

Abstract
Background:
Alternative mRNA splicing can be dysregulated in cancer, resulting in the generation of aberrant splice variants (SVs). Given the paucity of actionable genomic mutations in clear cell renal cell carcinoma (ccRCC), aberrant SVs may be an avenue to novel mechanisms of pathogenesis.
Objective:
To identify and characterize aberrant SVs enriched in ccRCC.
Design, setting, and participants:
Using RNA-seq data from the Cancer Cell Line Encyclopedia, we identified neojunctions uniquely expressed in ccRCC. Candidate SVs were then checked for expression across normal tissue in the Genotype-Tissue Expression Project and primary tumor tissue from The Cancer Genome Atlas (TCGA), Clinical Proteomic Tumor Analysis Consortium (CPTAC), and our institutional Total Cancer Care database.
Outcome measurements and statistical analysis:
Clinicopathologic, genomic, and survival data were available for all cohorts. Epigenetic data were available for the TCGA and CPTAC cohorts. Proteomic data were available for the CPTAC cohort. The association of aberrant SV expression with these variables was examined using the Kruskal-Wallis test, pairwise t test, Spearman correlation test, and Cox regression analysis.
Results and limitations:
Our pipeline identified 16 ccRCC-enriched SVs. EGFR, HPCAL1-SV and RNASET2-SV expression was negatively correlated with gene-specific CpG methylation. We derived a survival risk score based primarily on the expression of five SVs (RNASET2, FGD1, PDZD2, COBLL1, and PTPN14), which was consistent and applicable across multiple cohorts on multivariate analysis. The splicing factor RBM4, which modulates splicing of HIF-1α, exhibited significantly lower expression at the protein level in the high-risk group, as defined by our SV-based score.
Conclusions:
We describe 16 aberrant SVs enriched in ccRCC, many of which are associated with disease biology and/or clinical outcomes. This study provides a novel strategy for identifying and characterizing disease-specific aberrant SVs.
Patient summary:
We describe a method to identify disease targets and biomarkers using transcriptomic analysis beyond somatic mutations or gene expression. Kidney tumors express unique splice variants that may provide additional prognostic information following surgery.
Keywords: Aberrant splicing, Clear cell renal cell carcinoma, Epigenetics, Proteomics
1. Introduction
Alternative mRNA splicing is recognized as a key driver of proteomic diversity in humans [1,2]. Approximately 95% of multiexon human genes are alternatively spliced in a process characterized by high efficiency and fidelity in normal cells [3]. However, disease settings, such as cancer, can result in an altered transcriptomic landscape and expression of potentially novel, otherwise unexpressed, mRNA iso-forms [4,5]. These aberrant splice variants (SVs) can have functional consequences, such as driving cancer development or progression. One prominent example is the aberrant mRNA isoform ARV7, which expresses a constitutively active androgen receptor variant [6] and has been proved to be clinically impactful by predicting resistance to androgen-targeted therapies in metastatic prostate cancer [7].
The etiology of aberrant splicing may involve altered expression or mutations in splicing factors [8]. Furthermore, DNA methylation is thought to be one of several key epigenetic regulators of alternative splicing [9]. Since methylation patterns are altered in cancer, this process may also play a role in aberrant splicing [10]. Previous work has characterized a subset of renal cell carcinoma (RCC) tumors with increased DNA hypermethylation compared with normal tissues and even other RCC tumors [11]. This hypermethylation pattern was associated with decreased survival for all histologic subtypes of RCC, including clear cell RCC (ccRCC) [11,12].
Despite the widely known mutational alterations in sporadic ccRCC, a majority have not been proved to be clinically actionable. Aberrant mRNA splicing holds promise as a source of new pathogenic drivers, targets, and biomarkers. However, research to investigate aberrant splicing among sporadic ccRCC tumors has been limited [3,13]. Our group initially identified a novel, aberrant epidermal growth factor receptor (EGFR)-SV that is observed in ccRCC at a higher frequency than in other tumor types [14]. Here, we have expanded this effort to identify 16 SVs that are strongly enriched in ccRCC by implementing a pipeline starting from cancer cell line data and filtered through multiple publicly available datasets. We performed molecular, proteomic, and epigenetic characterization of these SVs in primary tumor tissue. Ultimately, we derived a unique SV-based survival signature that is consistent and applicable across multiple distinct ccRCC cohorts.
2. Patients and methods
2.1. SV identification and screening pipeline
Our identification and screening pipeline is outlined in Fig. 1 and described in the Supplementary material (Methods and results section).
Fig. 1 –
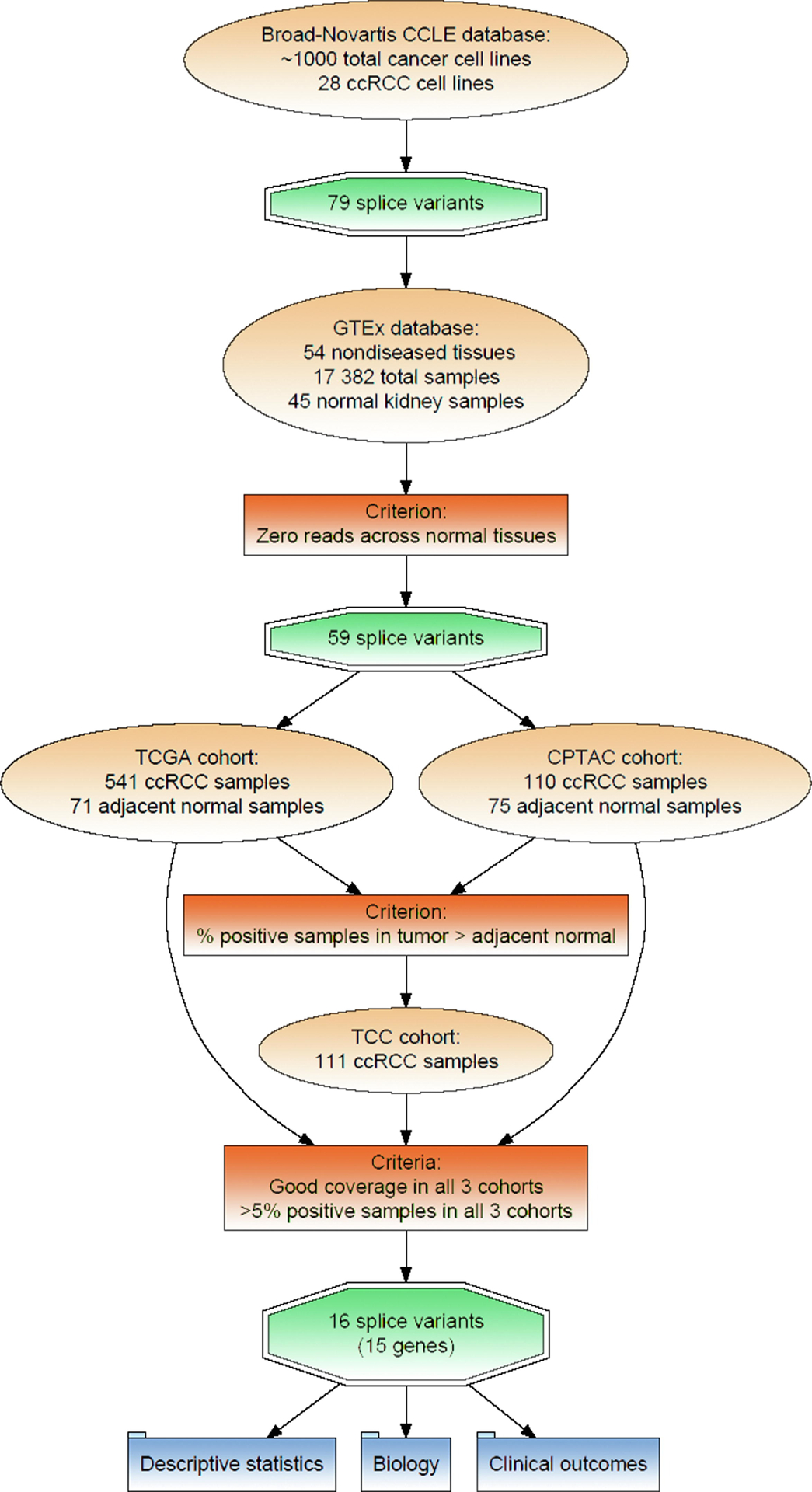
SV screening pipeline. CCLE = Cancer Cell Line Encyclopedia; ccRCC = clear cell renal cell carcinoma; CPTAC = Clinical Proteomic Tumor Analysis Consortium; SV = splice variant; TCC = Total Cancer Care; TCGA = The Cancer Genome Atlas.
2.2. Clinicopathologic and genomic mutation analysis
Statistical analysis was performed using R (version 4.1.1; R Foundation for Statistical Computing, Vienna, Austria). The Wilcoxon rank-sum test was used for statistical comparisons using the continuous splice fraction metric for SV expression. We used the chi-square test to assess for a statistically significant relationship with categorical SV detection. The p values were adjusted using the Bonferroni correction [15]. We used a logistic regression analysis to calculate odds ratios (ORs).
2.3. Survival outcomes and risk score development
We performed a univariate Cox regression analysis to determine the association of SV detection and SV expression with postsurgical survival outcomes in The Cancer Genome Atlas (TCGA), Total Cancer Care (TCC), and Clinical Proteomic Tumor Analysis Consortium (CPTAC) cohorts. The p values were adjusted using the Bonferroni correction. We then derived a survival risk score using the LASSO Cox regression analysis for variable selection, implementing the “glmnet” R package; all 16 SVs were initially included in this step. TCGA was selected as the training cohort given its largest sample size. A point-based risk score was calculated using a regression coefficient–based approach [16]. This score was validated on the TCC and CPTAC cohorts. Two separate risk scores were generated using either SV detection or SV expression; for the latter, values were normalized using TCGA as the reference distribution. Kaplan-Meier plots were generated using the “survminer” R package. A prognostic index (PI) was calculated using the TCGA dataset and assessed using a “calibration slope” on the TCC and CPTAC datasets according to the methods described previously for model validation [17]. We also calculated a Harrell’s C-index [18] using the “compareC” R package. Multivariate Cox regression analyses were performed to assess our SV-based risk scores while adjusting for pathologic stage, histologic grade, age, and gender.
2.4. DNA methylation and proteomic analysis
Please see the Supplementary material (Methods and results section).
3. Results
3.1. Screening, selection, and distribution pattern of ccRCC-enriched SVs
After implementation of our screening pipeline (Fig. 1), we identified 16 SVs spanning 15 genes (Table 1). UCSC genome tracks depicting the SVs are available in the Supplementary material. We previously described the identification and clinical significance of the EGFR-SV (EGFR_pr20CTF) [14]. We provide evidence of its expression as a protein in the Supplementary material (Methods and results section). To our knowledge, none of the remaining SVs have been described in the RCC literature. (See Fig. 2)
Table 1 –
Sixteen splice variants (SVs) spanning 15 genes selected by our pipeline to be enriched and prevalent in clear cell renal cell carcinoma
| SV | SV locusa | Gene name | Strand | Alternative splicing classification |
|---|---|---|---|---|
| PTPN14 | 1:214638300–214649697 | ENSG00000152104;PTPN14 | − | A5 |
| HPCAL1 | 2:10529703–10536960 | ENSG00000115756;HPCAL1 | + | A5 |
| COBLL1 | 2:165561615–165577187 | ENSG00000082438; COBLL1 | − | A5 |
| MCCC1 | 3:182756923–182757100 | ENSG00000078070;MCCC1 | + | A5 |
| SYNPO | 5:150019047–150027505 | ENSG00000171992;SYNPO | + | A5 |
| PDZD2 | 5:31995744–32000244 | ENSG00000133401;PDZD2 | + | A5 |
| RNASET2-beta | 6:167369679–167370715 | ENSG00000026297;RNASET2 | − | A5 |
| EGFR | 7:55259222–55259411 | ENSG00000146648;EGFR | + | A5 |
| FAM107B | 10:14664173–14709632 | ENSG00000065809;FAM107B | − | A3 |
| MVK | 12:110032624–110032832 | ENSG00000110921;MVK | + | A5 |
| MVK-beta | 12:110032708–110032832 | ENSG00000110921;MVK | + | A5 |
| SLC15A4 | 12:129299615–129307792 | ENSG00000139370;SLC15A4 | − | A5 |
| KDELC1 | 13:103444486–103445690 | ENSG00000134901;KDELC1 | − | ES |
| PARN | 16:14576684–14605072 | ENSG00000140694;PARN | − | A5 |
| GAL3ST1-gamma | 22:30953388–30959216 | ENSG00000128242;GAL3ST1 | − | Alt exon usage |
| FGD1 | X:54497920–54500960 | ENSG00000102302;FGD1 | − | A5 |
A3 = alternative 30-splice site; A5 = alternative 50-splice site; Alt exon usage = alternative exon usage; ES = exon skipping.
Loci are based on reference genome sequence hs37d5.
Fig. 2 –
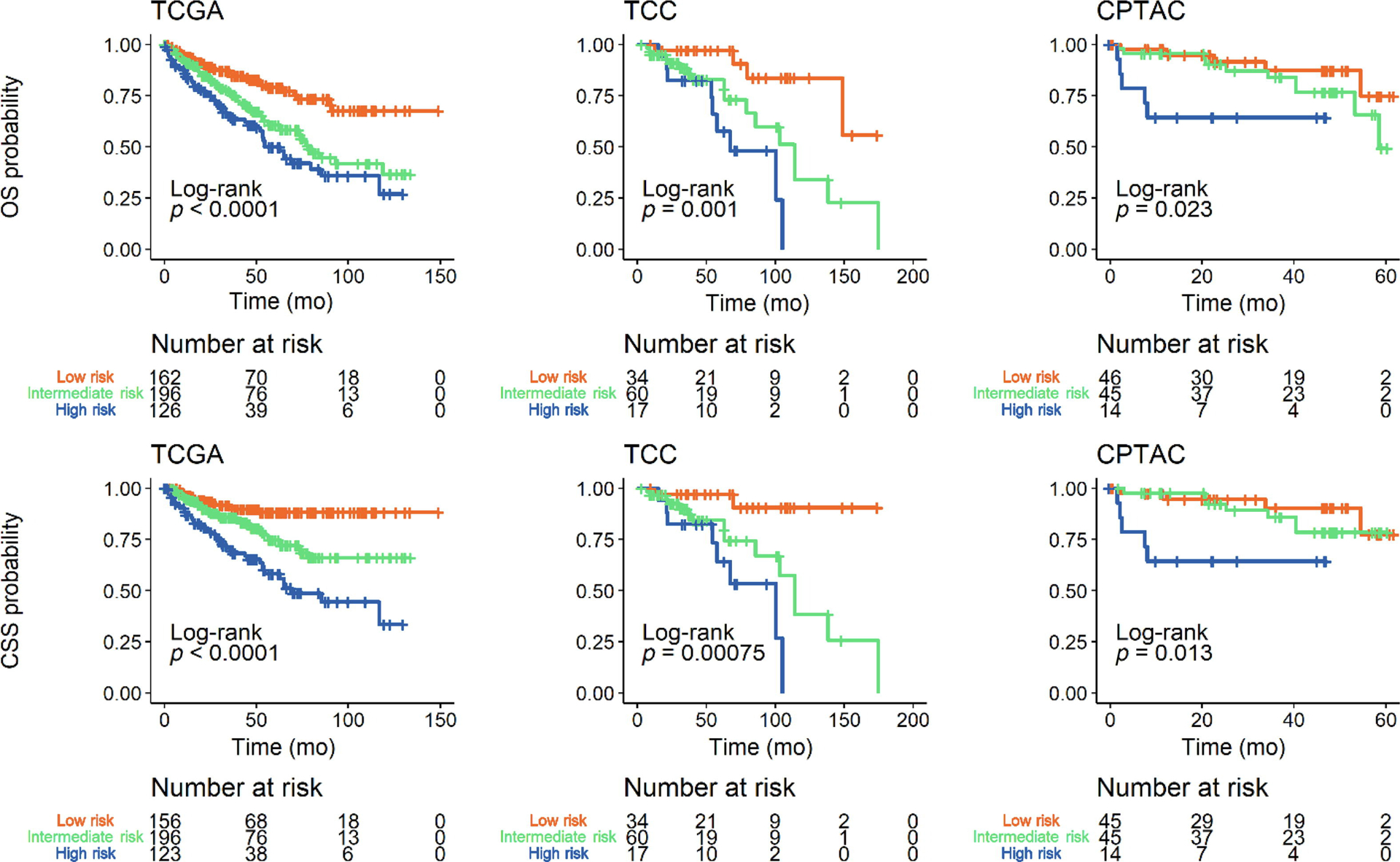
Kaplan-Meier plots stratifying overall survival (OS) and cancer-specific survival (CSS) based on a categorical splice variant (SV)-derived risk score. SV detection was used as a categorical variable to calculate a risk score for OS and CSS in datasets from The Cancer Genome Atlas (TCGA), Total Cancer Care (TCC), and Clinical Proteomics Tumor Analysis Consortium (CPTAC). For all cohorts: SV score ≤–3, low risk; SV score >–3 and <1, intermediate risk); and SV score ≥1, high risk.
The splice fraction distribution was relatively consistent across all cohorts (Supplementary Fig. 1). Within each cohort, the most prominently and consistently correlated splice fractions were seen between the two MVK SVs (Supplementary Fig. 2A–C). Otherwise, expression of each SV in our final list was relatively independent of the others.
3.2. Clinicopathologic and genomic associations of SV expression
The clinicopathologic characteristics of the TCGA, TCC, and CPTAC cohorts are summarized in Supplementary Tables 2–4. To elucidate potential links with disease biology, we screened for associations with important clinicopathologic covariates or recurrent genomic mutations in ccRCC (Supplementary Fig. 3 and 4). We used two SV metrics: continuous SV expression (splice fraction) and categorical SV detection (present vs absent; cutoff of >1% splice fraction).
Male gender was significantly associated with higher RNASET2beta-SV detection (OR 2.3–8.2) and expression (Supplementary Fig. 3A and 4A) in all cohorts. Higher histologic tumor grade was significantly associated with higher MVKbeta-SV expression in the TCGA and TCC cohorts (Supplementary Fig. 3B); a similar trend was noted in the CPTAC cohort but did not reach statistical significance. However, there was no significant association between histologic grade and SV detection in any cohort (Supplementary Fig. 4B). KDM5C genomic alteration was significantly associated with higher SYNPO-SV expression and detection (OR 6.0–11.3) and lower GAL3ST1-SV expression in the TCGA and CPTAC cohorts (Supplementary Fig. 3K and 4K); these differences did not reach statistical significance in the TCC cohort. SETD2 genomic alteration was significantly associated with higher SLC15A4-SV expression in the TCGA and TCC cohorts (Supplementary Fig. 3E); however, there was no association with SV detection (Supplementary Fig. 4E). Finally, BAP1 genomic alteration was significantly associated with higher FGD1-SV, MCCC1-SV, and MVKbeta-SV expression in the TCGA and CPTAC cohorts (Supplementary Fig. 3G); it was significantly associated with higher FGD1-SV detection in the CPTAC cohort (OR 9.6) but did not reach statistical significance in the TCGA or TCC cohort despite a similar trend (Supplementary Fig. 4G). We found that both SV expression and SV detection were not consistently associated with pathologic stage when compared across cohorts (Supplementary Figs. 3C and 4C).
3.3. Select SV expression is correlated with DNA methylation patterns
Given the purported links between DNA methylation and aberrant splicing, we examined for an association between SV expression and a global hypermethylation pattern, as defined for the TCGA and CPTAC cohorts in prior studies [11,19]. Hypermethylated ccRCC was associated with higher FGD1-SV, MCCC1-SV, MVK-SV, MVKbeta-SV, PARN-SV, and SLC15A4-SV expression in the TCGA cohort (Supplementary Fig. 5A); a similar trend was seen in the CPTAC cohort but did not achieve statistical significance. A significant association was also seen between FGD1-SV detection and hyper-methylation in the TCGA cohort (OR 3.1) but likewise did not reach significance in the CPTAC cohort (Supplementary Fig. 5B).
We also analyzed individual gene-specific DNA methylation probes in the CPTAC cohort, using the TCGA cohort as validation, by correlating the methylation of selected CpG probes with splice fraction. We identified three genes that demonstrated a negative correlation (Spearman ρ < −0.3) between individual CpG probes and splice fraction. Multiple CpG probes for EGFR showed a negative correlation between methylation level and splice fraction in both CPTAC and TCGA (Supplementary Fig. 6A–D). Gene structure methylation plots comparing tumors with adjacent normal tissue show that many of the probes are significantly hypomethylated in tumors compared with normal tissue (Supplementary Fig. 6E and 6F). Consistent with the splice fraction metric, we also show that categorical EGFR-SV detection is significantly associated with lower average methylation of these probes (Supplementary Fig. 6G and 6H). Similar trends were seen for HPCAL1 (see Supplementary Fig. 7A–H) and RNASET2 (see Supplementary Fig. 8A–H).
3.4. SV-based survival signature is consistent and applicable across all cohorts
We performed univariate Cox regression analyses to evaluate the association of SV detection and expression with postsurgical survival outcomes. Using a threshold adjusted p value of ≤0.1, PDZD2-SV detection was associated with improved survival outcomes in both the TCGA and the TCC cohort (Supplementary Fig. 9 and 10). COBLL1-SV and PTPN14-SV detection were associated with increased survival in the TCGA cohort but did not reach significance in the TCC cohort (Supplementary Fig. 9). Using the same threshold adjusted p value of ≤0.1, higher MCCC1-SV and PARN-SV expression was associated with worse survival outcomes in the TCGA and TCC cohorts, respectively (Supplementary Fig. 10).
A LASSO Cox regression analysis was then implemented to derive an SV-based risk score for overall survival; this was performed using either categorical (SV detection) or continuous (splice fraction) variables. The larger TCGA cohort was used as the training dataset; the TCC and CPTAC cohorts were validation datasets. The resulting coefficients from this analysis are shown in Table 2. For our SV risk model using categorical variables (SV detection), the score distribution in each cohort is shown in Supplementary Fig. 11. There was a high Harrell’s C-index in predicting cancer-specific survival (CSS) for the TCGA (0.67, confidence interval [CI] 0.62–0.73), TCC (0.71, CI 0.60–0.82), and CPTAC (0.70, CI 0.59–0.81) cohorts. The “calibration slopes” of the SV risk model’s PI for CSS were 1.15 (CI 0.54–1.76) and 1.19 (CI 0.33–2.04) in the TCC and CPTAC cohorts, respectively. The Kaplan-Meier curves further demonstrate good discrimination between low-, intermediate-, and high-risk overall survival and CSS outcomes consistently across all three cohorts (Fig. 2). These curves were directly compared in Supplementary Fig. 12 to provide a visual assessment of calibration. We then applied a multivariate Cox regression analysis adjusting for pathologic stage, histologic grade, age, and gender; our risk score remained statistically significant across all cohorts (Fig. 3).
Table 2 –
Final LASSO Cox regression coefficients trained on overall survival in the TCGA cohorta
| SV | Coefficient (categorical) | Coefficient (continuous) |
|---|---|---|
| COBLL1 | −0.23 | 0.00 |
| FAM107B | −0.04 | −0.63 |
| FGD1 | 0.33 | 0.32 |
| MCCC1 | 0.00 | 1.28 |
| PARN | 0.00 | 1.00 |
| PDZD2 | −0.25 | −0.64 |
| PTPN14 | −0.13 | 0.00 |
| RNASET2-beta | 0.19 | 0.59 |
| SLC15A4 | −0.01 | 0.00 |
SV = splice variant; TCGA = The Cancer Genome Atlas.
Two separate models were trained using SV detection (categorical) and SV expression (continuous).
Fig. 3 –
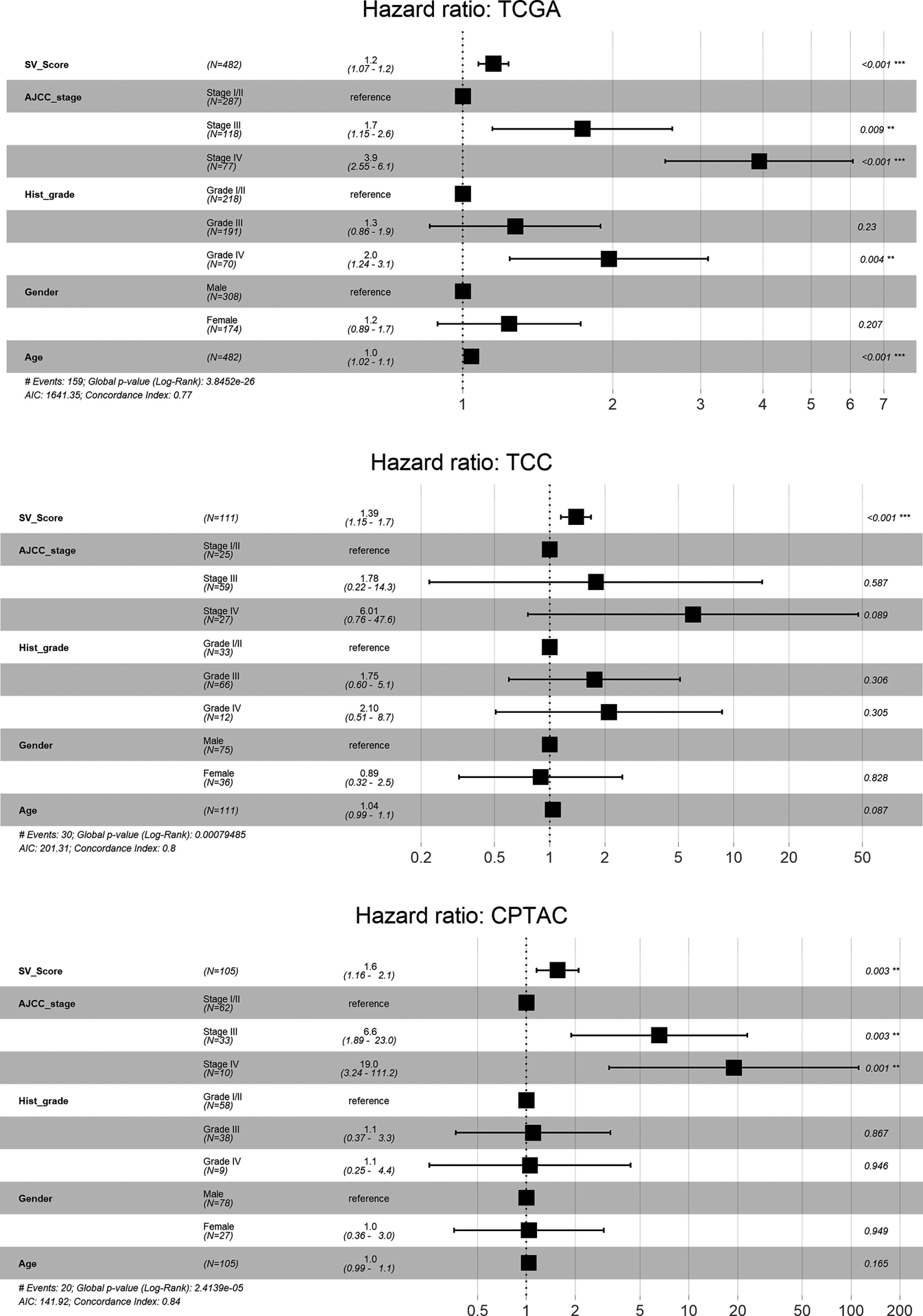
Multivariate Cox regression analysis of overall survival. The splice variant (SV)-derived risk score (SV_Score) calculated using SV detection as a categorical variable was adjusted for American Joint Committee on Cancer pathologic stage (AJCC_stage), histologic grade (Hist_grade), gender, and age. For clarity, SV_Score is a continuous model prediction score calculated using the regression coefficients in Table 2. Datasets are from The Cancer Genome Atlas (TCGA), Total Cancer Care (TCC), and Clinical Proteomics Tumor Analysis Consortium (CPTAC).
A similar analysis was performed to derive an SV risk model using continuous variables (splice fraction). The score distribution in each cohort is shown in Supplementary Fig. 11. The calibration slopes of the model’s PI for CSS were 1.76 (CI 0.95–2.58) and 0.22 (CI −0.32 to 0.76) in the TCC and CPTAC cohorts, respectively. Thus, discrimination was poor in the CPTAC cohort. The Kaplan-Meier curves demonstrate similar findings with good discrimination between low-, intermediate-, and high-risk overall survival and CSS outcomes in the TCGA and TCC cohorts only (Supplementary Fig. 13). On multivariate Cox regression analysis, this risk score remained statistically significant in the TCGA and TCC cohorts after adjustment for pathologic stage, histologic grade, age, and gender (Supplementary Fig. 14).
3.5. High-risk SV-based score is associated with proteomic and phosphoproteomic changes in splicing and cell–cell adhesion pathways
For the proteomic and phosphoproteomic analyses, patients in the CPTAC cohort were binned into low-, intermediate-, and high-risk groups based on their categorical SV-based risk score (see Fig. 2). RBM4B, a known RNA binding protein that suppresses exonic and intronic splicing [20], demonstrated significantly lower protein expression in the high-risk group than in both the low- and the intermediate-risk group (adjusted p = 0.004 and 0.015, respectively Supplementary material). Additionally, based on the BioPlex interaction network performed through Enrichr, the proteins (RBM28, TBL3, and NOP2) most strongly positively correlated with the categorical SV score likely interact with RBM4.
The splicing cofactor RBM5 was phosphorylated at significantly lower levels on S621 in the high-risk group than in both the low- and the intermediate-risk group (adjusted p = 0.038 and 0.027, respectively Supplementary material). When comparing the intermediate- and high-risk groups, the most significant difference was a decreased expression of ZC3H13 S1295-p Supplementary material, which may also have a role in alternative splicing [21]. Using a significance cutoff of p < 0.05, we saw decreased quantity of phosphoproteins involved in tight junction and cell-cell adhesion pathways; most of these changes were also seen between the low- and high-risk groups, but did not reach statistical significance Supplementary material. Differential protein and phosphoprotein expression between the low and intermediate-risk groups did not reach statistical significance Supplementary material.
4. Discussion
We present a novel pipeline to screen and select aberrant SVs enriched in ccRCC. Our analysis identified significant biological associations with SV expression, including gender, recurrent genomic mutations, and DNA methylation patterns. We ultimately derived a unique SV-based survival signature that was consistent across multiple distinct ccRCC cohorts. This process can widely be implemented for other tumor types.
Strategies to extract aberrant SVs from tumor tissue RNA-seq data have been described previously [22–24]. However, one of the main challenges in aberrant SV identification is sorting signal from noise; true aberrant splicing signals can be confounded by noise from inaccurate splice junction detection, low tumor purity, and normal alternative splicing. To address these challenges, our pipeline starts with cancer cell line data, which are the most controlled model system. The Cancer Cell Line Encyclopedia provides a high-volume, publicly available dataset that can broadly be applied as an initial step in SV identification for other tumor types; however, we acknowledge that it may not be all inclusive due to genetic drift inherent to cancer cell lines. The GTEx database provides a filter for SVs that may be expressed in normal kidney; inclusion of normal nonkidney samples increases the stringency of our filter and may enable us to rule out normal SVs expressed by other constituents of the tumor microenvironment. Whereas 25% of SVs identified in our initial screen were excluded through GTEx, 73% of the remaining SVs had inadequate coverage in at least one of the three distinct cohorts (TCGA, TCC, and CPTAC); this outcome may be attributed to variations in tumor cohorts, sample collection, and methodologies between studies. Overall, the validity and robustness of our selection process was reinforced by the similar splice fraction distribution across cohorts for each of our final aberrant SVs.
The next challenge was to determine whether aberrant SVs have any biological or clinical relevance. We approached this from two angles—on the one hand, SV detection alone has intrinsic relevance as these were selected to be specifically enriched in ccRCC tumor cells; on the other hand, SV expression is a continuous quantitative metric with potential to be informationally richer. Overall, we demonstrate good concordance in the association of clinicopathologic, genomic, and epigenetic variables with SV detection and expression. This may be explained by the nonsymmetric distribution and skew in splice fraction toward zero for almost all aberrant SVs (Supplementary Fig. 1). It is very reasonable that a nonlinear relationship exists between splice fraction and disease biology—akin to existing dichotomization of genetic mutations (wild type vs altered) rather than the use of variant allele frequency.
Post-translational histone modifications and DNA methylation act together to regulate gene expression by controlling accessibility of DNA for transcription and splice factors [25,26]. Methylation of histone tails is an active process mediated by lysine methyltransferases (KMTs) and histone-lysine demethylases (KDMs). Relevant to ccRCC, the SETD2 gene encodes for a KMT and the KDM5C gene encodes for a KDM. We found that alterations in these genes are associated with increased detection and expression of two of our identified aberrant SVs (SYNPO and SLC15A4). Additionally, aberrant SVs were associated with changes in DNA methylation. Higher EGFR-SV, HPCAL1-SV, and RNASET2beta-SV detection and expression were associated with hypomethylation of gene-specific CpG islands. Of note, CpG island methylation has been used as a prognostic marker of survival in ccRCC [27].
In our categorical SV-based model, survival was most associated with five SVs: FGD1-SV and RNASET2-SV expression was associated with unfavorable outcomes, and COBLL1-SV, PTPN14-SV, and PDZD2-SV expression was associated with favorable outcomes. Our SV-derived risk score was significantly associated with survival even after adjustment for key prognostic factors, including pathologic stage and histologic grade. It was able to even stratify survival in the CPTAC cohort, which thus far has immature follow up data on survival to only 60 mo. We also derived a continuous SV-based risk model, which selected most of the same SVs as the categorical model. Compared with the categorical model, there was poor discrimination in the CPTAC cohort; however, as mentioned above, the follow-up interval in this cohort is still very short. Regardless, this reinforces good concordance in analyses using either SV detection or SV expression.
Additionally, our proteomic and phosphoproteomic analysis of the highest SV-based risk group demonstrated significant changes in known splicing factors, specifically, RBM4B expression and RBM5 phosphorylation on S621. Consistent with our study, lower levels of RBM4 expression have been associated with poor cancer survival outcomes [20]. As a potentially pertinent connection to ccRCC, RBM4 has also been found to regulate the splicing profile of the HIF-1α gene in lung cancer [28]. Similarly, RBM5 is thought to be a tumor suppressor that is mostly phosphorylated under normal conditions [29]. Downregulated expression of this gene was identified as one of a set of 17 genes that constituted a molecular signature of metastasis in primary solid tumors [30]. In summary, these data highlight the potential of aberrant splicing to enhance our understanding of disease biology and clinical outcomes that heretofore remain relatively unexplored. Future studies are needed to clarify the mechanistic roles of aberrant SVs, implications in predicting response to systemic treatment, and utility as a bio-marker of disease recurrence or metastasis.
5. Conclusions
We implemented a novel screening pipeline starting with cancer cell line RNA-seq data to identify 16 unique ccRCC-enriched SVs. Several of these SVs were found to be significantly associated with molecular and epigenetic features of ccRCC. Additionally, our SV-based risk score was strongly associated with overall survival and CSS across multiple cohorts. This study provides a template for identifying and characterizing disease-specific aberrant SVs to aid in the discovery of new disease biology and biomarkers.
Supplementary Material
Funding/Support and role of the sponsor:
Support for this study was provided by the Kidney Cancer Association—Young Investigator Award. This work has been supported in part by a Moffitt Division of Quantitative Sciences Team Science Award (PAS Co-PI) and the Biostatistics and Bioinformatics, Proteomics and Metabolomics, and Shared Resources at the H. Lee Moffitt Cancer Center & Research Institute, a National Cancer Institute designated Comprehensive Cancer Center (P30-CA076292).
Footnotes
Financial disclosures: Andrew Chang certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: None.
References
- [1].Barash Y, Calarco JA, Gao W, et al. Deciphering the splicing code. Nature 2010;465:53–9. [DOI] [PubMed] [Google Scholar]
- [2].Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol 2014;15:108–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature 2008;456:470–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Danan-Gotthold M, Golan-Gerstl R, Eisenberg E, Meir K, Karni R, Levanon EY. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res 2015;43:5130–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lee SC, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med 2016;22:976–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res 2008;68:5469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Antonarakis ES, Lu C, Wang H, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014;371:1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang J, Manley JL. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov 2013;3:1228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lev Maor G, Yearim A, Ast G. The alternative role of DNA methylation in splicing regulation. Trends Genet 2015;31:274–80. [DOI] [PubMed] [Google Scholar]
- [10].Kulis M, Esteller M. DNA methylation and cancer. Adv Genet 2010;70:27–56. [DOI] [PubMed] [Google Scholar]
- [11].Ricketts CJ, De Cubas AA, Fan H, et al. The Cancer Genome Atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep 2018;23:3698. [DOI] [PubMed] [Google Scholar]
- [12].Chen F, Zhang Y, Senbabaoglu Y, et al. Multilevel genomics-based taxonomy of renal cell carcinoma. Cell Rep 2016;14:2476–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Piekielko-Witkowska A, Wiszomirska H, Wojcicka A, et al. Disturbed expression of splicing factors in renal cancer affects alternative splicing of apoptosis regulators, oncogenes, and tumor suppressors. PLoS One 2010;5:e13690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zaman S, Hajiran A, Coba GA, et al. Aberrant epidermal growth factor receptor RNA splice products are among the most frequent somatic alterations in clear cell renal cell carcinoma and are associated with a poor response to immunotherapy. Eur Urol Focus 2021;7:373–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bland JM, Altman DG. Multiple significance tests: the Bonferroni method. BMJ 1995;310:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Austin PC, Lee DS, D’Agostino RB, Fine JP. Developing points-based risk-scoring systems in the presence of competing risks. Stat Med 2018;37:1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Royston P, Altman DG. External validation of a Cox prognostic model: principles and methods. BMC Med Res Methodol 2013;13:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Harrell FE Jr, Califf RM, Pryor DB, Lee KL, Rosati RA. Evaluating the yield of medical tests. JAMA 1982;247:2543–6. [PubMed] [Google Scholar]
- [19].Clark DJ, Dhanasekaran SM, Petralia F, et al. Integrated proteogenomic characterization of clear cell renal cell carcinoma. Cell 2020;180:207. [DOI] [PubMed] [Google Scholar]
- [20].Wang Y, Chen D, Qian H, et al. The splicing factor RBM4 controls apoptosis, proliferation, and migration to suppress tumor progression. Cancer Cell 2014;26:374–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Horiuchi K, Kawamura T, Hamakubo T. Wilms’ tumor 1-associating protein complex regulates alternative splicing and polyadenylation at potential G-quadruplex-forming splice site sequences. J Biol Chem 2021;297:101248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cotto KC, Feng YY, Skidmore ZL, Griffith OL, Griffith M. Application of RegTools to TCGA samples for the identification of tumor-specific splice variants. Cancer Res 2019;79:2487. [Google Scholar]
- [23].Sun W, Duan T, Ye P, et al. TSVdb: a web-tool for TCGA splicing variants analysis. BMC Genomics 2018;19:405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].West S, Kumar S, Batra SK, Ali H, Ghersi D. Uncovering and characterizing splice variants associated with survival in lung cancer patients. PLoS Comput Biol 2019;15:e1007469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Vaissiere T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res 2008;659:40–8. [DOI] [PubMed] [Google Scholar]
- [26].Tian Y, Soupir A, Liu Q, et al. Novel role of prostate cancer risk variant rs7247241 on PPP1R14A isoform transition through allelic TF binding and CpG methylation. Hum Mol Genet 2022;31:1610–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wei JH, Haddad A, Wu KJ, et al. A CpG-methylation-based assay to predict survival in clear cell renal cell carcinoma. Nat Commun 2015;6:8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chang HL, Lin JC. SRSF1 and RBM4 differentially modulate the oncogenic effect of HIF-1 alpha in lung cancer cells through alternative splicing mechanism. Biochim Biophys Acta Mol Cell Res 2019;1866:118550. [DOI] [PubMed] [Google Scholar]
- [29].Shu Y, Rintala-Maki ND, Wall VE, et al. The apoptosis modulator and tumour suppressor protein RBM5 is a phosphoprotein. Cell Biochem Funct 2007;25:643–53. [DOI] [PubMed] [Google Scholar]
- [30].Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet 2003;33:49–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.