

Abstract
Background
Lung adenocarcinoma metastasizing to the brain results in a notable increase in patient mortality. The high incidence and its impact on survival presents a critical unmet need to develop an improved understanding of its mechanisms.
Methods
To identify genes that drive brain metastasis of tumor cells, we collected cerebrospinal fluid samples and paired plasma samples from 114 lung adenocarcinoma patients with brain metastasis and performed 168 panel-targeted gene sequencing. We examined the biological behavior of PMS2 (PMS1 Homolog 2)-amplified lung cancer cell lines through wound healing assays and migration assays. In vivo imaging techniques are used to detect fluorescent signals that colonize the mouse brain. RNA sequencing was used to compare differentially expressed genes between PMS2 amplification and wild-type lung cancer cell lines.
Results
We discovered that PMS2 amplification was a plausible candidate driver of brain metastasis. Via in vivo and in vitro assays, we validated that PMS2 amplified PC-9 and LLC lung cancer cells had strong migration and invasion capabilities.
The functional pathway of PMS2 amplification of lung cancer cells is mainly enriched in thiamine, butanoate, glutathione metabolism.
Conclusion
Tumor cells elevated expression of PMS2 possess the capacity to augment the metastatic potential of lung cancer and establish colonies within the brain through metabolism pathways.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12575-024-00238-1.
Keywords: Lung cancer, Brain metastasis, PMS2
Introduction
Brain metastasis (BM) is a significant contributor to morbidity and mortality, carrying an unfavorable prognosis [1]. Common primary tumors giving rise to BM include breast cancer, lung cancer, and melanoma. Among these, lung cancer exhibits the highest incidence of BM ranging from 20 to 56%, far beyond other malignancies [2]. The median survival post-metastasis varies widely, spanning from 3 to 27 months [3]. It highlights an unmet need for enhanced comprehension and novel treatments.
Understanding the mechanisms underlying brain metastasis is imperative due to its prevalence and adverse impact on survival. It highlights an unmet need for enhanced comprehension and novel treatments. Intriguingly, some patients manifest brain metastasis even while their extracranial disease remains controlled [3]. This clinical disparity is partly explained by insufficient systemic therapeutic penetration of the blood–brain barrier, but unfortunately most of the mechanisms are under water. The exploration is hindered by the fact that patients with BM often encounter limitations in undergoing surgical resection of primary tumors, and excising intracranial metastatic tumors poses significant challenges.
Observations suggest the presence of additional potentially oncogenic alterations in brain metastases, contributing to the therapeutic response divergence seen in some cases. The evolutionary process, marked by mutations, may serve as precursors to tumor development and subsequent metastasis [4]. Observations suggest the presence of additional potentially oncogenic alterations in brain metastases, contributing to the therapeutic response divergence seen in some cases. This underscores the complexity of the metastatic process and emphasizes the necessity for in-depth exploration of underlying mechanisms to pave the way for improved therapeutic interventions.
Patients with BM often face limitations in undergoing surgical resection of primary tumors, and the excision of intracranial metastatic tumors poses significant challenges. Consequently, obtaining specimens for histopathological or biomarker studies becomes exceedingly difficult. Recognizing this constraint, circulating cell-free DNA (cfDNA) emerges as a promising avenue in cancer management.
Previous investigations have underscored the substantial value of cfDNA in plasma or other body fluids for cancer diagnosis and treatment [5, 6]. Notably, the analysis of driver mutations through ctDNA in plasma demonstrates clinical utility, particularly in epidermal growth factor receptor (EGFR)-mutated non-small-cell lung cancer (NSCLC). Given the inherent inaccessibility to intracranial tumor tissues, cfDNA derived from brain malignancies has garnered increased attention as a valuable resource in cancer management. Herein, cerebrospinal fluid (CSF), in direct contact with brain neoplasms, emerges as a more suitable and less invasive avenue for diagnosing brain tumors, circumventing the need for risky surgical procedures [7, 8]
Furthermore, CSF liquid biopsy proves informative, especially in patients with multiple brain metastases, as it can detect mutations not observed in the primary tumor [4]. The study conducted by Wu highlights the significant value of CSF as a source for liquid biopsy, demonstrating its efficacy in detecting actionable mutations in leptomeningeal metastasis. This emphasizes the potential of CSF analysis as a valuable tool in the diagnosis and management of brain metastases [9].
The PMS2 gene, situated on chromosome 7p22 within a 16 kb region, comprises 15 exons and 862 codons [10]. Functionally, the PMS2 protein forms a heterodimer with the MLH1 protein, constituting the MutLα complex, which collaborates with the MutSα complex in the primary repair of single-nucleotide mismatches [11]. As one of the DNA mismatch repair (MMR) genes, PMS2 plays a crucial role in genomic stability. Notably, pathogenic heterozygous germline variants in PMS2 have been associated with Lynch Syndrome (LS) [12–16],while mutations in MMR genes are frequently detected in various solid tumors [17–19].
Moreover, PMS2 variants have been identified in primary brain tumors, with their presence reported in cerebrospinal fluid samples from glioma patients through exome sequencing [21–23]. The loss of PMS2 is also correlated with poorer overall survival (OS) and progression-free survival (PFS) in lower grade astrocytomas [22]. Immunohistochemical analysis indicating the loss of MLH1/PMS2 co-expression is associated with a lower tumor mutational burden (TMB) [24]. Additionally, the PMS2 gene is implicated in the apoptotic pathway [25, 26].
The association of the PMS2 gene with brain metastasis in non-small cell lung cancer has not been investigated to date. This comprehensive approach aims to shed light on the involvement of the PMS2 gene in the complex landscape of brain metastasis in non-small cell lung cancer.
Methods
Patients enrollment and samples collection
One hundred fourteen patients with brain metastases were enrolled in our study, who had all been admitted to the Guangdong Sanjiu Brain Hospital between January 2019 and January 2022. Brain parenchymal metastases have been evaluated by MRI, while leptomeningeal metastases were diagnosed with MRI or Cerebrospinal fluid cytologic testing. Brain metastasis is identified by clinicians and MRI images are evaluated by two radiologists. This research was approved by the ethics committee of Guangdong Sanjiu Brain Hospital and conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all patients. One hundred fourteen CSF samples acquisition was performed by the clinician through the lumbar puncture, 10 ml of CSF was collected during the procedure. Sixty-eight plasma samples were obtained within one week. 2 ml of CSF was used for the cytologic test, 8 ml of CSF and 8 ml of plasma were collected for NGS. 8 ml of CSF or plasma was collected in a standard ethylenediaminetetraacetic (EDTA) acid tube and incubated at room temperature for 2 h. The supernatant was transferred into a 15 ml centrifuge tube and was centrifuged at 16,000 g at 4 °C for 10 min. The supernatant was then transferred to a new tube and stored at − 80 °C for further analysis. All procedures were completed within two hours of sample collection.
DNA extraction and Library construction, target sequencing
DNA extraction, library construction, and targeted sequencing with a commercial panel of the 168-gene panel (Burning Rock Biotech, Guangzhou, China) followed with routine processing described in previous studies [9, 27]. In brief, cfDNA was recovered from 5 ml of CSF or plasma using the QIAamp Circulating Nucleic Acid kit by Qiagen (Valencia, California, US). The Qubit 2.0 Fluorimeter (Carlsbad, California, US) was used to assess the Quantification of cfDNA. At least 50 ng of cfDNA is required to construct NGS library. Fragments between 200 and 400 bp from the sheared cfDNA were selected using Agencourt AMPure beads (Beckman Coulter, California, US), then hybridized with capture probes baits. PCR amplification was performed after hybridization selection with magnetic beads. Target capture was performed using a 168-gene targeted panel. A bioanalyzer high-sensitivity DNA kit was used to assess the quality and size of the fragments. Indexed samples were sequenced on the Nextseq 500 sequencer (Illumina, Inc., California, US) with pair-end reads.
Cell culture
Seven human lung cancer cell lines, namely H1299, A549, H1650, HCC827, H3255, H1975, and PC-9, along with the human bronchial epithelial cell line BEAS-2B and the Lewis lung carcinoma cell line, were obtained from the American Type Culture Collection (ATCC). These cell lines were cultured in either Roswell Park Memorial Institute (RPMI) 1640 medium (Biosharp, China) or Dulbecco's Modified Eagle Medium (DMEM) (Biosharp, China), both supplemented with 10% fetal bovine serum (FBS, Pricella, China) and 100 U/ml of a streptomycin/penicillin combination (Gibco; Thermo Fisher Scientific, Inc.). The cultures were maintained in a humidified atmosphere containing 5% CO2 at 37 °C.
Lentivirus and transfections
A lentivirus overexpressing PMS2 was generated through lentiviral transduction utilizing the pcSLenti-EF1-EGFP-P2A-Puro-CMV-MCS-3xFLAG-WPRE vector provided by OBIO Technology (Shanghai) Corp., LTD. To establish a control, a lentivirus lacking the PMS2 construct (designated LV-control) was created using an empty vector, pcSLenti-EF1-EGFP-P2A-Puro-CMV-PMS2-3xFLAG-WPRE. Additionally, a lentivirus overexpressing luciferase was generated by lentiviral transduction employing the pSLenti-EF1-Luc2-P2A-BSR-CMV-MCS-WPRE vector. The transfection process followed the manufacturer's instructions, with polybrene serving as the transfection agent. This standardized procedure ensures the efficient introduction of genetic material into the target cells, facilitating the overexpression of PMS2 or luciferase as intended.
Wound healing assay
Transfected cells were seeded in 6-well plates at a concentration of 2.5 × 10^5 cells/mL. Subsequently, cell monolayers were mechanically wounded using a sterile pipette tip (10 μL) to create a gap. The cells were then washed with 1X phosphate-buffered saline (PBS), and the remaining cells were cultured continuously in serum-free medium for 24 h. Representative fields were photographed, and the migrated cells were quantified by counting. This standardized protocol ensures consistent and reproducible assessment of cell migration following the specified experimental conditions.
Transwell assay
Invasion assessment was conducted using 24-well Transwell inserts (8 μm aperture, BD Biosciences) pre-coated with Matrigel matrix (Corning, NY). Approximately 2 × 10^4 cells, suspended in serum-free medium, were seeded into the upper chambers, while the lower chambers were filled with DMEM containing 10% FBS. Cells were maintained at 37 °C throughout the experiment. After 24 h, non-invading cells on the upper side of the inserts were gently wiped off using a cotton swab. Subsequently, cells that had invaded through the Matrigel and reached the lower chambers were treated with 0.1% crystal violet and counted. This method provides a standardized approach for assessing cell invasion capabilities, ensuring reliable and reproducible results.
RNA extraction and real-time quantitative PCR
Total RNA extraction was conducted using TRIzol reagent. Subsequently, cDNA was synthesized utilizing the PrimeScript™ RT Master Mix cDNA Synthesis Kit (RR360A, TaKaRa, China), following the manufacturer’s protocol. Real-time PCR was performed with TB Green qPCR Master Mix (RR820A, Takara, China) on the QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA). For normalization, relative mRNA levels were assessed based on the expression of GADPH. The expression values were analyzed using the 2 − ΔΔCt method, a widely accepted relative quantitative approach in real-time PCR studies. The primer sequences used were as follows: PMS2 forward: 5′- CTGGATGCTGGTCCACTAA-3′, reverse: 5′- TGTGTGATGTTTCAGAGTTAAGCC-3′; GAPDH forward: 5′- ACAACTTTGGTATCGTGGAAGG -3′, reverse: 5′- GCCATCACGCCACAGTTTC -3′.
Western blot analysis
Cells were lysed by cold RIPA Lysis Buffer (EpiZyme, China) supplemented with protease inhibitor on ice. Samples were boiled with SDS/PAGE sample buffer for 10 min and then were separated on SDS-PAGE. Post transferring, PVDF membranes (Millipore) were blocked by 5% non-fat milk TBST buffer, followed by incubation with primary antibodies at 4 °C overnight. After incubation with horseradish-peroxidase-coupled secondary antibodies at room temperature for 2 h, immunoreactive bands were visualized using ECL kit (EpiZyme, China). Anti-PMS2 antibody (A4577) and anti-GAPDH (AC001) were purchased from ABclonal Technology Co., Ltd.
In vivo mouse studies
We purchased 22 five-week-old female nude mice and mice were randomly assigned to experimental group and control group for all the experiments. After one week’s adaption, mice were anesthetized and injected with lung cancer cells (PC-9 control and PMS2 amplificated PC-9 cells: 10^5/mouse) into the left cardiac ventricle as reported previously [28].
Bioluminescence imaging (BLI)
For in vivo imaging, mice were anesthetized using 3% isoflurane in 100% oxygen. Subsequently, they were injected with 300 mg/kg of the substrate D-luciferin. The mice were then placed in the IVIS Spectrum instrument (Tanon 6600) to monitor the systemic dissemination of tumor cells. Images were captured at 5-min intervals until the photon counts reached their peak.
DNA sequencing and analysis
DNA Sequencing data were mapped to the reference human genome (hg19) using Burrows-Wheeler Aligner version 0.7.10 [29]. Local alignment optimization, variant calling, and annotation were performed using GATK 3.2, MuTect, and VarScan. Full description was provided in a previous study [9].
RNA sequencing and differentially expressed genes analysis
The libraries were sequenced on the llumina Novaseq 6000 platform and 150 bp paired-end reads were generated. Raw reads of fastq format were firstly processed using fastp and the low quality reads were removed to obtain the clean reads. Then clean reads for each sample were retained for subsequent analyses. The clean reads were mapped to the reference genome using HISAT2. FPKM of each gene was calculated and the read counts of each gene were obtained by HTSeq-count. PCA analysis were performed to evaluate the biological duplication of samples. Differential expression analysis was performed using the DESeq2. Q value < 0.05 and foldchange > 2 or foldchange < 0.5 was set as the threshold for significantly differential expression gene (DEGs). Based on the hypergeometric distribution, KEGG pathway enrichment analysis of DEGs was performed to screen the significant enriched term, respectively.
Statistical analysis
Fisher’s exact test was used to compare the proportions between the two groups. Kaplan–Meier analysis was used to analyse the OS of each group and the log-rank test was performed to test survival differences between two groups. Besides, we performed all statistical analyses and plotting using the software package R version 4.2.3. The two-sided p < 0.05 was considered significant.
Results
Patients’ characteristics
We included 114 patients with brain metastases from non-small cell lung cancer, including 112 adenocarcinomas, 1 squamous cell carcinoma, and 1 adenosquamous cell carcinoma. Of the 114 patients included in the study, 28 had brain parenchymal metastases (BPM), including 18 who also had leptomeningeal metastases (LM). The rest of the patients presented with a combination of BPM and LM. The clinical characteristic of these patients was summarized in Supplemental Table 1.
PMS2 amplification identified in CSF samples
EGFR, TP53, CDKN2A, PMS2 and CDK4 are the top five most common mutations in cerebrospinal fluid samples from patients with brain metastases (Fig. 1A), while EGFR, TP53, MYC, ALK and BRINP3 are most frequently detected in plasma samples (Fig. 1B). Comparisons of the mutational landscape of CSF samples with that of plasma revealed that PMS2 gene mutations in tumor cells are identified in a copy-number amplified form in the cerebrospinal fluid, no mutations in the PMS2 gene were detected in the plasma. Besides, in previous article, similar result was reported that PMS2 is a high frequently mutated genes in CSF compared with primary lung tumor tissues [30]. We analyzed the genomic characteristics of 337 primary early-stage lung adenocarcinomas from patients in the TCGA M0 Stage LUAD cohort, revealing an absence of PMS2 alterations (Supplemental Fig. 1). These results suggest that PMS2 amplification is specifically detected in the cerebrospinal fluid of patients with brain metastases. We then divided patients in our cohort into PMS2 amplified and unamplified groups, the two group had a similar long-term follow-up overall survival (OS) (P = 0.57) (Supplement Fig. 2).
Fig. 1.
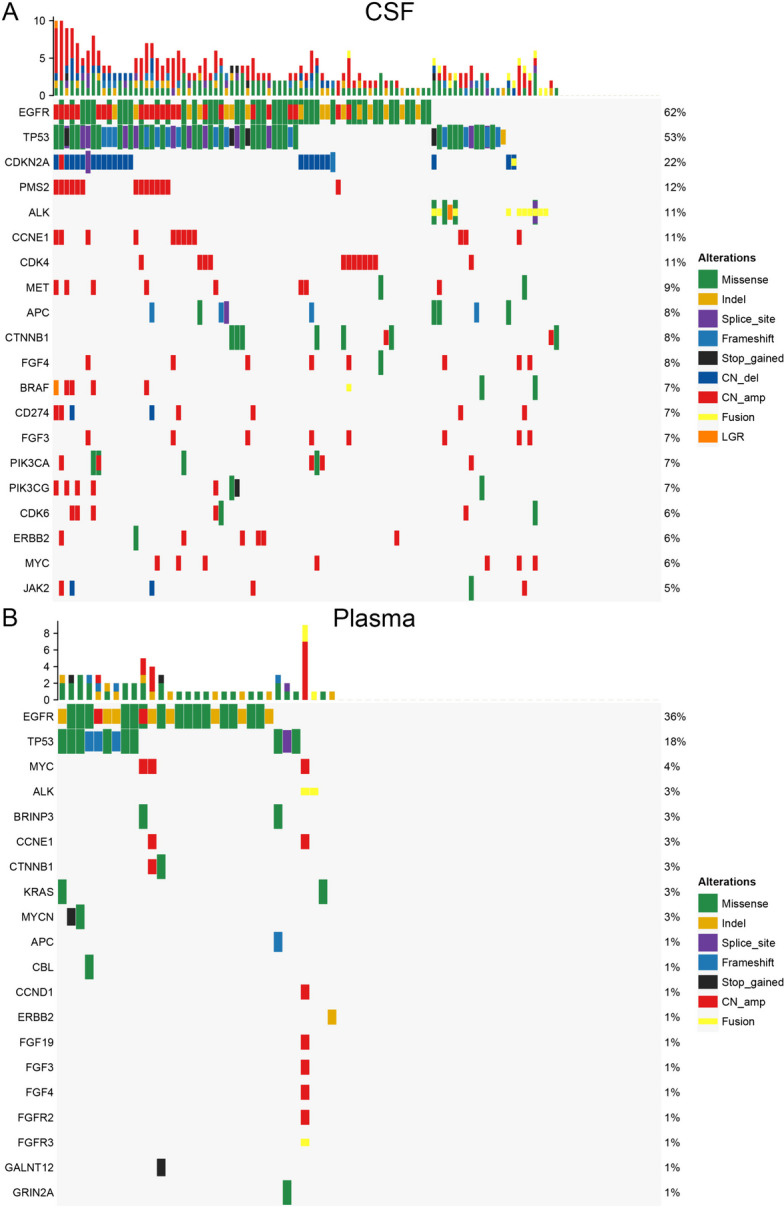
Genomic atlas of samples from patients with brain metastases. A genomic alternation detected in the cerebrospinal fluid. B genomic alternation detected in the plasma
PMS2 promotes the migration and invasion of lung cancer cells
Migration is a vital step for tumor cells to move from the primary lesion to distant organs. In order to investigate the effect of PMS2 gene on the migration ability of lung cancer cells, we screened lung cancer cell lines by PCR (Fig. 2A) and western blotting (Fig. 2B, Supplement Fig. 3A) and we found PC9 lung cancer cell lines with consistently low PMS2 expressed in at the transcriptional and protein levels. We further overexpressed PMS2 in PC9 cell lines by lentiviral infection system. At the same time, we introduced the PMS2 gene into a murine lung cancer LLC cell line (Fig. 2C,D, Supplement Fig. 3B). Through invasion experiments, we found that PMS2- amplificated tumor cells had a stronger invasion ability than control cells (Fig. 2E). Then we performed the wound healing assay, compared to the control cancer cell lines, PMS2-amplificated lung cancer cells had a stronger ability to migrate (Fig. 2F).
Fig. 2.

The expression level and cell migration and invasion abilities of PMS2 in cell lines. A The relative transcriptional expression level of PMS2 in in the human bronchial epithelial cell line BEAS-2B and seven lung cancer cell lines. B The protein expression levels of PMS2 in in the human bronchial epithelial cell line BEAS-2B and seven lung cancer cell lines. C PMS2 protein expression levels in transgenic cell lines. D Fluorescence photograph of lentivirus-infected lung cancer cell lines. E Transwell invasion assay and F Wound healing assay
PMS2 overexpression stimulates tumor cells colonization in mice brains
To investigate the role of PMS2 in brain metastasis, we tagged tumor cells with a dual luciferase labeling system to apply for tracking of tumor cells, and injected luciferase-labeled lung cancer cells into left ventricle of nude mice (Fig. 3A). We found that the mortality rate was higher in the PMS2 amplified group (Supplement Fig. 4). By quantifying the fluorescence signal by in vivo Bioluminescence imaging (BLI) and comparing the signal intensity between the two groups (Fig. 3B-C), we found that the brain signals were stronger in the experimental group, but there was no statistical difference (Supplement Fig. 5). This may be due to the higher mortality rate of tumor cells after colonization of tumor cells overexpressing PMS2 in the mouse brain. Next, we sacrificed the mice with fluorescent signals in the brain in the two groups, separated the whole brains of mice, and extracted tumor cells from the brains for further culture. RNA sequencing was performed on tumor cells in PMS2 amplification and control groups. We confirmed that expression level of PMS2 was elevated in PMS2 amplificated tumor cells (Fig. 3D). Functional enrichment analysis showed that the main pathways of the differentially differentiated genes between the two groups were concentrated in the thiamine, butanoate, glutathione metabolism (Fig. 3E). Furthermore, when conducting RNA sequencing on lung cancer cells before and after intracardiac injection without the transfer of the PMS2 gene, it was found that there were no enriched pathways related to metabolism (Supplement Fig. 6).
Fig. 3.

The Mechanism of PMS2 Promoting Brain Metastasis. A Xenograft Model Flow Diagram. B Bioluminescence imaging of PMS2 overexpressed mice. C Bioluminescence imaging of control mice. D Volcano plot of differentially expressed genes in tumor cells in the PMS2 overexpression group versus the control group. E Functional enrichment dot plot of differentially expressed genes
Discussion
The process and molecular mechanism underlying the metastasis and colonization of lung cancer tumor cells in the brain remain incompletely understood. Genetic divergence between brain metastases and primary tumors has been reported [31]. The formation of brain tumors results from a series of genetic changes, playing a pivotal role in tumor evolution [32]. Genomic analysis of brain metastases offers an opportunity to identify clinically informative alterations not detected in primary tumors sampled during routine clinical procedures [4]. Therefore, discovering new genes is crucial for a better understanding of tumor development and progression.
HBEGF, a ligand for EGFR, has been specifically identified in brain metastasis and demonstrated to enhance invasion in breast cancer cells. Another brain metastasis-specific gene, ST6GALNAC5, mediates the interaction of cancer cells with brain endothelial cells [33]. In this context, our study identified PMS2 as a potential driver gene for brain metastasis by sequencing cerebrospinal fluid and plasma and comparing genes with high-frequency mutations.
Approximately 4–5% of NSCLCs exhibit alterations in the genes constituting the mismatch repair (MMR) system [34]. The MMR system is crucial for DNA replication fidelity, involving seven known genes (MLH1, MLH3, MSH2, MSH6, PMS1, PMS2, and EPCAM) that recognize and repair single base–base mismatches or insertion–deletion loops [35, 36]. Mutations in these genes elevate susceptibility to various cancers [37, 38]. Loss of function in any MMR gene results in hypermutation and high microsatellite instability (MSI-H), causing errors in DNA replication and recombination [35, 39]. Dysregulation of mismatch repair gene expression, both loss and overexpression, can be detrimental to genomic stability, with loss-of-function mutations correlating with high TMB [40–42]. Overexpression of the PMS2 gene disrupts MMR function, establishing an additional carcinogenic mechanism leading to genetic instability and resistance to cytotoxic cancer therapy [43]. Our gain-of-function studies reveal that PMS2 overexpression increases the migration and invasion capabilities of cancer cells, crucial for the initiation of metastasis. Organ colonization emerged as a main rate-limiting step in the metastatic cascade, through BLI, our cell-derived xenograft mouse models validated that PMS2 overexpression increased the brain metastasis incidence, compared to the control group. These results demonstrate that PMS2 amplification is critical to brain metastasis formation by lung cancer cells.
The development of brain metastasis hinges on intricate interactions between cancer cells and the tumor microenvironment. The blood–brain barrier (BBB)/blood tumor barrier (BTB) imposes restrictions on nutrient access from circulation [44], resulting in a microenvironment characterized by hypoxia and depletion of essential metabolites, growth factors, and proteins [45]. Consequently, metastasized tumor cells undergo genetic and epigenetic changes to better adapt to this challenging environment [46–50]. Breast cancer cells that metastasize to the brain demonstrate a unique ability to utilize gluconeogenesis and oxidize branched-chain amino acids for growth, independent of glucose availability [51]. Likewise, limited microenvironmental serine and glycine results in selection of brain metastatic cells with increased dependency on de novo serine synthesis. Additionally, in response to limited microenvironmental serine and glycine, brain metastatic cells exhibit an increased dependency on de novo serine synthesis [52]. Brain-tropic breast cancer lines undergo reprogrammed lipid metabolism, including alterations in lipid transport, synthesis, and beta-oxidation [53–55]. Notably, lactate secretion by highly metastatic cells serves to limit innate immune surveillance and promotes significant metastases [56]. Overall, accumulating evidence underscores the crucial role of dynamic interplay between brain metastatic cancer cells and the surrounding immune microenvironment in the process of brain metastatic colonization [57–59].
It is reasonable to hypothesize that PMS2 amplification occurs in tumor cells after entering the brain tissue through blood circulation, enabling better adaptation to the brain microenvironment. Transcriptome sequencing of PMS2-amplificated tumor cells colonized in the brain, compared to tumor cells in the control group, reveals enrichment in metabolic pathways, particularly involving glutamine, thiamine, and methyl butyrate. These findings shed light on the functional implications of PMS2 amplification in tumor cells during brain metastatic colonization, providing insights into potential metabolic adaptations that contribute to the successful establishment of brain metastases.
Our data provide new insights into complex interactions between brain metastatic cancer cells and DNA mismatch genes during metastatic colonization in the brain. However, our study still had four shortcomings. First, samples of patients with brain metastases were collected retrospectively from only one hospital. Second, Lack of brain tumor tissue samples to verify the reliability of our cerebrospinal fluid results. Third, the number of gene panel tests is small and may miss some genes related to brain metastasis. Last, current models of brain metastasis are largely based on hematogenous cancer cell dissemination upon intra-arterial or cardiac injection, thereby bypassing spontaneous dissemination from primary site [60, 61].
Conclusion
In summary, this work demonstrated that somatic alterations of PMS2 contribute to brain metastases and provides compelling evidence that PMS2 upregulation is required for the tumor metastasizing to brain. We also suggested the potential of PMS2 targeting for therapeutic intervention for life-threatening brain metastases.
Supplementary Information
Additional file 1: Supplemental Table 1. Clinical characteristics of patients with brain metastases.
Additional file 2: Supplemental Figure 1. Waterfall diagram of gene mutations in tumor samples of M0 patients in TCGA database.
Additional file 3: Supplemental Figure 2. Overall survival plot of PMS2 amplification positive and negative patients.
Additional file 4: Supplemental Figure 3. The protein level of cell lines. (A) the ratio of PMS2 /GAPDH density in eight cell lines. (B) the ratio of PMS2 /β-actin density in transgenic lung cancer cell lines.
Additional file 5: Supplemental Figure 4. Overall survival of mice after intracardiac injection.
Additional file 6: Supplemental Figure 5. Histogram of fluorescence signal intensity in mouse brain.
Additional file 7: Supplemental Figure 6. Functional enrichment plot of PC9 cells before and after intracardiac injection.
Acknowledgements
We would like to thank Mr. Chengji Wang and Mr. Haijie Wang (Shanghai Laboratory Animal Research Center, Shanghai, China) for their technical support.
Abbreviations
- BM
Brain metastasis
- cfDNA
circulating cell-free DNA
- NSCLC
Non-small cell lung cancer
- EGFR
Epidermal growth factor receptor
- CNS
Central nervous system
- CSF
Cerebrospinal fluid
- MMR
Mismatch repair
- LS
Lynch Syndrome
- OS
Overall survival
- PFS
Progression-free survival
- TMB
Tumor mutational burden
- EDTA
Ethylenediaminetetraacetic
- BLI
Bioluminescence imaging
- MSI-H
High microsatellite instability
- BBB
Blood–brain barrier
- BTB
Blood tumor barrier
Authors’ contributions
Jianing Chen: Conceptualization; writing original draft; experiments operation; Data curation; formal analysis; Congli Hu: experiments operation; manuscript revision; Hainan Yang: Data curation; editing. Li Wang: Formal analysis; software; editing. Xiangling Chu: Data curation; review. Xin Yu: Data curation; formal analysis; editing. Shiji Zhang: Data curation; review. Xuefei Li: Investigation; review. Chao Zhao: Investigation; review. Lei Cheng: Investigation; review. Linbo Cai: Review; supervision; Lei Wen: Conceptualization; review; supervision; Chunxia Su: Project administration; supervision; funding acquisition.
Funding
This study was supported by the National Natural Science Foundation of China (grant numbers: 82072568,82373320), the Excellent Academic Leader of Shanghai “Science and Technology Innovation Action Plan” (grant number: 22XD1402500), the Shanghai Shenkang Hospital Development Center (grant number: SHDC12020110), and the Shanghai Shenkang development research physician project (grant number: SHDC2022CRD048).
Availability of data and materials
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
This research was approved by the ethics committee of Guangdong Sanjiu Brain Hospital and conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all patients.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Jianing Chen and Congli Hu contributed equally to this work.
Contributor Information
Lei Wen, Email: wenlei1998@sina.com.
Chunxia Su, Email: susu_mail@126.com.
References
- 1.Patel RR, Mehta MP. Targeted therapy for brain metastases: Improving the therapeutic ratio. Clin Cancer Res. 2007;13(6):1675–1683. doi: 10.1158/1078-0432.CCR-06-2489. [DOI] [PubMed] [Google Scholar]
- 2.Achrol AS, Rennert RC, Anders C, Soffietti R, Ahluwalia MS, Nayak L, et al. Brain metastases. Nat Rev Dis Prim. 2019;5(1):5. doi: 10.1038/s41572-018-0055-y. [DOI] [PubMed] [Google Scholar]
- 3.Brastianos PK, Curry WT, Oh KS. Clinical discussion and review of the management of brain metastases. J Natl Compr Canc Netw. 2013;11(9):1153–1164. doi: 10.6004/jnccn.2013.0133. [DOI] [PubMed] [Google Scholar]
- 4.Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, et al. Genomic characterization of brain metastases reveals branched evolution and potential therapeutic targets. Cancer Discov. 2015;5(11):1164–1177. doi: 10.1158/2159-8290.CD-15-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murtaza M, Dawson SJ, Tsui DWY, Gale D, Forshew T, Piskorz AM, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497(7447):108–112. doi: 10.1038/nature12065. [DOI] [PubMed] [Google Scholar]
- 6.Mok T, Wu YL, Lee JS, Yu CJ, Sriuranpong V, Sandoval-Tan J, et al. Detection and dynamic changes of EGFR mutations from circulating tumor DNA as a predictor of survival outcomes in NSCLC patients treated with first-line intercalated erlotinib and chemotherapy. Clin Cancer Res. 2015;21(14):3196–3203. doi: 10.1158/1078-0432.CCR-14-2594. [DOI] [PubMed] [Google Scholar]
- 7.Weston CL, Glantz MJ, Connor JR. Detection of cancer cells in the cerebrospinal fluid: current methods and future directions. Fluids Barriers CNS. 2011;8(1):14. doi: 10.1186/2045-8118-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shalaby T, Achini F, Grotzer MA. Targeting cerebrospinal fluid for discovery of brain cancer biomarkers. J Cancer Metastasis Treat. 2016;2016(2):176–187. [Google Scholar]
- 9.Li YS, Jiang BY, Yang JJ, Zhang XXC, Zhang Z, Ye JY, et al. Unique genetic profiles from cerebrospinal fluid cell-free DNA in leptomeningeal metastases of EGFR-mutant non-small-cell lung cancer: a new medium of liquid biopsy. Ann Oncol. 2018;29(4):945–952. doi: 10.1093/annonc/mdy009. [DOI] [PubMed] [Google Scholar]
- 10.Thompson E, Meldrum CJ, Crooks R, McPhillips M, Thomas L, Spigelman AD, et al. Hereditary non-polyposis colorectal cancer and the role of hPMS2 and hEXO1 mutations. Clin Genet. 2004;65(3):215–225. doi: 10.1111/j.1399-0004.2004.00214.x. [DOI] [PubMed] [Google Scholar]
- 11.Hegan DC, Narayanan L, Jirik FR, Edelmann W, Liskay RM, Glazer PM. Differing patterns of genetic instability in mice deficient in the mismatch repair genes Pms2, Mlh1, Msh2, Msh3 and Msh6. Carcinogenesis. 2006;27(12):2402–2408. doi: 10.1093/carcin/bgl079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alpert L, Pai RK, Srivastava A, McKinnon W, Wilcox R, Yantiss RK, et al. Colorectal Carcinomas With Isolated Loss of PMS2 Staining by Immunohistochemistry. Arch Pathol Lab Med. 2018;142(4):523–528. doi: 10.5858/arpa.2017-0156-OA. [DOI] [PubMed] [Google Scholar]
- 13.Bajwa-ten Broeke SW, Ballhausen A, Ahadova A, Suerink M, Bohaumilitzky L, Seidler F, et al. The coding microsatellite mutation profile of PMS2-deficient colorectal cancer. Exp Mole Pathol. 2021;122:104668. [DOI] [PubMed]
- 14.Borràs E, Pineda M, Cadiñanos J, del Valle J, Brieger A, Hinrichsen I, et al. Refining the role of pms2 in Lynch syndrome: germline mutational analysis improved by comprehensive assessment of variants. J Med Genet. 2013;50(8):552–563. doi: 10.1136/jmedgenet-2012-101511. [DOI] [PubMed] [Google Scholar]
- 15.Liccardo R, Della Ragione C, Mitilini N, De Rosa M, Izzo P, Duraturo F. Novel variants of unknown significance in the PMS2 gene identified in patients with hereditary colon cancer. Cancer Manage Res. 2019;11:6719–6725. doi: 10.2147/CMAR.S167348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugano K, Nakajima T, Sekine S, Taniguchi H, Saito S, Takahashi M, et al. Germline PMS2 mutation screened by mismatch repair protein immunohistochemistry of colorectal cancer in Japan. Cancer Sci. 2016;107(11):1677–1686. doi: 10.1111/cas.13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Starr J, Puebla G, McMillan J, Lewis JT, Kasi PM. Microsatellite instability-high, malignant insulinoma with brain metastasis. Cureus. 2021;13(8):e16969. [DOI] [PMC free article] [PubMed]
- 18.Lamba M, Wakeman C, Ebel R, Hamilton S, Frampton C, Kiesanowski M, et al. Associations Between Mutations in MSH6 and PMS2 and Risk of Surveillance-detected Colorectal Cancer. Clin Gastroenterol Hepatol. 2020;18(12):2768–2774. doi: 10.1016/j.cgh.2020.03.048. [DOI] [PubMed] [Google Scholar]
- 19.Ye SL, Wang HH, He KC, Peng M, Wang YH, Li YW, et al. Clinical characterization of mismatch repair gene-deficient metastatic castration-resistant prostate cancer. Front Oncol. 2020;10:533282. [DOI] [PMC free article] [PubMed]
- 20.Roberts ME, Jackson SA, Susswein LR, Zeinomar N, Ma XR, Marshall ML, et al. MSH6 and PMS2 germ-line pathogenic variants implicated in Lynch syndrome are associated with breast cancer. Genet Med. 2018;20(10):1167–1174. doi: 10.1038/gim.2017.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andrianova MA, Chetan GK, Sibin MK, McKee T, Merkler D, Narasinga R, et al. Germline PMS2 and somatic POLE exonuclease mutations cause hypermutability of the leading DNA strand in biallelic mismatch repair deficiency syndrome brain tumours. J Pathol. 2017;243(3):331–341. doi: 10.1002/path.4957. [DOI] [PubMed] [Google Scholar]
- 22.Yang RR, Li KKW, Zhang ZY, Chan AKY, Wang WW, Chan DTM, et al. Mismatch repair proteins PMS2 and MLH1 can further refine molecular stratification of IDH-mutant lower grade astrocytomas. Clin Neurol Neurosurg. 2021;208:106882. [DOI] [PubMed]
- 23.Krol I, Castro-Giner F, Maurer M, Gkountela S, Szczerba BM, Scherrer R, et al. Detection of circulating tumour cell clusters in human glioblastoma. Br J Cancer. 2018;119(4):487–491. doi: 10.1038/s41416-018-0186-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salem ME, Bodor JN, Puccini A, Xiu J, Goldberg RM, Grothey A, et al. Relationship between MLH1, PMS2, MSH2 and MSH6 gene-specific alterations and tumor mutational burden in 1057 microsatellite instability-high solid tumors. Int J Cancer. 2020;147(10):2948–2956. doi: 10.1002/ijc.33115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeng M, Narayanan L, Xu XXS, Prolla TA, Liskay RM, Glazer PM. Ionizing radiation-induced apoptosis via separate Pms2-and p53-dependent pathways. Can Res. 2000;60(17):4889–4893. [PubMed] [Google Scholar]
- 26.Fink D, Nebel S, Aebi S, Nehme A, Howell SB. Loss of DNA mismatch repair due to knockout of MSH2 or PMS2 results in resistance to cisplatin and carboplatin. Int J Oncol. 1997;11(3):539–542. doi: 10.3892/ijo.11.3.539. [DOI] [PubMed] [Google Scholar]
- 27.Mao XW, Zhang Z, Zheng XX, Xie FF, Duan FD, Jiang LY, et al. Capture-Based Targeted Targeted Ultradeep Sequencing in Paired Tissue and Plasma Samples Demonstrates Differential Subclonal ctDNA-Releasing Capability in Advanced Lung Cancer. J Thorac Oncol. 2017;12(4):663–672. doi: 10.1016/j.jtho.2016.11.2235. [DOI] [PubMed] [Google Scholar]
- 28.Palmieri D, Lockman PR, Thomas FC, Hua E, Herring J, Hargrave E, et al. Vorinostat Inhibits Brain Metastatic Colonization in a Model of Triple-Negative Breast Cancer and Induces DNA Double-Strand Breaks. Clin Cancer Res. 2009;15(19):6148–6157. doi: 10.1158/1078-0432.CCR-09-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng Z, Cui L, Li P, Ren N, Zhong Z, Tang Z, et al. Genomic comparison between cerebrospinal fluid and primary tumor revealed the genetic events associated with brain metastasis in lung adenocarcinoma. Cell Death Dis. 2021;12(10):935. doi: 10.1038/s41419-021-04223-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vassella E, Kashani E, Zens P, Kuendig A, Fung C, Scherz A, et al. Mutational profiles of primary pulmonary adenocarcinoma and paired brain metastases disclose the importance of KRAS mutations. Eur J Cancer. 2021;159:227–36. doi: 10.1016/j.ejca.2021.10.006. [DOI] [PubMed] [Google Scholar]
- 32.Collins VP. Brain tumours: classification and genes. J Neurol Neurosurg Psychiatry. 2004;75(Suppl 2):ii2–11. doi: 10.1136/jnnp.2004.040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bos PD, Zhang XHF, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459(7249):1005–U137. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao P, Li L, Jiang X, Li Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J Hematol Oncol. 2019;12:1–14. doi: 10.1186/s13045-019-0738-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beltran H. DNA Mismatch Repair in Prostate Cancer. J Clin Oncol. 2013;31(14):1782–4. doi: 10.1200/JCO.2012.48.4667. [DOI] [PubMed] [Google Scholar]
- 36.Latham A, Srinivasan P, Kemel Y, Shia J, Bandlamudi C, Mandelker D, et al. Microsatellite Instability Is Associated With the Presence of Lynch Syndrome Pan-Cancer. J Clin Oncol. 2019;37(4):286. doi: 10.1200/JCO.18.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aarnio M, Sankila R, Pukkala E, Salovaara R, Aaltonen LA, de la Chapelle A, et al. Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer. 1999;81(2):214–8. doi: 10.1002/(sici)1097-0215(19990412)81:2<214::aid-ijc8>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 38.Talseth-Palmer BA, McPhillips M, Groombridge C, Spigelman A, Scott RJ. MSH6 and PMS2 mutation positive Australian Lynch syndrome families: novel mutations, cancer risk and age of diagnosis of colorectal cancer. Hereditary Cancer Clin Pract. 2010;8:1–10. doi: 10.1186/1897-4287-8-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sedhom R, Antonarakis ES. Clinical implications of mismatch repair deficiency in prostate cancer. Future Oncol. 2019;15(20):2395–411. doi: 10.2217/fon-2019-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duval A, Hamelin R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: Toward a new concept of target genes for instability. Can Res. 2002;62(9):2447–54. [PubMed] [Google Scholar]
- 41.Peltomäki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol. 2003;21(6):1174–9. doi: 10.1200/JCO.2003.04.060. [DOI] [PubMed] [Google Scholar]
- 42.Zysman M, Saka A, Millar A, Knight J, Chapman W, Bapat B. Methylation of adenomatous polyposis coli in endometrial cancer occurs more frequently in tumors with microsatellite instability phenotype. Can Res. 2002;62(13):3663–6. [PubMed] [Google Scholar]
- 43.Gibson SL, Narayanan L, Hegan DC, Buermeyer AB, Liskay RM, Glazer PM. Overexpression of the DNA mismatch repair factor, PMS2, confers hypermutability and DNA damage tolerance. Cancer Lett. 2006;244(2):195–202. doi: 10.1016/j.canlet.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 44.Haddad-Tóvolli R, Dragano NRV, Ramalho AFS, Velloso LA. Development and function of the blood-brain barrier in the context of metabolic control. Front Neurosci. 2017;11:261080. [DOI] [PMC free article] [PubMed]
- 45.McGale EHF, Pye IF, Stonier C, Hutchinson EC, Aber GM. Studies of the inter-relationship between cerebrospinal fluid and plasma amino acid concentrations in normal individuals. J Neurochem. 1977;29(2):291–7. doi: 10.1111/j.1471-4159.1977.tb09621.x. [DOI] [PubMed] [Google Scholar]
- 46.Bergers G, Fendt S-M. The metabolism of cancer cells during metastasis. Nat Rev Cancer. 2021;21(3):162–80. doi: 10.1038/s41568-020-00320-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Elia I, Doglioni G, Fendt S-M. Metabolic hallmarks of metastasis formation. Trends Cell Biol. 2018;28(8):673–84. doi: 10.1016/j.tcb.2018.04.002. [DOI] [PubMed] [Google Scholar]
- 48.Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. 2020;368(6487):eaaw5473. [DOI] [PMC free article] [PubMed]
- 49.Lehuede C, Dupuy F, Rabinovitch R, Jones RG, Siegel PM. Metabolic plasticity as a determinant of tumor growth and metastasis. Can Res. 2016;76(18):5201–8. doi: 10.1158/0008-5472.CAN-16-0266. [DOI] [PubMed] [Google Scholar]
- 50.Li F, Simon MC. Cancer cells don't live alone: metabolic communication within tumor microenvironments. Dev Cell. 2020;54(2):183–95. doi: 10.1016/j.devcel.2020.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen J, Lee H-J, Wu X, Huo L, Kim S-J, Xu L, et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Can Res. 2015;75(3):554–65. doi: 10.1158/0008-5472.CAN-14-2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ngo B, Kim E, Osorio-Vasquez V, Doll S, Bustraan S, Liang RJ, et al. Limited environmental serine and glycine confer brain metastasis sensitivity to PHGDH inhibition. Cancer Discov. 2020;10(9):1352–73. doi: 10.1158/2159-8290.CD-19-1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cordero A, Kanojia D, Miska J, Panek WK, Xiao A, Han Y, et al. FABP7 is a key metabolic regulator in HER2+breast cancer brain metastasis. Oncogene. 2019;38(37):6445–60. doi: 10.1038/s41388-019-0893-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zou Y, Watters A, Cheng N, Perry CE, Xu K, Alicea GM, et al. Polyunsaturated fatty acids from astrocytes activate PPARγ signaling in cancer cells to promote brain metastasis. Cancer Discov. 2019;9(12):1720–35. doi: 10.1158/2159-8290.CD-19-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Santana-Codina N, Marcé-Grau A, Muixí L, Nieva C, Marro M, Sebastián D, et al. GRP94 is involved in the lipid phenotype of brain metastatic cells. Int J Mole Sci. 2019;20(16):3883. doi: 10.3390/ijms20163883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parida PK, Marquez-Palencia M, Nair V, Kaushik AK, Kim K, Sudderth J, et al. Metabolic diversity within breast cancer brain-tropic cells determines metastatic fitness. Cell Metab. 2022;34(1):90–105. doi: 10.1016/j.cmet.2021.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Quail DF, Joyce JA. The Microenvironmental landscape of brain tumors. Cancer Cell. 2017;31(3):326. doi: 10.1016/j.ccell.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schulz M, Michels B, Niesel K, Stein S, Farin H, Rodel F, et al. Cellular and molecular changes of brain metastases-associated myeloid cells during disease progression and therapeutic response. iScience. 2020;23(6):101178. [DOI] [PMC free article] [PubMed]
- 59.Klemm F, Mock A, Salamero-Boix A, Alekseeva T, Schaffer A, Schulz M, et al. Compensatory CSF2-driven macrophage activation promotes adaptive resistance to CSF1R inhibition in breast-to-brain metastasis. Nat Cancer. 2021;2(10):1086-1101. [DOI] [PubMed]
- 60.Boire A, Zou Y, Shieh J, Macalinao DG, Pentsova E, Massague J. Complement Component 3 Adapts the Cerebrospinal Fluid for Leptomeningeal Metastasis. Cell. 2017;168(6):1101–1113. [DOI] [PMC free article] [PubMed]
- 61.Valiente M, Van Swearingen AED, Anders CK, Bairoch A, Boire A, Bos PD, et al. Brain metastasis cell lines panel: a public resource of organotropic cell lines. Can Res. 2020;80(20):4314–23. doi: 10.1158/0008-5472.CAN-20-0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Supplemental Table 1. Clinical characteristics of patients with brain metastases.
Additional file 2: Supplemental Figure 1. Waterfall diagram of gene mutations in tumor samples of M0 patients in TCGA database.
Additional file 3: Supplemental Figure 2. Overall survival plot of PMS2 amplification positive and negative patients.
Additional file 4: Supplemental Figure 3. The protein level of cell lines. (A) the ratio of PMS2 /GAPDH density in eight cell lines. (B) the ratio of PMS2 /β-actin density in transgenic lung cancer cell lines.
Additional file 5: Supplemental Figure 4. Overall survival of mice after intracardiac injection.
Additional file 6: Supplemental Figure 5. Histogram of fluorescence signal intensity in mouse brain.
Additional file 7: Supplemental Figure 6. Functional enrichment plot of PC9 cells before and after intracardiac injection.
Data Availability Statement
No datasets were generated or analysed during the current study.