

Summary
LMTK3 is a brain-specific transmembrane serine/threonine protein kinase that acts as a scaffold for protein phosphatase-1 (PP1). Although LMKT3 has been identified as a risk factor for autism and epilepsy, its physiological significance is unknown. Here, we demonstrate that LMTK3 copurifies and binds to KCC2, a neuron-specific K+/Cl− transporter. KCC2 activity is essential for Cl−-mediated hyperpolarizing GABAAR receptor currents, the unitary events that underpin fast synaptic inhibition. LMTK3 acts to promote the association of KCC2 with PP1 to promote the dephosphorylation of S940 within its C-terminal cytoplasmic domain, a process the diminishes KCC2 activity. Accordingly, acute inhibition of LMTK3 increases KCC2 activity dependent upon S940 and increases neuronal Cl− extrusion. Consistent with this, LMTK3 inhibition reduced intrinsic neuronal excitability and the severity of seizure-like events in vitro. Thus, LMTK3 may have profound effects on neuronal excitability as an endogenous modulator of KCC2 activity.
Subject areas: Biochemistry, Molecular biology, Neuroscience, Omics, Proteomics
Graphical abstract
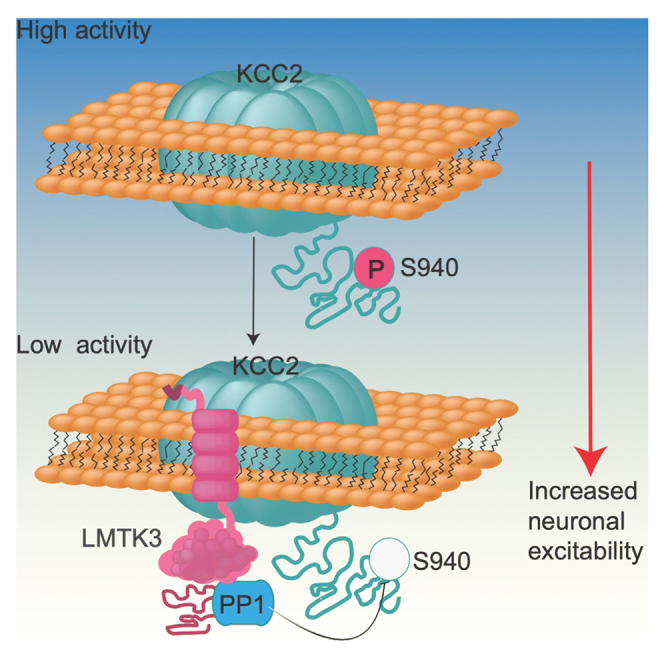
Highlights
-
•
Complexes of LMTK3 and KCC2 are found within the neuronal plasma membrane
-
•
LMTK3 acts as a scaffold to target protein phosphatase-1 (PP1) to KCC2
-
•
LMTK3 inhibits KCC2 by promoting dephosphorylation of serine 940 (S940)
-
•
Inhibiting LMTK3 reduces excitability by resorting KCC2-dependent neuronal Cl− extrusion
Biochemistry; Molecular biology; Neuroscience; Omics; Proteomics
Introduction
In the adult mammalian CNS, fast hyperpolarizing postsynaptic inhibition is mediated by type A γ-aminobutyric acid receptors (GABAARs), which are primarily permeable to Cl− and HCO3− anions. In mature neurons, this Cl− gradient is maintained by the neuron-specific solute transporter KCC2 (SLC12A5), which maintains the GABAAR reversal potential (EGABA) below the resting membrane potential (RMP) by extruding Cl− from the neuron. In contrast to this, EGABA values in developing neurons and epileptic neurons are more positive, which reflects increased intracellular Cl− accumulation, resulting in depolarizing GABAAR receptor currents, which contribute to net neuronal excitability.1,2,3,4 Consistent with its role in facilitating inhibitory neurotransmission, loss-of-function mutations in KCC2 in humans result in epilepsy, developmental delay, and premature mortality.5,6 Modifications in KCC2 activity are also thought to contribute to the acquired epilepsies, autism spectrum disorders, and neuropathic pain.4,7,8,9
It is emerging that there is not a simple relationship between KCC2 expression levels and its activity, rather it is subject to dynamic modulation via the phosphorylation of up to 11 residues within its intracellular, N- and C-termini.10,11,12 These covalent modifications act to potentiate or inhibit KCC2 activity depending on which residues are phosphorylated in addition to regulating transporter membrane trafficking.13,14,15,16,17 Significantly, PP1-dependent dephosphorylation of KCC2 on residues including serine 940 (S940) underpins KCC2 inactivation during neuronal hyperexcitability, a process that directly contributes to seizure severity and lethality.18,19,20
To begin to identify endogenous regulators of KCC2 activity, we employed affinity Blue Native PAGE coupled with mass spectroscopy to identify signaling molecules that form stable protein complexes with KCC2 in brain plasma membranes. This approach revealed that KCC2 is intimately associated with LMTK3 in the brain, a poorly understood brain-specific transmembrane kinase.21,22,23 Mechanistically, LMTK3 acts to target PP1 to KCC2 to mediate dephosphorylation of S940, leading to KCC2 inhibition. Accordingly, inhibition of LMTK3 increased KCC2 activity dependent on S940, reduced intracellular Cl accumulation, and reduced neuronal excitability and the severity of seizure-like events in vitro. Thus, our results suggest that LMTK3 may exert profound effects on neuronal excitability via its ability to modulate KCC2 phosphorylation and activity.
Results
KCC2 copurifies with LMTK3 from brain plasma membranes
To delineate endogenous mechanisms that regulate KCC2 activity in the brain, we have purified native stable protein complexes from the brain and examined their composition using quantitative mass spectroscopy. This non-biased approach revealed that high levels of the transmembrane brain-specific protein kinase LMTK3 copurified with KCC2.11 Structurally, LMTK3 consists of 1,460 amino acids with a small extracellular N-terminus, a single transmembrane domain, an adjacent catalytic domain, and large highly disordered cytoplasmic C-terminus that binds PP1, an interaction mediated by residues 1376–1382 in this enzyme (Figure 1).23 Consistent with its role in regulating excitability, copy-number variants of LMTK3 have been identified in humans that lead to developmental delay and epilepsy.24,25 Studies in cell lines suggest that LMTK3 regulates the activity of mitogen-activated protein kinase (ERK) and phosphorylates estrogen receptor-ɑ (ERɑ), increasing its stability, leading to agonist independent ERɑ signaling.26,27
Figure 1.

Examining the proteins that copurify with KCC2 from brain plasma membranes
(A) Schematic representation of structure of LMTK3. TM, transmembrane domain, AA, amino acid. The PP1 binding site is shown in blue.
(B) Solubilized brain membranes from 8- to 12-week-old mice were exposed to immobilized LMTK3 antibody or control IgG. After extensive washing, bound was eluted with SDS, subject to SDS-PAGE, and immunoblotted with KCC2 or LMTK3 antibodies. In, input.
(C) Material purified on IgG and LMTK3 was eluted in 2% Tween, subject to BN-PAGE, and stained with colloidal Coomassie Brilliant Blue (CCB). The regions of the gels indicated by red arrows were excised, digested with trypsin, and subject to LC-MS/MS.
(D) Significantly enriched proteins seen with LMTK3 antibody compared with IgG were then ranked via SiGi values. n = 4 purifications.
(E) Phosphorylation of individual amino acids within LMTK3 was examined by comparing the ratios of phosphorylated/dephosphorylated amino acids with A scores>18; n = 4 purifications. In all panels, data represent mean ± SEM.
To further assess the significance of LMTK3 as a KCC2 binding partner, solubilized brain plasma membranes were immunoprecipitated with LMTK3 antibodies subject to SDS-PAGE. LMTK3 migrated as a major species of ∼170 kDa in brain membranes, and this band was immunoprecipitated with LMTK3 antibody but not control immunoglobulin G (IgG) (Figure 1B). KCC2 migrated as a major species of 130 kDa, and this species coimmunoprecipitated with LMKT3 antibody but not control IgG (Figure 1B). To gain further insights into their association, we immunopurified LMTK3 from brain plasma membrane, eluted under native conditions, and subjected purified material to Blue Native PAGE visualized using Colloidal Coomassie (CC). A major species of 500 kDa was seen in material purified on LMTK3 antibodies but not control IgG (Figure 1C). This band was then excised together with the corresponding region from IgG, which were digested with trypsin and subject to liquid chromatography coupled with mass spectroscopy. Recovered proteins were ranked via their normalized spectral indices (SiGi), a widely accepted label-free method for quantifying mass spectroscopic data.28,29,30,31 Downstream analysis was limited to proteins that were significantly enriched compared with those recovered on control IgG and were detected in all replicates (Table S1), and of these, the 10 most abundant proteins recovered are indicated (Figure 1D). KCC2 copurified with LMTK3 in an approximate stoichiometry of 1:2 based on their respective SiGi values. High levels of the signaling proteins (ITPR1, CELR2, PTK7), components of the cytoskeleton (My18A, GCC2, CNTLN), and solute transporters (SLC4A4 and ATP9A) were associated with LMTK3.
To gain more insights into the nature of the interaction between LMTK3 and KCC2, we examined they were coexpressed in HEK-293 cells. Although we acknowledge the differences in cell environment of these cells compared with neurons, HEK-293 cells are useful to test protein-protein interactions. To do so, plasmids encoding and LMTK3 modified with the FLAG epitope and KCC2 were transiently expressed in HEK-293 cells13,32,33; 48 h later, cells were solubilized and subject to immunoprecipitation with FLAG or KCC2 antibodies. Bound material was subjected to SDS-PAGE and visualized using immunoblotting. Under these conditions, LMTK3 migrated as a major species of 180 kDa, whereas KCC2 exhibited a mass of 130 kDa (Figure S1A). Significantly, KCC2 was detected immunoprecipitating with LMTK3 when coexpressed in HEK-293 cells (Figure S1B). The subcellular distribution of the proteins was also compared using immunofluorescence and confocal microscopy. Colocalization of LMTK3 and KCC2 immunoreactivity was evident on or close to the plasma membrane (Figure S1C).
Collectively, these results demonstrate that LMTK3 and KCC2 are intimately associated in the brain. Our studies in HEK-293 cells further suggest that LMTK3 directly binds to KCC2.
LMTK3 exhibits constitutive activity in the brain
The cellular mechanisms that regulate LMTK3 activity in the brain are unknown, but studies in vitro suggest that phosphorylation of residues within its catalytic domain underpin its activation. Thus, we examined LMTK3 phosphorylation in the brain. To do so, LMTK3 was purified from brain plasma membranes and subject to SDS-PAGE. Immunoblotting confirmed the presence of a major band of 170 kDa in material purified with LMTK3 antibody but not control IgG. Parallel gels were also stained with CC, and the bands of interests were excised, digested with trypsin, and subject to LC-MS/MS. Sites of statistically significant phosphorylation were determined using the Ascore algorithm where A score <18, p < 0.05.34 Spectral counting was then used to examine their levels of phosphorylation by comparing the ratio of phospho/total peptides for individual serine, threonine, and tyrosine residues.11,28,29 LMTK3 was phosphorylated on multiple serine residues within its catalytic domain and within its intrinsically disordered C-terminus (Figures 1A and 1E). Significantly high levels of phosphorylation were seen for serine 232 (S232), the phosphorylation of which has been shown to be critical for the catalytic activity of LMTK3.22,23,35 In addition to S232, LMTK3 was phosphorylated on 12 other amino acids, but the significance of these covalent modifications for its activity, membrane trafficking, or stability is unknown.
Comparing the subcellular distribution of KCC2 and LMTK3
To further explore the significance of our biochemical experiments, we immunostained cultured hippocampal neurons with antibodies against LMTK3, KCC2, and MAP2 to identify dendritic branches (Figure 2A). Following confocal microscopy, the distribution of KCC2 and LMTK3 puncta were compared along MAP2-positive puncta using ImageJ.36 Both LMTK3 and KCC2 were distributed evenly and along the dendritic processes, and the fluorescent signal overlapped with 31 ± 2 colocalized puncta found per 100 μm of dendritic length (Figure 2B). We then performed immunohistochemistry on fixed brain tissue to investigate the extent of overlap of LMTK3 and KCC2 in the hippocampus in vivo, using LMTK3, KCC2, and MAP2 antibodies (Figure 2C). Quantification of the KCC2 and LMTK3 total stained area in high magnification images of the hippocampal stratum pyramidale revealed that 35 ± 1.4% of LMTK3 puncta on dendrites contained KCC2 immunoreactivity (Figure 2D). Thus, consistent with our biochemical studies, LMTK3 and KCC2 exhibit similar subcellular distributions in cultured neurons and in the brain.
Figure 2.

Comparing the subcellular distribution of LTK3 and KCC2
(A) 18–20 Div cultures were stained with LMTK3, KCC2, and MAP2 antibodies followed by confocal microscopy. A representative neuron is shown in the upper panel, and an enlargement of boxed area is shown below; the scale bar: 10 μm.
(B) The number of LMTK3-KCC2 colocalized clusters per 100 micron of dendrite was then determined; n = 17 neurons from 3 cultures.
(C) Brain sections were subject to immunostaining and confocal microscopy as outlined earlier.
(D) The percentage of LMTK3 puncta that contain KCC2 was then determined; n = 7 hippocampi from 3 animals; the scale bar: 30 μm. In all panels data represent mean ± SEM.
Ablating LMTK3 expression does not modify KCC2 expression levels or its subcellular distribution
As a means of determining if LMTK3 plays any gross role in regulating KCC2 expression levels or subcellular distribution, we used mice in which its expression has been constitutively ablated using homologous recombination.53 Immunoblotting revealed dose-dependent decreases in LMTK3 expression in LMTK3 +/− (Figure 3A; 42 ± 0.02%; p = 0.010) or LMTK3 −/− mice (Figure 3A; 5.07 ± 0.010%; p = 0.018). Consistent with this, LMTK3 antibody immunostaining was abolished in hippocampal slices from LMTK3 −/− mice compared with wild-type (WT) control (Figure 3B). To examine the consequences of ablating LMTK3 expression on the subcellular distribution KCC2, brain sections from both genotypes were immunostained with KCC2 antibodies. After normalization to WT, the intensity (97.6 ± 0.050%; p = 0.671) and area (104.5 ± 0.025%; p = 0.338) of KCC2 puncta were not modified in LMTK3 knockout (KO) mice (Figure 3C). Results of similar studies revealed that ablating LMTK3 expression did not modify KCC2 puncta intensity or area in dentate gyrus granule cells (Figure S2). Thus, ablating LMTK3 does not modify total expression levels of KCC2 or its subcellular distribution.
Figure 3.

Examining the effects of ablating LMTK3 on KCC2 expression levels and activity
(A) Brain extracts from WT (+/+), heterozygotes (−/+), and homozygous (−/−) mice were immunoblotted with LMTK3, KCC2, and GAPDH antibodies. The levels of LMTK3 and KCC2 expression were then compared with those in WT (100%) mice; n = 3; ∗p < 0.05.
(B) Brain sections from WT (+/+) and KO (LMTK3-KO) mice were stained with LMTK3 and MAP2 antibodies followed by confocal microscopy, and a representative image of CA1; scale bar: 20 μm.
(C) High-magnification confocal images of the stratum pyramidale (s.p.) of WT (+/+) and LMTK3-KO (−/−) stained with KCC2 antibody; scale bar: 10 μm. KCC2 fluorescence intensity and total stained area were then compared with those seen in WT (100%), 8–10 slices from 3 mice.
(D) Representative traces and I–V plots are shown for the polarity of currents induced by rapid application of muscimol in DGGCs in slices from WT and LMTK3-KO mice loaded with 32-mM Cl− at differing voltages.
(E) EGABA values and [Cl−]i were determined from the voltage ramps and then compared in DGGCs between genotypes; ∗p < 0.01; t test; n = 7–9 mice.
(F) Individual shifts in EGABA are shown for DGGCs from WT and KO mice following a 15-min exposure to the KCC2 inhibitor 11K. The magnitude in the EGABA shift (ΔEGABA) was then compared between genotypes. ∗p < 0.05; n = 7–9 mice. In all panels data represent mean ± SEM. Voltages are adjusted with a liquid junction potential value of −13 mV.
Ablating LMTK3 expression enhances KCC2 activity and decreases neuronal Cl− accumulation
To investigate the effects of LMTK3 inactivation on KCC2 activity, we performed whole-cell patch-clamp Cl− loading experiments on dentate gyrus granule cells (DGGCs) in acute hippocampal slices from adult LMTK3 control and KO mice. The steady-state GABAAR-mediated currents were measured at increasing holding potentials and were used to determine the reversal potential of GABA-induced currents (EGABA). To do so, the magnitude of muscimol (10 μM, a GABAAR-specific agonist) was at differing holding potentials using voltage ramps, for neurons loaded with 32 mM Cl− via the patch pipette. These measurements were made in the presence of bumetanide to inhibit NKCC1 and TTX to block neuronal depolarizations, conditions under which EGABA is primarily determined by KCC2 activity as detailed previously.16,37 Using this methodology, we found that EGABA was hyperpolarized in DGGCs from LMTK3-KO compared with WT mice (Figures 3D and 3E; −67.3 ± 2.18 mV and −60.5 ± _1.56 mV; p = 0.019). We used the Nernst equation to calculate [Cl−], between genotypes, which was reduced from 15.0 ± 0.86 mM in WT mice to 11.7 ± 0.86 mM in WT in LMTK3 KO mice (Figures 3D and 3E; p = 0.015). In both genotypes, a 15-min exposure to the specific KCC2 inhibitor VU046327138,39 (11K; 10 μM) resulted in depolarizing shifts in EGABA; however, this shift was significantly larger in the absence of LMTK3 (Figure 3F; WT = 11.1 ± 1.09 and KO = 14.9 ± 1.32 mV, respectively; p = 0.037). Collectively these recordings suggests that KCC2 activity is increased in LMTK3 KO mice.
LMTK3 promotes the association of PP1 with KCC2
Our results suggest that ablating LMTK3 expression leads to increased KCC2 activity without modifying its expression levels or grossly modifying its subcellular distribution (Figures 3 and S1). To begin to explore the underlying mechanism, we compared the protein networks associated with KCC2 in WT and LMTK3 KO mice. To do so, KCC2 was immunopurified from both genotypes, resolved by BN-PAGE. As measured using Coomassie staining, a major species of 300 kDa was seen for native KCC2 together with minor species at 600 and 800 kDa (Figure 4A), a result that is consistent with published studies.11 These bands were excised together with the corresponding regions of the IgG lanes, digested with trypsin, and subject to LC-MS/MS. Significantly enriched proteins relative to IgG controls were then identified and were ranked according to their SiGi’s, an accepted label-free method to quantify to LC-MS/MS data. Principal-component analysis (PCA) was then used to compare the proteins that copurified with KCC2 between genotypes as detailed previously.11,28,29,40
Figure 4.

Examining the effects of ablating LMTK3 on PP1 recruitment to KCC2 and its phosphorylation
(A) Solubilized brain membranes from 6- to 8-week-old WT and LMTK3-KO (KO) mice were exposed to immunopurified onKCC2 antibody or control IgG. Bound material was eluted in 2% Tween, subject to BN-PAGE, and stained with CCB. The regions of the gels indicated by the red arrows were excised, digested with trypsin, and subject to LC-MS/MS.
(B) Proteins that copurified with KCC2 from WT and KO mice were using neutral labeling, and their composition was compared using PCA for each replicate (n = 4/genotype). The proteins indicated are the principle proteins that contribute to the separation between genotypes.
(C) The phosphorylation of KCC2 was compared between genotypes using neutral labeling to determine the ratio of phosphorylated/dephosphorylated peptides for specific amino acids in the mature transporter. ∗p < 0.05; t test; n = 4 purifications.
(D) KCC2 was immunopurified from WT and KO mice. Purified material was then subject to SDS-PAGE and immunoblotted with KCC2, PP1, pS940, and pT1007 antibodies.
(E) The ratio of PP1/KCC2 immunoreactivity was determined and normalized to levels in WT; ∗p < 0.05; t test; n = 3 mice.
(F) Total brain lysates were immunoblotted with antibodies against PP1 and actin. The ratios of PP1/actin immunoreactivity were then determined and compared with WT; n = 4 mice.
(G) pS940/KCC2 and pT1007/KCC2 immunoreactivity were determined and normalized to levels in WT. ∗p < 0.05; t test; n = 3 mice. In all panels data represent mean ± SEM.
The proteins that copurified with KCC2 in the respective genotypes separated into 2 distinct groups, with greater variability being evident in WT (Figure 4B; Table S2). In addition to LMTK3, the major proteins that underpin this segregation were the catalytic subunit of PP1 and PP1M. This result is consistent with published studies revealing that LMTK3 acts as scaffold for PP1 to regulate substrate-specific dephosphorylation.22,23 Other proteins of significance were COA3, a putative regulator of cytochrome oxidase that is associated with neuropathy and ERK2.41,42
Given this deficit in PP1 association in LMTK3 KO mice, we compared basal phosphorylation of purified KCC2 between genotypes. The phosphorylation of individual residues with A scores >18 were then quantified using neutral labeling (Beausoleil, 2006; Smalley et al., 2020, Choi et al., 2022). In WT and LMTK3 KO mice, KCC2 was stoichiometrically phosphorylated on S25, S26, T902, T906, S932, T934, S940, T1007, and S1022, within its N- and C-terminal intracellular domains. Ablating LMTK3 expression significantly increased the phosphorylation of S940, (Figure 4C; WT 0.373 ± 0.004 and LMTK3KO 0.439 ± 0.020 Mol/Mol; p = 0.0170). Phosphorylation of the adjacent residues S932 (Figure 4C; WT 0.131 ± 0.023 and LMTK3 KO 0.289 ± 0.024, Mol/Mol; p = 0.0029) and T934 (Figure 4C; WT 0.144 ± 0.010 and LMTK3 KO 0.207 ± 0.018 Mol/Mol; p = 0.0208) was also increased in LMTK3 KO mice. Significantly, phosphorylation of T906 (p = 0.147) and T1007 (p = 0.319) covalent modifications that decrease KCC2 activity was not modified between genotypes (Figure 4C).
To explore the significance of our measurements with LC-MS/MS, immunopurified KCC2 was subject to SDS-PAGE and immunoblotted with antibodies against PP1 and KCC2 (Figure 4D). The ratio of PP1/KCC2 immunoreactivity was significantly reduced in LMTK3 KO mice to 28.6 ± 5.7% of that seen in WT (Figure 4E; p = 0.024); however, total PP1 levels were unaffected (Figure 4F; 104.3 ± 5.7%; p = 0.893). Immunoblotting with phospho-specific antibodies against pS940 and pT100714,43 revealed that S940 phosphorylation was significantly increased to 217.5 ± 21.7% of WT in LMTK3KO mice, whereas that of T1007 was comparable between genotypes (Figure 4G; p = 0.010). Thus, our LC-MS/MS and immunoblotting experiments suggest that LMTK3 acts to recruit PP1 to KCC2 to selectively modulate dephosphorylation of S940, S932, and T934.
Acute inhibition of LMTK3 enhances KCC2 activity and S940 phosphorylation
To further examine the role that LMK3 plays in regulating KCC2 activity, we employed a recently developed competitive inhibitor of this kinase, C28, that prevents ATP binding. In addition to this, prolonged exposure of cells to C28 for >24 h also induces LMTK3 ubiquitination and degradation.26 Thus, the effects that 1-h incubation with C28 (10 μM) or vehicle has on EGABA in DGGCs was then analyzed using patch-clamp recording. C28 exposure hyperpolarized EGABA (Figure 5A; V = −53.6 ± 1.12 and C28 = −72.2 ± 2.72 mV, respectively; p=0.00009) and [Cl−]i (Figure 5A; V = 20.2 ± 0.88 mM and C28 = 10.2 ± 1.16 mM; p = 0.00004) compared with controls performed with vehicle. To examine if C28’s effect on EGABA was mediated by LMTK3, similar measurements were performed on slices from LMTK3KO mice. Compared with WT mice, C28 did not significantly modify EGABA (Figure 5B; V = −58.8 ± 5.11 and C28–59.6 ± 4.12 mV; p = 0.920) or [Cl−] (Figure 5B; V = 17.4 ± 2.92 mM and C28 17.5 ± 2.73 mV; p = 0.978) in DGGCs from LMTK3 KO mice.
Figure 5.

Inhibition of LMTK3 enhances KCC2 activity and S940 phosphorylation
(A) Representative traces are shown for the polarity of currents induced by rapid application of muscimol in DGGCs in slices from WT mice loaded with 32-mM Cl− at differing voltages, which had been pretreated with V or 10-μM C28 for 1 h prior to recording. The recordings were used to determine EGABA and [Cl−], which were then compared between treatments. ∗p < 0.01; t test; n = 5–6 mice.
(B) Representative traces are shown for the polarity of currents induced by rapid application of muscimol in DGGCs in slices from LMTK3-KO mice loaded with 32-mM Cl− at differing voltages, which had been pretreated with V or 10-μM C28 for 1 h prior to recording. The recordings were used to determine EGABA and [Cl−], which were then compared between treatments. n = 5–6 mice.
(C) Acute hippocampal slices from WT mice were exposed to V or C28. SDS-soluble lysates were then immunoblotted with KCC2, pS940, pT1007, or actin antibodies. The ratio of pS940/KCC2 and pT1007/KCC2 immunoreactivity were then determined and normalized to values seen in V. ∗p < 0.05; t test; n = 4 mice. In all panels data represent mean ± SEM.
As an additional means of confirming the effects of C28 on neuronal Cl− homeostasis, we measured its effects on EGABA in 18–22 days in vitro (Div) neurons using gramicidin-perforated patch-clamp recording, a technique that does not disrupt endogenous neuronal Cl− gradients as detailed previously14,32,33 (Figure S3A). C28 hyperpolarized EGABA (Figure S3B; V = −74.8 ± 9.71 and C28–108.6 ± 2.35 mV; p = 0.012) and reduced [Cl−] (Figure S3C; V = 10.7 ± 2.99 mM and C28 2.58 ± 0.275 mM; p = 0.010).
In addition to KCC2 function, we evaluated if C28 impacts KCC2 phosphorylation using immunoblotting. C28 significantly increased S940 to 185.7 ± 32.2% of control (Figure 5C; p = 0.026) but not pT1007 phosphorylation (Figure 5C; 135.3 ± 54.7%; p = 0.2532). Collectively our studies suggest that C28 acts to increase KCC2 activity and S940 phosphorylation effects that are dependent upon LMTK3 expression.
The effects of C28 on KCC2 activity are dependent upon S940
To further evaluate the mechanism underlying the effects of LMTK3 on KCC2 activity, we use mice in which S940 has been mutated to an alanine (KCC2S940A). This mutation does not modify KCC2 expression levels in the adult hippocampus but increases the severity and lethality of kainate-induced seizures, and spontaneous seizures have also been reported in some cohorts of juvenile mutant mice.19,20,44 In contrast to WT mice (Figure 5A), C28 did not modify EGABA (Figure 6A; V = 52.7 ± 1.322 and C28–54.71 ± 2.716 mV, respectively; p = 0.562) or [Cl−] (Figure 6A; V = 20.87 ± 1.029 and C28 = 19.70 ± 2.094 mM, respectively; p = 0.658) in DGGCs from KCC2S940A mutant mice.
Figure 6.

Examining the role that S940 plays in mediating the effects of C28 on KCC2 activity
(A) Representative traces are shown for the polarity of currents induced by rapid application of muscimol in DGGCs in slices from S940A mice loaded with 32-mM Cl− at differing voltages, which had been pretreated with V or 10-μM C28 for 1 h prior to recording. The recordings were used to determine EGABA and [Cl−], which were then compared between treatments. n = 4–5 mice.
(B) Representative traces are shown for the polarity of currents induced by rapid application of muscimol in DGGCs in slices from S940A mice loaded with 32-mM Cl− at differing voltages, which had been pretreated with 10-mM WNK-463 (WNK) for 1 h prior to recording. The recordings were used to determine EGABA and [Cl−], which were then compared between treatments. ∗p < 0.01; t test; n = 4–5 mice. In all panels data represent mean ± SEM. Voltages are adjusted with a liquid junction potential value of −13 mV.
To control for the specificity of C28, we assessed the ability of the Pan WNK inhibitor WNK-463 to modulate KCC2 activity in KCC2S940A mice, as published studies have shown this agent increases KCC2 activity via a mechanism dependent upon T1007 (Lee et al., 2022). WNK-463 induced a significant hyperpolarizing shift in EGABA in DGGCs from KCC2S940A mice compared with vehicle (Figure 6B; V = −53.18 ± 1.322 and +WNK = −78.38 ± 1.882 mV, respectively; p = 0.00002) and the significantly reduced [Cl−] (Figure 6B; V = 20.87 ± 1.029 and WNK = 958 ± 0.534 mM; p = 0.0001). Collectively these results suggest that S940-specific contributes to the ability of C28 to modulate KCC2 activity.
Inhibiting LMTK3 decreases neuronal excitability
In mature neurons, deficits in KCC2 activity result in neuronal hyperexcitability that leads to the development of seizures. Our results suggest that LMTK3 exhibits constitutive activity in the brain and reduces KCC2 activity by promoting its phosphorylation. To examine the role that LMTK3 plays in regulating neuronal excitability in DGGCs, we used current clamp recording to compare action potentials generated in response to a series of 500-ms current injections from −80 to 200 pA in 20-pA stepwise increments45 (Figure 7A). These data were then used to generate input-output curves for slices treated with 10-μM C28 or vehicle, for 1 h prior to recording. With C28 treatment, neurons fired significantly fewer APs at a given level of current injection compared with the DMSO-treated neurons (Figure 7B; 140 pA, p = 0.028; 160 pA, p = 0.011; 180 pA, p = 0.003; 200 pA, p = 0.000003). Further analysis of membrane properties of DGGCs treated with DMSO or C28 shows no difference in resting membrane potential (Figure 7C; V = −82.9 ± 2.084 and C28–78.2 ± 2.786 mV; p = 0.242), input resistance (Figure 7C; V = 276.2 ± 22.34 and C28 290.9 ± 36.59 MΩ, respectively; p = 0.763), and rheobase (Figure 7C, V = 60.0 ± 10.95 pA and C28 62.86 ± 6.801 pA; unpaired t test; p = 0.820). The ability of C28 to reduce neuronal excitability without modifying the abovementioned intrinsic neuronal parameters is consistent with its ability to enhance KCC2 activity, leading to reduction in intracellular Cl− accumulation.
Figure 7.

Selective inhibition of LMTK3 results in reduced neuronal and network excitability
(A) Representative traces following current injection of DGGCs in WT slices exposed to V or 10-μM C28 for 1 h prior to experimentation.
(B) Input-output curves are shown that were generated for each treatment group, ∗p < 0.01; t test; n = 3–4 mice.
(C) Resting membrane potential, input resistance, and rheobase were compared between treatments. n = 3–4 mice.
(D) Representative traces are shown of field recordings from acute brain slices exposed to ASCF containing 4-AP pretreated with V or 10-μM C28 for 1 h prior to recording.
(E) The latency to first SLE and their duration, in addition to the latency to LRD, was then compared between treatments. ∗p < 0.05; t test; n = 4–5 mice. In all panels data represent mean ± SEM.
To test whether C28 reduces epileptiform activity, we examined C28’s effects on the development of seizure-like events (SLE) induced by 4-amino pyridine (4-AP).46 To do so, acute horizontal slices (400 μm) from WT mice were incubated with vehicle or C28 for 1 h followed by 4-AP (100 μM), and the development of SLEs was measured using extracellular field recording with an electrode placed in layer IV of the medial entorhinal cortex as detailed previously16,17 (Figure 7D). C28 did not modify the latency to 1st SLE (Figure 7E; V = 8.91 ± 0.275 and C28 8.82 ± 0.421 min; p = 0.848) or their duration (Figure 7E; V = 0.79 ± 0.290 and C28 0.94 ± 0.090 min; p = 0.601). However, C28 delayed the development of late recurrent discharges (LRDs) (Figure 7E, V = 13.68 ± 0.834 and C28 18.19 ± 1.633 min; p = 0.041). LRDs are believed to be analogous to drug seizures that develop in rodents and humans undergoing status epilepticus.47,48 Thus, inhibiting LMTK3 is sufficient to reduce neuronal excitability and the severity of SLE.
Discussion
LMTK3 is a poorly understood brain-specific transmembrane protein kinase, which has been implicated in regulating ERK activity, ERɑ signaling, and as a scaffold for PP1.23,26,27 Genome-wide association studies (GWAS) in humans and evidence from the study of LMTK3 knockout mice suggest that modifications in LMTK3 activity contribute to anxiety, autism spectrum disorders, and epilepsy.24,25 However, the effectors that mediate the effects of LMTK3 on neuronal excitability are unknown.
Here, we demonstrate that LMTK3 copurifies from the brain with KCC2, the principal mediator of Cl− extrusion employed by mature neurons, and its activity is a prerequisite for fast inhibitory neurotransmission mediated by GABAARs. Immunopurification coupled with quantitative mass spectroscopy revealed that LMTK3 and KCC2 exist as stable protein complexes in brain plasma membrane in an approximate stoichiometry of 2:1—stoichiometric levels of myo18, an unconventional myosin, that regulates vesicular trafficking49 and the electrogenic Na/bicarbonate transporter SLC4A4, an epilepsy risk gene50 copurified with LMTK3. Notably in addition to Cl−, GABAAR receptors are also permeable to bicarbonate,51 thus, the proximity of KCC2 and SLC4A4 may allow neurons to fine-tune the polarity of GABAAR currents. Significantly, PP1 and PP1M were present in native protein complexes enriched in LMTK3, consistent with the proposed role of LMTK3 in regulating the subcellular targeting of this enzyme to facilitate local substrate dephosphorylation. Immunostaining confirmed the close association of KCC2 and LMTK3 in cultured neurons and in hippocampal CA1 pyramidal neurons and DGGC.
To gain initial insights into the significance of the association between LMTK3 and KCC2, we employed LMTK3 KO mice. At 4–8 weeks after birth, these mice are viable but exhibit several behavioral deficits, compromised synaptic plasticity, and premature mortality after 14 weeks. Significantly, ablating LMTK3 expression does modify excitatory neurotransmission, the expression levels of AMPA-preferring ionotropic glutamate, or GABAAR.52,53 Our experiments revealed that ablating LMTK3 expression increase KCC2 activity without impacting its stability or subcellular distribution. To examine the underlying mechanism(s) we used quantitative proteomics, to evaluate the effect that ablating LMTK3 has on the proteins that copurify with KCC2. This revealed that ablating LMTK3 expression reduced the levels of PP1 and PP1M that are associated with KCC2 in the brain, effects that were confirmed using immunoblotting. Given this deficit in PP1 association, we compared the effects of ablating LMTK3 on KCC2 basal phosphorylation. In WT mice and LMTK3 mice, KCC2 was stoichiometrically phosphorylated on S25, S26, T902, T906, S932, T934, S940, T1007, and S1022, consistent with published studies.11 However, the phosphorylation of S932, T934, and S940 was specifically increased in LMTK3 mice. Significantly, we have previously shown that S940 is a PP1 substrate, and its dephosphorylation leads to KCC2 inhibition.13,16,19 Thus, our studies using LMTK3KO mice suggest that LMTK3 acts to target PP1 to KCC2 to regulate the dephosphorylation of S940 and the adjacent residues S932 in addition to T934. Significantly, the elevations in S940 phosphorylation seen in LMTK3KO mice are consistent with the enhanced levels of KCC2 activity seen in these mutant mice. However, in addition to this PP1 anchoring, LMTK3 may exert other effects on KCC2 activity or membrane trafficking via phosphorylation of other intimately associated proteins.
As an independent means of exploring the role LMTK3 plays in regulating KCC2 activity, we used C28, a recently developed rationally designed inhibitor of this kinase. C28 increases KCC2 activity together with S940 phosphorylation in WT but not LMTK3 KO. To directly evaluate the significance of S940 in mediating the effects of LMTK3, we assessed the effects of C28 on KCC2 activity in KCC2S940A mice. In contrast to WT mice, C28 was without effects on KCC2 activity in KCC2S940A mice. However, mutation of S940 did not impact the ability of WNK-463 to potentiate KCC2 activity, an effect that is dependent upon T1007 within the C-terminus of KCC2. Thus, our studies using LMTK3KO mice and C28 suggest that LMTK3 acts to reduce KCC2 activity by promoting dephosphorylation of S940. Recent studies have shown that KCC2 is enriched at inhibitory synapses and copurifies with synaptic GABAARs and the inhibitory scaffold gephyrin.28,29,54 Given these results, it will be of interest to test if LMTK3 also acts to modulate GABAAR phosphorylation and thereby the properties of inhibitory synaptic currents.29,55,56
Finally, deficits in KCC2 activity directly contribute to the gross increases in neuronal hyperexcitability that underpin the development and severity of seizures. Given the ability of LMTK3 to reduce KCC2 activity, we explore the effects that its inhibition has on neuronal excitability using the selective inhibitor C28. Inhibiting LMTK3 activity reduced the intrinsic excitability of DGGCs and the onset of LRDs induced by 4-AP in acute brain slices.
In summary, our results suggest that the brain-specific protein kinase LMTK3 may exert profound effects on neuronal excitability by regulating KCC2 activity and thereby the efficacy of synaptic inhibition.
Limitations of study
This study demonstrates that LMTK3, a brain-specific kinase, enhances neuronal excitability by reducing the activity of KCC2, the activity of which is a prerequisite for fast synaptic inhibition. Several limitations of our study require consideration. Our studies using LMTK3-KO verified the specificity of C28. However, its effects on KCC2 activity were assessed at a single dose. Thus, further studies are warranted to directly compare the efficacy of C28 to inhibit the activity of LMTK3 with its potency to increase KCC2 activity. Likewise, the potency of LMTK3 inhibition as a mechanism to reduce neuronal excitability also requires further in-depth pharmacological analysis. To ensure these findings are not model specific, testing the efficacy of LMTK3 inhibition to arrest seizure activity induced by other agents is essential. Although our experiments using KCC2-S940A mice provide strong evidence for the mechanism underlying the effects of LMTK3 on KCC2 activity, this kinase is likely to play pleiotropic roles in neuronal physiology. For example, LMTK3 has been implicated in modulating glutamate receptor trafficking and, in peripheral tissues, estrogen receptor signaling. Further studies using quantitative phosphoproteomics are required to identify specific LMKT3 substrates in the brain to provide further insights into this kinase’s role as a determinant of neuronal excitability.
Finally, in our study, we utilized the widely accepted method of whole-cell patch-clamp recording to measure Cl− extrusion, and the role that KCC2 plays in the process was validated using 11K, a highly selective inhibitor of this transporter. Alternatives to this functionally assay are optical chloride imaging using genetically encoded Cl− sensor proteins or perforated patch-clamp recording.18,54 In our experiments using cultured neurons, we confirmed the veracity of our results obtained using intracellular dialysis using perforated patch-clamp recording; however, this technique is technically challenging to perform in brain slices due to the very low rates of perforation.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| Mouse KCC2 | UC Davis/NIH NeuroMab Facility | Cat# N1/12; RRID: AB_2877330 | |
| Rabbit KCC2 | Millipore | Cat# 07-432; RRID: AB_310611 | |
| Mouse Actin | Sigma-Aldrich | Cat# A1978; RRID:AB_476692 | |
| Mouse GAPDH | Santa Cruz Biotechnology | Cat# sc-25778; RRID: AB_10167668 | |
| Rabbit KCC2 pS940 | Phosphosolutions | Cat# p1551-940; RRID: AB_2492213 | |
| Rabbit KCC2 pT1007 | Phosphosolutions | Cat# p1551-1007; RRID: AB_2716769 | |
| Rabbit LMTK3 | Atlas Antibodies | Cat# HPA077070; RRID: AB_2732330 | |
| Chicken MAP2 | Phosphosolutions | Cat# 1100-MAP2; RRID: AB_2492141 | |
| Mouse PP1 | Santa Cruz Biotechnology | Cat# sc-7482; RRID: AB_628177 | |
| Goat anti-guinea pig DyLight™ 405 | Jackson ImmunoResearch Labs | Cat# 106-475-003; RRID: AB_2337432 | |
| Goat anti-chicken Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11039; RRID: AB_2534096 | |
| Goat anti-rabbit Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-21428; RRID: AB_2535849 | |
| Goat anti-mouse Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21235; RRID: AB_2535804 | |
| Peroxidase AffiniPure donkey anti-rabbit IgG | Jackson ImmunoResearch Labs | Cat# 711-035-152; RRID: AB_10015282 | |
| Chemicals, peptides, and recombinant proteins | |||
| Kynurenic Acid | Tocris | 0223 | |
| Tetrodotoxin | Tocris | 2805/50 | |
| Bumetanide | Tocris | 1078/1 | |
| Muscimol | Tocris | 3108/50 | |
| DNQX | Tocris | 0189/10 | |
| VU0463271 | Tocris | 0219/1G | |
| 4-aminopyridine | Tocris | 0940/100 | |
| Software and algorithms | |||
| ImageJ | NIH | https://imagej.nih.gov/ij/ | |
| GraphPad Prism 8 | GraphPad Software | https://imagej.nih.gov/ij/ | |
| Axon pClamp 10.6.2 Molecular Devices https://www.moleculardevices.com/ | Molecular Devices | https://www.moleculardevices.com/ | |
| Mini Analysis6.0.3 | Synaptosoft | Synaptosoft Inc., Decatur, GA | |
| Proteome X change | ProteomeXchange | PXD050212 and 10.6019/PXD050212 | |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Professor Stephen Moss (Stephen.moss@tufts.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
The mass spectrometry datasets have been deposited at the ProteomeXchange Consortium partner repository (https://www.proteomexchange.org/). The DOIs are listed in the key resources table. Original immunoblotting and microscopy data reported in the paper will be shared by the lead contact upon request.
-
•
No original code was developed by this study.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental models and subject details
Animals
Animal housing protocols and surgical procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee of Tufts Medical Center (IACUC). C57/BL6 Mice (4-12weeks) were maintained at 22°C2 on a standard 12-h light/dark cycle with free access to food and water.
Cell lines
HEK-293 cells CRL-1573 were purchased from ATCC (https://www.atcc.org/products/crl-11268).
Method details
Mouse strains and genotyping
Lmtk3tm1Tya (MGI:5618022) and KCC2-S940A homozygous mice were generated from het x het breeding pairs were used for our experiments.19,53 Experiments were performed on brain slices prepared from 6 to 8 weeks old mutant mice and will type littermates of both sexes. Genotyping was performed according to recommended Sanger procedures on samples from ear biopsies from adult mice. PCRs were performed using the genotyping primers LMTK3 P1 (5′-CTCTCGTACCCCAGGAGTTTGAGAACCCGG-3′), LMTK3 P2 (5′-CTAGAGCGGCCGATTTAAATACGTGCTAGC-3′), LMTK3 P3 (5′-GGCTCCCAGGTTAAGATCTAGGCTGATGGC-3′), S940A P1 (5′-ATGAGTAGCAGATCCCATAGGCGAACC-3′), S940A P2 (5′-CTGCCAAGAGCCATTACTACAGTGGATG -3′).
Antibodies
The primary antibodies used in this study were the LMTK3 (rabbit, Sigma Aldrich HPA077070, WB 1:1000, ICC/IHC 1:500), KCC2 (mouse, Neuromab 75-013, WB 1:1000, ICC 1:500), KCC2 (rabbit, Millipore 07–432, WB 1:2000, ICC 1:500–1000), pS940 (rabbit, Phosphosolutions p1551-940, WB 1:1000), pT1007 (rabbit, Phosphosolutions p1551-1007, WB 1:1000), MAP2 (chicken, Phosphosolutions 1100-MAP2, ICC/IHC 1:500-1:1000, GAPDH (mouse, Santa Cruz sc-25778, WB 1:5000), and β-actin (mouse, Sigma A1978, WB 1:5000). The secondary antibodies used were the goat anti-guinea pig DyLight 405 (Jackson ImmunoResearch 106-475-003, IHC 1:500), goat anti-chicken Alexa Fluor 488 (Thermo Fisher, ICC/IHC 1:1000), goat anti-rabbit Alexa Fluor 555 (Thermo Fisher, ICC/IHC 1:1000), goat anti-mouse Alexa Fluor 647 (Thermo Fisher, ICC/IHC 1:1000), peroxidase AffiniPure donkey anti-rabbit IgG (Jackson ImmunoResearch 711-035-152, WB 1:3000), and peroxidase AffiniPure donkey anti-mouse IgG (Jackson ImmunoResearch 715-035-150, WB 1:3000).
Protein Purification and immunoprecipitation
Protein Purification was performed as described in57 Briefly, Protein A dynabeads (Thermo Fisher Scientific 10002D), Protein G dynabeads (Thermo Fisher Scientific 10004D), FLAG dynabeads (Pierce), or myc Dynabeads (Pierce) were used for the LMTK3 and KCC2 purifications, respectively. The dynabeads were washed 3X with 0.05% (v/v) Tween 20 in PBS (PBS/Tween 0.05%) and incubated on a rotating wheel, overnight at 4°C in PBS/Tween 0.05% with either LMTK3 (rabbit, Sigma Aldrich HPA077070) or KCC2 (mouse, Neuromab 75-013) antibodies. The crosslink reaction of the antibodies to the dynabeads was performed by washing twice with 0.2 M triethanolamine (TEA, pH 8.2) (Sigma 90279) and then incubating in TEA containing 40 mM dimethyl pimelimidate (DMP) (Sigma D8388) on a rotating wheel for 30 min at room temperature. The dynabeads were then incubated with 50 mM Tris (pH 7.5) on a rotating wheel for 15 min at room temperature. The dynabeads were washed 3X in PBS/Tween 0.05% before incubated with either total lysates or purified plasma membrane fractions in Triton lysis buffer (150 mM NaCl, 10 mM Tris, 0.5% (v/v) Triton X-100, pH 7.5) supplemented with mini cOmplete protease inhibitors (Roche/Millipore-Sigma 04693159001) and PhosSTOP phosphatase inhibitors (Roche/Millipore-Sigma 4906837001) in a rotating wheel overnight at 4°C. The Dynabeads were washed 3X with PBS/Tween 0.05% and eluted with either 2x sample Laemmli buffer (Sigma S3401) or soft elution buffer (0.2% (w/v) SDS, 0.1% (v/v) Tween 20, 50 mM Tris-HCl, pH 8.0) for SDS-PAGE and BN-PAGE respectively.
Blue native polyacrylamide gel electrophoresis (BN-PAGE)
BN-PAGE was carried out as previously described.11 Proteins were isolated in Triton Lysis Buffer (150 mM NaCl, 10 mM Tris, 0.5% (v/v) Triton X-100, pH 7.5) supplemented with mini cOmplete protease inhibitors (Roche/Millipore-Sigma 04693159001) and PhosSTOP phosphatase inhibitors (Roche/Millipore-Sigma 4906837001). The immunopurified were in soft elution buffer (0.2% (w/v) SDS, 0.1% (v/v) Tween 20, 50 mM Tris-HCl, pH 8.0). The samples were diluted in 4x NativePAGE sample buffer (Thermofisher Scientific BN2008) and loaded on a 4–16% NativePAGE Bis-Tris gels (Thermofisher Scientific, BN1002). The gels were run at 150 V for approximately 2 h using the 1X NativePAGE running buffer (Thermofisher Scientific BN2001) on the back compartment and the 1X “dark blue” running buffer (containing the NativePAGE Cathode Additive) on the front compartment of the Mini Gel Tank (Thermofisher Scientific A25977). For western blots, the “dark blue” cathode buffer was replaced with 1X “light-blue” cathode buffer after 10–15 min. Gels were either fixed and prepared for Coomassie staining or transferred to PVDF membranes (Sigma/Millipore IPVH00010) for immunoblotting. BN-PAGE gels were visualized using Colloidal Coomassie Blue.
Immunoblotting
After SDS-PAGE and BN-PAGE, proteins on the gels were transferred onto PVDF or nitrocellulose membranes using the Mini Blot Modules (Thermofisher Scientific B1000) for the Mini Gel Tank or BioRad Wet Transfer system using 1X NuPAGE transfer buffer (Thermofisher Scientific NP0006). The quality of the blot was confirmed by applying the Ponceau stain. The Ponceau stain was washed off and the membranes were blocked in 5% (w/v) milk or 5% (w/v) BSA in tris-buffered saline with 0.1% (v/v) Tween 20 (TBST) on a shaking plate for 1 h at room temperature. The membranes were probed with primary antibodies (see antibodies section) in TBST on a shaking plate overnight at 4°C. The membranes were washed multiple times with TBST and incubated with HRP-conjugated secondary antibodies (see antibodies section) on a shaking plate for 1 h at room temperature. Blots were developed and protein bands were visualized with Pierce ECL (ThermoFisher Scientific) and imaged using a ChemiDoc MP (Bio-Rad).
Primary neuronal cultures
Primary hippocampal neuronal cultures were prepared and cultured as previously described.28,29 Briefly, p1 mice of both sexes were anesthetized on ice and the brains were removed. The brains were dissected in Hank’s buffered salt solution (HBSS, Invitrogen) with 10 mM HEPES. The hippocampi were trypsinized and triturated to dissociate the neurons. Cells were counted using a hemocytometer and plated on poly-l-lysine-coated coverslips (for ICC and electrophysiology) or in 35 mm dishes (for biochemistry) at a density of 4 × 105 cells. At days in vitro 18 (DIV18), cells were exposed to either 0.1% (v/v) DMSO or 10 μM C28 (Giamas Lab, University of Sussex). The cells were harvested after 1 h and 48 h of drug treatment. Cells were either fixed using 4% PFA in PBS or lysed in RIPA buffer (50 mM Tris, 150 mM NaCl, 0.1% (w/v) SDS, 0.5% (w/v) sodium deoxycholate, and 1% (v/v) Triton X-100, pH 7.4) supplemented with mini cOmplete protease inhibitors (Roche/Millipore-Sigma 04693159001) and PhosSTOP phosphatase inhibitors (Roche/Millipore-Sigma 4906837001) for biochemistry.
Immunocytochemistry (IHC)
Adult animals were anesthetized with isoflurane and sacrificed by decapitation. For immunohistochemistry (IHC) the brains were dropped-fixed in 4% PFA in PBS solution overnight at 4°C. The brains were then cryo-protected in 30% sucrose/PBS solution overnight at 4°C before freezing at −80°C. The frozen brains were embedded in 30% sucrose/PBS solution, and 30 μm coronal brain slices were serially cryosectioned using a Microtome (Leica SM2000R). Free floating slices were either kept at in PBS at 4°C for short term storage or in cryoprotectant solution (30% (v/v) glycerol, 30% (v/v) ethylene glycol, 40% (v/v) 1× PBS) at −20°C for long-term storage. For biochemistry, the telencephalon was dissected out and the forebrain was lysed in either RIPA (for western blots) or Triton buffers (for co-IP experiments). IHC was performed as previously described.58 Free floating sections were washed with PBS and permeabilized for 4–5 h in block solution consisting of 1X PBS with 0.5% Triton X-100, 3% (w/v) Bovine Serum Albumin (BSA), 10% normal goat serum, and 0.2 M glycine. The sections were then incubated with primary antibody diluted in block solution overnight at 4°C. Slices were washed four to five times in PBS over 2 h and then incubated for 3–4 h with secondary antibody in block solution (1:500-1:1000) at room temperature. The slices were then washed four to five times in PBS for 2 h and mounted onto glass slides using ProLong Gold with DAPI (Thermo Fisher P36931) or Fluoromount G mounting media (Thermo Fisher 00-4958-02).
For analyzing colocalization, the Synapse Counter plugin for ImageJ was used. High-magnification images (1024 ′ 1024, zoom 4′) of the hippocampus were auto-thresholded using the Otsu Thresholding method. The rolling ball radius (background subtraction) and maximum filter parameters were set to 10 and 0, respectively. The acceptable particle size range was set to 10–400 square pixels. Default colocalization settings accepted 33–100% overlap between the fluorophores to be analyzed. Cluster analysis was done with Image Calculator and Analyze Particle to determine overlapping areas and total areas of colocalization.36
Whole-cell patch-clamp and current clamp recordings
Recordings were performed in dentate granule cells in the whole-cell configuration in coronal slices (350 μm) obtained from 7 to 9 week old mice. Recording electrodes (5–6 MΩ resistance) contained the following (in mM): 115 K-Gluconate, 30 KCl, 2 HEPES, 2 Mg-ATP, 2 Na-ATP, 0.4 Na-GTP, pH 7.4. Slices were continuously perfused with oxygenated aCSF containing kynurenic acid (3 mM), bumetanide (10 μM), and VU0463271 (10 μM) for selective inhibition of KCC2. A picospritzer pipette containing muscimol (5 μM), a GABAAR agonist, was placed in the molecular layer of the dentate gyrus to activate GABAAR mediated currents in cells held at voltages between −90 and −10 mV. Peak amplitude responses of the muscimol-activated current were plotted for each holding voltage and data fitted by linear regression analysis. The reversal potential of GABA responses (EGABA) was obtained from the x-intercept value of the fit. Voltages were corrected offline with a liquid junction potential value of 13.2 mV. Current-clamp recordings were performed in dentate granule cells (DGGCs) in the whole-cell configuration in coronal slices (350 μm) obtained from 8 to 9 week old mice. Recording electrodes (5–6 MΩ resistance) contained the following (in mM): 130 K-Gluconate, 10 KCl, 4 NaCl, 10 HEPES, 0.1 EGTA, 2 Mg-ATP, 0.3 Na-ATP, pH 7.25. The number of actional potentials in response to a series of 500-ms current injections from −80 to 200 pA in 20-pA step increment were recorded and plotted to input-output curves.16,17
Perforated patch clamp recordings
Recordings were performed on DIV 18 rat corticohippocampal neuronal cultures at 34°C, in bath saline containing the following (in mM): 140 NaCl, 2.5 KCl, 2.5 CaCl2, 2.5 MgCl2, 10 HEPES, and 11 glucose, pH 7.4. Tetrodotoxin (300 nM) was applied to block activity-dependent shifts in EGABA, bumetanide (10 μM) to inhibit NKCC1, DNQX (20 μM) to block glutamatergic excitatory synaptic transmission, and muscimol (1 μM) to activate GABAARs. All compound solutions were applied through a three-barrel 700 μM applicator (Warner Instruments) positioned just above the cell. Recording electrodes (5–6 MΩ resistance) contained the following (in mM): 140 KCl and 10 HEPES, pH 7.4 and gramicidin (50 μg/mL). Experiments began once membrane perforation reached a series resistance <50 MΩ. EGABA values were obtained by calculating the reversal potential of leak-subtracted muscimol-activated currents recorded during 10 mV or 20 mV positive going voltage ramps over 1 s periods. Intracellular Cl− concentrations were back-calculated using EGABA measurements and the Nernst equation: ECl– = RT/zF ′ ln[Cl−]o/[Cl−]I.18,19,45
Extracellular field recordings
Horizontal slices (400 μm) were placed in an RC-27L dual perfusion recording chamber (Warner Instruments) perfused with oxygenated aCSF (2–3 mL/min) for 5 min before perfusion with oxygenated control artificial CSF (aCSF) composed of (in mM): 126 NaCl, 2.5 KCl, 0.5 CaCl2, 26 NaHCO3, 1.25 NaH2PO4, 2 MgCl2, 1.5 Na-pyruvate, and 10 glucose (equilibrated with 95% O2 and 5% CO2, or aCSF with reduce divalent cations (1.6 mM CaCl2 and 0.8 mM MgCl2) and 100 μM 4-aminopyridine (4-AP). aCSF filled electrodes (<1 MΩ resistance) were placed in layer III of the medial entorhinal cortex. Data were acquired at 10 kHz on a Multiclamp 700B amplifier (Molecular Devices) with Clampex 10 acquisition software (Molecular Devices). Recordings were analyzed on Clampfit (Molecular Devices) and traces were high-pass filtered at 1 Hz for event analysis. Seizure like events (SLEs) were defined as events exhibiting tonic-clonic discharges that had a duration of 10 s or longer. Late recurrent discharges (LRDs) were defined as periods of seizure activity with interictal durations of less than 20 s.16,17,48
LC-MS/MS data analysis
Quantitative label-free proteomic analysis was conducted as previously detailed.11,28 Briefly, excised gel bands were processed in a series of dehydration and rehydration steps with acetonitrile, 50 mM ammonium bicarbonate solution containing sequencing-grade trypsin (Promega) and wash repeated with ammonium bicarbonate solution to remove excess trypsin. Peptide extraction was carried out with a 50% acetonitrile and 1% formic acid wash, dehydrated in a speed-vac for 1 h, and reconstituted in HPLC solvent A containing 2.5% acetonitrile and 0.1% formic acid. Extracted peptides were run through a nanoscale reverse-phase HPLC capillary column containing C18 silica beads and a gradient formed between solvent A (97.5% water, 2.5% acetonitrile, and 0.1% formic acid) and solvent B (2.5% water, 97.5% acetonitrile, and 0.1% formic acid). Eluted peptides were subjected to Nanospray ionization (NSI) and subsequently entered to an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (Thermo Scientific). MS1 parameters used were 70K resolution, scan range of m/z 85–2000, charge-state screening set to +2 and +5, and centroid acquisition mode with precursor ion isolation window of 2 m/z and 35% normalized collision energy. The Top 10 scan mode was used to detect and isolate the most intense eluting peptides and fragmentation by Higher-energy C-trap dissociation (HCD). Analysis of MS2 ions were carried out by an Orbitrap mass spectrometer at 17.5K resolution and dynamic exclusion settings to produce tandem mass spectrum of specific fragment ions for each peptide. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository (https://www.proteomexchange.org/) with the dataset identifier PXD050212 and 10.6019/PXD050212".
Quantification and statistical analysis
GraphPad Prism software (Version 9.2.0) was used for statistical analysis. Averaged data are expressed as mean ± standard error of the mean (SEM). Student’s t test was used to assess statistical significance between two groups, where p values <0.05 was considered significant using GraphPad. One-Way analysis of variance (ANOVA) and Tukey’s post hoc test was used to compare three or more groups. SIGI values for each significantly enriched protein detected by LMTK3 immunopurification were compared to non-immune IgG purifications by Welch’s t-test. EGABA values were determined by linear regression fit analysis using GraphPad Prism software.
Acknowledgments
S.J.M. and P.D. are supported by the National Institutes of Health (NIH)—National Institute of Neurological Disorders and Stroke Grants, NS108378, NS101888, NS103865, and NS111338 and NIH—National Institute of Mental Health Grant MH118263. G.K. was funded by the AstraZeneca Postdoctoral Program.
Author contributions
N.C., G.K., J.L.S., and S.J.M. contributed to conceptualization of the project and wrote the paper. N.C., G.K., and J.L.S. performed experiments. C.B. and J.D. provided and managed the animal colonies. K.M. and T.Y. developed and provided the LMTK3KO mice. G.G. supplied the C28 compound. N.J.B., T.Z.D., and P.A.D. provided resources and developed methodology. J.L.S., G.G., and S.J.M. edited the paper.
Declaration of interests
The authors declare no competing interests.
Published: March 15, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109512.
Supplemental information
References
- 1.Rivera C., Voipio J., Payne J.A., Ruusuvuori E., Lahtinen H., Lamsa K., Pirvola U., Saarma M., Kaila K. The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397:251–255. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- 2.Williams J.R., Sharp J.W., Kumari V.G., Wilson M., Payne J.A. The neuron-specific K-Cl cotransporter, KCC2. Antibody development and initial characterization of the protein. J. Biol. Chem. 1999;274:12656–12664. doi: 10.1074/jbc.274.18.12656. [DOI] [PubMed] [Google Scholar]
- 3.Kaila K., Price T.J., Payne J.A., Puskarjov M., Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat. Rev. Neurosci. 2014;15:637–654. doi: 10.1038/nrn3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Virtanen M.A., Uvarov P., Mavrovic M., Poncer J.C., Kaila K. The Multifaceted Roles of KCC2 in Cortical Development. Trends Neurosci. 2021;44:378–392. doi: 10.1016/j.tins.2021.01.004. [DOI] [PubMed] [Google Scholar]
- 5.Duy P.Q., David W.B., Kahle K.T. Identification of KCC2 Mutations in Human Epilepsy Suggests Strategies for Therapeutic Transporter Modulation. Front. Cell. Neurosci. 2019;13:515. doi: 10.3389/fncel.2019.00515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saitsu H., Watanabe M., Akita T., Ohba C., Sugai K., Ong W.P., Shiraishi H., Yuasa S., Matsumoto H., Beng K.T., et al. Impaired neuronal KCC2 function by biallelic SLC12A5 mutations in migrating focal seizures and severe developmental delay. Sci. Rep. 2016;6 doi: 10.1038/srep30072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mavrovic M., Uvarov P., Delpire E., Vutskits L., Kaila K., Puskarjov M. Loss of non-canonical KCC2 functions promotes developmental apoptosis of cortical projection neurons. EMBO Rep. 2020;21 doi: 10.15252/embr.201948880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huberfeld G., Wittner L., Clemenceau S., Baulac M., Kaila K., Miles R., Rivera C. Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J. Neurosci. 2007;27:9866–9873. doi: 10.1523/JNEUROSCI.2761-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kahle K.T., Khanna A.R., Duan J., Staley K.J., Delpire E., Poduri A. The KCC2 Cotransporter and Human Epilepsy: Getting Excited About Inhibition. Neuroscientist. 2016;22:555–562. doi: 10.1177/1073858416645087. [DOI] [PubMed] [Google Scholar]
- 10.Kahle K.T., Deeb T.Z., Puskarjov M., Silayeva L., Liang B., Kaila K., Moss S.J. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends Neurosci. 2013;36:726–737. doi: 10.1016/j.tins.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smalley J.L., Kontou G., Choi C., Ren Q., Albrecht D., Abiraman K., Santos M.A.R., Bope C.E., Deeb T.Z., Davies P.A., et al. Isolation and Characterization of Multi-Protein Complexes Enriched in the K-Cl Co-transporter 2 From Brain Plasma Membranes. Front. Mol. Neurosci. 2020;13 doi: 10.3389/fnmol.2020.563091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moore Y.E., Kelley M.R., Brandon N.J., Deeb T.Z., Moss S.J. Seizing Control of KCC2: A New Therapeutic Target for Epilepsy. Trends Neurosci. 2017;40:555–571. doi: 10.1016/j.tins.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 13.Lee H.H.C., Walker J.A., Williams J.R., Goodier R.J., Payne J.A., Moss S.J. Direct protein kinase C-dependent phosphorylation regulates the cell surface stability and activity of the potassium chloride cotransporter KCC2. J. Biol. Chem. 2007;282:29777–29784. doi: 10.1074/jbc.M705053200. [DOI] [PubMed] [Google Scholar]
- 14.Lee H.H.C., Deeb T.Z., Walker J.A., Davies P.A., Moss S.J. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat. Neurosci. 2011;14:736–743. doi: 10.1038/nn.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rinehart J., Vázquez N., Kahle K.T., Hodson C.A., Ring A.M., Gulcicek E.E., Louvi A., Bobadilla N.A., Gamba G., Lifton R.P. WNK2 kinase is a novel regulator of essential neuronal cation-chloride cotransporters. J. Biol. Chem. 2011;286:30171–30180. doi: 10.1074/jbc.M111.222893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore Y.E., Deeb T.Z., Chadchankar H., Brandon N.J., Moss S.J. Potentiating KCC2 activity is sufficient to limit the onset and severity of seizures. Proc. Natl. Acad. Sci. USA. 2018;115:10166–10171. doi: 10.1073/pnas.1810134115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee K.L., Abiraman K., Lucaj C., Ollerhead T.A., Brandon N.J., Deeb T.Z., Maguire J., Moss S.J. Inhibiting with-no-lysine kinases enhances K+/Cl- cotransporter 2 activity and limits status epilepticus. Brain. 2022;145:950–963. doi: 10.1093/brain/awab343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deeb T.Z., Lee H.H.C., Walker J.A., Davies P.A., Moss S.J. Hyperpolarizing GABAergic transmission depends on KCC2 function and membrane potential. Channels. 2011;5:475–481. doi: 10.4161/chan.5.6.17952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silayeva L., Deeb T.Z., Hines R.M., Kelley M.R., Munoz M.B., Lee H.H.C., Brandon N.J., Dunlop J., Maguire J., Davies P.A., Moss S.J. KCC2 activity is critical in limiting the onset and severity of status epilepticus. Proc. Natl. Acad. Sci. USA. 2015;112:3523–3528. doi: 10.1073/pnas.1415126112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sullivan B.J., Kipnis P.A., Carter B.M., Shao L.R., Kadam S.D. Targeting ischemia-induced KCC2 hypofunction rescues refractory neonatal seizures and mitigates epileptogenesis in a mouse model. Sci. Signal. 2021;14 doi: 10.1126/scisignal.abg2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mórotz G.M., Bradbury N.A., Caluseriu O., Hisanaga S.I., Miller C.C.J., Swiatecka-Urban A., Lenz H.J., Moss S.J., Giamas G. A revised nomenclature for the lemur family of protein kinases. Commun. Biol. 2024;7:57. doi: 10.1038/s42003-023-05671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giamas G., Filipović A., Jacob J., Messier W., Zhang H., Yang D., Zhang W., Shifa B.A., Photiou A., Tralau-Stewart C., et al. Kinome screening for regulators of the estrogen receptor identifies LMTK3 as a new therapeutic target in breast cancer. Nat. Med. 2011;17:715–719. doi: 10.1038/nm.2351. [DOI] [PubMed] [Google Scholar]
- 23.Wendler F., Purice T.M., Simon T., Stebbing J., Giamas G. The LMTK-family of kinases: Emerging important players in cell physiology and pathogenesis. Biochim. Biophys. Acta, Mol. Basis Dis. 2021;1867 doi: 10.1016/j.bbadis.2018.12.023. [DOI] [PubMed] [Google Scholar]
- 24.Telias M. Molecular Mechanisms of Synaptic Dysregulation in Fragile X Syndrome and Autism Spectrum Disorders. Front. Mol. Neurosci. 2019;12:51. doi: 10.3389/fnmol.2019.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Willsey A.J., Sanders S.J., Li M., Dong S., Tebbenkamp A.T., Muhle R.A., Reilly S.K., Lin L., Fertuzinhos S., Miller J.A., et al. Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell. 2013;155:997–1007. doi: 10.1016/j.cell.2013.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ditsiou A., Cilibrasi C., Simigdala N., Papakyriakou A., Milton-Harris L., Vella V., Nettleship J.E., Lo J.H., Soni S., Smbatyan G., et al. The structure-function relationship of oncogenic LMTK3. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abc3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stebbing J., Filipović A., Giamas G. Lemur tyrosine kinase-3 (LMTK3) in cancer and evolution. Oncotarget. 2011;2:428–429. doi: 10.18632/oncotarget.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smalley J.L., Cho N., Ng S.F.J., Choi C., Lemons A.H.S., Chaudry S., Bope C.E., Dengler J.S., Zhang C., Rasband M.N., et al. Spectrin-beta 2 facilitates the selective accumulation of GABAA receptors at somatodendritic synapses. Commun. Biol. 2023;6:11. doi: 10.1038/s42003-022-04381-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi C., Smalley J.L., Lemons A.H.S., Ren Q., Bope C.E., Dengler J.S., Davies P.A., Moss S.J. Analyzing the mechanisms that facilitate the subtype-specific assembly of gamma-aminobutyric acid type A receptors. Front. Mol. Neurosci. 2022;15 doi: 10.3389/fnmol.2022.1017404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakamura Y., Morrow D.H., Nathanson A.J., Henley J.M., Wilkinson K.A., Moss S.J. Phosphorylation on Ser 359 of the α2 subunit in GABA type A receptors down-regulates their density at inhibitory synapses. J. Biol. Chem. 2020;295:12330–12342. doi: 10.1074/jbc.RA120.014303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakamura Y., Morrow D.H., Modgil A., Huyghe D., Deeb T.Z., Lumb M.J., Davies P.A., Moss S.J. Proteomic Characterization of Inhibitory Synapses Using a Novel pHluorin-tagged gamma-Aminobutyric Acid Receptor, Type A (GABAA), alpha2 Subunit Knock-in Mouse. J. Biol. Chem. 2016;291:12394–12407. doi: 10.1074/jbc.M116.724443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conway L.C., Cardarelli R.A., Moore Y.E., Jones K., McWilliams L.J., Baker D.J., Burnham M.P., Bürli R.W., Wang Q., Brandon N.J., et al. Ethylmaleimide increases KCC2 cotransporter activity by modulating transporter phosphorylation. J. Biol. Chem. 2017;292:21253–21263. doi: 10.1074/jbc.M117.817841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jarvis R., Josephine Ng S.F., Nathanson A.J., Cardarelli R.A., Abiraman K., Wade F., Evans-Strong A., Fernandez-Campa M.P., Deeb T.Z., Smalley J.L., et al. Direct activation of KCC2 arrests benzodiazepine refractory status epilepticus and limits the subsequent neuronal injury in mice. Cell Rep. Med. 2023;4 doi: 10.1016/j.xcrm.2023.100957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beausoleil S.A., Villén J., Gerber S.A., Rush J., Gygi S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 35.Agnarelli A., Lauer Betrán A., Papakyriakou A., Vella V., Samuels M., Papanastasopoulos P., Giamas C., Mancini E.J., Stebbing J., Spencer J., et al. The Inhibitory Properties of a Novel, Selective LMTK3 Kinase Inhibitor. Int. J. Mol. Sci. 2023;24 doi: 10.3390/ijms24010865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kontou G., Josephine Ng S.F., Cardarelli R.A., Howden J.H., Choi C., Ren Q., Rodriguez Santos M.A., Bope C.E., Dengler J.S., Kelley M.R., et al. KCC2 is required for the survival of mature neurons but not for their development. J. Biol. Chem. 2021;296 doi: 10.1016/j.jbc.2021.100364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hines D.J., Contreras A., Garcia B., Barker J.S., Boren A.J., Moufawad El Achkar C., Moss S.J., Hines R.M. Human ARHGEF9 intellectual disability syndrome is phenocopied by a mutation that disrupts collybistin binding to the GABAA receptor alpha2 subunit. Mol. Psychiatry. 2022;27:1729–1741. doi: 10.1038/s41380-022-01468-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Delpire E., Baranczak A., Waterson A.G., Kim K., Kett N., Morrison R.D., Daniels J.S., Weaver C.D., Lindsley C.W. Further optimization of the K-Cl cotransporter KCC2 antagonist ML077: development of a highly selective and more potent in vitro probe. Bioorg. Med. Chem. Lett. 2012;22:4532–4535. doi: 10.1016/j.bmcl.2012.05.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sivakumaran S., Cardarelli R.A., Maguire J., Kelley M.R., Silayeva L., Morrow D.H., Mukherjee J., Moore Y.E., Mather R.J., Duggan M.E., et al. Selective inhibition of KCC2 leads to hyperexcitability and epileptiform discharges in hippocampal slices and in vivo. J. Neurosci. 2015;35:8291–8296. doi: 10.1523/JNEUROSCI.5205-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carlyle B.C., Kitchen R.R., Kanyo J.E., Voss E.Z., Pletikos M., Sousa A.M.M., Lam T.T., Gerstein M.B., Sestan N., Nairn A.C. A multiregional proteomic survey of the postnatal human brain. Nat. Neurosci. 2017;20:1787–1795. doi: 10.1038/s41593-017-0011-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ostergaard E., Weraarpachai W., Ravn K., Born A.P., Jønson L., Duno M., Wibrand F., Shoubridge E.A., Vissing J. Mutations in COA3 cause isolated complex IV deficiency associated with neuropathy, exercise intolerance, obesity, and short stature. J. Med. Genet. 2015;52:203–207. doi: 10.1136/jmedgenet-2014-102914. [DOI] [PubMed] [Google Scholar]
- 42.Buscà R., Pouysségur J., Lenormand P. ERK1 and ERK2 Map Kinases: Specific Roles or Functional Redundancy? Front. Cell Dev. Biol. 2016;4:53. doi: 10.3389/fcell.2016.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conway L.C., Cardarelli R.A., Moore Y.E., Jones K., McWilliams L.J., Baker D.J., Burnham M.P., Bürli R.W., Wang Q., Brandon N.J., et al. N-Ethylmaleimide increases KCC2 cotransporter activity by modulating transporter phosphorylation. J. Biol. Chem. 2017;292:21253–21263. doi: 10.1074/jbc.M117.817841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moore Y.E., Conway L.C., Wobst H.J., Brandon N.J., Deeb T.Z., Moss S.J. Developmental Regulation of KCC2 Phosphorylation Has Long-Term Impacts on Cognitive Function. Front. Mol. Neurosci. 2019;12:173. doi: 10.3389/fnmol.2019.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deeb T.Z., Nakamura Y., Frost G.D., Davies P.A., Moss S.J. Disrupted Cl(-) homeostasis contributes to reductions in the inhibitory efficacy of diazepam during hyperexcited states. Eur. J. Neurosci. 2013;38:2453–2467. doi: 10.1111/ejn.12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heuzeroth H., Wawra M., Fidzinski P., Dag R., Holtkamp M. The 4-Aminopyridine Model of Acute Seizures in vitro Elucidates Efficacy of New Antiepileptic Drugs. Front. Neurosci. 2019;13:677. doi: 10.3389/fnins.2019.00677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dreier J.P., Zhang C.L., Heinemann U. Phenytoin, phenobarbital, and midazolam fail to stop status epilepticus-like activity induced by low magnesium in rat entorhinal slices, but can prevent its development. Acta Neurol. Scand. 1998;98:154–160. doi: 10.1111/j.1600-0404.1998.tb07286.x. [DOI] [PubMed] [Google Scholar]
- 48.Kelley M.R., Deeb T.Z., Brandon N.J., Dunlop J., Davies P.A., Moss S.J. Compromising KCC2 transporter activity enhances the development of continuous seizure activity. Neuropharmacology. 2016;108:103–110. doi: 10.1016/j.neuropharm.2016.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Buschman M.D., Field S.J. MYO18A: An unusual myosin. Adv. Biol. Regul. 2018;67:84–92. doi: 10.1016/j.jbior.2017.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gil-Perotín S., Jaijo T., Verdú A.G., Rubio P., Mazón M., Gasqué-Rubio R., Díaz S. Epilepsy, status epilepticus, and hemiplegic migraine coexisting with a novel SLC4A4 mutation. Neurol. Sci. 2021;42:3647–3654. doi: 10.1007/s10072-020-04961-x. [DOI] [PubMed] [Google Scholar]
- 51.Kaila K., Voipio J. Postsynaptic fall in intracellular pH induced by GABA-activated bicarbonate conductance. Nature. 1987;330:163–165. doi: 10.1038/330163a0. [DOI] [PubMed] [Google Scholar]
- 52.Montrose K., Kobayashi S., Manabe T., Yamamoto T. Lmtk3-KO Mice Display a Range of Behavioral Abnormalities and Have an Impairment in GluA1 Trafficking. Neuroscience. 2019;414:154–167. doi: 10.1016/j.neuroscience.2019.06.033. [DOI] [PubMed] [Google Scholar]
- 53.Inoue T., Hoshina N., Nakazawa T., Kiyama Y., Kobayashi S., Abe T., Yamamoto T., Manabe T., Yamamoto T. LMTK3 deficiency causes pronounced locomotor hyperactivity and impairs endocytic trafficking. J. Neurosci. 2014;34:5927–5937. doi: 10.1523/JNEUROSCI.1621-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Al Awabdh S., Donneger F., Goutierre M., Séveno M., Vigy O., Weinzettl P., Russeau M., Moutkine I., Lévi S., Marin P., Poncer J.C. Gephyrin Interacts with the K-Cl Cotransporter KCC2 to Regulate Its Surface Expression and Function in Cortical Neurons. J. Neurosci. 2022;42:166–182. doi: 10.1523/JNEUROSCI.2926-20.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pribiag H., Stellwagen D. TNF-alpha downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABA(A) receptors. J. Neurosci. 2013;33:15879–15893. doi: 10.1523/JNEUROSCI.0530-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakamura Y., Darnieder L.M., Deeb T.Z., Moss S.J. Regulation of GABAARs by phosphorylation. Adv. Pharmacol. 2015;72:97–146. doi: 10.1016/bs.apha.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wobst H.J., Wesolowski S.S., Chadchankar J., Delsing L., Jacobsen S., Mukherjee J., Deeb T.Z., Dunlop J., Brandon N.J., Moss S.J. Cytoplasmic Relocalization of TAR DNA-Binding Protein 43 Is Not Sufficient to Reproduce Cellular Pathologies Associated with ALS. Front. Mol. Neurosci. 2017;10:46. doi: 10.3389/fnmol.2017.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kelley M.R., Cardarelli R.A., Smalley J.L., Ollerhead T.A., Andrew P.M., Brandon N.J., Deeb T.Z., Moss S.J. Locally Reducing KCC2 Activity in the Hippocampus is Sufficient to Induce Temporal Lobe Epilepsy. EBioMedicine. 2018;32:62–71. doi: 10.1016/j.ebiom.2018.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The mass spectrometry datasets have been deposited at the ProteomeXchange Consortium partner repository (https://www.proteomexchange.org/). The DOIs are listed in the key resources table. Original immunoblotting and microscopy data reported in the paper will be shared by the lead contact upon request.
-
•
No original code was developed by this study.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.