocyte differentiation and maturation173. Indeed, neural stem/progenitor cell differentiation into oligodendrocytes is affected by interactions between APOE and LDLR-related protein-1174. Together, these findings raise the possibility that alterations in the differentiation and cholesterol metabolism of oligodendrocytes may causally contribute to the link between APOE4 and AD (Fig. 5b), although this hypothesis requires further testing.
APOE4 in disease-associated oligodendrocyte subtypes
snRNA-seq analyses have identified subpopulations of oligodendrocytes that are enriched in brains of patients with neurodegenerative diseases and related mouse models, termed disease-associated oligodendrocytes (DAO)175 (Fig. 5b). In a variety of pathological conditions, including animal models of multiple sclerosis, amyloidosis, tauopathy and aging brains, DAO show increased expression of immune-related genes175, suggesting neuroinflammation as a common driver of DAO formation175.
Selective neuronal ablation of APOE in PS19-APOE4 mice identified a specific subtype of DAO (neuronal APOE4-promoted DAO or nE4-DAO)24. nE4-DAO had increased expression of genes encoding APOE, heat shock protein, calmodulin, or ubiquitin B, and reduced expression of genes encoding MBP or myelin-associated oligodendrocyte basic protein, as compared to other oligodendrocyte subpopulations24. The number of nE4-DAO was negatively related to hippocampal volume and MBP coverage area and positively related to the extent of tau pathology24. Removal of neuronal APOE4 diminished the number of nE4-DAO, demonstrating their dependence on neuronal APOE4 effects. Interestingly, the protective APOE-R136S mutation markedly reduced the number of DAO in PS19-APOE4 mice28.
Taken together, these findings suggest that oligodendrocytes respond to APOE4 effects from other types of brain cells, especially neurons (Fig. 3c), but are also directly impacted by the APOE isoforms that they express (Fig. 5b). The detrimental effects of APOE4, either directly or indirectly, on oligodendrocytes can lead to myelin impairments in AD.
Vascular cell APOE4 in AD pathogenesis
Expression and regulation of APOE in vascular cells
Multiple vascular cell types, including endothelial cells, pericytes, and smooth muscle cells, express APOE39,176–182 (Fig. 5c). Several transcription factors, including C/EBPβ and NFAT, may regulate APOE expression in these cells178. Pericytes seem to secrete large amounts of APOE that they produce181 and are located in a position in which they can affect the neurovascular junction. Interestingly, both cultured APOE4 hiPSC-derived pericytes and pericytes in the hippocampus of human APOE4 carriers expressed higher levels of APOE than APOE3 pericytes178. Though APOE from vascular cells has been studied much less than APOE produced by other CNS cell types, it is possible that it may also affect brain health and disease in an isoform-dependent manner (Fig. 5c,d).
Vascular cell APOE4 in stress and AD
APOE4 produced by vascular cells has detrimental effects on endothelial cell function, pericyte mobility, Aβ-driven cytotoxicity and BBB integrity176–178,183 (Fig. 5d). Human endothelial cells co-cultured with pericytes from APOE4-KI mice exhibited reduced extracellular matrix protein production, impaired formation of normal tube-like structures and decreased barrier formation compared to those cultured with APOE3 pericytes183. In cultures of primary human brain vascular pericytes in which endogenous APOE3 was 90% knocked down via small interfering RNA treatment, addition of exogenous APOE3 led to typical levels of pericyte migration, whereas cells treated with APOE4 had aberrantly high pericyte mobility176. Conceivably, this effect could contribute to BBB dysfunction in AD, as the BBB becomes increasingly vulnerable when pericytes migrate away towards sites of pathology176. By increasing pericyte mobility, APOE4 may promote leakiness of this important barrier. In addition, APOE4 increases and activates the BBB-degrading pro-inflammatory cyclophilin A (CYPA)–matrix metalloproteinase 9 (MMP9) pathway in pericytes, leading to breakdown of the BBB via degradation of BBB tight junction proteins36,184. Increased activity of this pathway in the CSF of human APOE4 carriers was associated with high baseline levels of soluble platelet-derived growth factor receptor beta (PDGFRB), a marker of pericyte injury linked to BBB breakdown, and this was predictive of future cognitive decline35.
APOE isoforms may also differentially affect the susceptibility of pericytes to Aβ (Fig. 5d). When treated with exogenous Aβ, primary human APOE4-expressing brain pericytes showed higher levels of cell death than their APOE3 counterparts177. Similarly, in a 3-dimensional in vitro BBB model that included hiPSC-derived pericyte-like cells, amyloid deposition was highly elevated only in cultures containing APOE4 pericytes178. Inhibition of NFAT–calcineurin signaling in pericytes decreased their APOE4 expression and reduced amyloid accumulation in this model178. These findings suggest a role for pericyte-derived APOE4 in cerebral amyloid angiopathy (CAA) and pinpoint calcineurin–NFAT signaling as a potential therapeutic target. APOE4 hiPSC-derived endothelial cells exhibited increased binding of platelets and higher secretion of cytokines and Aβ compared to endothelial cells expressing APOE3180, indicating that APOE4 may exert detrimental effects also on this cell type (Fig. 5d).
Beyond these in vitro models, human APOE4 and APOE3 have been expressed in ApoE-KO mice under the control of the smooth muscle cell-specific promoter SM22alpha in vivo185. In this model, smooth muscle cell-produced APOE4 reduced cerebral blood flow, decreased synaptic plasticity, and increased astrocyte reactivity185. These mice also exhibited increased anxiety-like behavior and impaired spatial learning185.
APOE4 in disease-associated vascular cell subtypes
In a transcriptomic study, pericytes demonstrated a number of differentially expressed genes in APOE4 versus APOE3 mice at young and old ages34. These included genes critical to cell adhesion, BBB function, inflammatory response, DNA damage and apoptosis34. Transcriptomic and protein analyses of hiPSC-derived pericyte-like cells revealed that, relative to APOE3, APOE4 was associated with increased expression of genes related to protein targeting to the membrane and endoplasmic reticulum and decreased expression of genes related to mitosis and cell cycle progression178. In post-mortem brain tissues from individuals with AD, pericytes showed altered expression of genes related to blood flow and vasoconstriction182, while pericytes from APOE4 carriers also had downregulation of genes involved in cell migration regulation, transport across the BBB, and cell junction maintenance186. In contrast to non-carriers, endothelial cells of APOE4 carriers had increased expression of interferon genes182, and decreased expression of genes related to transport across the BBB and cell junction organization186. In conjunction with the findings from cell culture and animal models, these single-cell data suggest that vascular cell APOE4 could contribute to AD pathogenesis through multiple mechanisms (Fig. 5d).
Cell type-specific APOE4 cascade model of AD
Based on the literature reviewed above, it is evident that, in AD pathogenesis, the APOE4 produced in each of several CNS cell types (astrocytes, neurons, microglia, oligodendrocytes and vascular cells) exerts distinct roles, probably in a spatiotemporally specific and interactive manner.
Thus, we propose a cell type-specific APOE4 cascade model (Fig. 6) to help explain how the different cellular sources of APOE4 may act in a stepwise and interactive manner to drive AD pathogenesis. During the first stage (neuronal initiation), stress and aging trigger increased APOE4 expression in neurons, leading to neuronal dysfunction and the appearance of nE4-DAN. The dysfunctional neurons may start to overproduce and release proinflammatory molecules, such as MHC1 molecules and HMGB1. Simultaneously, dysfunctional neurons may also increase their Aβ production and secretion and accumulate pathological p-tau intracellularly. During the second stage (glial response), astrocytes and microglia become activated in response to proinflammatory molecules released by dysfunctional neurons and to the elevated levels of pathological Aβ and p-tau assemblies. As this stage evolves, disease-associated glia (such as DAA, DAM, LDAM, IRM and MHC2-M) in which APOE expression and secretion are increased start to appear, disrupting the BBB and further promoting the formation and spread of Aβ and tau pathologies. The combination of these processes, in concert with elevated APOE4 expression in neurons, oligodendrocytes and vascular cells, ultimately triggers the third stage (degeneration), which is characterized by the loss of synapses, neurons, and myelin. These cellular impairments and degeneration further augment the expression of APOE4 in neurons and other cell types, promoting vicious cycles and ultimately leading to the fourth stage (AD dementia).
Fig. 6: Cell type-specific APOE4 cascade model of AD and related therapeutic strategies.
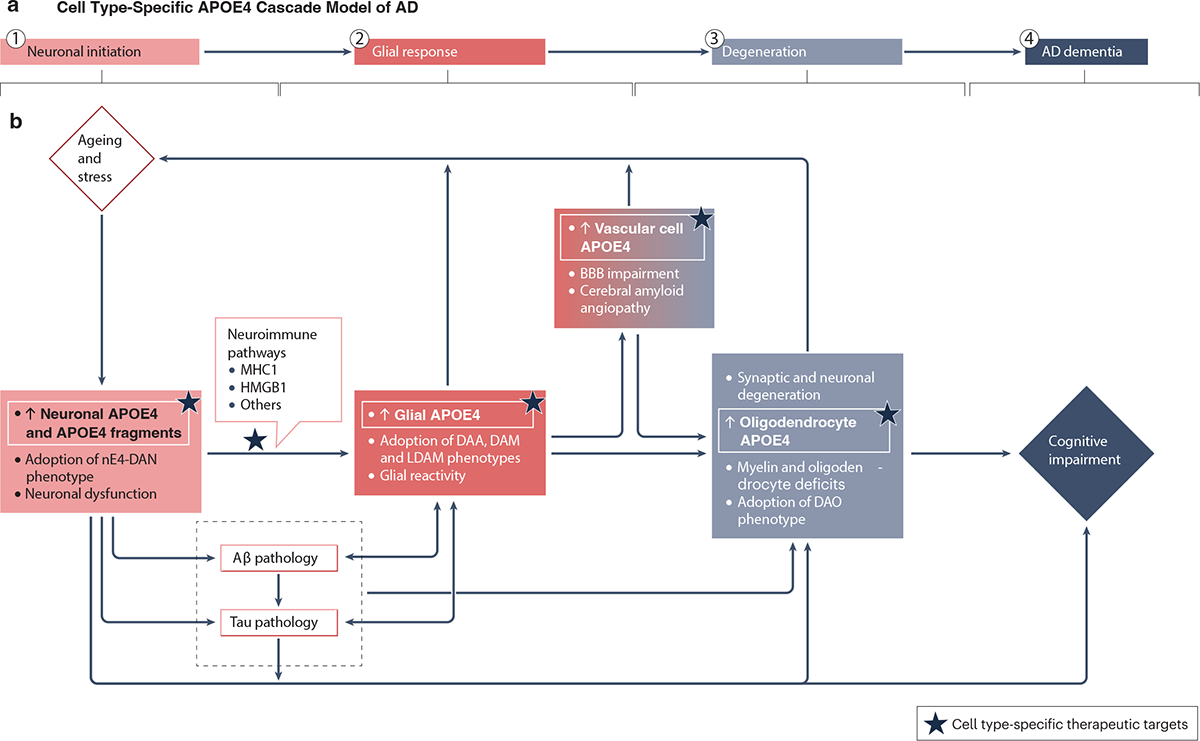
a, Overview of the four stages of the cascade model. b, During the first stage (neuronal initiation), stress and aging trigger increased neuronal expression of the E4 isoform of apolipoprotein E (APOE4), leading to neuronal dysfunction and the appearance of neuronal APOE4-promoted disease-associated neurons (nE4-DAN). The dysfunctional neurons may start to overproduce and release proinflammatory molecules (such as major histocompatibility complex class 1 (MHC1) molecules and high mobility group box 1 (HMGB1)) and increase their amyloid β (Aβ) production and secretion and intracellular accumulation of pathological p-tau. During the second stage (glial response), astrocytes and microglia become activated in response to proinflammatory molecules released by dysfunctional neurons and to the elevated levels of pathological Aβ and p-tau assemblies. As this stage evolves, disease-associated glia (such as disease-associated astrocytes (DAA), disease-associated microglia (DAM), lipid-droplet-accumulating microglia (LDAM), interferon responsive microglia (IRM), and antigen-presenting microglia (MHC2-M)) with increased APOE expression and secretion start to appear, disrupting the BBB and further promoting the formation and spread of Aβ and tau pathologies. The combination of these processes, in concert with elevated APOE4 expression in neurons, oligodendrocytes, and vascular cells, ultimately triggers the third stage (degeneration), which is characterized by the loss of synapses, neurons, and myelin. These cellular impairments and degeneration further augment the expression of APOE4 in neurons and other cell types, promoting vicious cycles and, ultimately, leading to the fourth stage (AD dementia). Based on the proposed cascade model, cell type-specific APOE4-guided AD therapeutic targets are highlighted, as indicated by stars.
Additional studies are needed to further test this model and to address the following questions: How is APOE4 expression regulated in neurons, astrocytes, microglia, oligodendrocytes and vascular cells? What protease is responsible for neuron-specific APOE4 cleavage? How does APOE4 in dysfunctional neurons promote the neuronal production and release of proinflammatory molecules? How exactly do the distinct cellular sources of APOE4 affect amyloid and tau pathologies? What are the mechanisms underlying the formation of disease-associated glial subtypes? To what extent is the emergence of these glial subtypes pathogenic or adaptive? Do they cause neurodegeneration, BBB impairments, and oligodendrocyte and/or myelin deficits? Answering these questions will help further elucidate the cell type-specific roles of APOE4 in AD pathogenesis and foster the development of novel therapeutics for this challenging condition.
Potential therapeutics targeting APOE4
Although there has been progress on AD therapeutic development with the recent US Food and Drug Administration (FDA) approval of Lecanemab and Aducanumab (anti-Aβ monoclonal antibodies), APOE4 carriers have unfortunately been found to be at high risk for developing serious side effects, such as amyloid-related imaging abnormalities (ARIA), to these therapies187,188. In addition, APOE genotype seems to also differentially modulate the amyloid lowering efficacy and microglial response to anti-Aβ immunotherapy in an AD mouse model189. Given the high proportion of APOE4 carriers in the AD population, there is an urgent need to devise therapeutic strategies that take into account the APOE4 carrier status, such as therapies directly targeting the detrimental effects of APOE4 expression.
A key question in developing APOE4-targeting therapies has been and continues to be whether APOE4 differs from the other APOE isoforms primarily through loss-of-function mechanisms or through gain-of-function mechanisms in AD-relevant contexts. In hiPSC-derived neuronal cultures, APOE-deficient neurons resembled those expressing APOE3, whereas APOE4-expressing neurons showed a range of AD-relevant pathological phenotypes, suggesting an adverse gain-of-function at least for APOE4 produced by neurons85. In vivo studies also support a gain-of-function mechanism for APOE4 produced by neurons or other cell types. Genetically reducing APOE4 by 50% markedly lowered amyloid pathology in mutant human APP transgenic mice with hemizygous APOE4 expression190,191. Furthermore, ablation of APOE4 specifically in neurons or astrocytes protected against tau pathology, gliosis, and degeneration in PS19-APOE4 mice24,43. Human case studies also support the conclusion of predominant gain-of-function effects of APOE4. For example, a recent preprint has reported that an APOE3/4 heterozygote with the APOE4 allele-specific loss-of-function mutation did not develop AD at old age192 and an older study reported a similar effect in one person with complete APOE deficiency193. Likewise, genetic studies revealed that APOE4 carriers with European ancestry have higher APOE4 expression levels in various types of CNS cells than those with African ancestry and, interestingly, the former have a relatively higher AD risk than the latter194,195. Together, these findings highlight the potential therapeutic benefit of reducing APOE4 levels. Indeed, anti-APOE immunotherapies and antisense oligonucleotides (ASO) targeting APOE are in development. In amyloidosis mouse models, such therapies efficiently inhibited the formation of amyloid plaques when initiated before the onset of pathology196,197. Treatment with an APOE-ASO also reduced tau pathology, neuroinflammation, and neurodegeneration in a tauopathy mouse model198.
Based on the cascade model we proposed above (Fig. 6), one could also envision directing therapeutic interventions at APOE4 in specific cell types, which may yield more precise outcomes and help avoid unexpected side effects from broader APOE reductions (Box 1). Potential AD-relevant targets include neuronal APOE4 and its proteolytic cleavage as well as the APOE4-dependent neuronal production and release of proinflammatory molecules, astrocytic and microglial APOE4, and oligodendrocytic or vascular cell APOE4. The rapidly evolving development of viral vectors and gene editing [G] approaches199,200 may also enable the isoform-specific deletion or conversion of APOE4 in neurons or other CNS cell types. To monitor the effects of APOE4-targeting therapies and for the stratification of AD patient populations in general, it will also be important to develop better APOE-related biomarkers. Potential examples include the detection and quantification of neuron-derived APOE4 fragments and of APOE4-induced molecules released from specific CNS cell types, such as MHC1 molecules and HMGB1, in the CSF and plasma.
Box 1: Cell type-specific therapeutic APOE4 targeting: strategies, advantages and challenges.
Targeting the E4 isoform of apolipoprotein E (APOE4) produced by neurons may be an effective way to reduce Alzheimer disease (AD) risk (Fig. 6b) because neuronal APOE4 promotes all major pathological hallmarks of AD24,75,76,85,98,125 and may also drive APOE4 expression in other cell types24. Reducing neuronal APOE4 through targeted gene editing, neuron-specific CRISPR-interference (CRISPRi) delivery or by blocking neuronal APOE4 proteolysis could limit the initiation of the pathological cascade shown in Fig. 6. To have the greatest therapeutic impact, however, suppression of neuronal APOE4 would probably have to begin in early stages of disease development. Highlighting the potential of targeting neuronal APOE4, treatment of APOE4-expressing human induced pluripotent stem cell (hiPSC)-derived neurons with an APOE4 structure corrector201,202 decreased neurotoxic APOE4 fragments, p-tau levels, and amyloid β (Aβ) production and secretion and reduced the loss of GABAergic neurons in a dose-dependent manner85. In an in vivo study, selective removal of neuronal APOE4 protected against tau pathology, gliosis, neuronal dysfunction, neurodegeneration and oligodendrocyte and/or myelin deficits in PS19-APOE4 mice24. Additionally, targeting neuronal APOE4-mediated inhibitory neuron loss, especially somatostatin (SST)-expressing interneuron loss, by stem cell-based replacement therapy is another avenue to pursue203.
Targeting APOE4-induced neuronal production or release of proinflammatory molecules, such as high mobility group box 1 (HMGB1) and major histocompatibility complex class 1 (MHC1) molecules, should also be considered, as this strategy could block downstream glial responses to APOE4-dependent neuronal dysfunctions and pathologies and prevent the escalation of pathogenic neuroinflammatory processes. Relevant therapeutics could be monoclonal antibodies or small molecule inhibitors.
Targeting astrocytic APOE4 could also prove to be valuable, as astrocytic APOE4 correlates with gliosis, Aβ accumulation, blood-brain barrier (BBB) vulnerability, and neurodegeneration43,51,54,70–74. However, astrocytic APOE4 does not seem to exhibit a correlation to tau pathology or neuronal dysfunction that is as strong as that of neuronal APOE4 and does not seem to contribute to neural network hyperexcitability or myelin deficits. There is also evidence that reducing astrocytic APOE4 might increase cerebral amyloid angiopathy (CAA)44. Additionally, since astrocytes normally produce and secrete high levels of APOE that serves a host of homeostatic functions, it is conceivable that targeting astrocytic APOE4 could result in some side effects, which warrants further studies.
Targeting microglial APOE4 may counteract the progression of tau and amyloid pathologies as well as inflammation-related neurodegenerative processes. Like neurons, microglia increase their APOE4 production in response to stress and disease39,47,138–140. Although microgliosis appears to happen downstream of neuronal APOE4 effects, targeting microglial APOE4 and inflammation could synergize with strategies targeting neuronal APOE4. Targeting oligodendrocytic APOE4 to protect against oligodendrocyte impairment and myelin loss, or targeting vascular cell APOE4 to alleviate pericyte and vascular dysfunction and BBB disruption, could also be beneficial.
With recent advances in gene editing technologies, gene therapy should also be considered to target APOE4, including correction of APOE4 to APOE3, APOE2, APOE-R136S, or APOE-KO in a cell type-specific manner. Since APOE2 markedly reduces AD risk compared to both APOE4 and APOE3204–206, adeno-associated viruses have been used to deliver this isoform into the brains of APP/PS1/APOE4 mice, which reduced amyloid plaque burdens207. Further studies are warranted to investigate how increasing APOE2 expression in specific CNS cell types affects other key AD pathologies, including tau pathology and neurodegeneration. In a similar vein, understanding the mechanisms through which rare protective variants of APOE, such as APOE-R136S, confer protection against AD pathology could also lead to the development of novel therapeutic approaches208.
Summary
Recent scientific advances have revealed the importance of cell type-specific roles of APOE4 in AD pathogenesis. APOE4 produced in various CNS cell types, such as neurons, astrocytes, microglia, oligodendrocytes, and vascular cells, exert differential effects on amyloid and tau pathology, altered lipid metabolism, glial reactivity, neurodegeneration, myelin deficits, and blood-brain barrier dysfunction, likely in a spatiotemporal and interactive manner. The cell type-specific APOE4 cascade model that we outline above poses neuronal APOE4 as a critical upstream initiator and driver of AD pathogenesis, instigating glial response and subsequent degeneration. This model also indicates that targeting specific cellular sources of APOE4 may provide new therapeutic strategies for the prevention or treatment of AD.
Acknowledgments
This work was supported by grants R01AG071697, RF1AG076647, R01AG065540, and P01AG073082 from the National Institutes of Health to YH. The authors would like to thank Drs. R. W. Mahley, L. Mucke, and A. Yang for their critical review of the manuscript. We also thank J. Carroll for assistance with figure preparation, S. Ordway for editorial assistance, and Huang lab members for providing feedback on the review.
Glossary:
- APOE fragments
Neurotoxic fragments of the APOE protein that are generated as a result of APOE expression in neurons, with more fragments being generated from APOE4, due to its unique conformation, than from APOE3 via neuron-specific proteolysis
- Cytokines
Classes of small secreted proteins that serve to signal and activate immune functions, including triggering of glial responses in the CNS
- Dementia
a general term for loss of memory, language, problem-solving and other thinking abilities that are severe enough to interfere with daily life. Diseases grouped under the general term dementia are caused by abnormal brain changes, with Alzheimer disease being the most common cause
- Disease-associated gene signatures
refers to specific patterns or sets of genes that are consistently found to be altered or expressed differently in a disease context compared to physiological contexts. These gene signatures are often identified through various transcriptomic analyses, such as microarray or RNA sequencing experiments
- Exosome
small, membrane-bound vesicles that are released by cells into the extracellular environment. These vesicles play a crucial role in intercellular communication by facilitating the transfer of various molecules between cells
- Gene editing
a type of genetic engineering by which specific changes can be made to DNA, including inserting, deleting, or altering DNA sequences
- Gliosis
the proliferation or hypertrophy of microglia and/or astrocytes in response to stressors, insults, injury, or diseases
- Human induced pluripotent stem cells (hiPSCs)
a type of pluripotent stem cell derived from adult somatic cells that have been genetically reprogrammed to an embryonic stem (ES) cell-like state through the forced expression of genes and factors important for maintaining the defining properties of ES cells
- Lysosomes
Lysosomes are membrane-bound organelles that are part of the endo-lysosomal system within cells and play a crucial role in cellular digestion and waste removal. Lysosomes contain various enzymes that are capable of breaking down different types of macromolecules, including proteins, lipids, nucleic acids, and carbohydrates
- Mild cognitive impairment (MCI)
an early stage of memory loss or other cognitive ability loss (such as language or visual/spatial perception) in individuals who maintain the ability to independently perform most activities of daily living. For neurodegenerative diseases, such as Alzheimer disease, MCI can be an early stage of the disease continuum
- Pericytes
contractile cells located at intervals along the walls of capillaries, playing a role in blood vessel formation, maintenance of the blood–brain barrier, regulation of immune cell entry to the central nervous system and control of brain blood flow
- Single-nucleus RNA-sequencing (snRNA-seq)
a molecular biology technique used to analyze the gene expression profiles of individual cell nuclei within a complex tissue or organ. Traditional RNA sequencing methods usually require intact cells, but snRNA-seq allows researchers to study gene expression in individual nuclei, providing insights into cellular diversity and heterogeneity within tissues
- Tauopathy
refers to a group of clinically, biochemically, and morphologically heterogeneous neurodegenerative diseases, including Alzheimer disease, characterized by the abnormal hyperphosphorylation of microtubule-associated protein, tau, that leads to the formation of neurofibrillary tangles in the brain
Footnotes
Competing interests
YH is a co-founder and scientific advisory board member of GABAeron, Inc. Other authors declare no competing financial interests.
References
- 1.Huang Y & Mucke L Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Long JM & Holtzman DM Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179, 312–339 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sperling RA et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc 7, 280–292 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albert MS et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7, 270–279 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vermunt L et al. Duration of preclinical, prodromal, and dementia stages of Alzheimer’s disease in relation to age, sex, and APOE genotype. Alzheimers Dement 15, 888–898 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gustavsson A et al. Global estimates on the number of persons across the Alzheimer’s disease continuum. Alzheimers Dement 19, 658–670 (2023). [DOI] [PubMed] [Google Scholar]
- 7.Sherrington R et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760 (1995). [DOI] [PubMed] [Google Scholar]
- 8.Goate A et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706 (1991). [DOI] [PubMed] [Google Scholar]
- 9.Corder EH et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet 7, 180–184 (1994). [DOI] [PubMed] [Google Scholar]
- 10.Corder EH et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923 (1993). [DOI] [PubMed] [Google Scholar]
- 11.Mahley RW & Rall SC Apolipoprotein E: far more than a lipid transport protein. Annu. Rev. Genomics Hum. Genet 1, 507–537 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Weisgraber KH Apolipoprotein E: structure-function relationships. Adv. Protein Chem 45, 249–302 (1994). [DOI] [PubMed] [Google Scholar]
- 13.Huang Y & Mahley RW Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol. Dis 72 Pt A, 3–12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farrer LA et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA 278, 1349–1356 (1997). [PubMed] [Google Scholar]
- 15.Genin E et al. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol. Psychiatry 16, 903–907 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Y-Y et al. The Proportion of APOE4 carriers among non-demented individuals: a pooled analysis of 389,000 community-dwellers. J. Alzheimers Dis. JAD 81, 1331–1339 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Ward A et al. Prevalence of apolipoprotein E4 genotype and homozygotes (APOE e4/4) among patients diagnosed with Alzheimer’s disease: a systematic review and meta-analysis. Neuroepidemiology 38, 1–17 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Zhong N & Weisgraber KH Understanding the association of apolipoprotein E4 with Alzheimer disease: clues from its structure. J. Biol. Chem 284, 6027–6031 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu C-C, Kanekiyo T, Xu H & Bu G Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol 9, 106–118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koutsodendris N, Nelson MR, Rao A & Huang Y Apolipoprotein E and Alzheimer’s Disease: Findings, Hypotheses, and Potential Mechanisms. Annu. Rev. Pathol. Mech. Dis 17, 73–99 (2022). [DOI] [PubMed] [Google Scholar]
- 21.Martens YA et al. ApoE Cascade Hypothesis in the pathogenesis of Alzheimer’s disease and related dementias. Neuron 110, 1304–1317 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Y et al. Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc. Natl. Acad. Sci 98, 8838–8843 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi Y et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549, 523–527 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koutsodendris N et al. Neuronal APOE4 removal protects against tau-mediated gliosis, neurodegeneration and myelin deficits. Nat. Aging 3, 275–296 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serrano-Pozo A, Das S & Hyman BT APOE and Alzheimer’s disease: advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol 20, 68–80 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iannucci J, Sen A & Grammas P Isoform-specific effects of apolipoprotein E on markers of inflammation and toxicity in brain glia and neuronal cells in vitro. Curr. Issues Mol. Biol 43, 215–225 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haney MS et al. APOE4/4 is linked to damaging lipid droplets in Alzheimer’s microglia. bioRxiv 2023.07.21.549930. 10.1101/2023.07.21.549930 (2023). [DOI] [Google Scholar]
- 28.Nelson MR et al. The APOE-R136S mutation protects against APOE4-driven Tau pathology, neurodegeneration and neuroinflammation. Nat. Neurosci 10.1038/s41593-023-01480-8 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marschallinger J et al. Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat. Neurosci 23, 194–208 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parhizkar S & Holtzman DM APOE mediated neuroinflammation and neurodegeneration in Alzheimer’s disease. Semin. Immunol 59, 101594 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blanchard JW et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 611, 769–779 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheng GW-Y et al. Apolipoprotein E ε4 mediates myelin breakdown by targeting oligodendrocytes in sporadic Alzheimer disease. J. Neuropathol. Exp. Neurol 81, 717–730 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi Y et al. Overexpressing low-density lipoprotein receptor reduces tau-associated neurodegeneration in relation to apoE-linked mechanisms. Neuron 109, 2413–2426.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barisano G et al. A ‘multi-omics’ analysis of blood-brain barrier and synaptic dysfunction in APOE4 mice. J. Exp. Med 219, e20221137 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montagne A et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 581, 71–76 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Montagne A et al. APOE4 accelerates advanced-stage vascular and neurodegenerative disorder in old Alzheimer’s mice via cyclophilin A independently of amyloid-β. Nat. Aging 1, 506–520 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raulin A-C et al. ApoE in Alzheimer’s disease: pathophysiology and therapeutic strategies. Mol. Neurodegener 17, 72 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamazaki Y, Zhao N, Caulfield TR, Liu C-C & Bu G Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat. Rev. Neurol 15, 501–518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu Q et al. Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J. Neurosci 26, 4985–4994 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pitas RE, Boyles JK, Lee SH, Foss D & Mahley RW Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim. Biophys. Acta BBA - Lipids Lipid Metab 917, 148–161 (1987). [DOI] [PubMed] [Google Scholar]
- 41.Boyles JK, Pitas RE, Wilson E, Mahley RW & Taylor JM Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J. Clin. Invest 76, 1501–1513 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knoferle J et al. Apolipoprotein E4 produced in GABAergic interneurons causes learning and memory deficits in mice. J. Neurosci 34, 14069–14078 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang C et al. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 109, 1657–1674.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiong M et al. Astrocytic APOE4 removal confers cerebrovascular protection despite increased cerebral amyloid angiopathy. Mol. Neurodegener 18, 17 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xia Y et al. C/EBPβ is a key transcription factor for APOE and preferentially mediates ApoE4 expression in Alzheimer’s disease. Mol. Psychiatry 26, 6002–6022 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wynne ME et al. APOE expression and secretion are modulated by mitochondrial dysfunction. eLife 12, e85779 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lanfranco MF, Sepulveda J, Kopetsky G & Rebeck GW Expression and secretion of apoE isoforms in astrocytes and microglia during inflammation. Glia 69, 1478–1493 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harris FM et al. Astroglial regulation of apolipoprotein E expression in neuronal cells: implications for Alzheimer’s disease. J. Biol. Chem 279, 3862–3868 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Cashikar AG et al. Regulation of astrocyte lipid metabolism and ApoE secretion by the microglial oxysterol, 25-hydroxycholesterol. J. Lipid Res 64, 100350 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mhatre-Winters I, Eid A, Han Y, Tieu K & Richardson JR Sex and APOE genotype alter the basal and induced inflammatory states of primary astrocytes from humanized targeted replacement mice. ASN NEURO 15, 17590914221144549 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Leeuw SM et al. APOE2, E3, and E4 differentially modulate cellular homeostasis, cholesterol metabolism, and inflammatory response in isogenic iPSC-derived astrocytes. Stem Cell Rep 17, 110–126 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chung W-S et al. Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc. Natl. Acad. Sci. U. S. A 113, 10186–10191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fernandez CG, Hamby ME, McReynolds ML & Ray WJ The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer’s disease. Front. Aging Neurosci 11, 14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin Y-T et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98, 1141–1154.e7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Staurenghi E et al. ApoE3 vs. ApoE4 astrocytes: a detailed analysis provides new insights into differences in cholesterol homeostasis. Antioxidants 11, 2168 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao J et al. APOE ε4/ε4 diminishes neurotrophic function of human iPSC-derived astrocytes. Hum. Mol. Genet 26, 2690–2700 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nishitsuji K, Hosono T, Nakamura T, Bu G & Michikawa M Apolipoprotein E regulates the integrity of tight junctions in an isoform-dependent manner in an in vitro blood-brain barrier model. J. Biol. Chem 286, 17536–17542 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bell RD et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 485, 512–516 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arnaud L et al. APOE4 drives inflammation in human astrocytes via TAGLN3 repression and NF-κB activation. Cell Rep 40, 111200 (2022). [DOI] [PubMed] [Google Scholar]
- 60.Tcw J et al. Cholesterol and matrisome pathways dysregulated in astrocytes and microglia. Cell 185, 2213–2233.e25 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Windham IA et al. APOE traffics to astrocyte lipid droplets and modulates triglyceride saturation and droplet size. bioRxiv. 2023.04.28.538740. 10.1101/2023.04.28.538740 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen Z-P et al. Lipid-accumulated reactive astrocytes promote disease progression in epilepsy. Nat. Neurosci 26, 542–554 (2023). [DOI] [PubMed] [Google Scholar]
- 63.Fang W et al. APOE4 genotype exacerbates the depression-like behavior of mice during aging through ATP decline. Transl. Psychiatry 11, 507 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Farmer BC et al. APOE4 lowers energy expenditure in females and impairs glucose oxidation by increasing flux through aerobic glycolysis. Mol. Neurodegener 16, 62 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Williams HC et al. APOE alters glucose flux through central carbon pathways in astrocytes. Neurobiol. Dis 136, 104742 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee H et al. ApoE4-dependent lysosomal cholesterol accumulation impairs mitochondrial homeostasis and oxidative phosphorylation in human astrocytes. Cell Rep 42, 113183 (2023). [DOI] [PubMed] [Google Scholar]
- 67.Li X et al. Astrocytic ApoE reprograms neuronal cholesterol metabolism and histone-acetylation-mediated memory. Neuron 109, 957–970.e8 (2021). [DOI] [PubMed] [Google Scholar]
- 68.Huang S, Zhang Z, Cao J, Yu Y & Pei G Chimeric cerebral organoids reveal the essentials of neuronal and astrocytic APOE4 for Alzheimer’s tau pathology. Signal Transduct. Target. Ther 7, 1–10 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen Y, Durakoglugil MS, Xian X & Herz J ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc. Natl. Acad. Sci. U. S. A 107, 12011–12016 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jackson RJ et al. APOE4 derived from astrocytes leads to blood–brain barrier impairment. Brain 145, 3582–3593 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Simonovitch S et al. Impaired autophagy in APOE4 astrocytes. J. Alzheimers Dis 51, 915–927 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Fagan AM et al. Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer’s disease. Neurobiol. Dis 9, 305–318 (2002). [DOI] [PubMed] [Google Scholar]
- 73.Holtzman DM et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 97, 2892–2897 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mahan TE et al. Selective reduction of astrocyte apoE3 and apoE4 strongly reduces Aβ accumulation and plaque-related pathology in a mouse model of amyloidosis. Mol. Neurodegener 17, 13 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brecht WJ et al. Neuron-specific apolipoprotein E4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J. Neurosci 24, 2527–2534 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tesseur I et al. Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am. J. Pathol 156, 951–964 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Saroja SR, Gorbachev K, TCW J, Goate AM & Pereira AC Astrocyte-secreted glypican-4 drives APOE4-dependent tau hyperphosphorylation. Proc. Natl. Acad. Sci. U. S. A 119, e2108870119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Habib N et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci 23, 701–706 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mathys H et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang C, Sun B, Zhou Y, Grubb A & Gan L Cathepsin B degrades amyloid-β in mice expressing wild-type human amyloid precursor protein. J. Biol. Chem 287, 39834–39841 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mucke L et al. Astroglial expression of human α1-antichymotrypsin enhances Alzheimer-like pathology in amyloid protein precursor transgenic Mice. Am. J. Pathol 157, 2003–2010 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dupont-Wallois L et al. ApoE synthesis in human neuroblastoma cells. Neurobiol. Dis 4, 356–364 (1997). [DOI] [PubMed] [Google Scholar]
- 83.Soulié C et al. Synthesis of apolipoprotein E (ApoE) mRNA by human neuronal-type SK N SH-SY 5Y cells and its regulation by nerve growth factor and ApoE. Neurosci. Lett 265, 147–150 (1999). [DOI] [PubMed] [Google Scholar]
- 84.DeKroon RM & Armati PJ The endosomal trafficking of apolipoprotein E3 and E4 in cultured human brain neurons and astrocytes. Neurobiol. Dis 8, 78–89 (2001). [DOI] [PubMed] [Google Scholar]
- 85.Wang C et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat. Med 24, 647–657 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee H et al. Cell-type-specific regulation of APOE and CLU levels in human neurons by the Alzheimer’s disease risk gene SORL1. Cell Rep 42, 112994 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Einstein G et al. Intraneuronal ApoE in human visual cortical areas reflects the staging of Alzheimer disease pathology. J. Neuropathol. Exp. Neurol 57, 1190–1201 (1998). [DOI] [PubMed] [Google Scholar]
- 88.Xu P-T et al. Specific regional transcription of apolipoprotein E in human brain neurons. Am. J. Pathol 154, 601–611 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xu P-T et al. Regionally specific neuronal expression of human APOE gene in transgenic mice. Neurosci. Lett 246, 65–68 (1998). [DOI] [PubMed] [Google Scholar]
- 90.Zalocusky KA et al. Neuronal ApoE upregulates MHC-I expression to drive selective neurodegeneration in Alzheimer’s disease. Nat. Neurosci 24, 786–798 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xu Q et al. Intron-3 retention/splicing controls neuronal expression of apolipoprotein E in the CNS. J. Neurosci 28, 1452–1459 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mahley RW & Huang Y Apolipoprotein E: from atherosclerosis to Alzheimer’s disease and beyond. Curr. Opin. Lipidol 10, 207–217 (1999). [DOI] [PubMed] [Google Scholar]
- 93.Weisgraber KH & Mahley RW Human apolipoprotein E: the Alzheimer’s disease connection. FASEB J 10, 1485–1494 (1996). [DOI] [PubMed] [Google Scholar]
- 94.Li G et al. GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell 5, 634–645 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mahley RW & Huang Y Apolipoprotein e sets the stage: response to injury triggers neuropathology. Neuron 76, 871–885 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen Z et al. Human-lineage-specific genomic elements are associated with neurodegenerative disease and APOE transcript usage. Nat. Commun 12, 2076 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Watts JA et al. A common transcriptional mechanism involving R-loop and RNA abasic site regulates an enhancer RNA of APOE. Nucleic Acids Res 50, 12497–12514 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang Z-H et al. Neuronal ApoE4 stimulates C/EBPβ activation, promoting Alzheimer’s disease pathology in a mouse model. Prog. Neurobiol 209, 102212 (2022). [DOI] [PubMed] [Google Scholar]
- 99.Buttini M et al. Expression of human apolipoprotein E3 or E4 in the brains of Apoe−/− mice: isoform-specific effects on neurodegeneration. J. Neurosci 19, 4867–4880 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Raber J et al. Apolipoprotein E and cognitive performance. Nature 404, 352–354 (2000). [DOI] [PubMed] [Google Scholar]
- 101.Raber J et al. Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc. Natl. Acad. Sci. U. S. A 95, 10914–10919 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cakir Z et al. Quantitative Proteomic Analysis Reveals apoE4-Dependent Phosphorylation of the Actin-Regulating Protein VASP. Mol. Cell. Proteomics 22, 100541 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jang S-S et al. Neuronal apoE4 induces early hyperexcitability in select populations of hippocampal neurons by altering Nell2 expression. bioRxiv. 2023.08.28.555153. 10.1101/2023.08.28.555153 (2023). [DOI] [Google Scholar]
- 104.Nuriel T et al. Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat. Commun 8, 1464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gillespie AK et al. Apolipoprotein E4 causes age-dependent disruption of slow gamma oscillations during hippocampal sharp-wave ripples. Neuron 90, 740–751 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jones EA, Gillespie AK, Yoon SY, Frank LM & Huang Y Early hippocampal sharp-wave ripple deficits predict later learning and memory impairments in an Alzheimer’s disease mouse model. Cell Rep 29, 2123–2133.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Andrews-Zwilling Y et al. Apolipoprotein E4 causes age- and Tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci 30, 13707–13717 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Leung L et al. Apolipoprotein E4 causes age- and sex-dependent impairments of hilar GABAergic interneurons and learning and memory deficits in mice. PloS One 7, e53569 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yin J et al. Effect of ApoE isoforms on mitochondria in Alzheimer disease. Neurology 94, e2404–e2411 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mahley RW, Weisgraber KH & Huang Y Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc. Natl. Acad. Sci. U. S. A 103, 5644–5651 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mahley RW Apolipoprotein E4 targets mitochondria and the mitochondria-associated membrane complex in neuropathology, including Alzheimer’s disease. Curr. Opin. Neurobiol 79, 102684 (2023). [DOI] [PubMed] [Google Scholar]
- 112.Chen H-K et al. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J. Biol. Chem 286, 5215–5221 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Orr AL et al. Neuronal apolipoprotein E4 expression results in proteome-wide alterations and compromises bioenergetic capacity by disrupting mitochondrial function. J. Alzheimers Dis 68, 991–1011 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Buttini M et al. Cellular source of apolipoprotein E4 determines neuronal susceptibility to excitotoxic injury in transgenic mice. Am. J. Pathol 177, 563–569 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cataldo AM et al. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol 157, 277–286 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ji Z-S et al. Apolipoprotein E4 potentiates amyloid beta peptide-induced lysosomal leakage and apoptosis in neuronal cells. J. Biol. Chem 277, 21821–21828 (2002). [DOI] [PubMed] [Google Scholar]
- 117.Wadhwani AR, Affaneh A, Van Gulden S & Kessler JA Neuronal apolipoprotein E4 increases cell death and phosphorylated Tau release in Alzheimer disease. Ann. Neurol 85, 726–739 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yamauchi Y et al. Role of the N- and C-terminal domains in binding of apolipoprotein E isoforms to heparan sulfate and dermatan sulfate: a surface plasmon resonance study. Biochemistry 47, 6702–6710 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rauch JN et al. Tau internalization is regulated by 6-O sulfation on heparan sulfate proteoglycans (HSPGs). Sci. Rep 8, 6382 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stopschinski BE et al. Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus α-synuclein and β-amyloid aggregates. J. Biol. Chem 293, 10826–10840 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Holmes BB & Diamond MI Prion-like properties of Tau protein: the importance of extracellular Tau as a therapeutic target. J. Biol. Chem 289, 19855–19861 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Arboleda-Velasquez JF et al. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report. Nat. Med 25, 1680–1683 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sepulveda-Falla D et al. Distinct tau neuropathology and cellular profiles of an APOE3 Christchurch homozygote protected against autosomal dominant Alzheimer’s dementia. Acta Neuropathol. (Berl.) 144, 589–601 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shi Y et al. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J. Exp. Med 216, 2546–2561 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rao Antara. Microglia depletion reduces human neuronal APOE4-driven pathologies in a chimeric Alzheimer’s disease model. bioRxiv 2023.11.10.566510. 10.1101/2023.11.10.566510 (2023). [DOI] [Google Scholar]
- 126.Harris FM et al. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc. Natl. Acad. Sci 100, 10966–10971 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bien-Ly N et al. C-terminal-truncated apolipoprotein (apo) E4 inefficiently clears amyloid-beta (Abeta) and acts in concert with Abeta to elicit neuronal and behavioral deficits in mice. Proc. Natl. Acad. Sci. U. S. A 108, 4236–4241 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rohn TT Proteolytic cleavage of apolipoprotein E4 as the keystone for the heightened risk associated with Alzheimer’s disease. Int. J. Mol. Sci 14, 14908–14922 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Koutsodendris N et al. APOE4-promoted gliosis and degeneration in tauopathy are ameliorated by pharmacological inhibition of HMGB1 release. Cell Rep 42, 113252 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tanaka H et al. YAP-dependent necrosis occurs in early stages of Alzheimer’s disease and regulates mouse model pathology. Nat. Commun 11, 507 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Foo H et al. Interaction between APOE-ɛ4 and HMGB1 is associated with widespread cortical thinning in mild cognitive impairment. J. Neurol. Neurosurg. Psychiatry 89, 225–226 (2018). [DOI] [PubMed] [Google Scholar]
- 132.Wan Z et al. TLR4-HMGB1 signaling pathway affects the inflammatory reaction of autoimmune myositis by regulating MHC-I. Int. Immunopharmacol 41, 74–81 (2016). [DOI] [PubMed] [Google Scholar]
- 133.Grundtman C et al. Effects of HMGB1 on in vitro responses of isolated muscle fibers and functional aspects in skeletal muscles of idiopathic inflammatory myopathies. FASEB J 24, 570–578 (2010). [DOI] [PubMed] [Google Scholar]
- 134.Brase L et al. Single-nucleus RNA-sequencing of autosomal dominant Alzheimer disease and risk variant carriers. Nat. Commun 14, 2314 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mathys H et al. Single-cell atlas reveals correlates of high cognitive function, dementia, and resilience to Alzheimer’s disease pathology. Cell 186, 4365–4385.e27 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Horvath AA et al. Subclinical epileptiform activity accelerates the progression of Alzheimer’s disease: A long-term EEG study. Clin. Neurophysiol 132, 1982–1989 (2021). [DOI] [PubMed] [Google Scholar]
- 137.Vossel KA et al. Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann. Neurol 80, 858–870 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Olah M et al. A transcriptomic atlas of aged human microglia. Nat. Commun 9, 539 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rangaraju S et al. Quantitative proteomics of acutely-isolated mouse microglia identifies novel immune Alzheimer’s disease-related proteins. Mol. Neurodegener 13, 34 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Keren-Shaul H et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017). [DOI] [PubMed] [Google Scholar]
- 141.Lee S et al. APOE modulates microglial immunometabolism in response to age, amyloid pathology, and inflammatory challenge. Cell Rep 42, 112196 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Grubman A et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci 22, 2087–2097 (2019). [DOI] [PubMed] [Google Scholar]
- 143.Krasemann S et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mancuso R et al. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain 142, 3243–3264 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Voloboueva LA, Emery JF, Sun X & Giffard RG Inflammatory response of microglial BV-2 cells includes a glycolytic shift and is modulated by mitochondrial glucose-regulated protein 75/mortalin. FEBS Lett 587, 756–762 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Huynh T-PV et al. Lack of hepatic apoE does not influence early Aβ deposition: observations from a new APOE knock-in model. Mol. Neurodegener 14, 37 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Victor MB et al. Lipid accumulation induced by APOE4 impairs microglial surveillance of neuronal-network activity. Cell Stem Cell 29, 1197–1212.e8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sienski G et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci. Transl. Med 13, eaaz4564 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Liu C-C et al. Cell-autonomous effects of APOE4 in restricting microglial response in brain homeostasis and Alzheimer’s disease. Nat. Immunol 24, 1854–1866 (2023). [DOI] [PubMed] [Google Scholar]
- 150.Wang N et al. Opposing effects of apoE2 and apoE4 on microglial activation and lipid metabolism in response to demyelination. Mol. Neurodegener 17, 75 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Fote GM et al. Isoform-dependent lysosomal degradation and internalization of apolipoprotein E requires autophagy proteins. J. Cell Sci 135, jcs258687 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Machlovi SI et al. APOE4 confers transcriptomic and functional alterations to primary mouse microglia. Neurobiol. Dis 164, 105615 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Overmyer M et al. Reactive microglia in aging and dementia: an immunohistochemical study of postmortem human brain tissue. Acta Neuropathol. (Berl.) 97, 383–392 (1999). [DOI] [PubMed] [Google Scholar]
- 154.Egensperger R, Kösel S, Eitzen U & Graeber MB Microglial activation in Alzheimer disease: association with APOE genotype. Brain Pathol 8, 439–447 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Vitek MP, Brown CM & Colton CA APOE genotype-specific differences in the innate immune response. Neurobiol. Aging 30, 1350–1360 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Muth C, Hartmann A, Sepulveda-Falla D, Glatzel M & Krasemann S Phagocytosis of apoptotic cells is specifically upregulated in ApoE4 expressing microglia in vitro. Front. Cell. Neurosci 13, 181 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yang Y et al. APOE3, but not APOE4, bone marrow transplantation mitigates behavioral and pathological changes in a mouse model of Alzheimer disease. Am. J. Pathol 183, 905–917 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yin Z et al. APOE4 impairs the microglial response in Alzheimer’s disease by inducing TGFβ-mediated checkpoints. Nat. Immunol 11, 1839–1853 (2023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Najm R et al. In vivo chimeric Alzheimer’s disease modeling of apolipoprotein E4 toxicity in human neurons. Cell Rep 32, 107962 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lau S-F et al. The VCAM1–ApoE pathway directs microglial chemotaxis and alleviates Alzheimer’s disease pathology. Nat. Aging 10, 1219–1236 (2023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gratuze M et al. TREM2-independent microgliosis promotes tau-mediated neurodegeneration in the presence of ApoE4. Neuron 111, 202–219.e7 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Asai H et al. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci 18, 1584–1593 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Clayton K et al. Plaque associated microglia hyper-secrete extracellular vesicles and accelerate tau propagation in a humanized APP mouse model. Mol. Neurodegener 16, 18 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hopp SC et al. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J. Neuroinflammation 15, 269 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ferrari-Souza JP et al. APOE ε4 associates with microglial activation independently of Aβ plaques and tau tangles. Sci. Adv 9, eade1474 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chen X et al. Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature 615, 668–677 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Serrano-Pozo A et al. Effect of APOE alleles on the glial transcriptome in normal aging and Alzheimer’s disease. Nat. Aging 1, 919–931 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Frigerio CS et al. The major risk factors for Alzheimer’s disease: age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep 27, 1293–1306.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gerrits E et al. Distinct amyloid-β and tau-associated microglia profiles in Alzheimer’s disease. Acta Neuropathol. (Berl.) 141, 681–696 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Serrano-Pozo A, Gómez-Isla T, Growdon JH, Frosch MP & Hyman BT A phenotypic change but not proliferation underlies glial responses in Alzheimer disease. Am. J. Pathol 182, 2332–2344 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Perlmutter LS, Scott SA, Barrón E & Chui HC MHC class II-positive microglia in human brain: association with Alzheimer lesions. J. Neurosci. Res 33, 549–558 (1992). [DOI] [PubMed] [Google Scholar]
- 172.Chen M, Xie M, Peng C & Long S The absorption of apolipoprotein E by damaged neurons facilitates neuronal repair. Cell Biol. Int 43, 623–633 (2019). [DOI] [PubMed] [Google Scholar]
- 173.Mok KK-S et al. Apolipoprotein E ε4 disrupts oligodendrocyte differentiation by interfering with astrocyte-derived lipid transport. J. Neurochem 165, 55–75 (2023). [DOI] [PubMed] [Google Scholar]
- 174.Safina D et al. Low-density lipoprotein receptor-related protein 1 is a novel modulator of radial glia stem cell proliferation, survival, and differentiation. Glia 64, 1363–1380 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Kenigsbuch M et al. A shared disease-associated oligodendrocyte signature among multiple CNS pathologies. Nat. Neurosci 25, 876–886 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Casey CS et al. Apolipoprotein E inhibits cerebrovascular pericyte mobility through a RhoA protein-mediated pathway. J. Biol. Chem 290, 14208–14217 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wilhelmus MMM et al. Apolipoprotein E genotype regulates amyloid-β cytotoxicity. J. Neurosci 25, 3621–3627 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Blanchard JW et al. Reconstruction of the human blood–brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat. Med 26, 952–963 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Majack RA et al. Expression of apolipoprotein E by cultured vascular smooth muscle cells is controlled by growth state. J. Cell Biol 107, 1207–1213 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Rieker C et al. Apolipoprotein E4 expression causes gain of toxic function in isogenic human induced pluripotent stem cell-derived endothelial cells. Arterioscler. Thromb. Vasc. Biol 39, e195–e207 (2019). [DOI] [PubMed] [Google Scholar]
- 181.Bruinsma IB et al. Apolipoprotein E protects cultured pericytes and astrocytes from D-Aβ1–40-mediated cell death. Brain Res 1315, 169–180 (2010). [DOI] [PubMed] [Google Scholar]
- 182.Yang AC et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 603, 885–892 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yamazaki Y et al. ApoE in brain pericytes regulates endothelial function in an isoform-dependent manner by modulating basement membrane components. Arterioscler. Thromb. Vasc. Biol 40, 128–144 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Halliday MR et al. Accelerated pericyte degeneration and blood–brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J. Cereb. Blood Flow Metab 36, 216–227 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Yamazaki Y et al. Vascular apoE4 impairs behavior by modulating glio-vascular function. Neuron 109, 438–447.e6 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sun N et al. Single-nucleus multiregion transcriptomic analysis of brain vasculature in Alzheimer’s disease. Nat. Neurosci 26, 970–982 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Honig LS et al. ARIA in patients treated with lecanemab (BAN2401) in a phase 2 study in early Alzheimer’s disease. Alzheimers Dement. Transl. Res. Clin. Interv 9, e12377 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Salloway S et al. Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients With Early Alzheimer Disease. JAMA Neurol 79, 13–21 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Pankiewicz JE et al. APOE Genotype Differentially Modulates Effects of Anti-Aβ, Passive Immunization in APP Transgenic Mice. Mol. Neurodegener 12, 12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Bien-Ly N, Gillespie AK, Walker D, Yoon SY & Huang Y Reducing human apolipoprotein E levels attenuates age-dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. J. Neurosci 32, 4803–4811 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kim J et al. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. J. Neurosci 31, 18007–18012 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chemparathy A et al. APOE loss-of-function variants: Compatible with longevity and associated with resistance to Alzheimer’s Disease pathology. MedRxiv 2023.07.20.23292771. doi: 10.1101/2023.07.20.23292771 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Mak ACY et al. Effects of the absence of apolipoprotein e on lipoproteins, neurocognitive function, and retinal function. JAMA Neurol 71, 1228–1236 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Rajabli F et al. Ancestral origin of ApoE ε4 Alzheimer disease risk in Puerto Rican and African American populations. PLoS Genet 14, e1007791 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Griswold AJ et al. Increased APOE ε4 expression is associated with the difference in Alzheimer’s disease risk from diverse ancestral backgrounds. Alzheimers Dement. 17, 1179–1188 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Huynh T-PV et al. Age-dependent effects of apoE reduction using antisense oligonucleotides in a model of β-amyloidosis. Neuron 96, 1013–1023.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Kim J et al. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Aβ amyloidosis. J. Exp. Med 209, 2149–2156 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Litvinchuk A et al. Apolipoprotein E4 reduction with antisense oligonucleotides decreases neurodegeneration in a tauopathy model. Ann. Neurol 89, 952–966 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Bulcha JT, Wang Y, Ma H, Tai PWL & Gao G Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther 6, 53 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zhao Z, Anselmo AC & Mitragotri S Viral vector‐based gene therapies in the clinic. Bioeng. Transl. Med 7, e10258 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Brodbeck J et al. Structure-dependent impairment of intracellular apolipoprotein E4 trafficking and its detrimental effects are rescued by small-molecule structure correctors. J. Biol. Chem 286, 17217–17226 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Chen H-K et al. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J. Biol. Chem 287, 5253–5266 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Tong LM et al. Inhibitory interneuron progenitor transplantation restores normal learning and memory in ApoE4 knock-in mice without or with Aβ accumulation. J. Neurosci 34, 9506–9515 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lefterov I et al. APOE2 orchestrated differences in transcriptomic and lipidomic profiles of postmortem AD brain. Alzheimers Res. Ther 11, 113 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Li Z, Shue F, Zhao N, Shinohara M & Bu G APOE2: protective mechanism and therapeutic implications for Alzheimer’s disease. Mol. Neurodegener 15, 63 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Reiman EM et al. Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat. Commun 11, 667 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Zhao L et al. Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer’s disease mouse models. Neurobiol. Aging 44, 159–172 (2016). [DOI] [PubMed] [Google Scholar]
- 208.Marino C et al. APOE Christchurch-mimetic therapeutic antibody reduces APOE-mediated toxicity and tau phosphorylation. Alzheimers Dement. J. Alzheimers Assoc (2023) doi: 10.1002/alz.13436. [DOI] [PMC free article] [PubMed] [Google Scholar]