

This cohort study examines the overall survival, genomic profile, immune profile, and genomic ancestries of patients with KRAS-mutated gallbladder cancer or cholangiocarcinoma.
Key Points
Question
What is the value of KRAS allelic variants in biliary tract cancers (BTCs)?
Findings
In this cohort study of 7457 patients with BTCs, KRAS allelic variants were common but prevalence was considerably higher among patients with perihilar and extrahepatic cholangiocarcinomas. The most common allelic variants were G12D, G12V, and Q61H, with G12D having the highest median overall survival compared with G12V and Q61H.
Meaning
Findings from this study suggest that KRAS allelic variants within BTCs are potentially actionable genomic alterations.
Abstract
Importance
Biliary tract cancers (BTCs) contain several actionable molecular alterations, including FGFR2, IDH1, ERBB2 (formerly HER2), and KRAS. KRAS allelic variants are found in 20% to 30% of BTCs, and multiple KRAS inhibitors are currently under clinical investigation.
Objectives
To describe the genomic landscape, co–sequence variations, immunophenotype, genomic ancestry, and survival outcomes of KRAS-mutated BTCs and to calculate the median overall survival (mOS) for the most common allelic variants.
Design, Setting, and Participants
This retrospective, multicenter, pooled cohort study obtained clinical and next-generation sequencing data from multiple databases between January 1, 2017, and December 31, 2022. These databases included Princess Margaret Cancer Centre, MD Anderson Cancer Center, Foundation Medicine, American Association for Cancer Research Project GENIE, and cBioPortal for Cancer Genomics. The cohort comprised patients with BTCs who underwent genomic testing.
Main Outcome and Measure
The main outcome was mOS, defined as date of diagnosis to date of death, which was measured in months.
Results
A total of 7457 patients (n = 3773 males [50.6%]; mean [SD] age, 63 [5] years) with BTCs and genomic testing were included. Of these patients, 5813 had clinical outcome data available, in whom 1000 KRAS-mutated BTCs were identified. KRAS allelic variants were highly prevalent in perihilar cholangiocarcinoma (28.6%) and extrahepatic cholangiocarcinoma (36.1%). Thirty-six KRAS allelic variants were identified, and the prevalence rates in descending order were G12D (41%), G12V (23%), and Q61H (8%). The variant G12D had the highest mOS of 25.1 (95% CI, 22.0-33.0) months compared with 22.8 (95% CI, 19.6-31.4) months for Q61H and 17.8 (95% CI, 16.3-23.1) months for G12V variants. The majority of KRAS-mutated BTCs (98.9%) were not microsatellite instability–high and had low tumor mutational burden (ranging from a median [IQR] of 1.2 (1.2-2.5) to a mean [SD] of 3.3 [1.3]). Immune profiling through RNA sequencing of KRAS and NRAS–mutated samples showed a pattern toward a more immune-inflamed microenvironment with higher M1 macrophage activation (0.16 vs 0.12; P = .047) and interferon-γ expression compared with wild-type tumors. The G12D variant remained the most common KRAS allelic variant in all patient ancestries. Patients with admixed American ancestry had the highest proportion of G12D variant (45.0%).
Conclusions and Relevance
This cohort study found that KRAS allelic variants were relatively common and may be potentially actionable genomic alterations in patients with BTCs, especially perihilar cholangiocarcinoma and extrahepatic cholangiocarcinoma. The findings add to the growing data on genomic and immune landscapes of KRAS allelic variants in BTCs and are potentially of value to the planning of specific therapies for this heterogeneous patient group.
Introduction
Biliary tract cancers (BTCs) are malignant neoplasms that emerge from the epithelium of the biliary system and are the second most common type of hepatobiliary cancer worldwide. A heterogenous group of cancers, BTCs consist of gallbladder cancer (GBC); intrahepatic cholangiocarcinoma (IHCC); and extrahepatic cholangiocarcinoma (EHCC), which includes perihilar cholangiocarcinoma (PHCC) and distal cholangiocarcinoma. The incidence and death rates of BTCs are increasing globally, and this disease remains a major health care burden.1 Additionally, BTCs have an overall poor outcome and a low 5-year survival rate of 5% to 15%.2,3,4
Surgery is the only curative treatment option for BTCs, but the majority of patients are diagnosed with metastatic or unresectable disease.5 For unresectable disease, the median overall survival (mOS) is between 2.5 and 4.5 months for the untreated cohort6,7 and approximately 1 year for the treated cohort.8,9 In the phase 3 TOPAZ-1 (Durvalumab or Placebo in Combination With Gemcitabine/Cisplatin in Patients With 1st Line Advanced Biliary Tract Cancer) clinical trial, the addition of the immune checkpoint inhibitor (ICI) durvalumab to gemcitabine and cisplatin vs chemotherapy alone increased mOS from 11.3 months to 12.9 months, with higher 2-year survival rates in patients with metastatic BTCs (23.6% vs 11.5%).8
Actionable molecular alterations have been found in approximately 30% of BTCs and include fibroblast growth factor receptor 2 (FGFR2) fusions, isocitrate dehydrogenase 1 (IDH1) sequence variations, v-raf murine sarcoma viral oncogene homologue B1 (BRAF) V600E sequence variations, and Erb-b2 receptor tyrosine kinase 2 (ERBB2; formerly HER2) amplifications.10,11 The frequency of these molecular alterations varies according to the anatomical location of the primary tumor. The IDH1 and FGFR2 alterations are predominantly found in IHCC, whereas ERBB2 amplifications are often found in EHCC and GBC.11,12,13,14 The most common sequence variation found in BTCs includes tumor protein p53 (TP53), cyclin-dependent kinase inhibitor 2A and 2B (CDKN2A and CDKN2B), AT-rich interactive domain-containing protein 1A (ARID1A), and Kirsten rat sarcoma viral oncogene homologue (KRAS).11,15
KRAS allelic variants are found in 20% to 30% of BTCs and have been associated with a more aggressive tumor phenotype and shortened survival.16,17,18 Multiple KRAS inhibitors are currently under investigation for KRAS-mutated tumors. In the phase 2 KRYSTAL-1 (Phase 1/2 Study of MRTX849 in Patients With Cancer Having a KRAS G12C Mutation) trial evaluating the KRAS G12C inhibitor adagrasib in patients with advanced solid tumors harboring a G12C variant, 12 patients with BTCs had an objective response rate (ORR) of 41.7%, a disease control rate (DCR) of 91.7%, a median progression-free survival of 8.6 months, and a mOS of 15.1 months.19 In addition, a post hoc exploratory analysis of the TOPAZ-1 trial in the combination immunotherapy and chemotherapy arm found that KRAS-mutated BTCs were more commonly found in long-term survivors who lived more than 18 months after randomization.20
Given the ongoing clinical development of molecular inhibitors targeting KRAS allelic variants and the potential synergistic property of these variants with immunotherapy, we sought to further describe the molecular and clinical features of KRAS-mutated BTCs. In this study, our objective was to describe the genomic landscape, co–sequence variations, immunophenotype, genomic ancestry, and survival outcomes of KRAS-mutated BTCs and to calculate the mOS for the most common allelic variants.
Methods
We conducted a retrospective, multicenter, pooled cohort study of patients with BTCs using data from multiple databases. Biliary tract tumors underwent next-generation sequencing (NGS) at Princess Margaret Cancer Centre and MD Anderson Cancer Center between January 1, 2017, and December 31, 2022. Patients consented to medical record review and genomic profiling of their tumor tissue at each institution. The study was conducted in accordance with the Declaration of Helsinki.21 The Princess Margaret Cancer Centre and MD Anderson Cancer Center Institutional Review Boards approved the study protocols, which allowed for the retrieval of clinical, pathologic, and molecular data of patients, who signed an informed written consent or whose consent was waived. The WIRB-Copernicus Group (formerly Western Institutional Review Board) approved the use of Foundation Medicine database in this study. We followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline.
We also extracted targeted NGS and clinical data from the American Association for Cancer Research (AACR) Project Genomics Evidence Neoplasia Information Exchange (GENIE) registry, version 13.0, which encompassed 148 268 patients across 111 cancer types at 19 cancer research institutions across the world. The AACR Project GENIE is one of the largest fully public cancer genomic datasets released to date,22 and it contains clinical sequencing information and a limited set of clinical information, including self-reported race and ethnicity, sex, age at sequencing, cancer type sequenced using the OncoTree hierarchy, and the sample type sequenced (primary or metastatic). Additional patients were included from the following cBioPortal for Cancer Genomics databases: Memorial Sloan Kettering–IMPACT (Integrated Mutation Profiling of Actionable Cancer Targets) Clinical Sequencing Cohort,23 Memorial Sloan Kettering MetTropism (Metastatic Events and Tropisms),24 China Pan-cancer,25 and University of Michigan Metastatic Solid Cancers.26
All data were deidentified via the Health Insurance Portability and Accountability Act Safe Harbor method and incorporated into 1 database to minimize bias. Each database was unique in that each had its own set of patients and there was no overlap in database information. Patients were classified on the basis of their site of disease, such as GBC, IHCC, PHCC, or EHCC. Patients classified with EHCC primarily had distal cholangiocarcinoma. For the majority of patients included, a complete treatment history (both types and lines of therapy) was unknown, but most of the data were likely collected before the routine access to immunotherapy or ICIs to treat BTCs. Data from the Foundation Medicine database were used for genomic ancestry, immune profiling, and biomarker analyses. All quantitative variables were used as whole values to the nearest 10th decimal place and not grouped.
Specimen Selection and Information Collection
At Princess Margaret Cancer Centre and MD Anderson Cancer Center, clinicopathologic information was retrieved from electronic medical records and included patient’s age, self-reported race and ethnicity, clinical stage at the time of diagnosis, histologic tumor subtype, tumor grade, T stage, N stage, and overall survival. The diagnosis of BTCs was confirmed by a board-certified pathologist with a subspecialty in gastrointestinal pathology. A representative formalin-fixed, paraffin-embedded (FFPE) block was chosen from each tumor specimen for performance of NGS at Princess Margaret Cancer Centre and MD Anderson Cancer Center.
Genetic Analysis
Testing was performed on both tumor tissue and a patient-specific normal control (blood) to ensure all variant calls were somatic in nature. For the AACR Project GENIE database, all participating centers committed to providing (1) sequence variation, copy number, and gene fusion data; (2) a minimal clinical dataset of 12 data elements; and (3) a detailed accounting of the genomic regions analyzed by each assay and the specimens to which each assay was applied.
For the Princess Margaret Cancer Centre database, 2 genomic panels were used: a custom hybridization capture panel (SureSelect; Agilent) of 555 cancer-relevant genes sequenced on a NextSeq series sequencing system (Illumina), and a commercial 161-gene amplicon DNA and RNA panel (Oncomine Comprehensive Assay v3; Thermo Fisher Scientific) sequenced on the NGS platform (Ion S5 XL; Thermo Fisher Scientific).
For the MD Anderson Cancer Center database, the MD Anderson Mutation Analysis Precision Panel is a custom high-throughput NGS-based chemiluminescence immunoassay that uses targeted hybridization-based capture technology for detection of sequence variations in 610 genes (single-nucleotide variations, insertions and deletions); copy number variants in 583 genes; select gene rearrangements in 34 genes (fusions); and selected genomic immune-oncology signatures, including microsatellite instability (MSI) and tumor mutational burden (TMB) in DNA isolated from FFPE tumor tissue and cytologic specimens. Data analysis was performed in house by the MD Anderson Mutation Analysis Precision Panel bioinformatics pipeline, which relies on the dual-duplex molecular barcoding for consensus analysis to reduce sequencing artifacts and achieve greater sensitivity and positive predictive value.
For the Foundation Medicine database,27 comprehensive genomic profiling was performed using an adaptor-ligation or hybrid capture–based assay of coding DNA extracted from FFPE primary or metastatic colorectal cancer tumors and sequenced to high, uniform median coverage (>500 × ). Coding exons of the KRAS gene were selected and analyzed for base substitutions, short insertions and deletions, copy number alterations, and rearrangements.
RNA Sequencing and Computation Analysis
RNA sequencing was performed using methods that have been previously described.28 The Estimation of Stromal and Immune cells in Malignant Tumours Using Expression data (ESTIMATE)29 algorithm was applied to assess stromal and immune cell infiltration as well as to calculate an ESTIMATE score, which is the sum of the 2 based on RNA expression data. The Cell-Type Identification By Estimating Relative Subsets of RNA Transcripts (CIBERSORT)30 computational method was used to assess values of 22 immune cell types with the mRNA expression data and to conduct immune cell profiling analysis. The immuno-oncology gene interaction maps package (ImogiMap, a bioinformatics tool and resources for interactions between oncogenic events and immune checkpoints associated with immunotherapy responses31) was used to calculate scores for epithelial to mesenchymal transition,32 vascularization,33 interferon (IFN)-γ expression score, and a T-cell inflammation gene expression signature.34
Clinical Outcomes and Immunophenotype
The mOS based on KRAS allelic variant subtype, co–sequence variation, and primary site of tumor origin was obtained from the Princess Margaret Cancer Centre, MD Anderson Cancer Center, AACR Project GENIE, and cBioPortal for Cancer Genomics databases. Overall survival was defined as date of diagnosis to date of death, measured in months. Patients who were lost to follow-up or still alive were censored.
Immune biomarkers, including programmed death ligand 1 (PD-L1), MSI, TMB, and genome-wide loss of heterozygosity (gLOH), were derived from the Foundation Medicine database. Ancestry data were based on the 5 superpopulations from the 1000 Genomes Project: Africans, admixed Americans, East Asians, Europeans, and South Asians.35
Statistical Analysis
For the primary outcome, descriptive statistics of cohort demographics, KRAS allelic variants, concurrent genetic aberrations, and immune biomarkers (PD-L1, TMB, MSI, and gLOH) were summarized. For survival analysis, log-rank, Wilcoxon, and Kaplan-Meier tests were conducted to compare time to death (or last seen) for each group. The designated threshold of statistical significance was P = .05, and no adjustment was made for multiple comparisons. All statistical analyses were conducted using SAS Studio, version 3.81 (SAS Institute Inc).
Results
A total of 7457 patients with BTCs and genomic testing (n = 3773 males [50.6%], n = 3684 females [49.4%]; mean [SD] age, 63 [5] years) were included in the study, of whom 5813 had clinical outcome data available. Among these patients, 2499 were identified from the AACR Project GENIE, 2034 from cBioPortal for Cancer Genomics, 1644 from Foundation Medicine, 1082 from MD Anderson Cancer Center, and 198 from Princess Margaret Cancer Centre databases (Table 1). The majority of the patients had a diagnosis of cholangiocarcinoma (89%) vs GBC, and IHCC was the largest represented subtype (59%).
Table 1. Breakdown of Databases in Each Section of the Results.
| Result section | Databases |
|---|---|
| KRAS allelic variants, allelic frequency, and codriver variations | Princess Margaret Cancer Centre |
| MD Anderson Cancer Center | |
| AACR Project GENIE | |
| cBioPortal for Cancer Genomics | |
| Survival analysis | Princess Margaret Cancer Centre |
| MD Anderson Cancer Center | |
| AACR Project GENIE | |
| cBioPortal for Cancer Genomics | |
| Immune profiling | Foundation Medicine |
| MD Anderson Cancer Center | |
| Ancestry | Foundation Medicine |
Abbreviations: AACR, American Association for Cancer Research; GENIE, Genomics Evidence Neoplasia Information Exchange.
Within the AACR Project GENIE, the patient cohort consisted of 220 males (53%) and 195 females (47%), with a mean (SD) age at diagnosis of 61 (6) years. The predominant race and ethnicity reported was White (303 [73%]), followed by Asian (25 [6%]) and Black (17 [4%]). The majority of genetic testing and DNA sequencing was completed from biopsies of the primary tumor site (228 [55%]), and 125 patients (30%) had 2 or fewer variants.
KRAS Allelic Variants, Allelic Frequency, and Codriver Variations
Within the clinical cohort (n = 5813), 1000 patients had a KRAS allelic variant, with an overall prevalence of 17.2%. The prevalence of KRAS allelic variants was higher in patients with EHCC (36.1%) and PHCC (28.6%) than in those with IHCC (11.8%) and GBC (7.6%) (Figure 1A). The most common KRAS allelic variant was G12D (39.5%), and the most common co–sequence variation was TP53 (except in PHCC, which was G12V) and suppressor of mothers against decapentaplegic 4 (SMAD4). The prevalence of a G12D allelic variant was 50.9% in patients with EHCC, 44.8% in patients with IHCC, and 33.3% in patients with GBC.
Figure 1. Genomic Profiling for KRAS Allelic Variants in Biliary Tract Cancers.

A, Extrahepatic cholangiocarcinoma (EHCC) and perihilar cholangiocarcinoma (PHCC) had the highest prevalence of KRAS allelic variants. B, KRAS copy number alterations and counts were obtained from The American Association for Cancer Research Project Genomics Evidence Neoplasia Information Exchange. C, G12D and G12V composed the majority of KRAS allelic variants in biliary tract cancer. GBC indicates gallbladder cancer; IHCC, intrahepatic cholangiocarcinoma; VUS, variant of uncertain significance.
Based on reported data from the AACR Project GENIE, the most common KRAS somatic variation type was a missense variant and diploid for copy number alterations (Figure 1B). Thirty-six KRAS allelic variants were identified, and the prevalence rates in descending order were as follows: G12D (41%), G12V (23%), Q61H (8%), G12C (6%), G12A (5%), G13D (4%), and G12R (4%) (Figure 1C).
Survival Analysis
In the survival analysis, patients with localized and advanced disease were included. Patients with the G12D allele subtype had the highest mOS at 25.1 (95% CI, 22.0-33.0) months, followed by those with Q61H (22.8 months; 95% CI, 19.6-31.4 months) and G12V (17.8 months; 95% CI, 16.3-23.1 months) variants (P = .02) (Figure 2A).
Figure 2. Overall Survival for KRAS Allelic Variants in Biliary Tract Cancers.

Plus signs indicate censored patients.
In the survival analysis of KRAS codriver variations, there was no significant survival difference. The mOS was 20.0 (95% CI, 15.9-23.0) months for TP53, 21.5 (95% CI, 14.0-37.2) months for SMAD4, 20.4 (95% CI, 14.0-27.0) months for CDK2NA, and 20.9 (95% CI, 11.6-42.6) months for additional KRAS allelic variants (P = .70).
In a covariance analysis of KRAS allelic variants and tumor site, there was no difference in IHCC. The mOS was lower in patients with Q61H variants in PHCC and patients with G12V variants in EHCC and GBC (Figure 2B).
Immune Profiling
A total of 1644 patients with BTCs and KRAS allelic variants were identified from the Foundation Medicine database for immune profiling and biomarker analysis (Table 2). The 3 most common KRAS allelic variants (G12D, G12V, and Q61H) were analyzed. The majority of KRAS-mutated tumors (98.9%) tested for MSI were not MSI-High. The MSI-high status was 1.5% for G12D, 1.4% for G12V, and 0.5% for Q61H. The TMB ranged from a median (IQR) of 1.2 (1.2-2.5) to a mean (SD) of 3.3 (1.3), with the higher values in patients with GBC, particularly G12V and Q61H variants, and lowest values in patients with EHCC. The highest PD-L1 positivity was among patients with GBC (24 [51%]) compared with those with IHCC (101 [37%]) and EHCC (33 [34%]). Low gLOH was common among all groups (314 of 359 [87%] in IHCC, 60 of 64 [94%] in EHCC, and 45 of 50 [90%] in GBC).
Table 2. Immunophenotype of KRAS Allelic Variants in Biliary Tract Cancers by Site.
| Characteristic | IHCC, No. (%) | EHCC, No. (%) | GBC, No. (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| G12D | G12V | Q61H | G12D | G12V | Q61H | G12D | G12V | Q61H | |
| Total No. | 633 | 333 | 128 | 215 | 141 | 30 | 104 | 45 | 15 |
| Sex | |||||||||
| Males | 349 (55) | 170 (51) | 68 (53) | 118 (55) | 84 (60) | 20 (67) | 33 (22) | 22 (49) | 5 (33) |
| Females | 284 (45) | 163 (49) | 60 (47) | 97 (45) | 57 (40) | 10 (33) | 71 (68) | 23 (51) | 10 (67) |
| Median (IQR) age, y | 64 (25 to ≥89) | 65 (23 to ≥89) | 68 (28 to 85) | 66 (31 to ≥89) | 64 (34 to ≥89) | 64.5 (31 to 86) | 67 (34 to ≥89) | 68 (41 to ≥89) | 67 (30 to 84) |
| GA or tumor, No. | 4.3 | 4.3 | 4.4 | 4.1 | 4.2 | 3.8 | 5.4 | 5 | 4.9 |
| TMB | |||||||||
| No. | 633 | 333 | 128 | 215 | 141 | 30 | 104 | 45 | 15 |
| Median (IQR) | 1.2 (0-2.5) | 1.2 (0.9-2.6) | 1.2 (0-2.5) | 1.2 (0-2.5) | 1.2 (0-2.1) | 1.2 (0-2.5) | 1.5 (0-3.8) | 2.5 (1.2-3.8) | 2.5 (1.7-4.4) |
| Mean (SD) | 2.1 (3.0) | 2.2 (2.8) | 2.4 (4.1) | 1.8 (3.0) | 1.6 (3.7) | 1.5 (1.5) | 2.7 (3.2) | 3.3 (5.3) | 3.1 (2.0) |
| ≥10 variants/Mb | 16 (3) | 7 (1) | 3 (2) | 3 (1) | 2 (1) | 0 | 5 (5) | 2 (4) | 0 |
| ≥20 variants/Mb | 3 (1) | 2 (1) | 3 (2) | 1 (1) | 1 (1) | 0 | 1 (1) | 1 (2) | 0 |
| PD-L1 IHCC | |||||||||
| No. | 158 | 87 | 28 | 53 | 34 | 10 | 29 | 12 | 6 |
| Negativity | 99 (63) | 59 (67) | 13 (46) | 31 (58) | 25 (73) | 8 (80) | 15 (52) | 5 (42) | 3 (50) |
| Low | 46 (29) | 24 (28) | 12 (43) | 19 (36) | 9 (27) | 2 (20) | 9 (31) | 6 (50) | 1 (17) |
| High | 13 (8) | 4 (5) | 3 (11) | 3 (6) | 0 | 0 | 5 (17) | 1 (8) | 2 (33) |
| MSI | |||||||||
| No. | 608 | 320 | 124 | 204 | 137 | 28 | 101 | 45 | 13 |
| MSI-High | 7 (1) | 2 (1) | 2 (2) | 1 (1) | 2 (2) | 0 | 3 (3) | 1 (2) | 0 |
Abbreviations: EHCC, extrahepatic cholangiocarcinoma; GA, genomic alterations; GBC, gallbladder cancer; IHCC, intrahepatic cholangiocarcinoma; Mb, megabase; PD-L1, programmed death 1 ligand 1; MSI, microsatellite instability; TMB, tumor mutational burden.
We also assessed immune profiling through RNA sequencing of KRAS and NRAS–mutated samples obtained at MD Anderson Cancer Center and used CIBERSORT30 to conduct immune cell profiling analysis (Figure 3). We found that KRAS and NRAS–mutated tumors had an RNA signature that favored M1 macrophage activation over wild-type tumors (0.16 vs 0.12; P = .047) (Figure 3A). The distribution of immune-oncology markers in KRAS and NRAS–mutated vs wild-type samples was analyzed using the gene expression signature scores within the ImogiMap. We found that KRAS and NRAS tumors had a pattern toward a more immune-inflamed microenvironment with a higher IFN-γ expression score (Figure 3E).
Figure 3. Immune Profile of KRAS Allelic Variants in Biliary Tract Cancers.
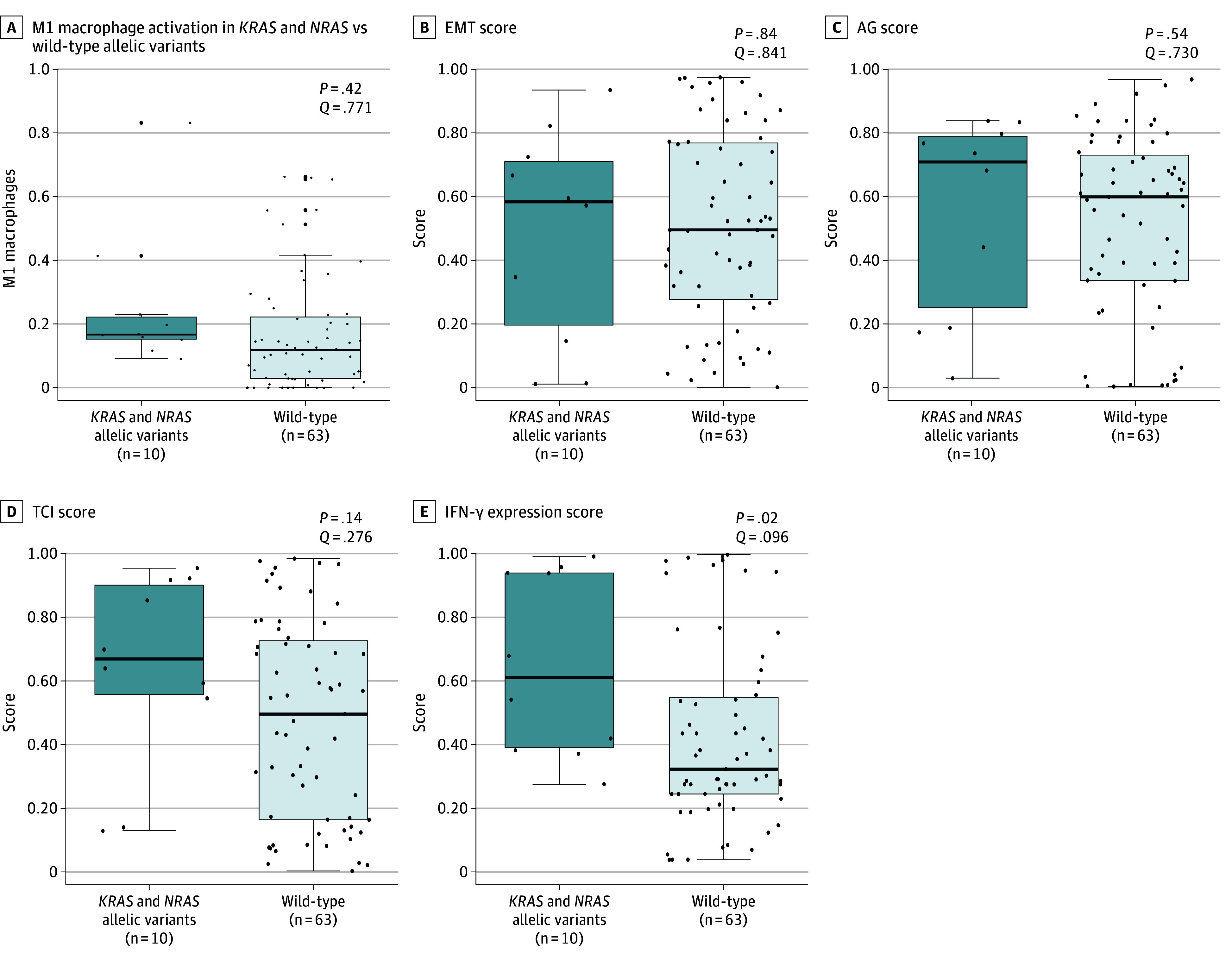
Horizontal line inside boxes represent medians; whiskers represent SDs; and dots represent individual sample score. EMT indicates epithelial to mesenchymal transition; IFN-γ, interferon gamma; TCI, T-cell inflammation.
Ancestry
Using the Foundation Medicine database, we also identified the frequency of KRAS allelic variants based on genomic ancestry assessment. We found that the G12D variant remained the most common KRAS allelic variant in all patient ancestries. Patients with admixed American ancestry had the highest proportion of G12D of all the ancestries we examined (45%). Patients with East Asian and admixed American ancestries had lower proportions of the G12V variant compared with other patient ancestries (eg, 25% [East Asian] and 29% [admixed American] vs 32% [European]). Patients with East Asian and South Asian ancestries had higher proportions of the Q61H variant compared with other patient ancestries (eg, 14% [East Asian] and 11% [South Asian] vs 6% [Africans]).
Discussion
Herein, we described the genomic and immune landscapes, clinical outcomes, and ancestries of KRAS-mutated BTCs. We identified 7457 patients with BTCs and 2644 patients (1000 from clinical cohort; 1644 from Foundation Medicine database) with KRAS allelic variants. The majority of large-sequencing studies and clinical trials for BTCs have primarily focused on identifying and targeting driver sequence variations in IHCC. Multiple FGFR and IDH1 inhibitors are currently available in North America for the treatment of IHCC. Fewer therapeutic options, however, are available for patients with GBC and EHCC. In this cohort study, we found that KRAS allelic variants were prevalent in IHCC, EHCC, and GBCs and highly prevalent in PHCC (28.6%) and EHCC (36.1%). In reviewing genomic ancestry and immune microenvironment, we found that G12D was the most common variant in all patient ancestries but that the frequencies of other KRAS variants varied between ancestries. Patients with East Asian and admixed American ancestries had lower proportions of G12V compared with other patient ancestries.
The predominant KRAS allelic variant was G12D (39.5%), a finding consistent with previously reported results.18,36,37 Additionally, BTCs contained a range of KRAS alleles, including G12V, Q61H, G12A, G12C, and G13D. The G12V and Q61H variants had lower mOS compared with G12D, which is consistent with previous reports.38 Survival analysis of the KRAS allelic variants and their codriver variation (TP53, SMAD4, CDKN2A, or additional KRAS variants) was not statistically significant, suggesting that the presence of a KRAS oncogenic driver variation operates independent of other possible sequence variations and is associated with outcome.
Recent studies with KRAS inhibitors have shown promising results. In the BTC cohort of the KRYSTAL-1 trial targeting KRAS G12C–mutated cancers, patients treated with the KRAS G12C inhibitor adagrasib had an ORR of 41.7%, DCR of 91.7%, median progression-free survival of 8.6 months, and mOS of 15.1 months.21 However, the G12C variant was found in only approximately 6% of all KRAS-mutated BTCs.39 A number of pan-KRAS inhibitors are currently under development.37,40,41
This study highlighted the potential for combining immunotherapy with KRAS inhibitors in KRAS-mutated BTCs. The results showed that GBC had a relative higher mean and median TMB and proportion of PD-L1 high expression, which are favorable factors for ICI therapy response in KRAS-mutated GBC.42 In a 2023 study by Jeong et al,43 patients with KRAS allelic variants and PD-L1 positivity had a longer progression-free survival than patients with KRAS allelic variants and PD-L1 negativity. Preclinically, KRAS inhibitors have been associated with promotion of a proinflammatory tumor microenvironment, increased major histocompatibility complex class I protein expression, and synergies with immunotherapy to enhance antitumor activity.44,45,46 Moreover, KRAS inhibition has played a role in decreased myeloid-derived suppressor cells and increased classically activated M1-polarized macrophages, dendritic cells, cluster of differentiation (CD) 4+ cells, and CD8+ T cells, which are associated with tumor sensitivity to immune checkpoint inhibition.45
Preliminary data in the present study also suggest that BTCs with RAS sequence variation may be associated with an inflammatory tumor microenvironment. Clinically, in the KRYSTAL-7 trial, the combination of the KRAS G12C inhibitor adagrasib with the ICI pembrolizumab demonstrated an ORR of 49% and a DCR of 89% in patients with KRAS G12C–mutated advanced non–small cell lung cancer.47
Limitations
This study has limitations. First, the sample was primarily composed of a North American population. We tried to overcome this limitation by including patients from the AACR Project GENIE, which includes institutions from the UK, France, Sweden, Netherlands, Spain, China, and Korea. However, a more global population may not be fully represented for interpreting the results. To our knowledge, BTCs have a growing incidence worldwide, and future studies need to include a more robust global population to support the present research. Second, each database used a different data processing and variant calling algorithm. However, given that these databases were all large cohort cancer genomic projects at major institutions within relatively the same time frame, we believe these differences were minor and did not affect the identification of a KRAS allelic variant nor the most common allelic variants. Third, there were fewer patients with CIBERSORT and ImogiMap analyses. Fourth, only the 3 most common KRAS allelic variants in this study (G12D, G12V, and Q61H) were further characterized and examined due to feasibility. Fifth, clinical data from this large cohort were limited, and the association of systemic immunotherapy with ICIs could not be assessed. Further investigation into the other KRAS allelic variants may unlock additional potential patterns, biomarkers, or treatment options.
Conclusions
In this cohort study of patients with BTCs, KRAS allelic variants were relatively common and may be potentially actionable genomic alterations, especially in PHCC and EHCC. The most common KRAS allelic variant within the North American population was G12D, which has a more favorable mOS profile than G12V or Q61H. Findings from this study add to the growing data on comprehensive genomic and immune landscapes of KRAS allelic variants in BTCs and are potentially of value to the planning of specific therapies for this heterogeneous patient group.
Data Sharing Statement
References
- 1.Ouyang G, Liu Q, Wu Y, et al. The global, regional, and national burden of gallbladder and biliary tract cancer and its attributable risk factors in 195 countries and territories, 1990 to 2017: a systematic analysis for the Global Burden of Disease Study 2017. Cancer. 2021;127(13):2238-2250. doi: 10.1002/cncr.33476 [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17-48. doi: 10.3322/caac.21763 [DOI] [PubMed] [Google Scholar]
- 3.Turkes F, Carmichael J, Cunningham D, Starling N. Contemporary tailored oncology treatment of biliary tract cancers. Gastroenterol Res Pract. 2019;2019:7698786. doi: 10.1155/2019/7698786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Charbel H, Al-Kawas FH. Cholangiocarcinoma: epidemiology, risk factors, pathogenesis, and diagnosis. Curr Gastroenterol Rep. 2011;13(2):182-187. doi: 10.1007/s11894-011-0178-8 [DOI] [PubMed] [Google Scholar]
- 5.Valle JW, Kelley RK, Nervi B, Oh DY, Zhu AX. Biliary tract cancer. Lancet. 2021;397(10272):428-444. doi: 10.1016/S0140-6736(21)00153-7 [DOI] [PubMed] [Google Scholar]
- 6.Glimelius B, Hoffman K, Sjödén PO, et al. Chemotherapy improves survival and quality of life in advanced pancreatic and biliary cancer. Ann Oncol. 1996;7(6):593-600. doi: 10.1093/oxfordjournals.annonc.a010676 [DOI] [PubMed] [Google Scholar]
- 7.Sharma A, Dwary AD, Mohanti BK, et al. Best supportive care compared with chemotherapy for unresectable gall bladder cancer: a randomized controlled study. J Clin Oncol. 2010;28(30):4581-4586. doi: 10.1200/JCO.2010.29.3605 [DOI] [PubMed] [Google Scholar]
- 8.Oh DY, Ruth He A, Qin S, et al. Durvalumab plus gemcitabine and cisplatin in advanced biliary tract cancer. NEJM Evid. 2022;1(8):a2200015. doi: 10.1056/EVIDoa2200015 [DOI] [PubMed] [Google Scholar]
- 9.Valle J, Wasan H, Palmer DH, et al. ; ABC-02 Trial Investigators . Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362(14):1273-1281. doi: 10.1056/NEJMoa0908721 [DOI] [PubMed] [Google Scholar]
- 10.Berchuck JE, Facchinetti F, DiToro DF, et al. The clinical landscape of cell-free DNA alterations in 1671 patients with advanced biliary tract cancer. Ann Oncol. 2022;33(12):1269-1283. doi: 10.1016/j.annonc.2022.09.150 [DOI] [PubMed] [Google Scholar]
- 11.Mody K, Jain P, El-Refai SM, et al. Clinical, genomic, and transcriptomic data profiling of biliary tract cancer reveals subtype-specific immune signatures. JCO Precis Oncol. 2022;6:e2100510. doi: 10.1200/PO.21.00510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chakrabarti S, Kamgar M, Mahipal A. Targeted therapies in advanced biliary tract cancer: an evolving paradigm. Cancers (Basel). 2020;12(8):2039. doi: 10.3390/cancers12082039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mondaca S, Schultz N, Roa JC, et al. Clinical and genomic characterization of ERBB2-altered gallbladder cancer. J Clin Oncol. 2022;40(16 suppl):4114. doi: 10.1200/JCO.2022.40.16_suppl.4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carotenuto M, Sacco A, Forgione L, Normanno N. Genomic alterations in cholangiocarcinoma: clinical significance and relevance to therapy. Explor Target Antitumor Ther. 2022;3(2):200-223. doi: 10.37349/etat.2022.00079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Javle M, Bekaii-Saab T, Jain A, et al. Biliary cancer: utility of next-generation sequencing for clinical management. Cancer. 2016;122(24):3838-3847. doi: 10.1002/cncr.30254 [DOI] [PubMed] [Google Scholar]
- 16.Procopio F, Branciforte B, Nappo G, Di Tommaso L, Lleo A, Torzilli G. Meta-analysis on prognostic value of KRAS mutation in resected mass-forming cholangiocarcinoma. Eur J Surg Oncol. 2022;48(7):1455-1463. doi: 10.1016/j.ejso.2022.03.005 [DOI] [PubMed] [Google Scholar]
- 17.Yokoyama M, Ohnishi H, Ohtsuka K, et al. KRAS mutation as a potential prognostic biomarker of biliary tract cancers. Jpn Clin Med. 2016;7:33-39. doi: 10.4137/JCM.S40549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou SL, Xin HY, Sun RQ, et al. Association of KRAS variant subtypes with survival and recurrence in patients with surgically treated intrahepatic cholangiocarcinoma. JAMA Surg. 2022;157(1):59-65. doi: 10.1001/jamasurg.2021.5679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yaeger R, Spira AI, Pelster M, et al. KRYSTAL-1: activity and safety of adagrasib (MRTX849) in patients with advanced solid tumors harboring a KRASG12C mutation. J Clin Oncol. 2023;41(36 suppl):425082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bouattour M, Valle JW, Vogel A, et al. Characterization of long-term survivors in the TOPAZ-1 study of durvalumab or placebo plus gemcitabine and cisplatin in advanced biliary tract cancer. J Clin Oncol. 2023;41(4)(suppl):531. doi: 10.1200/JCO.2023.41.4_suppl.531 [DOI] [Google Scholar]
- 21.Pant S, Yaeger R, Spira AI, et al. KRYSTAL-1: activity and safety of adagrasib (MRTX849) in patients with advanced solid tumors harboring a KRAS G12C mutation. J Clin Oncol. 2023;41(suppl 36):425082. doi: 10.1200/JCO.2023.41.36_suppl.425082 [DOI] [Google Scholar]
- 22.AACR Project GENIE Consortium . AACR Project GENIE: powering precision medicine through an international consortium. Cancer Discov. 2017;7(8):818-831. doi: 10.1158/2159-8290.CD-17-0151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23(6):703-713. doi: 10.1038/nm.4333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen B, Fong C, Luthra A, et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell. 2022;185(3):563-575.e11. doi: 10.1016/j.cell.2022.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wu L, Yao H, Chen H, et al. Landscape of somatic alterations in large-scale solid tumors from an Asian population. Nat Commun. 2022;13(1):4264. doi: 10.1038/s41467-022-31780-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson DR, Wu YM, Lonigro RJ, et al. Integrative clinical genomics of metastatic cancer. Nature. 2017;548(7667):297-303. doi: 10.1038/nature23306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023-1031. doi: 10.1038/nbt.2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kwong LN, De Macedo MP, Haydu L, et al. Biological validation of RNA sequencing data from formalin-fixed paraffin-embedded primary melanomas. JCO Precis Oncol. Published online June 14, 2018. doi: 10.1200/PO.17.00259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoshihara K, Shahmoradgoli M, Martínez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612. doi: 10.1038/ncomms3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling tumor infiltrating immune cells with CIBERSORT. Methods Mol Biol. 2018;1711:243-259. doi: 10.1007/978-1-4939-7493-1_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bozorgui B, Kong EK, Luna A, Korkut A. Mapping the functional interactions at the tumor-immune checkpoint interface. Commun Biol. 2023;6(1):462. doi: 10.1038/s42003-023-04777-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mak MP, Tong P, Diao L, et al. A patient-derived, pan-cancer EMT signature identifies global molecular alterations and immune target enrichment following epithelial-to-mesenchymal transition. Clin Cancer Res. 2016;22(3):609-620. doi: 10.1158/1078-0432.CCR-15-0876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Masiero M, Simões FC, Han HD, et al. A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell. 2013;24(2):229-241. doi: 10.1016/j.ccr.2013.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ayers M, Lunceford J, Nebozhyn M, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930-2940. doi: 10.1172/JCI91190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Auton A, Brooks LD, Durbin RM, et al. ; 1000 Genomes Project Consortium . A global reference for human genetic variation. Nature. 2015;526(7571):68-74. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Dell MR, Huang JL, Whitney-Miller CL, et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012;72(6):1557-1567. doi: 10.1158/0008-5472.CAN-11-3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee JK, Sivakumar S, Schrock AB, et al. Comprehensive pan-cancer genomic landscape of KRAS altered cancers and real-world outcomes in solid tumors. NPJ Precis Oncol. 2022;6(1):91. doi: 10.1038/s41698-022-00334-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carrot-Zhang J, Chambwe N, Damrauer JS, et al. ; Cancer Genome Atlas Analysis Network . Comprehensive analysis of genetic ancestry and its molecular correlates in cancer. Cancer Cell. 2020;37(5):639-654.e6. doi: 10.1016/j.ccell.2020.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moffat GT, Hu ZI, De Armas AD, Ross JS, Javle MM, Knox JJ. Characterizing KRAS allele variants within biliary tract cancers. J Clin Oncol. 2023;41(16)(suppl):4088. doi: 10.1200/JCO.2023.41.16_suppl.4088 [DOI] [Google Scholar]
- 40.Hofmann MH, Gerlach D, Misale S, Petronczki M, Kraut N. Expanding the reach of precision oncology by drugging all KRAS mutants. Cancer Discov. 2022;12(4):924-937. doi: 10.1158/2159-8290.CD-21-1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Plangger A, Rath B, Stickler S, et al. Cytotoxicity of combinations of the pan-KRAS SOS1 inhibitor BAY-293 against pancreatic cancer cell lines. Discov Oncol. 2022;13(1):84. doi: 10.1007/s12672-022-00550-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kato S, Fujiwara Y, Hong DS. Targeting KRAS: crossroads of signaling and immune inhibition. J Immunother Precis Oncol. 2022;5(3):68-78. doi: 10.36401/JIPO-22-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jeong SY, Hong JY, Park JO, et al. The efficacy of immune checkpoint inhibitors in biliary tract cancer with KRAS mutation. Therap Adv Gastroenterol. Published online May 5, 2023. doi: 10.1177/17562848231170484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217-223. doi: 10.1038/s41586-019-1694-1 [DOI] [PubMed] [Google Scholar]
- 45.Briere DM, Li S, Calinisan A, et al. The KRASG12C inhibitor MRTX849 reconditions the tumor immune microenvironment and sensitizes tumors to checkpoint inhibitor therapy. Mol Cancer Ther. 2021;20(6):975-985. doi: 10.1158/1535-7163.MCT-20-0462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kemp SB, Cheng N, Markosyan N, et al. Efficacy of a small-molecule inhibitor of KrasG12D in immunocompetent models of pancreatic cancer. Cancer Discov. 2023;13(2):298-311. doi: 10.1158/2159-8290.CD-22-1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jänne PA, Smit EF, de Marinis F, et al. Preliminary safety and efficacy of adagrasib with pembrolizumab in treatment-naïve patients with advanced non-small cell lung cancer (NSCLC) harboring a KRASG12C mutation. Immuno-Oncology and Technology. 2022;16(1)(suppl):100360. doi: 10.1016/j.iotech.2022.100360 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Sharing Statement