

Abstract

Canthin-4-one is synthesized via a six-step procedure starting from commercially available 3-amino-4-bromopyridine in 26% overall yield. 3-Amino-4-bromopyridine is initially converted to 8-bromo-1,5-naphthyridin-4(1H)-one. O-Methylation, intermolecular Pd-catalyzed C–C coupling, and demethylation afford the key intermediate, 8-(2-chlorophenyl)-1,5-naphthyridin-4(1H)-one, for which intramolecular C–N coupling completes the synthesis of the canthin-4-one skeleton. Ten canthin-4-one analogues were prepared in addition to the parent compound. With minor modifications, the synthesis also applies to the synthesis of two series of isocanthin-4-ones.
Introduction
Canthin-4-ones represent the lesser-known family of canthine alkaloids,1 the most well-known of which are the canthin-6-ones.2 More than 60 natural analogues of canthin-6-ones have been isolated to date, but only four natural analogues of canthin-4-ones are known: tuboflavine,3 norisotuboflavine,4 isotuboflavine,4 and oxoproopaline H5 (Figure 1). The latter was only recently isolated from a mutant ΔstnK4 of Streptomyces flocculus CGMCC4, and its structure elucidated by extensive one- and two-dimensional nuclear magnetic resonance (NMR).5
Figure 1.

Structures of canthin-6-one and canthin-4-one and natural analogues of canthin-4-one.
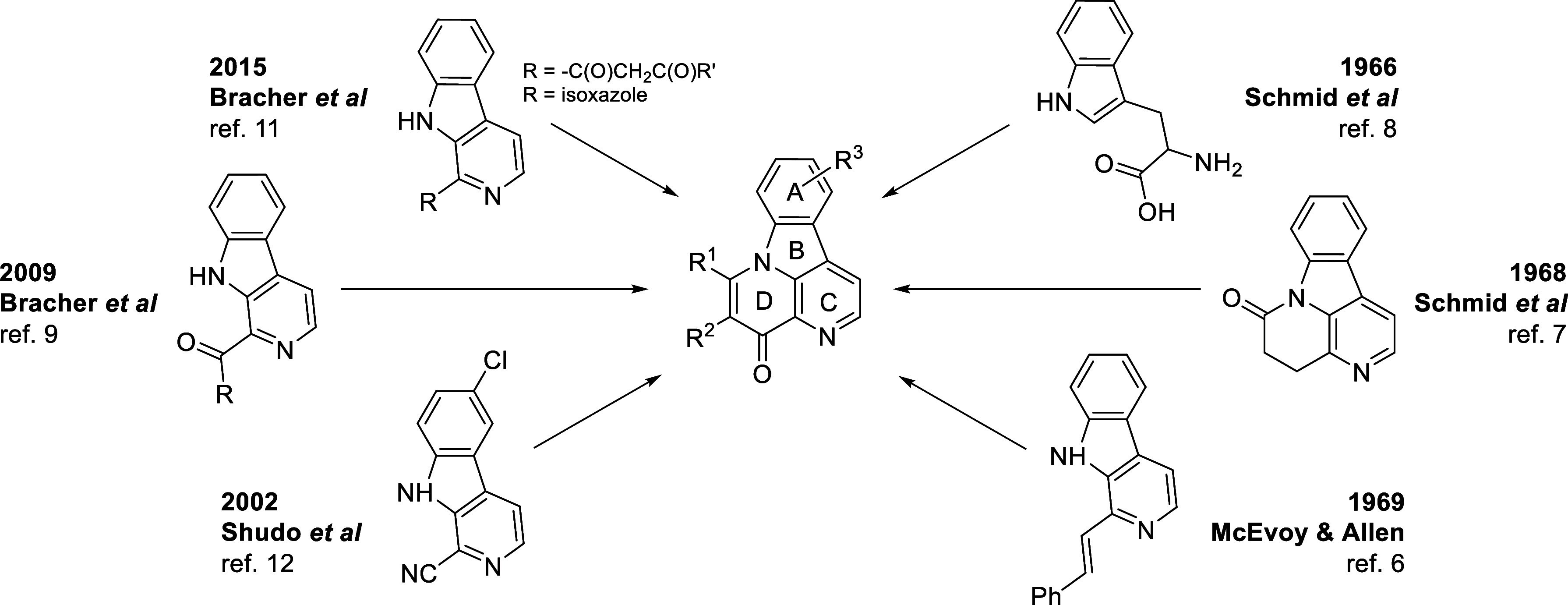
Several syntheses for norisotuboflavine,6 isotuboflavine,7 and tuboflavine8 have been reported. In 1966, Schmid et al. reported a four-step synthesis for tuboflavine, starting from tryptophan in 0.3% overall yield,7 while in 1968, they reported the syntheses of norisotuboflavine and isotuboflavine,6 in three steps starting from 4,5-dihydrocanthin-6-one in <1% overall yield. An alternative five-step synthesis for norisotuboflavine was also reported by McEvoy and Allen, starting from the natural product benzalharman with an overall yield of <1%.6 More recently, in 2009, Bracher reported a one-step, moderate- to good-yielding synthesis by reaction of carbolines with Bredereck’s reagent.9 The same approach was used for the synthesis of 5-methoxycanthin-4-one,10 which was mistakenly identified as a new alkaloid named drymaritin isolated from Drymaria diandra,11 which was then confirmed to be the already known alkaloid cordatanine (4-methoxycanthin-6-one).12 In 2015, Bracher et al. reported two new methods for the syntheses of norisotuboflavine and tuboflavine.13 The first involved the reaction of 1-acetyl-β-carboline with either N-acetylbenzotriazole or N-propanoylbenzotriazole, which, via the in situ formation of 1,3-diketone intermediates, afforded in one step norisotuboflavine and isotuboflavine, in 34% and 16% yields, respectively. The second approach involved a four-step sequence starting from 1-bromo-β-carboline giving norisotuboflavine and isotuboflavine, in 50% and 47% yields, respectively. While these routes (Scheme 1) provided the natural products in yields higher than those of the prior syntheses, they lacked versatility. Multistep routes to synthetic canthin-4-ones starting from 9H-pyrido[3,4-b]indole-1-carbonitrile were also reported in the patent literature but with low overall yields.14 A high-yielding synthesis of chiral S-(−)-5,6-dihydrocanthin-4-ones was recently reported by Batra et al.15
Scheme 1. Literature Syntheses of Canthin-4-ones.
While scarce, both natural and synthetic canthin-4-ones have potential in medicinal chemistry, with antimicrobial16 and phosphodiesterase-inhibitory14 activities reported to date. Nevertheless, due to the lack of good general syntheses, there is limited access to structurally diverse canthin-4-one libraries, which are needed to probe structure–activity relationships. In addition, all of the known routes are based on the construction of the D ring via β-carboline precursors.
In 2017, we reported the synthesis of 3-deazacanthin-4-ones via construction of the B ring,17 starting from 8-haloquinol-4-ones using sequential Suzuki–Miyaura and C–N coupling reactions. This route allowed diversification of the A ring thanks to commercially accessible 2-halo(het)aryl boronic acids, esters, and trifluoroborates. The method was originally introduced by our group for the synthesis of canthin-6-ones.18 To apply this synthesis to canthin-4-ones, we needed access to unknown 8-bromo-1,5-naphthyridin-4(1H)-one (2) (Scheme 2). Our initial efforts to synthesize this scaffold via the thermolysis of the corresponding ylidene 1 gave only intractable tar. Herein, we report the successful thermolysis of aza-ylidene 1 and the subsequent synthesis of canthin-4-ones.
Scheme 2. Synthesis of 8-Bromo-1,5-naphthyridin-4(1H)-one (2).

Reagents and conditions: (i) Meldrum’s acid (1.1 equiv), CH(OEt)3 (2.0 equiv), MeCN, reflux, 1.75 h, 91%; (ii) Ph2O (0.08 mol/L), 250 °C, 8.5 min (2, 12%; 3, 4%); (iii) Ph2O (0.03 mol/L), 250 °C, 0.5 min (2, 52%; 3, 0%).
Furthermore, we extend the method to structurally related isocanthin-4-ones.
Results and Discussion
Ylidene 1, needed to access the desired 8-bromo-1,5-naphthyridin-4(1H)-one (2), was prepared according to literature procedures from commercially available 3-amino-4-bromopyridine in 91% yield. Initial attempts to thermolyze ylidene 1 under conditions analogous to those used for the synthesis of 8-bromoquinol-4-one 2 (∼0.61 mol/L at 220 °C, 5 min)17 led to the formation of intractable black solids. Dilution of the reaction mixture gave intractable black solids along with naphthyridinone 2 in 12% yield and side product 3 in 4% yield (Scheme 2).
The structural elucidation of side product 3 was not immediately obvious (see section S1 of the Supporting Information), but its formation suggested further dilution of the reaction mixture and/or shortening of the reaction time. After a series of trial-and-error experiments, the optimum concentration of the reaction was found, 0.03 mol/L, at ∼250 °C for 0.5 min. The reaction performed at a 1.83 mmol scale gave the desired naphthyridinone 2 in 52% yield. Attempts to improve this yield further were unsuccessful. Initial attempts to create an efficient C–C coupling at position C-8 of 8-bromonaphthyridinone 2 involved the reaction of naphthyridinone 2 with 2-chlorophenylboronic acid (1.5 equiv), using two sets of conditions: (i) Pd(dppf)Cl2·DCM (5 mol %), K2CO3 (2 equiv), and dioxane/H2O (75:25) or (ii) XPhos Pd G4 (2.5 mol %), K2CO3 (2 equiv), and MeOH. Both sets of conditions were investigated under conventional heating or microwave (MW) irradiation, under an air or Ar atmosphere, but failed to give a complete reaction [20–30% of the starting material consumed by thin layer chromatography (TLC)]. Tentatively, this was attributed to poisoning of the Pd catalyst with naphthyridinone 2 acting as a bidentate ligand to form complex 4. Complexes of Pd with bidentate ligands with a similar structural motif are reported in the literature.19
To resolve this, O-methylated analogue 5 was prepared using MeI and K2CO3 in DMF (Scheme 3). Gratifyingly, the reaction of methoxynaphthyridine 5 with 2-chlorophenylboronic acid (1.5 equiv), Pd(dppf)Cl2·DCM (5 mol %), and K2CO3 (2 equiv) in a dioxane/H2O (75:25) solvent heated at reflux gave the desired product 6 in high yield. The loading of the catalyst could be decreased to 1.25 mol % without any detrimental effect on the reaction time or yield. O-Demethylation of methoxynaphthyridine 6 using aqueous HCl in dioxane gave 7, and subsequent C–N coupling gave the desired canthin-4-one 8a (76%) (18% over seven steps, starting from commercial aminopyridine). The last three steps (Suzuki coupling, demethylation, and C–N coupling) were telescoped, affording canthin-4-one 8a in an improved yield of 80% versus 54% over the three steps, and a 26% overall yield from 3-amino-4-bromopyridine.
Scheme 3. Synthesis of Canthin-4-one 8a from 8-Bromo-1,5-naphthyridin-4(1H)-one (2).

Reagents and conditions: (i) MeI (1.2 equiv), K2CO3 (1.2 equiv), DMF, ∼20 °C, 16 h, 70%; (ii) 2-ClC6H4B(OH)2 (1.5 equiv), K2CO3 (2 equiv), Pd(dppf)Cl2·DCM (1.25 mol %), dioxane/H2O (75:25), ∼88 °C, 8 min, 74%; (iii) concentrated HCl/H2O (50:50), dioxane, ∼101 °C, 40 min, 96%; (iv) K2CO3 (2 equiv), CuI (10 mol %), N,N′-dimethyl-trans-1,2-cyclohexanediamine (DMCDA) (20 mol %), dioxane/H2O (75:25), ∼88 °C, 5.5 h, 76%; (v) (a) 2-ClC6H4B(OH)2 (1.5 equiv), K2CO3 (2 equiv), Pd(dppf)Cl2·DCM (1.25 mol %), dioxane/H2O (75:25), ∼88 °C, 12 min; (b) concentrated HCl, dioxane/H2O (75:25), ∼88 °C, 2 h; (c) K2CO3 (10 equiv), CuI (10 mol %), DMCDA (20 mol %), dioxane/H2O (75:25), ∼88 °C, 12 h, 80%.
With a successful synthesis for canthin-4-one 8a, we applied this method to synthesize isocanthin-4-ones. Translocating the nitrogen atom in the C ring can give two possible isocanthin-4-ones, 9a and 10a. Of these, only isocanthin-4-one 9a and four of its derivatives have appeared in a patent.14
As in the case of canthin-4-one, the literature synthesis of isocanthin-4-one 9a involves the construction of the D ring. Fortunately, the required naphthyridinone 11 needed for the synthesis of isocanthin-4-one 9a via our approach was known.20 The Suzuki–Miyaura coupling of naphthyridinone 11 with 2-chlorophenylboronic acid, using our standard conditions, led to considerable unreacted starting material (TLC). Increasing the Pd loading from 5 to 25 mol % or performing the reaction under MW radiation20 led to a complete reaction; however, in both cases, product 13 was obtained as a mixture with protodechlorinated 8-phenylnaphthyridinone 12, which was difficult to separate (Scheme 4). Protodechlorinated product 12 was synthesized in pure form by reaction of the naphthyridinone with phenylboronic acid, which allowed its full characterization (see section S2). Interestingly, with phenylboronic acid the reaction was performed successfully under conventional heating and 5 mol % Pd(dppf)Cl2·DCM giving the desired product in 71% yield. Further development of the reaction with 2-chlorophenylboronic acid was pursued using the MW conditions, owing to the lower catalyst loading and cleaner reaction mixture observed under these conditions. The C–N coupling of key intermediate 13 was also performed under MW radiation. The two-step reaction sequence, Suzuki–Miyaura coupling followed by C–N coupling, was telescoped to afford the desired isocanthin-4-one 9a in 69% yield.
Scheme 4. Synthesis of Isocanthin-4-one 9a.

Reagents and conditions: (i) 2-ClC6H4B(OH)2 (1.5 equiv), Pd(dppf)Cl2·DCM (5 mol %), K2CO3 (2 equiv), dioxane/H2O (75:25), MW (T = 120 °C, 150 W, ∼50 psi), 45 min; (ii) K2CO3 (2 equiv), CuI (10 mol %), DMCDA (20 mol %), dioxane/H2O (75:25), MW (T = 120 °C, 150 W, ∼50 psi), 6 h; (iii) (a) 2-ClC6H4B(OH)2 (1.5 equiv), Pd(dppf)Cl2·DCM (5 mol %), K2CO3 (2 equiv), dioxane/H2O (75:25), MW (T = 120 °C, 150 W, ∼50 psi), 45 min; (b) K2CO3 (2 equiv), CuI (10 mol %), DMCDA (20 mol %), dioxane/H2O (75:25), MW (T = 120 °C, 150 W, ∼50 psi), 6 h, 69%.
The telescoped reaction was also performed on a larger scale using conventional heating conditions and an increased level of the Pd catalyst (25 mol %) to give the desired isocanthin-4-one 9a in 60% yield.
In a similar manner, 8-bromo-1,7-naphthyridin-4(1H)-one (15) could give us access to isocanthin-4-one 10a. Naphthyridinone 15 was not reported in the literature, and like naphthyridinone 2, our initial attempts to thermolyze ylidene 14 led to complex mixtures. Nevertheless, applying our “optimized” high-dilution thermolysis conditions described above to ylidene 14 gave the desired product 15 in 85% yield.
Our standard Suzuki–Miyaura coupling conditions worked well for this analogue giving a complete reaction within 10 min, without the need for MW irradiation or increased Pd loading. As in the case of naphthyridinone 11, the reaction led to a difficult to separate mixture of the desired product 17 and protodechlorinated side product 16. Performing the C–N coupling in one pot gave the desired isocanthin-4-one 10a in 77% yield (Scheme 5).
Scheme 5. Synthesis of Isocanthin-4-one 10a.

Reagents and conditions: (i) Meldrum’s acid (1.1 equiv), CH(OEt)3 (1.2 equiv), MeCN, ∼82 °C, 5 h, 75%; (ii) Ph2O, ∼250 °C, 0.5 min, 85%; (iii) 2-ClC6H4B(OH)2 (1.5 equiv), Pd(dppf)Cl2·DCM (5 mol %), K2CO3 (2 equiv), dioxane/H2O (75:25), ∼88 °C, 15 min; (iv) K2CO3 (2 equiv), CuI (10 mol %), DMCDA (20 mol %), H2O (1 equiv), dioxane/H2O (75:25), ∼88 °C, 12 h, 77%.
With three optimized procedures in hand for the synthesis of canthin-4-one 8a and isocanthin-4-ones 9a and 10a, a variety of 2-chlorophenylboronic acids were subjected to our optimized reaction conditions (Table 1). Sterically hindered 6-substituted (6-MeO and 6-Cl) 2-chlorophenylboronic acids failed in the Suzuki–Miyaura coupling. Both electron-releasing and -withdrawing substituents were tolerated in the synthesis of canthinones 8 and isocanthinones 10. In the synthesis of isocanthinones 9, the reaction was more sensitive to electron-withdrawing substituents, which afforded lower yields. In total, 28 new substituted analogues were prepared.
Table 1. Analogues of Canthin-4-one 8 and Isocanthin-4-ones 9 and 10.

| entry | conditiona | ArB(OH)2 (equiv) | FGb | first step (min) | yield (%) |
|---|---|---|---|---|---|
| 1 | A | 1.5 | 4-Me | 10 | 8b (74) |
| 2 | A | 1.5 | 4-MeO | 10 | 8c (35) |
| 3 | A | 3.0 | 4-F3C | 10 | 8d (67) |
| 4 | A | 1.5 | 4-Cl | 20 | 8e (35) |
| 5 | A | 1.5 | 4-F | 10 | 8f (89) |
| 6 | A | 1.5 | 5-MeO | 10 | 8g (69) |
| 7 | A | 3.0 | 5-F3CO | 10 | 8h (67) |
| 8 | A | 3.0 | 5-F3C | 20 | 8i (63) |
| 9 | A | 3.0 | 5-Cl | 10 | 8j (61) |
| 10 | A | 1.5 | 5-F | 10 | 8k (63) |
| 11 | B | 1.5 | 4-Me | 45 | 9b (88) |
| 12 | B | 3.0 | 4-MeO | 45 | 9c (68) |
| 13 | B | 3.0 | 4-F3C | 45 | 9d (49) |
| 14 | B | 1.5 | 4-F | 45 | 9e (36) |
| 15 | B | 1.5 | 5-MeO | 45 | 9f (85) |
| 16 | B | 3.0 | 5-F3CO | 45 | 9g (26) |
| 17 | B | 1.5 | 5-F | 45 | 9h (23) |
| 18 | C | 1.5 | 3-Cl | 10 | 10b (47) |
| 19 | C | 1.5 | 4-Me | 15 | 10c (78) |
| 20 | C | 1.5 | 4-MeO | 5 | 10d (81) |
| 21 | C | 3.0 | 4-F3C | 50 | 10e (47) |
| 22 | C | 3.0 | 4-Cl | 12 | 10f (71) |
| 23 | C | 1.5 | 4-F | 60 | 10g (67) |
| 24 | C | 1.5 | 5-MeO | 15 | 10h (66) |
| 25 | C | 1.5 | 5-F3CO | 20 | 10i (62) |
| 26 | C | 3.0 | 5-Cl | 10 | 10j (65) |
| 27 | C | 1.5 | 5-F | 6 | 10k (70) |
Condition A (one pot): 5 (1 equiv), PdCl2(dppf)·DCM (1.25 mol %), K2CO3 (2 equiv), dioxane/H2O (75:25), ∼88 °C; (ii) concentrated HCl, ∼88 °C, 2 h; (iii) K2CO3 (10 equiv), CuI (10 mol %), DMCDA (20 mol %), ∼88 °C, 12 h. Condition B (one pot): (i) 11 (1 equiv), PdCl2(dppf)·DCM (1.25 mol %), K2CO3 (2 equiv), dioxane/H2O (75:25), MW (150 W, T = 120 °C, ∼60 psi), 45 min; (ii) K2CO3 (2 equiv), CuI (10 mol %), DMCDA (20 mol %), MW (150 W, T = 120 °C, ∼60 psi), 6 h. Condition C (one pot): (i) 15 (1 equiv), PdCl2(dppf)·DCM (5 mol %), K2CO3 (2 equiv), dioxane/H2O (75:25), ∼88 °C; (ii) K2CO3 (2 equiv), CuI (10 mol %), DMCDA (20 mol %), ∼88 °C, 12 h.
Functional group.
Conclusions
A new and efficient method for accessing canthin-4-ones via the construction of the B ring was developed. The parent canthin-4-one was prepared in six steps from commercially available 3-amino-4-bromopyridine, with the last three steps telescoped in one pot (26% overall yield). A key step was the synthesis of the previously unknown 8-bromo-1,5-naphthyridin-4(1H)-one (2). Suzuki–Miyaura coupling with commercially available 2-chlorophenylboronic acids followed by Cu-catalyzed C–N coupling afforded the canthin-4-one skeleton. The method was general with 10 analogues prepared in good yields (35–89%). The methodology was also applied for the synthesis of two aza isomers of the canthin-4-one. In this way, a library of 19 isocanthin-4-ones was also prepared (23–88% yields).
Experimental Section
General Methods and Materials
All chemicals were commercially available except those whose synthesis is described. All volatiles were removed under reduced pressure. The DCM/NH3 solvent mixture was prepared by extracting aqueous NH4OH (∼500 mL) with DCM (4 × 500 mL). The DCM layers were combined, dried (Na2SO4), filtered, and stored in an amber glass bottle. Dioxane was distilled from CaH2 before use. The combined DCM extracts were dried (Na2SO4), filtered, and stored in a glass bottle. All microwave experiments were performed in a CEM Discover Microwave Reactor with an external surface sensor in sealed vials. For conventional heating of the reaction mixtures, aluminum heating blocks were used. All reaction mixtures and column eluents were monitored by TLC using commercial aluminum-backed TLC plates (Kieselgel 60 F254). The plates were observed under ultraviolet (UV) light at 254 and 365 nm. The technique of dry flash chromatography was used throughout for all non-TLC scale chromatographic separations using silica gel 60 (<0.063 mm).21 Melting points were determined using a PolyTherm-A, Wagner & Munz, Kofler hot-stage microscope apparatus or using a TA Instruments DSC Q1000 instrument with samples hermetically sealed in aluminum pans under an argon atmosphere [using heating rates of 5 °C/min (DSC mp listed by onset and peak values)]. Samples were purified by bulk recrystallization from hot, filtered saturated solutions slowly cooled to room temperature (unless otherwise stated). Recrystallization solvents are listed after melting points. UV–visible spectra were recorded using a Shimadzu UV-1900 spectrophotometer, and inflections are identified by the abbreviation “inf”. Infrared spectra were recorded on a Shimadzu FTIR-NIR Prestige-21 spectrometer fitted with a Pike Miracle Ge ATR accessory, and strong, medium, and weak peaks are represented by s, m, and w, respectively. 1H and 13C NMR spectra were recorded, as indicated, on a Bruker Avance 300 machine at 300 and 75 MHz, respectively, or on a Bruker Avance 500 machine at 500 and 125 MHz, respectively. Deuterated solvents were used for homonuclear lock, and the signals are referenced to the deuterated solvent peaks. Attached proton test (APT) NMR studies were used for the assignment of the 13C peaks C (quaternary), CH2, CH3, and CH4 as C (s), C (d), C (t), and C (q), respectively. Mass spectrometry data were recorded with a matrix-assisted laser desorption/ionization time of flight (MALDI-TOF) mass spectrometer (positive mode) on a Bruker Autoflex III Smartbeam instrument or with the Agilent 1260 Infinity II Preparative LC/MSD System. Elemental analysis was performed using a Euro-Vector EA3000 CHN elemental analyzer. 8-Bromo-1,6-naphthyridin-4(1H)-one (11) was prepared according to a literature procedure (see the Supporting Information).20
Synthesis of Meldrum’s Acid Ylidenes
5-{[(4-Bromopyrid-3-yl)amino]methylene}-2,2-dimethyl-1,3-dioxane-4,6-dione (1)
To a solution of 3-amino-4-bromopyridine (3.06 g, 17.7 mmol) in MeCN (50 mL) were added Meldrum’s acid (2.93 g, 20.3 mmol) and CH(OEt)3 (6.0 mL, 36.0 mmol), and the mixture was heated to ∼82 °C (reflux) for 1.75 h. Then the solvent was removed in vacuo, and the residue was recrystallized to give compound 1 (5.79 g, 91%) as beige-pink needles: mp (hot stage) 161.5–163.0 °C (EtOH); mp (DSC) onset 164.61 °C, peak max 165.25 °C (EtOH); Rf = 0.48 (80:20 DCM/t-BuOMe); λmax (DCM, nm) 267 inf (log ε = 3.85), 271 inf (3.87), 290 inf (4.08), 300 inf (4.17), 326 (4.41); vmax (ATR, cm–1) 1730m (C=O), 1678m, 1607s, 1555m, 1433m, 1317m, 1312m, 1269s, 1234m, 1221m, 1206m, 1184m, 1145m, 1065m, 1018m, 1009m, 935m, 891w, 853w, 816m, 799m, 787m, 752w, 727m; 1H NMR (CDCl3, 500 MHz) δ 11.56 (1H, d, J = 13.0 Hz), 8.683 (1H, s), 8.678 (1H, d, J = 13.5 Hz), 8.32 (1H, d, J = 5.5 Hz), 7.62 (1H, d, J = 5.5 Hz), 1.77 (6H, s); 13C{1H} NMR (CDCl3, 125 MHz) δ 165.4 (s), 163.0 (s), 152.0 (d), 147.8 (d), 139.1 (d), 134.2 (s), 128.2 (d), 124.6 (s), 105.8 (s), 90.1 (s), 27.4 (q); m/z (MALDI-TOF) 329 (81Br, MH+, 31%), 327 (79Br, MH+, 30), 297 (23), 271 (90), 269 (100), 227 (71), 225 (82). Anal. Calcd for C12H11BrN2O4: C, 44.06; H, 3.39; N, 8.56. Found: C, 44.01; H, 3.47; N, 8.43.
5-{[(2-Bromopyrid-3-yl)amino]methylene}-2,2-dimethyl-1,3-dioxane-4,6-dione (14)
To a solution of 3-amino-2-bromopyridine (1.2 g, 6.8 mmol) in anhydrous MeCN (25 mL) were added Meldrum’s acid (1.07 g, 7.5 mmol) and CH(OEt)3 (1.4 mL, 8.2 mmol), and the mixture was heated to ∼82 °C (reflux) for 5 h. The solvent was evaporated in vacuo, and the solid residue was recrystallized to give compound 14 (1.66 g, 75%) as pale pink needles: mp (hot stage) 170.6–172.0 °C (EtOH); mp (DSC) onset 173.6 °C, peak max 173.9 °C (EtOH); decomposition (DSC) onset 191.6 °C, peak max 196.4 °C; Rf = 0.50 (90:10 DCM/t-BuOMe); λmax (DCM, nm) 287 inf (log ε = 4.27), 292 (4.31), 335 (4.63); vmax (ATR, cm–1) 1721m (C=O), 1686m, 1618m, 1560m, 1479w, 1445m, 1400s, 1379m, 1319m, 1275m, 1227m, 1204m, 1146w, 1051m, 1022m, 1009w, 991m, 930m, 893w, 847w, 799m, 785m, 763w, 731m; 1H NMR (CDCl3, 500 MHz) δ 11.62 (1H, br d, J = 13.5 Hz), 8.60 (1H, d, J = 13.5 Hz), 8.28 (1H, dd, J = 5.0, 1.5 Hz), 7.69 (1H, dd, J = 8.0, 1.5 Hz), 7.40 (1H, dd, J = 8.0, 4.5 Hz), 1.77 (6H, s); 13C{1H} NMR (125 MHz, CDCl3) δ 165.1 (s), 163.2 (s), 151.4 (d), 147.1 (d), 134.4 (s), 134.2 (s), 124.1 (d), 124.0 (d), 105.8 (s), 90.4 (s), 27.4 (q); m/z (ESI) 329 (81Br, MH+, 42%), 327 (79Br, MH+, 44%), 303 (98%), 301 (100%). Anal. Calcd for C12H11BrN2O4: C, 44.06; H, 3.39; N, 8.56. Found: C, 43.97; H, 3.45; N, 8.26.
Synthesis of 8-Halonaphthyridinones
8-Bromo-1,5-naphthyridin-4(1H)-one (2) (optimized conditions)
Ph2O (60 mL) was heated to ∼250 °C (external temperature), and 5-{[(4-bromopyrid-3-yl)amino]methylene}-2,2-dimethyl-1,3-dioxane-4,6-dione (1) (600 mg, 1.83 mmol) was added in one portion. After 0.5 min, the reaction mixture was immediately removed from the heat source (metal heating block) and after 1 min poured into cold n-hexane (−40 °C) (1000 mL). The mixture was cooled to approximately −40 °C for 2 h, and the precipitate was filtered, washed with n-pentane, and recrystallized to give compound 2 (214.8 mg, 52%) as beige microcrystalline powder: mp (hot stage) 158.0–160.0 °C (PhH); Rf = 0.13 (82:18 DCM-NH3/EtOH); λmax (EtOH, nm) 205 (log ε = 4.53), 234 inf (4.19), 241 (4.34), 248 (4.34), 273 inf (3.81), 279 (3.80), 287 inf (3.74), 321 inf (3.86), 333 inf (4.00), 341 (4.03); νmax (ATR, cm–1) 3200–2800w br (NH), 1620m (C=O), 1603m, 1572m, 1551m, 1504s, 1435w, 1400m, 1290m, 1240w, 1209m, 1198m, 1121w, 1084w, 1065w, 898w, 852w, 826m, 810m, 787m; 1H NMR (CD3OD, 500 MHz) δ 8.52 (1H, d, J = 5.0 Hz), 8.05–8.03 (2H, m), 6.55 (1H, d, J = 7.0 Hz); 13C{1H} NMR (CD3OD, 125 MHz) δ 178.4 (s), 147.8 (d), 142.6 (d), 141.7 (s), 137.3 (s), 131.8 (d), 125.8 (s), 113.1 (d); m/z (ESI) 249 (81Br, MH+ + Na, 100%), 247 (79Br, MH+ + Na, 93%), 227 (81Br, MH+, 55%), 225 (79Br, MH+, 55%). Anal. Calcd for C8H5BrN2O: C, 42.70; H, 2.24; N, 12.45. Found: C, 42.56; H, 2.62; N, 12.39.
8′-Bromo-4H-[1,4′-bi(1,5-naphthyridine)]-4,8(5H)-dione (3)
Ph2O (20 mL) was heated to ∼250 °C (external temperature), and ylidene 1 (200 mg, 0.61 mmol) was added in one portion. After 2 min, the reaction mixture was left to cool to <80 °C, and the solution was poured into cold n-hexane (−40 °C) (1000 mL). The mixture was cooled to approximately −40 °C for 2 h, and the precipitate was filtered. The collected solid was washed with n-pentane and recrystallized to give compound 3 as beige powder (29 mg, 13%): decomposition (DSC) onset 307.1 °C, peak max 316.0 °C (PhMe/EtOH); Rf = 0.71 (78:22 EtOH/DMSO); λmax (DMSO, nm) 302 inf (log ε = 4.02), 320 inf (4.16), 329 (4.21), 340 (4.24); vmax (ATR, cm–1) 3280–2960w br (NH), 1599s (C=O), 1574s, 1547m, 1518w, 1483m, 1466m, 1385m, 1337w, 1292m, 1273m, 1244w, 1225m, 1188s, 1099w, 1086w, 1055w, 1038w, 986w, 945m, 868m, 837m, 814m, 789m, 777m; 1H NMR (DMSO-d6, 500 MHz) δ 11.96 (1H, br s), 8.62 (1H, d, J = 4.5 Hz), 8.23 (1H, d, J = 4.8 Hz), 7.991 (1H, d, J = 4.8 Hz), 7.990 (1H, d, J = 7.5 Hz), 7.77 (1H, d, J = 7.2 Hz), 6.46 (1H, d, J = 7.8 Hz), 6.03 (1H, d, J = 7.2 Hz); 13C{1H} NMR (DMSO-d6, 125 MHz) δ 171.8 (s, C=O), 171.2 (s, C=O), 152.4 (d), 151.0 (d), 149.6 (s), 144.2 (d), 140.9 (s), 140.7 (s), 137.7 (d), 135.5 (s), 132.8 (s), 132.1 (s), 128.9 (d), 121.5 (d), 113.9 (d), 110.9 (d); m/z (MALDI-TOF) 393 (81Br, M+ + Na, 29%), 391 (79Br, M+ + Na, 25%), 371 (81Br, M+, 92%), 369 (79Br, M+, 100%). Anal. Calcd for C16H9BrN4O2: C, 52.06; H, 2.46; N, 15.18. Found: C, 51.89; H, 2.53; N, 15.24.
8-Bromo-1,7-naphthyridin-4(1H)-one (15)
Ph2O (60 mL) was heated to ∼250 °C (external temperature) and supplemented with 5-{[(2-bromopyridin-3-yl)amino]methylene}-2,2-dimethyl-1,3-dioxane-4,6-dione (14) (600 mg, 1.83 mmol), and the mixture heated for 0.5 min, cooled to ∼20 °C, and filtered to remove insoluble solids. The filtrate was poured into n-hexane (800 mL) and cooled to approximately −40 °C for 0.5 h, and the formed precipitate was collected by filtration and recrystallized to give compound 15 as beige needles (352 mg, 85%): mp (DSC) onset 135.6 °C, peak max 161.0 °C (PhMe); Rf = 0.26 (90:10 DCM-NH3/EtOH); λmax (DCM, nm) 209 (log ε = 4.26), 223 inf (4.06), 255 (3.88), 281 (3.95), 292 (3.93), 315 inf (3.91), 322 inf (3.59), 332 (3.58), 346 (3.49); vmax (ATR, cm–1) 3280–2800w br (NH), 1684w, 1620m, 1593m, 1535m, 1508s, 1441m, 1406w, 1371w, 1319m, 1290m, 1246m, 1200m, 1184m, 1123m, 1080m, 1055m, 899m, 814m; 1H NMR (DMSO-d6, 300 MHz) δ 11.49 (1H, br s), 8.25 (1H, d, J = 5.1 Hz), 7.95–7.94 (2H, m), 6.24 (1H, d, J = 7.5 Hz); 13C{1H} NMR (DMSO-d6, 75 MHz) δ 175.5 (s), 141.8 (d), 141.3 (d), 134.6 (s), 133.5 (s), 131.5 (s), 118.3 (d), 111.0 (d); m/z (ESI) 227 (81Br, MH+, 99%), 225 (79Br, MH+, 100%). Anal. Calcd for C8H5BrN2O: C, 42.70; H, 2.24; N, 12.45. Found: C, 42.83; H, 2.30; N, 12.34.
Synthesis of Canthin-4-one (stepwise procedure)
4-Bromo-8-methoxy-1,5-naphthyridine (5)
8-Bromo-1,5-naphthyridin-4(1H)-one (2) (269.0 mg, 1.20 mmol) was dissolved in anhydrous DMF (4 mL), and K2CO3 (198.2 mg, 1.43 mmol) was added in one portion, followed by MeI (90 μL, 1.45 mmol). The mixture was left to stir at ∼20 °C for 14 h. PhMe (20 mL) was added, and DMF was azeotropically distilled off in vacuo. This process was repeated multiple times; additional PhMe (∼20 mL) was added and removed in vacuo until no DMF remained. The solid residue was suspended in DCM, adsorbed onto silica, and chromatographed (95:5 DCM/t-BuOMe) to give compound 5 (201.2 mg, 70%) as colorless needles: mp (hot stage) 133.0–134.0 °C (c-hexane); Rf = 0.33 (80:20 DCM/t-BuOMe); λmax (DCM, nm) 280 inf (log ε = 3.94), 288 (3.99), 299 inf (3.88), 309 inf (3.65); vmax (ATR, cm–1) 3069w and 3024m (aryl C–H), 2945w and 2847w (alkyl C–H), 1593m, 1582m, 1551m, 1497s, 1466m, 1447m, 1393m, 1369m, 1346w, 1302s, 1261w, 1229w, 1194w, 1136m, 1098m, 1007m, 966w, 860m, 839m, 777s, 723w; 1H NMR (500 MHz, CDCl3) δ 8.92 (1H, d, J = 5.5 Hz), 8.69 (1H, d, J = 4.5 Hz), 7.99 (1H, d, J = 4.5 Hz), 7.05 (1H, d, J = 5.0 Hz), 4.15 (3H, s); 13C{1H} NMR (125 MHz, CDCl3) δ 162.3 (s), 152.9 (d), 149.2 (d), 142.8 (s), 137.9 (s), 136.1 (s), 128.9 (d), 104.5 (d), 58.9 (q); m/z (MS-ESI) 241 (81Br, MH+, 100%), 239 (79Br, MH+, 96%). Anal. Calcd for C9H7BrN2O: C, 45.22; H, 2.95; N, 11.72. Found: C, 45.31; H, 3.08; N, 11.56.
4-(2-Chlorophenyl)-8-methoxy-1,5-naphthyridine (6)
A mixture of 4-bromo-8-methoxy-1,5-naphthyridine (5) (45.0 mg, 0.2 mmol), 2-chlorophenylboronic acid (46.9 mg, 0.3 mmol), K2CO3 (55.3 mg, 0.4 mmol), and PdCl2(dppf)·DCM (2.0 mg, 1.25 mol %) in dioxane/H2O (75:25) was heated to ∼88 °C (reflux) for 8 min. After completion of the reaction (as determined by TLC), the mixture was left to cool to ∼20 °C, diluted with DCM (20 mL), dried (Na2SO4), and filtered, and the volatiles were removed under reduced pressure. The remaining residue was redissolved in DCM (10 mL) and adsorbed onto silica. Chromatography (90:10 DCM/t-BuOMe) gave compound 6 as colorless needles (44.1 mg, 74%): mp (hot stage) 158.0–160.0 °C (c-hexane/PhMe); Rf = 0.75 (90:10 DCM/t-BuOMe); λmax (DCM, nm) 278 inf (log ε = 4.16), 286 (4.21), 293 inf (4.18), 305 inf (4.00); vmax (ATR, cm–1) 3015w (aryl C–H), 2988w and 2938w (alkyl C–H), 1595m, 1580m, 1562m, 1510m, 1481m, 1462m, 1427m, 1400m, 1371m, 1350w, 1304m 1292m, 1246w, 1219m, 1155w, 1177w, 1113m, 1092m, 1059m, 1040m, 993m, 955w, 862m, 835m, 816m, 764s, 735m, 710m; 1H NMR (CDCl3, 500 MHz) δ 9.01 (1H, d, J = 4.5 Hz), 8.81 (1H, d, J = 5.5 Hz), 7.59 (1H, d, J = 4.0 Hz), 7.56–7.54 (1H, m), 7.43–7.39 (3H, m), 6.99 (1H, d, J = 5.0 Hz), 4.16 (3H, s); 13C{1H} NMR (CDCl3, 125 MHz) δ 162.1 (s), 152.2 (d), 149.2 (d), 146.9 (s), 143.0 (s), 137.5 (s), 136.6 (s), 133.4 (s), 131.5 (d), 129.9 (d, 2 × CH), 126.7 (d), 125.8 (d), 103.6 (d), 56.6 (q); m/z (ESI) 273 (37Cl, MH+, 31%), 271 (35Cl, MH+, 100%). Anal. Calcd for C15H11ClN2O: C, 66.55; H, 4.10; N, 10.35. Found: C, 66.43; H, 4.15; N, 10.29.
8-(2-Chlorophenyl)-1,5-naphthyridin-4(1H)-one (7)
To a solution of 4-(2-chlorophenyl)-8-methoxy-1,5-naphthyridine (6) (128.3 mg, 0.47 mmol) in dioxane (2.5 mL) was added concentrated HCl/H2O (50:50) (3.6 mL), and the mixture was heated to ∼101 °C (reflux) until the starting material had been completely consumed (45 min, as determined by TLC). The mixture was left to cool to ∼20 °C, and the mixture was poured onto crushed ice, neutralized with saturated aqueous NaHCO3, and extracted with DCM (10 × 30 mL). The combined organic extracts were dried (Na2SO4), filtered, evaporated to dryness, and recrystallized to give compound 7 as colorless microcrystalline powder (117.0 mg, 96%): mp (DSC) onset 265.7 °C, peak max 268.0 °C (PhCl); Rf = 0.67 (82:18 DCM-NH3/EtOH); λmax (EtOH, nm) 244 inf (log ε = 4.30), 247 (4.31), 332 inf (4.00), 339 (4.03); vmax (ATR, cm–1) 3362–2641w br (NH), 1622s, 1601s, 1582m, 1518s, 1477m, 1408m, 1335w, 1290m, 1202s, 1182m, 1159w, 1123w, 1107w, 1065w, 1038m, 910w, 866w, 829s, 760m, 750m; 1H NMR (CD3OD, 500 MHz) δ 8.79 (1H, d, J = 4.0 Hz), 7.87 (1H, d, J = 7.0 Hz), 7.65 (1H, dd, J = 8.0, 1.0 Hz), 7.59–7.55 (2H, m) 7.53 (1H, ddd, J = 7.5, 7.5, 1.0 Hz), 7.47 (1H, dd, J = 7.5, 2.0 Hz); 13C{1H} NMR (CD3OD, 125 MHz) δ 179.7 (s), 147.9 (d), 141.9 (d), 141.5 (s), 140.5 (s), 136.4 (s), 134.4 (s), 134.3 (s), 132.5 (d, 2 × CH), 131.4 (d), 129.0 (d), 128.9 (d), 112.6 (d); m/z (ESI) 281 (37Cl, MH+ + Na, 17%), 279 (35Cl, MH+ + Na, 49%), 259 (37Cl, MH+, 33%), 257 (35Cl, MH+, 100%). Anal. Calcd for C14H9ClN2O: C, 65.51; H, 3.53; N, 10.91. Found: C, 65.72; H, 3.28; N, 10.78.
4H-Indolo[3,2,1-de][1,5]naphthyridin-4-one (canthin-4-one) (8a)
To a stirred solution of naphthyridinone 7 (77.1 mg, 0.3 mmol) in dioxane/H2O (75:15, 3 mL) was added K2CO3 (83.1 mg, 0.6 mmol). A premixed solution of CuI (5.7 mg, 10 mol %), DMCDA (9.45 μL, 20 mol %), and H2O (3 μL, 0.17 mmol) in dioxane (300 μL) was added to the reaction mixture and heated to ∼88 °C (reflux) for 12 h. The reaction mixture was left to cool to ∼20 °C, diluted with DCM, dried (Na2SO4), filtered, and evaporated to dryness. The remaining residue was redissolved in DCM (10 mL), adsorbed onto silica, and chromatographed (98:2 DCM-NH3/t-BuOMe) to give compound 8a as pale yellow needles (50.0 mg, 76%): mp (hot stage) 263.9–265.3 °C (PhMe) [lit.14 mp 264 °C (n-heptane/DCM)]; Rf = 0.42 (50:50 THF/EtOAc); λmax (DCM, nm) 256 inf (log ε = 4.26), 261 (4.33), 286 (4.31), 292 inf (4.25), 311 inf (3.65), 319 inf (3.60), 388 inf (3.99), 396 (4.00); vmax (ATR, cm–1) 3075w and 3049w (aryl C–H), 1643m, 1620s, 1557s, 1504s, 1470m, 1441m, 1327m, 1294m, 1267m, 1223m, 1194m, 1159m, 1113m, 1078w, 1028m, 976w, 941w, 856m, 826m, 808m, 746s; 1H NMR (CDCl3, 500 MHz) δ 9.04 (1H, d, J = 5.0 Hz), 8.26 (1H, d, J = 7.5 Hz), 8.15 (1H, d, J = 7.5 Hz), 8.11 (1H, d, J = 4.5 Hz), 7.75–7.70 (2H, m), 7.53–7.43 (1H, m), 6.67 (1H, d, J = 7.5 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.4 (s), 147.3 (d), 139.11 (s), 139.08 (s), 135.0 (s), 133.7 (s), 131.5 (d), 131.1 (d), 125.0 (d), 124.7 (s), 124.0 (d), 118.7 (d), 117.6 (d), 111.0 (d); m/z (MALDI-TOF) 243 (M+ + Na, 100%), 221 (M+, 22%). Anal. Calcd for C14H8N2O: C, 76.35; H, 3.66; N, 12.72. Found: C, 76.42; H, 3.54; N, 12.81.
Synthesis of Canthin-4-one 8a (one-pot procedure)
A mixture of 4-bromo-8-methoxy-1,5-naphthyridine (5) (71.7 mg, 0.3 mmol), 2-chlorophenylboronic acid (70.4 mg, 0.45 mmol), K2CO3 (82.9 mg, 0.6 mmol), and PdCl2(dppf)·DCM (3.1 mg, 1.25 mol %) in dioxane/H2O (75:25, 6 mL) was heated to ∼88 °C (reflux) for 12 min until starting material 6 had been completely consumed (as determined by TLC). The reaction mixture was left to cool to ∼20 °C; concentrated HCl (300 μL) was added, and the mixture was heated back to ∼88 °C (reflux). After 2 h, the reaction mixture was left to cool to ∼20 °C and K2CO3 (414.6 mg, 3.00 mmol) was added carefully. The reaction mixture was stirred until the effervescence ceased and the K2CO3 was completely dissolved. A premixed solution of CuI (5.7 mg, 0.03 mmol, 10 mol %), DMCDA (9.45 μL, 0.06 mmol, 20 mol %), and H2O (3 μL, 0.17 mmol) in dioxane (300 μL) was added, and the reaction mixture was heated to ∼88 °C (reflux) for 12 h. The reaction mixture was left to cool to ∼20 °C, diluted (DCM, 15 mL), dried (Na2SO4), filtered, and evaporated to dryness. The residue was redissolved in DCM (10 mL) and adsorbed onto silica. Chromatography gave 4H-indolo[3,2,1-de][1,5]naphthyridin-4-one (canthin-4-one) (8a) as pale yellow needles (52.9 mg, 80%) identical to those described above.
Synthesis of 4H-Indolo[3,2,1-ij][1,6]naphthyridin-4-one (9a)
8-Phenyl-1,6-naphthyridin-4(1H)-one (12)
A mixture of 8-bromo-1,6-naphthyridin-4(1H)-one (11) (112.5 mg, 0.5 mmol), K2CO3 (138.5 mg, 1.0 mmol), phenylboronic acid (94.0 mg, 0.75 mmol) and Pd(dppf)Cl2·DCM (20.0 mg, 5 mol %) in dioxane/H2O (75:25, 5 mL) was heated to ∼88 °C (reflux) for 12 min until the 8-bromo-1,6-naphthyridin-4(1H)-one (11) had been consumed (as determined by TLC). The reaction mixture was then allowed to cool to ∼20 °C, diluted (DCM, 15 mL), dried (Na2SO4), filtered, and adsorbed onto silica gel. Chromatography (THF) gave compound 12 as colorless needles (83.9 mg, 75%): mp (DSC) onset 253.6 °C, peak max 254.5 °C (PhCl); Rf = 0.30 (60:40 DCM/THF; λmax (DCM, nm) 280 inf (log ε = 3.82), 289 (3.88), 319 (4.16); vmax (ATR, cm–1) 3250–2650w br (NH), 1641s, 1589s, 1568m, 1493s, 1451w, 1435w, 1310m, 1275w, 1248w, 1209m, 1138m, 1088w, 1043m, 1024m, 916w, 853m, 843m, 829m, 808m, 791m, 770s, 704m; 1H NMR (DMSO-d6, 500 MHz) δ 9.22 (1H, s), 8.48 (1H, s), 7.78 (1H, d, J = 7.5 hz), 7.61–7.53 (5H, m), 6.21 (1H, d, J = 7.5 Hz); 13C{1H} NMR (DMSO-d6, 125 MHz) δ 176.9 (s), 149.6 (d), 148.4 (d), 141.9 (s), 141.4 (d), 133.3 (s), 129.6 (d), 129.2 (d), 128.7 (d), 125.5 (s), 120.3 (s), 112 (d); m/z (ESI) 261 (M+ + K, 16%), 245 (M+ + Na, 22%), 223 (MH+, 100%). Anal. Calcd for C14H10N2O: C, 75.66; H, 4.54; N, 12.60. Found: C, 75.80; H, 4.32; N, 12.73.
4H-Indolo[3,2,1-ij][1,6]naphthyridin-4-one (9a) (one-pot procedure, MW conditions)
A glass vessel tube used as a microwave reactor was charged with naphthyridinone 11 (45.0 mg, 0.2 mmol), 2-chlorophenylboronic acid (46.9 mg, 0.3 mmol), K2CO3 (55.3 mg, 0.4 mmol), and PdCl2(dppf)·DCM (8.2 mg, 5 mol %) in dioxane/H2O (75:25, 2 mL). The tube was capped, and the reaction performed in the microwave reactor at ∼120 °C (P = 150 W; pressure = 50 psi) for 45 min. The mixture was left to cool to ∼20 °C, and K2CO3 (55.3 mg, 0.4 mmol) and a premixed solution of CuI (3.8 mg, 10 mol %) and DMCDA (6.3 μL, 20 mol %) in dioxane/H2O (75:25, 300 μL) were added. The tube was capped and placed back in the microwave reactor at 120 °C (P = 150 W; pressure = 50 psi) for 6 h. The mixture was left to cool to ∼20 °C, diluted (DCM, 20 mL), dried (Na2SO4), filtered, and evaporated to dryness. The residue was dissolved in DCM (20 mL) and adsorbed onto silica. Chromatography (50:50 EtOAc/THF) gave compound 9a as beige needles (30.4 mg, 69%): mp (hot stage) 237.0–238.0 °C (PhMe); Rf = 0.50 (50:50 EtOAc/THF); λmax (DCM, nm) 233 (log ε = 3.29), 263 (4.36), 280 inf (4.08), 285 inf (4.12), 287 (4.14), 292 inf (4,09), 318 (3.63), 359 (4.01), 366 inf (4.00), 369 inf (4.00); vmax (ATR, cm–1) 3100w, 3067w, and 3032w (aryl C–H), 1649s (C=O), 1618m, 1597m, 1562m, 1503m, 1472m, 1454m, 1425w, 1379w, 1337m, 1285m, 1229m, 1200m, 1167m, 1159m, 1125w, 1088m, 1024m, 1016m, 955w, 903w, 872m, 833m, 800m, 789m, 745s; 1H NMR (CDCl3, 500 MHz) δ 9.46 (1H, s), 9.40 (1H, s), 8.30 (1H, d, J = 8.0 Hz), 8.16 (1H, d, J = 8.0 Hz), 7.72 (1H, d, J = 8.5 Hz), 7.65 (1H, ddd, J = 7.8, 7.8, 1.0 Hz), 7.51 (1H, ddd, J = 7.8, 7.8, 1.0 Hz), 6.60 (1H, d, J = 8.0 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.2 (s), 146.1 (d), 144.3 (d), 142.2 (s), 138.3 (s), 132.6 (d), 129.1 (d), 125.2 (d), 124.4 (s), 123.0 (d), 121.7 (s), 118.6 (s), 118.1 (d), 110.7 (d); m/z (MALDI-TOF) 243 (M+ + Na, 100%), 221 (MH+, 56%), 220 (M+, 34%). Anal. Calcd for C14H8N2O: C, 76.35; H, 3.66; N, 12.72. Found: C, 76.49; H, 3.58; N, 12.80.
4H-Indolo[3,2,1-ij][1,6]naphthyridin-4-one (9a) (one-pot procedure, conventional heating)
A mixture of naphthyridinone 11 (225.1 mg, 1.0 mmol), 2-chlorophenylboronic acid (235.0 mg, 1.5 mmol), K2CO3 (277.0 mg, 2.0 mmol), and PdCl2(dppf)·DCM (210 mg, 25 mol %) in dioxane/H2O (75:25, 10 mL) was heated to ∼88 °C (reflux) for 10 min until the starting naphthyridinone 11 had been completely consumed (as determined by TLC). The reaction mixture was left to cool to room temperature (rt); K2CO3 (277.0 mg, 2.0 mmol) was added, followed by a premixed solution of CuI (19 mg, 0.1 mmol, 10 mol %) and DMCDA (31.5 μL, 0.2 mmol, 20 mol %) in dioxane/H2O (75:25, 2 mL), and the mixture was heated to ∼88 °C (reflux) for 12 h. The reaction mixture was left cool to rt, diluted (DCM, 50 mL), dried (Na2SO4), filtered, and evaporated to dryness. The residue was dissolved in DCM and adsorbed onto silica. Chromatography (50:50 EtOAc/THF) gave compound 9a as beige needles (132.0 mg, 60%) identical to those described above.
Synthesis of 4H-Indolo[3,2,1-ij][1,7]naphthyridin-4-one (10a)
8-Phenyl-1,7-naphthyridin-4(1H)-one (16)
A mixture of 8-bromo-1,7-naphthyridin-4(1H)-one (15) (45.0 mg, 0.2 mmol), K2CO3 (55.4 mg, 0.4 mmol), phenylboronic acid (36.6 mg, 0.3 mmol), and PdCl2(dppf)·DCM (8.2 mg, 5 mol %) in dioxane/H2O (75:25, 1 mL) was heated to ∼88 °C (reflux) for 10 min. After the starting material 15 had been completely consumed (as determined by TLC), the reaction mixture was diluted (DCM, 15 mL), dried (Na2SO4), filtered, and evaporated in vacuo to dryness. The residue was absorbed onto silica and chromatographed (96:4 DCM-NH3/EtOH) to give compound 16 (40.4 mg, 91%) as beige prisms: mp (DSC) onset 206.9 °C, peak max 208.3 °C (n-pentane/EtOAc); Rf = 0.51 (50:50 EtOAc/THF); λmax (DCM, nm) 282 (log ε = 3.77), 292 (3.83), 339 (4.14), 344 inf (4.14); vmax (ATR, cm–1) 3200–2800w br (NH), 1626m, 1607m, 1582m, 1531m, 1514s, 1493m, 1450m, 1441m, 1402w, 1294m, 1267m, 1227w, 1190m, 1072w, 1030w, 1022w, 970w, 908w, 856w, 814m, 768m, 719m; 1H NMR (CDCl3, 500 MHz) δ 9.18 (1H, br s), 8.57 (1H, d, J = 5.5 Hz), 8.06 (1H, d, J = 5.0 Hz), 7.68–7.65 (3H, m), 7.55 (2H, dd, J = 7.5, 7.5 Hz), 7.49 (1H, t, J = 7.4 Hz), 6.29 (1H, d, J = 7.5 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 177.8 (s), 150.9 (s), 142.9 (d), 138.8 (d), 136.1 (s), 132.9 (s), 131.2 (s), 130.0 (d), 129.7 (d), 129.0 (d), 117.8 (d), 111.5 (d); m/z (ESI) 261 (M+ + K, 38%), 245 (M+ + Na, 50%), 223 (M+, 100%). Anal. Calcd for C14H10N2O: C, 75.66; H, 4.54; N, 12.60. Found: C, 75.70; H, 4.48; N, 12.53.
4H-Indolo[3,2,1-ij][1,7]naphthyridin-4-one (10a) (one-pot procedure)
A mixture of naphthyridinone 15 (225.1 mg, 1.0 mmol), 2-chlorophenylboronic acid (235.0 mg, 1.5 mmol), K2CO3 (277.0 mg, 2.0 mmol), and PdCl2(dppf)·DCM (5 mol %) in dioxane/H2O (75:25, 10 mL) was heated to ∼88 °C (reflux) for 15 min until the starting naphthyridinone 15 had been completely consumed (as determined by TLC). The reaction mixture was left to cool to rt; K2CO3 (277.0 mg, 2.0 mmol) was added, followed by a premixed solution of CuI (19 mg, 0.1 mmol, 10 mol %), DMCDA (31.5 μL, 0.2 mmol, 20 mol %), and H2O (18 μL, 1.0 mmol), in dioxane (1.0 mL), and the mixture was heated to ∼88 °C (reflux) for 12 h. The reaction mixture was left cool to rt, diluted (DCM, 50 mL), dried (Na2SO4), filtered, and evaporated to dryness. The residue was dissolved in DCM and adsorbed onto silica. Chromatography (70:30 EtOAc/THF) gave compound 10a as beige needles (170.0 mg, 77%): mp (hot stage) 237.0–238.0 °C (PhMe); Rf = 0.59 (50:50 EtOAc/THF); λmax (DCM, nm) 243 inf (log ε = 4.21), 262 (4.09), 281 inf (4.20), 288 inf (4.31), 292 inf (4.34), 295 (4.36), 332 inf (3.85), 341 (3.88), 389 (4.24); vmax (ATR, cm–1) 3050w (aryl C–H), 1659m, 1643m, 1614s (C=O), 1587m, 1572w, 1541s, 1499s, 1460m, 1433m, 1393w, 1354m, 1317s, 1290m, 1265m, 1202m, 1186m, 1152m, 1111w, 1098m, 1070w, 1030m, 947w, 868w, 824m, 812m, 797m, 712m; 1H NMR (CDCl3, 500 MHz) δ 8.90 (1H, d, J = 5.5 Hz), 8.34 (1H, d, J = 8.0 Hz), 8.27 (1H, d, J = 8.0 Hz), 7.99 (1H, d, J = 5.5 Hz), 7.70 (1H, d, J = 8.0 Hz), 6.67 (1H, ddd, J = 7.5, 7.5, 1.0 Hz), 7.72 (1H, ddd, J = 7.5, 7.5, 1.0 Hz), 6.53 (1H, d, J = 7.5 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.5 (s), 148.4 (s), 146.2 (d), 139.6 (s), 132.6 (d), 131.5 (s), 130.4 (d), 127.1 (s), 125.7 (s), 125.3 (d), 122.8 (d), 116.2 (d), 115.6 (d), 110.7 (d); m/z (MALDI-TOF) 244 (MH+ + Na, 66%), 243 (M+ + Na, 95%), 221 (MH+, 100%), 220 (M+, 81%). Anal. Calcd for C14H8N2O: C, 76.35; H, 3.66; N, 12.72. Found: C, 76.23; H, 3.72; N, 12.85.
Analogues of Canthin-4-ones 8 and Isocanthin-4-ones 9 and 10
Canthin-4-ones 8 (general procedure)
A mixture of 4-bromo-8-methoxy-1,5-naphthyridine (5) (71.7 mg, 0.3 mmol), K2CO3 (82.8 mg, 0.6 mmol), PdCl2(dppf)·DCM (3.0 mg, 1.25 mol %), and the appropriate 2-chlorophenylboronic acid (1.5 or 3 equiv) in dioxane/H2O (75:25, 6 mL) was heated to ∼88 °C (reflux) until the starting material had been completely consumed (as determined by TLC). The mixture was left to cool to rt; concentrated HCl (300 μL) was added in one portion, and the mixture heated again to reflux for 2 h. Then the mixture was left to cool to rt, and K2CO3 (414.6 mg, 3.0 mmol) was added portionwise. The mixture was left to stir for a few minutes until the effervescence ceased and K2CO3 dissolved; the color changed to deep brown. A premixed mixture of CuI (5.7 mg, 10 mol %) and DMCDA (9.45 μL, 20 mol %) in dioxane/H2O (75:25, 1.0 mL) was added, and the reaction mixture heated to ∼88 °C (reflux). After 12 h, the mixture was left to cool to ∼20 °C, diluted (DCM, 50 mL), dried (Na2SO4), filtered, and evaporated to dryness. The remaining residue was dissolved in DCM, adsorbed onto silica, and chromatographed to give 4H-indolo[3,2,1-de][1,5]naphthyridin-4-one 8.
9-Methyl-4H-indolo[3,2,1-de][1,5]naphthyridin-4-one (8b)
2-Chloro-4-methyl-phenylboronic acid (76.8 mg, 0.45 mmol) gave compound 8b (50:50 EtOAc/THF) as beige needles (52.4 mg, 74%): mp (hot stage) 264–266 °C; mp (DSC) onset 269.1 °C, peak max 271.1 °C (EtOH); Rf = 0.40 (60:40 EtOAc/THF); λmax (DCM, nm) 257 inf (log ε = 4.46), 262 (4.51), 287 inf (4.51), 293 (4.57), 321 (4.04), 329 inf (4.04), 389 (4.25); vmax (ATR, cm–1) 3045w (aryl C–H), 2924w (alkyl C–H), 1620s (C=O), 1557m, 1508s, 1472m, 1431m, 1375w, 1325w, 1290m, 1223m, 1204m, 1161m, 1125m, 1084w, 1024w, 866m, 827m, 812m, 785m, 746w, 731w; 1H NMR (CDCl3, 500 MHz) δ 9.01 (1H, d, J = 4.5 Hz), 8.22 (1H, d, J = 8.0 Hz), 8.05 (1H, d, J = 5.0 Hz), 8.0 (1H, d, J = 7.5 Hz), 7.51 (1H, s), 7.31 (1H, d, J = 8.0 Hz), 2.61 (3H, s); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.5 (s), 147.3 (d), 142.3 (s), 139.6 (s), 139.0 (s), 135.3 (s), 133.9 (s), 131.4 (d), 126.2 (d), 123.6 (d), 122.2 (s), 118.3 (d), 117.5 (d), 111.3 (d), 22.5 (q); m/z (ESI) 257 (M+ + Na, 20%), 235 (MH+, 100%). Anal. Calcd for C15H10N2O: C, 76.91; H, 4.30; N, 11.96. Found: C, 77.02; H, 4.26; N, 11.74.
9-Methoxy-4H-indolo[3,2,1-de][1,5]naphthyridin-4-one (8c)
2-Chloro-4-methoxyphenylboronic acid (83.9 mg, 0.45 mmol) gave compound 8c (chromatography eluent, 50:50 EtOAc/THF) as pale yellow needles (26.5 mg, 35%): mp (DSC) onset 298.2 °C, peak max 301.0 °C, decomposition peak max 302.9 °C (EtOH); Rf = 0.18 (THF); λmax (DCM, nm) 259 inf (log ε = 4.26), 264 (4.29), 284 inf (4.28), 293 (4.45), 299 (4.48), 345 (4.10), 373 (4.16); vmax (ATR, cm–1) 3032w (aryl C–H), 1641m, 1607s (C=O), 1557m, 1510s, 1477m, 1470m, 1454m, 1441m, 1431m, 1329m, 1310m, 1275w, 1227s, 1192m, 1175m, 1163m, 1115m, 1069w, 976w, 868m, 853m, 826m, 802m, 793m; 1H NMR (CDCl3, 500 MHz) δ 8.98 (1H, d, J = 4.5 Hz), 8.20 (1H, d, J = 8.0 Hz), 8.00 (1H, d, J = 8.5 Hz), 7.99 (1H, d, J = 4.5 Hz), 7.18 (1H, d, J = 2.0 Hz), 7.04 (1H, dd, J = 8.5, 2.0 Hz), 6.65 (1H, d, J = 8.0 Hz), 3.99 (3H, s); 13C{1H} NMR (DMSO-d6, 125 MHz) δ 178.2 (s), 162.3 (s), 146.8 (d), 140.8 (s), 137.9 (s), 135.0 (s), 133.6 (d), 133.3 (s), 124.9 (d), 116.54 (s), 116.47 (d), 112.4 (d), 97.3 (d), 56.1 (q); m/z (ESI) 273 (M+ + Na, 20%), 251 (MH+, 100%). Anal. Calcd for C15H10N2O2: C, 71.99; H, 4.03; N, 11.19. Found: C, 72.03; H, 4.16; N, 11.12.
9-(Trifluoromethyl)-4H-indolo[3,2,1-de][1,5]naphthyridin-4-one (8d)
2-Chloro-4-(trifluoromethyl)phenylboronic acid (201.9 mg, 0.9 mmol) gave compound 8d (chromatography eluent, 80:20 DCM/THF) as beige needles (57.9 mg, 67%): mp (DSC) onset 302.8 °C, peak max 303.9 °C (PhCl); Rf = 0.44 (30:70 DCM/EtOAc); λmax (DCM, nm) 255 inf (log ε = 4.28), 258 (4.30), 282 (4.30), 288 (4.30), 297 (4.21), 316 (3.52), 368 inf (3.81), 385 inf (4.06), 398 (4.18); vmax (ATR, cm–1) 3063w and 3034w (aryl C–H), 1649m, 1627m, 1557m, 1510m, 1474m, 1450m, 1435m, 1336s, 1302m, 1225m, 1177s, 1109s, 1061s, 1024m, 999w, 982w, 961w, 908w, 851w, 831m, 818s, 795m, 758w, 741m, 725m; 1H NMR (CDCl3, 500 MHz) δ 9.13 (1H, d, J = 5.0 Hz), 8.34–8.31 (2H, m), 8.22 (1H, d, J = 4.5 Hz), 8.018 (1H, s), 7.78 (1H, d, J = 8.0 Hz), 6.73 (1H, d, J = 7.5 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.1 (s), 147.5 (d), 139.3 (s), 138.7 (s), 135.6 (s), 133.0 (q, 2JCF = 32.5 Hz), 132.4 (s), 131.3 (d), 127.5 (s), 124.5 (d), 123.8 (q, 1JCF = 271.4 Hz), 121.7 (q, 3JCF = 3.5 Hz), 119.4 (d), 118.3 (d), 108.5 (q, 3JCF = 3.8 Hz); m/z (ESI) 311 (M+ + Na, 48%), 289 (MH+, 100%). Anal. Calcd for C15H7F3N2O: C, 62.51; H, 2.45; N, 9.72. Found: C, 62.63; H, 2.57; N, 9.80.
9-Chloro-4H-indolo[3,2,1-de][1,5]naphthyridin-4-one (8e)
2,4-Dichlorophenylboronic acid (85.9 mg, 0.45 mmol) gave compound 8e (chromatography eluent, 50:50 THF/EtOAc) as yellow plates (26.7 mg, 35%): mp (hot stage) 272.0–274.0 °C (EtOAc); Rf = 0.44 (30:70 DCM/EtOAc); λmax (DCM, nm) 257 inf (log ε = 4.18), 261 (4.23), 288 inf (4.30), 292 (4.33), 318 inf (3.75), 328 (3.97), 364 inf (3.78), 382 inf (3.97), 391 (4.01); vmax (ATR, cm–1) 3084w and 3040w (aryl C–H), 1649m, 1643m, 1620s, 1612s, 1574w, 1555m, 1503s, 1449m, 1431m, 1321m, 1298m, 1279m, 1223m, 1184m, 1159w, 1121m, 1099m, 1076m, 1067m, 1022m, 961w, 935w, 924w, 878m, 827m, 802m, 791m, 745w; 1H NMR (CDCl3, 500 MHz) δ 9.06 (1H, d, J = 4.5 Hz), 8.21 (1H, d, J = 8.0 Hz), 8.11 (1H, d, J = 4.5 Hz), 8.09 (1H, d, J = 8.5 Hz), 7.74 (1H, d, J = 1.5 Hz), 7.49 (1H, dd, J = 8.5, 1.5 Hz), 6.68 (1H, d, J = 8.0 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.2 (s), 147.5 (d), 139.7 (s), 139.2 (s), 137.2 (s), 135.4 (s), 132.9 (s), 131.2 (d), 125.4 (d), 124.7 (d), 123.2 (s), 118.7 (d), 118.1 (d), 111.7 (d); m/z (ESI) 257 (37Cl, MH+, 33%), 255 (35Cl, MH+, 100%). Anal. Calcd for C14H7ClN2O: C, 66.03; H, 2.77; N, 11.00. Found: C, 66.07; H, 2.69; N, 10.85.
9-Fluoro-4H-indolo[3,2,1-de][1,5]naphthyridin-4-one (8f)
2-Chloro-4-fluorophenylboromic acid (78.5 mg, 0.45 mmol) gave compound 8f (chromatography eluent, 75:25 DCM/THF) (63.6 mg, 89%) as yellow needles: mp (DSC) onset 309.0 °C, peak max 310.7 °C (PhCl); Rf = 0.39 (50:50 EtOAc/THF); λmax (DCM, nm) 288 inf (log ε = 3.81), 294 (3.89), 303 (3.89), 321 (3.25), 394 inf (3.60), 402 (3.63); vmax (ATR, cm–1) 3034w (aryl C–H), 1641m, 1614m, 1580m, 1555m, 1503s, 1468m, 1443m, 1422m, 1298m, 1321m, 1269m, 1223m, 1159m, 1188m, 1130w, 1101w, 1078m, 1024w, 943m, 878m, 823s, 795m, 758m; 1H NMR (CDCl3, 500 MHz) δ 8.98 (2H, d, J = 5.0 Hz), 8.92 (1H, d, J = 8.0 Hz), 8.46 (1H, d, J = 5.0 Hz), 8.43 (1H, dd, J = 8.5, 5.5 Hz), 8.17 (1H, dd, J = 9.5, 2.0 Hz), 7.40 (1H, ddd, J = 8.0, 8.0, 2.0 Hz), 6.54 (1H, d, J = 8.0 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.4 (s), 164.8 (d, 1JCF = 251.9 Hz), 147.6 (s), 140.3 (d, 3JCF = 12.1 Hz), 139.0 (s), 135.9 (s), 133.2 (s), 125.4 (d, 3JCF = 10.2 Hz), 121.0 (d, 4JCF = 2.2 Hz), 118.6 (d), 118.1 (d), 113.0 (d, 2JCF = 24.1 Hz), 99.3 (d, 2JCF = 27.9 Hz); m/z (ESI) 261 (M+ + Na, 74%), 239 (MH+, 100%). Anal. Calcd for C14H7FN2O: C, 70.59; H, 2.96; N, 11.76. Found: C, 70.47; H, 2.90; N, 11.69.
10-Methoxy-4H-indolo[3,2,1-de][1,5]naphthyridin-4-one (8g)
2-Chloro-5-methoxyphenylboromic acid (83.9 mg, 0.45 mmol) gave compound 8g (chromatography eluent, THF) as yellow needles (52.1 mg, 69%): mp (DSC) onset 257.0 °C, peak max 258.3 °C (EtOH); Rf = 0.58 (THF); λmax (DCM, nm) 243 (log ε = 3.98), 252 (4.09), 268 (4.30), 275 inf (4.19), 307 inf (4.28), 311 (4.30), 327 inf (3.85), 410 (3.90), 422 inf (3.86); vmax (ATR, cm–1) 3076w (aryl C–H), 1639m, 1632m, 1612s, 1593m, 1553m, 1504s, 1485s, 1429m, 1323m, 1283m, 1223s, 1190m, 1119m, 1036m, 947w, 822m; 1H NMR (CDCl3, 500 MHz) δ 9.04 (1H, d, J = 4.8 Hz), 8.20 (1H, d, J = 7.8 Hz), 8.09 (1H, d, J = 4.8 Hz), 7.63–7.61 (2H, m), 7.29 (1H, dd, J = 8.8, 2.6 Hz), 6.65 (1H, d, J = 7.7 Hz), 3.97 (3H, s); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.2 (s), 157.6 (s), 147.1 (d), 139.3 (s), 135.4 (s), 133.8 (s), 133.5 (s), 131.6 (d), 125.8 (s), 118.7 (d), 118.6 (d), 117.2 (d), 111.7 (d), 107.5 (d), 56.2 (q); m/z (ESI) 273 (M+ + Na, 35%), 251 (MH+, 100%). Anal. Calcd for C15H10N2O2: C, 71.99; H, 4.03; N, 11.19. Found: C, 72.03; H, 3.97; N, 11.05.
10-(Trifluoromethoxy)-4H-indolo[3,2,1-de][1,5]naphthyridin-4-one (8h)
2-Chloro-5-(trifluoromethoxy)phenylboronic acid (216.3 mg, 0.9 mmol) gave compound 8h (chromatography eluent, EtOAc) as yellow needles (60.9 mg, 67%): mp (DSC) onset 267.2 °C, peak max 267.8 °C (EtOH); Rf = 0.70 (THF); λmax (DCM, nm) 255 inf (log ε = 4.43), 259 (4.47), 285 inf (4.46), 289 (4.49), 295 inf (4.43), 318 inf (3.79), 388 inf (4.22), 397 (4.26); vmax (ATR, cm–1) 3103w and 3038w (aryl C–H), 1643m, 1620m, 1601m, 1553m, 1504m, 1476m, 1449w, 1429m, 1329w, 1265s, 1221m, 1194m, 1153m, 1094w, 1074w, 1051w, 1026w, 962w, 935w, 914w, 875m, 858m, 835m, 793w, 760w, 731w; 1H NMR (CDCl3, 500 MHz) δ 9.09 (1H, d, J = 4.5 Hz), 8.27 (1H, d, J = 8.0 Hz), 8.15 (1H, d, J = 5.0 Hz), 8.03 (1H, d, J = 1.0 Hz), 7.77 (1H, d, J = 9.0 Hz), 7.60 (1H, dd, J = 8.8, 2.2 Hz), 6.69 (1H, d, J = 8.0 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.1 (s), 147.4 (d), 146.4 (s), 139.3 (s), 137.3 (s), 135.6 (s), 132.7 (s), 131.5 (d), 125.8 (s), 124.4 (d), 120.7 (q, 1JCF = 258.3 Hz), 119.1 (d), 118.1 (d), 117.0 (d), 112.0 (d); m/z (ESI) 327 (M+ + Na, 25%), 305 (MH+, 100%). Anal. Calcd for C15H7F3N2O2: C, 59.22; H, 2.32; N, 9.21. Found: C, 59.30; H, 2.19; N, 9.34.
10-(Trifluoromethyl)-4H-indolo[3,2,1-de][1,5]naphthyridin-4-one (8i)
2-Chloro-5-(trifluoromethyl)phenylboronic acid (201.9 mg, 0.9 mmol) gave compound 8i (chromatography eluent, 50:50 EtOAc/THF) as beige needles (86.7 mg, 63%): mp (DSC) onset 311.4 °C, peak max 312.4 °C (EtOH); Rf = 0.44 (50:50 EtOAc/THF); λmax (DCM, nm) 253 inf (log ε = 4.16), 258 (4.20), 282 (4.28), 285 (4.28), 293 inf (4.13), 316 (3.42), 380 inf (4.00), 390 (4.10); vmax (ATR, cm–1) 3059w and 3017w (aryl C–H), 1649m, 1626s, 1560m, 1504m, 147m, 1427m, 1335s, 1298m, 1275m, 1221m, 1198m, 1148m, 1121m, 1078m, 1061m, 1026m, 945m, 899m, 883m, 826m, 723m; 1H NMR (CDCl3, 500 MHz) δ 9.13 (1H, d, J = 4.8 Hz), 8.46 (1H, s), 8.32 (1H, d, J = 7.8 Hz), 8.21 (1H, d, J = 4.8 Hz), 8.00 (1H, d, J = 8.5 Hz), 7.87 (1H, J = 8.5 Hz), 6.73 (1H, d, J = 7.8 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.2 (s), 147.6 (d), 140.7 (s), 139.2 (s), 135.6 (s), 132.7 (s), 131.4 (d), 128.1 (q, 3JCF = 3.6 Hz), 127.4 (q, 2JCF = 33.1 Hz), 124.8 (s), 124.0 (q, 1JCF = 272.2 Hz), 121.5 (q, 3JCF = 3.8 Hz), 119.1 (d), 118.5 (d), 111.5 (d); m/z (ESI) 327 (M+ + K, 8%), 311 (M+ + Na, 67%), 289 (MH+, 100%). Anal. Calcd for C15H7F3N2O: C, 59.22; H, 2.32; N, 9.21. Found: C, 59.30; H, 2.51; N, 9.19.
10-Chloro-4H-indolo[3,2,1-de][1,5]naphthyridin-4-one (8j)3
2,5-Dichlorophenylboronic acid (171.7 mg, 0.9 mmol) gave compound 8j (chromatography eluent, 50:50 EtOAc/THF) as pale yellow needles (53.1 mg, 61%): mp (DSC) onset 321.8 °C, peak max 323.0 (EtOH); Rf = 0.39 (50:50 EtOAc/THF); λmax (DCM, nm) 258 inf (log ε = 4.15), 262 (4.18), 288 inf (4.14), 294 (4.23), 302 (4.22), 304 inf (4.22), 322 (3.58), 403 (3.96); vmax (ATR, cm–1) 3088w and 3028w (aryl C–H), 1649s, 1632m, 1620s, 1614s, 1580m, 1553m, 1505s, 1468m, 1443m, 1422m, 1321m, 1298m, 1271m, 1225m, 1190m, 1159m, 1128m, 1105w, 1080m, 1026m, 941m, 880m, 826s; 1H NMR (CDCl3, 500 MHz) δ 9.09 (1H, d, J = 4.5 Hz), 8.24 (1H, d, J = 7.8 Hz), 8.16 (1H, s), 8.13 (1H, d, J = 4.6 Hz), 7.71–7.67 (2H, m), 6.70 (1H, d, J = 7.7 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.1 (s), 147.4 (d), 139.2 (s), 137.4 (s), 135.3 (s), 132.7 (s), 131.4 (d), 131.1 (d), 130.8 (s), 126.0 (s), 124.0 (d), 119.0 (d), 117.9 (d), 112.0 (d); m/z (ESI) 279 (37Cl, M+ + Na, 37%), 277 (35Cl, M+ + Na, 100%), 257 (37Cl, MH+, 10%), 255 (35Cl, MH+, 28%). Anal. Calcd for C14H7ClN2O: C, 66.03; H, 2.77; N, 11.00. Found: C, 66.11; H, 2.65; N, 10.98.
10-Fluoro-4H-indolo[3,2,1-de][1,5]naphthyridin-4-one (8k)
2-Chloro-5-fluorophenylboronic acid (78.5 mg, 0.45 mmol) gave compound 8k (chromatography eluent, 50:50 EtOAc/THF) as yellow needles (45.1 mg, 63%): mp (DSC) onset 298.1 °C, peak max 301.6 °C (PhCl); Rf = 0.63 (THF); λmax (DCM, nm) 249 inf (log ε = 4.16), 256 inf (4.28), 261 (4.33), 287 inf (4.26), 300 (4.30), 321 (3.72), 393 inf (4.05), 403 (4.06); vmax (ATR, cm–1) 3049w (aryl C–H), 1643m, 1618m, 1593m, 1555m, 1506m, 1483m, 1445m, 1430m, 1329m, 1300m, 1273m, 1229m, 1190m, 1152m, 1121w, 1084w, 1024m, 953m, 912m, 862m, 849m, 824s, 793w, 756m; 1H NMR (CDCl3, 500 MHz) δ 9.08 (1H, d, J = 5.0 Hz), 8.24 (1H, d, J = 7.5 Hz), 8.12 (1H, d, J = 5.0 Hz), 7.86 (1H, dd, J = 7.5, 2.5 Hz), 7.70 (1H, dd, J = 9.0, 4.0 Hz), 7.46 (1H, ddd, J = 9.0, 9.0, 2.5 Hz), 6.68 (1H, d, J = 8.0 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.1 (s), 160.2 (d, 1JCF = 244.7 Hz), 147.3 (d), 139.4 (s), 135.6 (s), 135.4 (s), 133.1 (d, 4JCF = 3.6 Hz), 131.5 (d), 126.0 (d, 3JCF = 9.1 Hz), 119.0 (d), 118.6 (d, 2JCF = 25.7 Hz), 117.7 (d), 112.0 (d, 3JCF = 9.1 Hz), 110.6 (d, 2JCF = 24.8 Hz); m/z (ESI) 261 (M+ + Na, 48%), 239 (MH+, 100%). Anal. Calcd for C14H7FN2O: C, 70.59; H, 2.96; N, 11.76. Found: C, 70.63; H, 2.70; N, 11.82.
4H-Indolo[3,2,1-ij][1,6]naphthyridin-4-ones (9) (general procedure)
A glass vessel tube used as the microwave reactor was charged with 8-bromo-1,6-naphthyridin-4(1H)-one (45.0 mg, 0.2 mmol), K2CO3 (55.3 mg, 0.4 mmol), PdCl2(dppf)·DCM (8.2 mg, 0.01 mmol, 5 mol %), and the appropriate 2-chlorophenylboronic acid (1.5 or 3 equiv) in dioxane/H2O (75:25, 2 mL). The tube was capped, and the reaction performed in the microwave reactor at 120 °C (P = 150 W; pressure = 50 psi) for 45 min. The mixture was left to cool to rt; then K2CO3 (55.3 mg, 0.4 mmol) and a premixed solution of CuI (3.8 mg, 10 mol %) and DMCDA (5.4 μL, 20 mol %) in dioxane/H2O (75:25, 500 μL) were added to the reaction mixture, and the mixture was placed back into the microwave reactor for 6 h. The mixture was left to cool to rt, diluted (DCM, 20 mL), dried (Na2SO4), filtered, and evaporated to dryness. The remaining residue was dissolved in DCM and adsorbed onto silica, and chromatography gave 4H-indolo[3,2,1-ij][1,6]naphthyridin-4-one 9.
9-Methyl-4H-indolo[3,2,1-ij][1,6]naphthyridin-4-one (9b)
2-Chloro-4-methylphenylboronic acid (51.1 mg, 0.3 mmol) gave compound 9b (chromatography eluent, 70:30 DCM/THF) as colorless needles (61.9 mg, 88%): mp (hot stage) 253.5–254.5 °C (c-hexane/EtOH); Rf = 0.37 (94:6 EtOAc/THF); λmax (DCM, nm) 266 (log ε = 3.62), 293 (3.84), 332 inf (3.66), 236 (4.18), 245 inf (4.21), 358 (3.84), 366 inf (3.89); vmax (ATR, cm–1) 3075w and 3048w (aryl C–H), 1655m, 1649s, 1626s, 1560m, 1541w, 1504s, 1468m, 1418m, 1375m, 1327m, 1288m, 1229m, 1211s, 1179w, 1165m, 1121m, 964w, 907m, 872m, 854m, 843m, 808s, 785w, 721w; 1H NMR (CDCl3, 500 MHz) δ 9.13 (1H, d, J = 4.8 Hz), 8.46 (1H, s), 8.32 (1H, d, J = 7.8 Hz), 8.21 (1H, d, J = 4.8 Hz), 8.00 (1H, d, J = 8.5 Hz), 7.87 (1H, J = 8.5 Hz), 6.73 (1H, d, J = 7.8 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.2 (s), 147.6 (d), 140.7 (s), 139.2 (s), 135.6 (s), 132.7 (s), 131.4 (d), 128.1 (q, 3JCF = 3.6 Hz), 127.4 (q, 2JCF = 33.1 Hz), 124.8 (s), 124.0 (q, 1JCF = 272.2 Hz), 121.5 (q, 3JCF = 3.8 Hz), 119.1 (d), 118.5 (d), 111.5 (d); m/z (ESI) 273 (M+ + K, 5%), 257 (M+ + Na, 30%), 235 (MH+, 100%). Anal. Calcd for C15H10N2O: C, 76.91; H, 4.30; N, 11.96. Found: C, 77.08; H, 4.12; N, 12.04.
9-Methoxy-4H-indolo[3,2,1-ij][1,6]naphthyridin-4-one (9c)
2-Chloro-4-methoxyphenylboronic acid (111.8 mg, 0.6 mmol) gave compound 9c (chromatography eluent, EtOAc) as beige microcrystalline powder (51.5 mg, 68%): mp (DSC) 296.3 °C, peak max 297.8 °C (PhMe); Rf = 0.51 (50:50 EtOAc/THF); λmax (DCM, nm) 248 inf (log ε = 4.12), 257 inf (3.99), 269 inf (4.22), 273 (4.26), 297 inf (4.23), 301 (4.27), 352 (4.15); vmax (ATR, cm–1) 3072w and 3034w (aryl C–H), 1657m, 1643m, 1612s, 1597m, 1555m, 1510s, 1474m, 1452m, 1416w, 1377w, 1331w, 1302m, 1226s, 1192m, 1169m, 984w, 903w, 872m, 818m, 745w; 1H NMR (DMSO-d6, 500 MHz) δ 9.44 (1H, s), 9.13 (1H, s), 9.02 (1H, d, J = 7.5 Hz), 8.20 (1H, d, J = 8.5 Hz), 7.84 (1H, d, J = 2.0 Hz), 7.10 (1H, dd, J = 8.5, 2.5 Hz), 6.50 (1H, d, J = 7.5 Hz), 3.92 (3H, s); 13C{1H} NMR (CDCl3, 125 MHz) δ 178.1 (s), 160.8 (s), 143.4 (d), 143.2 (d), 141.6 (s), 139.9 (s), 135.1 (d), 123.8 (d), 121.5 (s), 117.8 (s), 116.7 (d), 116.2 (s), 112.3 (d), 97.4 (d), 56.0 (q); m/z (ESI) 289 (M+ + K, 10%), 273 (M+ + Na, 40%), 251 (MH+, 100%). Anal. Calcd for C15H10N2O2: C, 71.99; H, 4.03; N, 11.19. Found: C, 72.05; H, 3.86; N, 11.30.
9-(Trifluoromethyl)-4H-indolo[3,2,1-ij][1,6]naphthyridine-4-one (9d)
2-Chloro-4-(trifluoromethyl)phenylboronic acid (67.3 mg, 0.6 mmol) gave compound 9d (chromatography eluent, 70:30 DCM/EtOAc) as beige needles (28.3 mg, 49%): mp (hot stage) 261.0–263.5 °C (PhMe); Rf = 0.50 (94:6 EtOAc/THF); λmax (DCM, nm) 248 (log ε = 4.16), 260 (4.24), 279 (4.12), 287 (4.14), 293 inf (4.06), 310 inf (3.58), 346 inf (3.81), 355 (3.95), 370 (3.95); vmax (ATR, cm–1) 3066w (aryl C–H), 1665m, 1649m, 1624m, 1585m, 1560, 1504m, 1468m, 1456m, 1416w, 1385w, 1364w, 1337m, 1325m, 1292m, 1267m, 1223m, 1200m, 1163s 1136m, 1121s, 1057s, 1022m, 970, 920m, 891m, 826m; 1H NMR (CDCl3, 500 MHz) δ 9.54 (2H, br s), 8.36 (1H, d, J = 7.5 Hz), 8.33 (1H, d, J = 8.5 Hz), 8.01 (1H, s), 7.82 (1H, d, J = 8.0 Hz), 6.67 (1H, d, J = 7.5 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 178.8 (s), 143.0 (s), 137.9 (s), 132.3 (d), 131.3 (q, 2JCF = 41.7 Hz), 127.3 (s), 123.9 (q, 1JCF = 275.0 Hz), 123.5 (d), 122.1 (q, 3JCF = 3.7 Hz), 118.8 (d), 118.7 (s), 108.2 (q, 3JCF = 4.1 Hz); m/z (ESI) 289 (MH+, 100%). Anal. Calcd for C15H7F3N2O: C, 62.51; H, 2.45; F, 19.77; N, 9.72. Found: C, 62.37; H, 2.58; N, 19.63.
9-Fluoro-4H-indolo[3,2,1-ij][1,6]naphthyridin-4-one (9e)
2-Chloro-4-fluorophenylboronic acid (52.3 mg, 0.3 mmol) gave compound 9e (chromatography eluent. 70:30 DCM/EtOAc) as beige needles (25.9 mg, 36%): mp (DSC) onset 275.6 °C, peak max 276.6 °C (PhCl); Rf = 0.47 (94:6 EtOAc/THF); λmax (DCM, nm) 263 (log ε = 4.34), 286 inf (4.18), 291 (4.20), 328 inf (3.77), 352 (4.07), 359 inf (4.04); vmax (ATR, cm–1) 3086w and 3063w (aryl C–H), 1665s, 1649m, 1620s, 1601m, 1541w, 1505s, 1456m, 1423m, 1375w, 1335m, 1288m, 1267m, 1227m, 1206s, 1173m, 1153m, 1082m, 1022m, 986m, 872m, 845m, 827m, 804m, 783w; 1H NMR (CDCl3, 500 MHz) δ 8.90 (1H, d, J = 5.5 Hz), 8.33 (1H, dd, J = 8.5, 5.5 Hz), 8.21 (1H, d J = 7.5 Hz), 7.99 (1H, d, J = 5.5 Hz), 7.42 (1H, dd, J = 8.5, 2.0 Hz), 7.26 (1H, ddd, J = 8.5, 8.5, 2.2 Hz), 6.55 (1H, d, J = 8.0 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.2 (s), 164.5 (d, 1JCF = 250.0 Hz), 147.3 (s), 145.9 (d), 140.7 (d, 3JCF = 12.5 Hz), 132.4 (d), 132.2 (s), 127.2 (s), 124.6 (d, 3JCF = 12.5 Hz), 121.6 (s), 116.8 (d), 115.4 (d), 113.2 (d, 2JCF = 25.0 Hz), 98.0 (d, 2JCF = 25.0 Hz); m/z (ESI) 261 (M+ + Na, 100%), 239 (MH+, 80%). Anal. Calcd for C14H7FN2O: C, 70.59; H, 2.96; N, 11.76. Found: C, 70.63; H, 3.08; N, 11.89.
10-Methoxy-4H-indolo[3,2,1-ij][1,6]naphthyridin-4-one (9f)
2-Chloro-5-methoxyphenylboronic acid (55.9 mg, 0.3 mmol) gave compound 9f (chromatography eluent, 90:10 EtOAc/THF) as yellow needles (42.7 mg, 85%): mp (hot stage) 229.0–230.5 °C; Rf = 0.32 (94:6 EtOAc/THF); λmax (DCM, nm) 270 (log ε = 3.56), 305 (3.79), 310 inf (3.92), 384 (3.92); vmax (ATR, cm–1) 3360w (aryl C–H), 1657s, 1651m, 1620m, 1614m, 1585s, 1551m, 1503sm 1485s, 1454w, 1435m, 1422m, 1375w, 1337w, 1283s, 1238m, 1215s, 1155m, 1136m, 1119m, 1092m, 1036m, 957m, 91-m, 849m, 820s, 785m, 752w; 1H NMR (CDCl3, 500 MHz) δ 9.48 (1H, br s), 9.39 (1H, br s), 8.23 (1H, d, J = 8.0 Hz), 7.63 (1H, d, J = 2.5 Hz), 7.60 (1H, d, J = 8.5 Hz), 7.20 (1H, dd, J = 9.0, 2.5 Hz), 6.57 (1H, d, J = 7.5 Hz), 3.97 (3H, s); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.0 (s), 157.9 (s), 146.2 (d), 144.2 (d), 142.5 (s), 132.69 (d), 132.67 (s), 125.6 (s), 117.7 (d), 116.6 (d), 111.4 (d), 106.8 (d), 56.3 (q), two C (s) resonances missing; m/z (ESI) 273 (M+ + Na, 37%), 251 (MH+, 100%). Anal. Calcd for C15H10N2O2: C, 71.99; H, 4.03; N, 11.19. Found: C, 72.03; H, 4.11; N, 11.06.
10-(Trifluoromethoxy)-4H-indolo[3,2,1-ij][1,6]naphthyridin-4-one (9g)
2-Chloro-5-(trifluoromethoxy)phenylboronic acid (144.2 mg, 0.6 mmol) gave compound 9g (chromatography eluent, 80:20 DCM/THF) as beige microcrystalline powder (15.8 mg, 26%): mp (DSC) onset 245.0 °C, peak max 246.9 °C; Rf = 0.52 (94:6 EtOAc/THF); λmax (DCM, nm) 260 (log ε = 3.81), 280 inf (3.76), 284 inf (3.81), 288 (3.88), 294 inf (3.85), 313 inf (3.32), 358 (3.71), 370 (3.70); vmax (ATR, cm–1) 1659s, 1649m, 1628m, 1614m, 1585m, 1562m, 1543w, 1526w, 1501m, 1477m, 1425m, 1300s, 1285s, 1267s, 1227s, 1207s, 1150s, 1138s, 1094m, 1018w, 966m, 945w, 880m, 831m, 812m; 1H NMR (CDCl3, 500 MHz) δ 9.55 (1H, br s), 9.49 (1H, s), 8.29 (1H, d, J = 8.0 Hz), 8.05 (1H, d, J = 1.0 Hz), 7.75 (1H, d, J = 9.0 Hz), 7.53 (1H, d, J = 9.0 Hz), 6.63 (1H, d J = 8.0 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 178.8 (s), 146.7 (d), 146.54 (s), 146.52 (s), 144.8 (d), 142.9 (s), 136.5 (s), 132.5 (d), 125.6 (s), 122.4 (d), 120.7 (q, 1JCF = 262.5 Hz), 118.6 (d), 116.1 (d), 111.6 (d), one C (s) resonance missing; m/z (ESI) 305 (MH+, 28%), 295 (100%). Anal. Calcd for C15H7F3N2O2: C, 59.22; H, 2.32; N, 9.21. Found: C, 59.47; H, 2.18; N, 9.16.
10-Fluoro-4H-indolo[3,2,1-ij][1,6]naphthyridin-4-one (9h)
2-Chloro-5-fluorophenylboronic acid (52.3 mg, 0.3 mmol) gave compound 9h (chromatography eluent, 80:20 DCM/THF) as microcrystalline powder (11 mg, 23%): mp (DSC) onset 333.7 °C, onset 335.4 °C (PhMe); Rf = 0.50 (94:6 EtOAc/THF); λmax (DCM, nm) 251 inf (log ε = 4.04), 263 (4.27), 284 inf (4.00), 292 (4.12), 300 (4.09), 314 inf (3.59), 350 inf (3.74), 364 (3.93), 372 inf (3.90); vmax (ATR, cm–1) 3045w (aryl C–H), 1655s (C=O), 1651s, 1620m, 1587m, 1560m, 1503s, 1479s, 1439m, 1427m, 1375m, 1337m, 1290m, 1275m, 1229m, 1202s, 1148m, 1111m, 1092m, 1020m, 961w, 926m, 876m, 816m; 1H NMR (DMSO-d6, 500 MHz) δ 9.59 (1H, s), 9.23 (1H, s), 9.02 (1H, d, J = 8.0 Hz), 8.26 (1H, dd, J = 8.5, 2.5 Hz), 8.20 (1H, dd, J = 9.0, 4.5 Hz), 7.58 (1H, ddd, J = 9.0, 9.0, 2.5 Hz), 6.51 (1H, d, J = 8.0 Hz); 13C{1H} NMR (DMSO-d6, 125 MHz) δ 177.8 (s), 159.7 (d, 1JCF = 275.0 Hz), 145.3 (d), 144.9 (d), 142.0 (s), 135.3 (d), 124.8 (s), 124.9 (d, 3JCF = 10.7 Hz), 120.9 (s), 117.9 (s), 116.7 (d), 116.3 (d, 2JCF = 25.0 Hz), 113.5 (d, 3JCF = 12.5 Hz), 109.7 (d, 2JCF = 25.0 Hz); m/z (ESI) 261 (M+ + Na, 79%), 239 (MH+, 100%). Anal. Calcd for C14H7FN2O: C, 70.59; H, 2.96; N, 11.76. Found: C, 70.64; H, 3.07; N, 11.93.
4H-Indolo[3,2,1-ij][1,7]naphthyridin-4-ones (10) (general procedure)
A mixture of 8-bromo-1,7-naphthyridin-4(1H)-one (15) (67.5 mg, 0.3 mmol), K2CO3 (83.1 mg, 0.6 mmol), PdCl2(dppf)·DCM (12.0 mg, 0.015 mmol, 5 mol %), and the appropriate 2-chlorophenylboronic acid (1.5 or 3 equiv) in dioxane/H2O (75:25, 2 mL) was heated at ∼88 °C (reflux) until the starting material had been completely consumed (as determined by TLC) (see Table 1). Then the mixture was left to cool to rt; K2CO3 (83.1 mg, 0.6 mmol) and a premixed solution of CuI (5.7 mg, 10 mol %) and DMCDA (9.45 μL, 20 mol %) in dioxane/H2O (75:25, 300 μL) were added to the reaction mixture, and the mixture was heated back to reflux for 12 h. The mixture was left to cool to rt, diluted (DCM, 20 mL), dried (Na2SO4), filtered, and evaporated to dryness. The remaining residue was dissolved in DCM, adsorbed onto silica, and chromatographed to give 4H-indolo[3,2,1-ij][1,7]naphthyridin-4-one 10.
8-Chloro-4H-indolo[3,2,1-ij][1,7]naphthyridin-4-one (10b)
2,3-Dichlorophenylboronic acid (74.2 mg, 0.45 mmol) gave compound 10b (chromatography eluent, DCM-NH3) as yellow needles (35.7 mg, 47%): mp (hot stage) 235.0–237.0 °C (EtOH); Rf = 0.37 (50:50 DCM/EtOAc); λmax (DCM, nm) 249 (log ε = 3.98), 263 (3.95), 284 (4.11), 293 (4.32), 330 inf (4.38), 337 (3.71), 387 inf (4.12), 395 (4.15); vmax (ATR, cm–1) 3125w and 3084w (aryl C–H), 1657m, 1649m, 1628s, 1574m, 1547m, 1485m, 1441s, 1406m, 1350m, 1335m, 1306m, 1285m, 1271w, 1246m, 1182s, 1159m, 1138m, 1105w, 1076w, 1061m, 1038m, 945m, 899m, 822m, 816m, 810m, 772m, 754m, 739m, 731m; 1H NMR (DMSO-d6, 500 MHz) δ 9.24 (1H, d, J = 8.0 Hz), 8.94 (1H, d, J = 5.5 Hz), 8.28 (1H, dd, J = 7.5, 1.0 Hz), 7.97 (1H, d, J = 5.0 Hz), 7.81 (1H, dd, J = 8.0, 1.0 Hz), 7.55 (1H, dd, J = 8.0, 8.0 Hz), 6.46 (1H, d, J = 8.0 Hz); 13C{1H} NMR (DMSO-d6, 125 MHz) δ 177.8 (s), 146.40 (s), 146.38 (d), 136.5 (d), 134.9 (s), 131.8 (d), 131.4 (s), 127.6 (s), 126.7 (s), 126.2 (d), 120.8 (d), 118.3 (s), 115.6 (d), 115.5 (d); m/z (ESI) 257 (37Cl, MH+, 35%), 255 (35Cl, MH+, 100%). Anal. Calcd for C14H7ClN2O: C, 66.03; H, 2.77; N, 11.00. Found: C, 66.19; H, 2.56; N, 11.23.
9-Methyl-4H-indolo[3,2,1-ij][1,7]naphthyridin-4-one (10c)
2-Chloro-4-methylphenylboronic acid (76.7 mg, 0.45 mmol) gave compound 10c (55.1 mg, 78%) as pale yellow needles: mp (hot stage) 249.0–251.0 °C (EtOH); Rf = 0.50 (30:70 DCM/EtOAc); λmax (DCM, nm) 243 inf (log ε = 4.22), 251 inf (4.19), 260 inf (4.05), 291 inf (4.31), 298 (4.46), 343 inf (4.03), 359 inf (4.09), 386 (4.32); vmax (ATR, cm–1) 3105w and 3055w (aryl C–H), 2907w (alkyl C–H), 1661m, 1624m, 1614s, 1585m, 1543s, 1508m, 1477m, 1452m, 1395m, 1350m, 1306m, 1273w, 1217w, 1196s, 1167m, 1132m, 1094m, 1028w, 860w, 802s, 787m, 746w; 1H NMR (CDCl3, 500 MHz) δ 8.84 (1H, d, J = 5.5 Hz), 8.23 (1H, d, J = 7.5 Hz), 8.21 (1H, d, J = 8.0 Hz), 7.96 (1H, d, J = 5.5 Hz), 7.48 (1H, s), 7.32 (1H, d, J = 8.0 Hz), 6.51 (1H, d, J = 7.5 Hz), 2.60 (3H, s); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.4 (s), 148.3 (s), 145.5 (d), 141.7 (s), 140.1 (s), 132.5 (d), 131.6 (s), 127.1 (s), 126.6 (d), 122.9 (s), 122.7 (d), 116.2 (d), 115.1 (d), 111.1 (d); m/z (ESI) 257 (M+ + Na, 45%), 235 (MH+, 100%). Anal. Calcd for C15H10N2O: C, 76.91; H, 4.30; N, 11.96. Found: C, 77.03; H, 4.16; N, 11.78.
9-Methoxy-4H-indolo[3,2,1-ij][1,7]naphthyridine-4-one (10d)
2-Chloro-4-methoxyphenylboronic acid (83.9 mg, 0.45 mmol) gave compound 10d (61.0 mg, 81%) as yellow needles: mp (hot stage) 269.5–270.0 °C (PhMe); Rf = 0.56 (50:50 DCM/THF); λmax (DCM, nm) 256 (log ε = 4.28), 266 inf (4.23), 288 inf (4.38), 297 (4.52), 303 (4.51), 375 inf (4.54), 383 (4.57); vmax (ATR, cm–1) 3107w, 3061w and 3042w (aryl C–H), 1940w and 2839w (alkyl C–H), 1612s (C=O), 1584m, 1547s, 1504m, 1477m, 1454m, 1435s, 1400m, 1354w, 1287m, 1279m, 1223s, 1194s, 1175m, 1130m, 1096m, 1072w, 1036m, 1016m, 968w, 864m, 843w, 808s, 745w; 1H NMR (CDCl3, 500 MHz) δ 8.81 (1H, d, J = 5.0 Hz), 8.21–8.20 (2H, m), 7.89 (1H, d, J = 5.5 Hz), 7.15 (1H, d, J = 2.0 Hz), 7.05 (1H, dd, J = 8.5, 2.0 Hz), 6.5 (1H, d, J = 7.5 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.6 (s), 162.4 (s), 148.5 (s), 146.0 (d), 141.4 (s), 132.4 (d), 131.7 (s), 126.7 (s), 123.7 (d), 118.7 (s), 116.2 (d), 114.2 (d), 111.9 (d), 96.6 (d), 58.2 (q); m/z (ESI) 273 (M+ + Na, 40%), 251 (MH+, 100%). Anal. Calcd for C15H10N2O2: C, 71.99; H, 4.03; N, 11.19. Found: C, 72.05; H, 4.14; N, 11.03.
9-(Trifluoromethyl)-4H-indolo[3,2,1-ij][1,7]naphthyridin-4-one (10e)
2-Chloro-4-(trifluoromethyl)phenylboronic acid (201.9 mg, 0.9 mmol) gave compound 10e (chromatography eluent, 80:20 DCM/THF) as yellow needles (40.6 mg, 47%): mp (DSC) onset 332.3 °C, peak max 333.4 °C (EtOH); Rf = 0.43 (50:50 DCM/EtOAc); λmax (DCM, nm) 263 (log ε = 3.80), 292 (4.29), 296 (4.32), 327 (3.80), 333 (3.81), 365 (3.90), 242 (4.17), 384 (4.19), 395 (4.29); vmax (ATR, cm–1) 3046w (aryl C–H), 1659m, 1649m, 1622m, 1551m, 1504m, 1479m, 1454m, 1439m, 1400m, 1327s, 1283m, 1194m, 1171m, 1159s, 1113s, 1074m, 1055m, 1028w, 968w, 893m, 818s, 725w; 1H NMR (DMSO-d6, 500 MHz) δ 9.17 (1H, d, J = 8.0 Hz), 8.99 (1H, d, J = 5.5 Hz), 8.77 (1H, s), 8.50 (1H, d, J = 8.5 Hz), 8.01 (1H, d, J = 5.5 Hz), 7.89 (1H, d, J = 8.0 Hz), 6.53 (1H, d, J = 7.5 Hz); 13C{1H} NMR (DMSO-d6, 125 MHz) δ 178.4 (s), 146.4 (d), 146.2 (s), 139.3 (s), 135.4 (d), 131.8 (s), 130.1 (q, 2JCF = 29.2 Hz), 127.8 (s), 126.5 (s), 124.2 (q, 1JCF = 270 Hz), 122.6 (d), 121.3 (q, 3JCF = 3.6 Hz), 116.1 (d), 115.5 (d), 110.2 (q, 3JCF = 3.9 Hz); m/z (ESI) 289 (MH+, 100%). Anal. Calcd for C15H7F3N2O: C, 62.51; H, 2.45; N, 9.72. Found: C, 63.08; H, 2.67; N, 9.83.
9-Chloro-4H-indolo[3,2,1-ij][1,7]naphthyridin-4-one (10f)
2,4-Dichlorophenylboronic acid (171.7 mg, 0.9 mmol) gave compound 10f (chromatography eluent, 90:10 EtOAc/THF) as beige cotton fibers (54.5 mg, 71%): mp (DSC) onset 326.5 °C, peak max 327.7 °C (EtOH); Rf = 0.64 (30:70 DCM/EtOAc); λmax (DCM, nm) 242 (log ε = 4.14), 263 (3.88), 283 inf (4.06), 290 inf (4.24), 294 inf (4.29), 298 (4.45), 335 inf (3.89), 347 inf (3.95), 380 inf (4.23), 387 (4.27); vmax (ATR, cm–1) 3055w (aryl C–H), 1663m, 1618m, 1587m, 1545m, 1499m, 1431m, 1395m, 1352m, 1306m, 1279m, 1261w, 1194m, 1123m, 1074m, 1063m, 1030w, 966w, 808s; 1H NMR (DMSO-d6, 500 MHz) δ 8.99 (1H, d, J = 7.5 Hz), 8.89 (1H, d, J = 5.5 Hz), 8.43 (1H, d, J = 2.0 Hz), 8.25 (1H, d, J = 8.5 Hz), 7.91 (1H, d, J = 5.5 Hz), 7.57 (1H, dd, J = 8.0, 2.0 Hz), 8.48 (1H, d, J = 8.0 Hz); 13C{1H} NMR (DMSO-d6, 125 MHz) δ 178.4 (s), 146.7 (s), 146.1 (d), 140.3 (s), 135.2 (d), 134.8 (s), 131.2 (s), 126.3 (s), 124.9 (d), 123.4 (s), 123.0 (d), 115.5 (d), 115.2 (d), 113.0 (d); m/z (ESI) 279 (37Cl, M+ + Na, 11%), 277 (35Cl, M+ + Na, 24%), 257 (37Cl, MH+, 40%), 255 (35Cl, MH+, 100%). Anal. Calcd for C14H7ClN2O: C, 66.03; H, 2.77; N, 11.00. Found: C, 66.16; H, 2.49; N, 11.12.
9-Fluoro-4H-indolo[3,2,1-ij][1,7]naphthyridin-4-one (10g)
2-Chloro-4-fluorophenylboronic acid (78.5 mg, 0.45 mmol) gave compound 10g (chromatography eluent, 80:20 DCM/THF) as beige needles (47.7 mg, 67%): mp (hot stage) 277.6–279.8 °C (EtOH); Rf = 0.48 (80:20 DCM/THF); λmax (DCM, nm) 242 (log ε = 4.17), 262 (3.96), 286 (4.26), 291 (4.31), 295 (4.44), 353 inf (4.00), 373 inf (4.25), 381 (4.29); vmax (ATR, cm–1) 3055w (aryl C–H), 1659m, 1649m, 1620s, 1587m, 1547m, 1505s, 1477m, 1447m, 1393w, 1350m, 1317m, 1281m, 1196s, 1171m, 1111w, 1090m, 1024w, 980w, 864m, 833m, 806s, 789w; 1H NMR (CDCl3, 500 MHz) δ 8.90 (2H, d, J = 5.5 Hz), 8.32 (1H, dd, J = 8.5, 5.5 Hz), 8.21 (1H, d, J = 8.0 Hz), 7.98 (1H, d, J = 5.5 Hz), 7.41 (1H, dd, J = 8.0, 2.0 Hz), 8.25 (1H, ddd, J = 8.5, 8.5, 2.0 Hz), 6.55 (1H, d, J = 8.0 Hz); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.4 (s), 164.3 (d, 1JCF = 250 Hz), 147.6 (s), 146.5 (d), 140.6 (d, 3JCF = 11.5 Hz), 132.3 (d), 132.0 (s), 127.0 (s), 124.2 (d, 3JCF = 10.4 Hz), 122.0 (s), 116.7 (d), 115.3 (d), 113.1 (d, 2JCF = 25.0 Hz), 99.0 (d, 2JCF = 25.0 Hz); m/z (ESI) 239 (MH+, 100%). Anal. Calcd for C14H7FN2O: C, 70.59; H, 2.96; N, 11.76. Found: C, 70.50; H, 2.79; N, 11.63.
10-Methoxy-4H-indolo[3,2,1-ij][1,7]naphthyridin-4-one (10h)
2-Chloro-5-methoxyphenylboronic acid (83.9 mg, 0.45 mmol) gave compound 10h (chromatography eluent, 70:30 DCM/THF) as yellow needles (50.1 mg, 66%): mp (hot stage) 236.0–237.2 °C (EtOH); Rf = 0.34 (80:20 DCM/THF); λmax (DCM, nm) 243 inf (log ε = 4.28), 271 (4.06), 280 inf (4.06), 291 inf (4.14), 308 inf (4.41), 311 (4.43), 342 (4.17), 407 (4.10); vmax (ATR, cm–1) 3084w and 3024w (aryl C–H), 2955w and 2932w (alkyl C–H), 1647m, 1618m, 1593m, 1541s, 1497m, 1485s, 1464m, 1433m, 1395m, 1356m, 1315m, 1285w, 1263m, 1227s, 1200m, 1188m, 1146m, 1121m, 1096m, 1040m, 943w, 912m, 860w, 810s, 795m; 1H NMR (CDCl3, 500 MHz) δ 8.88 (1H, d, J = 5.5 Hz), 8.21 (1H, d, J = 7.5 Hz), 8.00 (1H, d, J = 5.5 Hz), 7.83 (1H, d, J = 2.5 Hz), 7.59 (1H, d, J = 8.5 Hz), 7.23 (1H, dd, J = 8.5, 2.5 Hz), 6.50 (1H, d, J = 7.5 Hz), 3.96 (3H, s); 13C{1H} NMR (CDCl3, 125 MHz) δ 179.3 (s), 157.9 (s), 148.4 (s), 145.9 (d), 134.0 (s), 132.7 (d), 132.0 (s), 127.3 (s), 126.8 (s), 118.6 (d), 115.8 (d), 115.7 (d), 111.5 (d), 105.7 (d), 56.2 (q); m/z (ESI) 251 (MH+, 100%). Anal. Calcd for C15H10N2O2: C, 71.99; H, 4.03; N, 11.19. Found: C, 72.06; H, 3.96; N, 11.22.
10-(Trifluoromethoxy)-4H-indolo[3,2,1-ij][1,7]naphthyridin-4-one (10i)
2-Chloro-5-(trifluoromethoxy)phenylboronic acid (110.4 mg, 0.45 mmol) gave compound 10i (chromatography eluent, 30:70 DCM/EtOAc) as pale yellow needles (56.7 mg, 62%): mp (hot stage) 197.6–199.0 °C (EtOH); Rf = 0.63 (30:70 DCM/EtOAc); λmax (DCM, nm) 240 inf (log ε = 4.10), 261 (3.83), 291 inf (4.23), 297 (4.29), 330 inf (3.81), 337 (3.82), 385 inf (4.15), 392 (4.16); vmax (ATR, cm–1) 3030w (aryl C–H), 1657m, 1622m, 1589m, 1549m, 1495m, 1464m, 1431w, 1404w, 1354m, 1333m, 1248m, 1194s, 1173s, 1098m, 1024w, 962w, 920w, 876m, 816m; 1H NMR (DMSO-d6, 500 MHz) δ 9.01 (1H, d, J = 8.0 Hz), 8.89 (1H, d, J = 5.5 Hz), 8.29 (1H, d, J = 9.0 Hz), 9.19 (1H, s), 7.92 (1H, d, J = 5.0 Hz), 7.78 (1H, d, J = 8.0 Hz), 6.45 (1H, d, J = 7.5 Hz); 13C{1H} NMR (DMSO-d6, 125 MHz) δ 178.2 (s), 146.4 (s), 146.0 (d), 145.1 (s), 138.0 (s), 135.2 (d), 131.6 (s), 126.4 (s), 125.7 (s), 123.5 (d), 120.2 (q, 1JCF = 258.3 Hz), 115.7 (d), 115.3 (d), 114.6 (d), 113.9 (d); m/z (ESI) 305 (MH+, 100%). Anal. Calcd for C15H7F3N2O2: C, 59.22; H, 2.32; N, 9.21. Found: C, 59.36; H, 2.19; N, 9.28.
10-Chloro-4H-indolo[3,2,1-ij][1,7]naphthyridine-4-one (10j)
2,5-Dichlorophenylboronic acid (171.7 mg, 0.9 mmol) gave compound 10j (chromatography eluent, 30:70 DCM/EtOAc) as yellow needles (46.7 mg, 65%): mp (hot stage) 290.0–291.0 °C (EtOH); Rf = 0.59 (30:70 DCM/EtOAc); λmax (DCM, nm) 244 inf (log ε = 3.99), 264 (3.84), 296 inf (4.22), 301 (4.32), 332 inf (3.82), 338 (3.83), 392 (4.10), 396 inf (4.09); vmax (ATR, cm–1) 3086w and 3026w (aryl C–H), 2924w (alkyl C–H), 1657m, 1620s, 1584m, 1549m, 1497m, 1468m, 1452m, 1423m, 1396m, 1352m, 1317m, 1288w, 1269w, 1260w, 1192m, 1157w, 1132w, 1101w, 1080m, 1065w, 1026w, 901m, 883m, 812s; 1H NMR (DMSO-d6, 500 MHz) δ 9.02 (1H, d, J = 8.0 Hz), 8.91 (1H, d, J = 5.5 Hz), 8.29 (1H, d, J = 2.0 Hz), 8.23 (1H, d, J = 8.5 Hz), 7.95 (1H, d, J = 5.5 Hz), 7.82 (1H, dd, J = 8.0, 2.0 Hz), 8.47 (1H, d, J = 7.5 Hz); 13C{1H} NMR (DMSO-d6, 125 MHz) δ 178.2 (s), 146.4 (s), 146.0 (d), 138.2 (s), 135.2 (d), 131.3 (s), 130.1 (d), 129.3 (s), 126.4 (s), 126.1 (s), 121.3 (d), 115.6 (d), 115.3 (d), 114.0 (d); m/z (ESI) 255 (MH+, 100%). Anal. Calcd for C14H7ClN2O: C, 66.03; H, 2.77; N, 11.00. Found: C, 66.11; H, 2.68; N, 11.09.
10-Fluoro-4H-indolo[3,2,1-ij][1,7]naphthyridin-4-one (10k)
2-Chloro-5-fluorophenylboronic acid (78.5 mg, 0.45 mmol) gave compound 10k (chromatography eluent, 30:70 DCM/EtOAc) as yellow needles (49.8 mg, 70%): mp (DSC) onset 312.4 °C, peak max 313.0 °C (EtOH); Rf = 0.51 (30:70 DCM/EtOAc); λmax (DCM, nm) 233 inf (log ε = 4.04), 241 inf (4.04), 263 (3.73), 284 inf (3.91), 295 inf (4.10), 301 (4.17), 332 inf (3.77), 336 (3.77), 390 (3.99), 397 (3.98); vmax (ATR, cm–1) 3042w (aryl C–H), 1659m, 1649m, 1618m, 1545m, 1497m, 1479m, 1462m, 1454m, 1429m, 1396w, 1354w, 1337m, 1317m, 1294w, 1283w, 1263m, 1196s, 1144w, 1096w, 1024m, 918m, 905m, 822m, 806s; 1H NMR (DMSO-d6, 500 MHz) δ 9.02 (1H, d, J = 7.5 Hz), 8.91 (1H, d, J = 5.5 Hz), 8.25 (1H, dd, J = 8.0, 4.0 Hz), 8.10 (1H, d, J = 8.0 Hz), 7.95 (1H, d, J = 5.5 Hz), 7.66 (1H, dd, J = 8.0, 8.0 Hz), 8.46 (1H, d, J = 8.0 Hz); 13C{1H} NMR (DMSO-d6, 125 MHz) δ 178.1 (s), 159.6 (d, 1JCF = 250.0 Hz), 146.9 (s), 145.8 (d), 136.1 (s), 135.3 (d), 131.6 (s), 126.5 (s), 126.1 (d, 3JCF = 12.5 Hz), 117.6 (d, 2JCF = 25.0 Hz), 115.6 (d), 115.0 (d), 114.0 (d, 3JCF = 12.5 Hz), 108.2 (d, 2JCF = 25.0 Hz); m/z (ESI) 239 (MH+, 100%). Anal. Calcd for C14H7FN2O: C, 70.59; H, 2.96; N, 11.76. Found: C, 70.50; H, 2.85; N, 11.89.
Acknowledgments
P.A.K. and M.K. thank the Research and Innovation Foundation for funding (POST-DOC/0718/0005).
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.4c00440.
Discussion of reaction optimization for the synthesis of compound 2, other experimental procedures, crystallographic data for S4, and NMR spectra for all new compounds (PDF)
Author Contributions
P.A.K. and M.K. conceived the work. M.K. performed all of the synthetic work, characterized all of the compounds, and wrote and edited the manuscript. A.K. collected the X-ray data. P.A.K. edited the manuscript. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Dedication
This paper is dedicated to the memory of Dr Nikos Chronakis.
Supplementary Material
References
- Showalter H. D. H. Progress in the Synthesis of Canthine Alkaloids and Ring-Truncated Congeners. J. Nat. Prod. 2013, 76, 455–467. 10.1021/np300753z. [DOI] [PubMed] [Google Scholar]
- a Dai J.; Li N.; Wang J.; Schneider U. Fruitful Decades for Canthin-6-ones from 1952 to 2015: Biosynthesis, Chemistry, and Biological Activities. Molecules 2016, 21, 493. 10.3390/molecules21040493. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Farouil L.; Sylvestre M.; Fournet A.; Cebrián-Torrejón G. Review on canthin-6-one alkaloids: Distribution, chemical aspects and biological activities. Eur. J. Med. Chem. Rep. 2022, 5, 100049 10.1016/j.ejmcr.2022.100049. [DOI] [Google Scholar]
- Kump C.; Seibl J.; Schmid H. Über die Struktur des Tuboflavins. 4. Mitteilung über Pleiocarpa-Alkaloide. Helv. Chim. Acta 1963, 46, 498–505. 10.1002/hlca.19630460211. [DOI] [Google Scholar]
- Achenbach H.; Biemann K. Isotuboflavine and Norisotuboflavine. Two New Alkaloids Isolated from Pleiocarpa mutica Benth. J. Am. Chem. Soc. 1965, 87, 4177–4181. 10.1021/ja01096a030. [DOI] [PubMed] [Google Scholar]
- Xiaozheng W.; Jing W.; Fei W.; Xinyue X.; Tingting H.; Shuangjun L. A new β-carboline alkaloid from the Streptomyces flocculus CGMCC4.1223 mutant ΔstnK4. Tetrahedron 2023, 130, 133170 10.1016/j.tet.2022.133170. [DOI] [Google Scholar]
- McEvoy F. J.; Allen G. R. Synthesis of norisotuboflavine. J. Org. Chem. 1969, 34, 4199–4201. 10.1021/jo01264a110. [DOI] [Google Scholar]
- Rosenkranz H. J.; Schmid H. Notiz über die Synthese von Iso-, Noriso- und Desäthyl-tuboflavin. Helv. Chim. Acta 1968, 51, 565–568. 10.1002/hlca.19680510324. [DOI] [Google Scholar]
- Rosenkranz H. J.; Botyos G.; Schmid H. Synthese von Tuboflavin, 4-Äthyl-canthin-6-on und Canthin-6-on. Liebigs Ann. Chem. 1966, 691, 159–164. 10.1002/jlac.19666910120. [DOI] [Google Scholar]
- Puzik A.; Bracher F. A convenient approach to the canthin-4-one ring system: Total synthesis of the alkaloids tuboflavine and norisotuboflavine. J. Heterocycl. Chem. 2009, 46, 770–773. 10.1002/jhet.126. [DOI] [Google Scholar]
- Wetzel I.; Allmendinger L.; Bracher F. Revised Structure of the Alkaloid Drymaritin. J. Nat. Prod. 2009, 72, 1908–1910. 10.1021/np900515b. [DOI] [PubMed] [Google Scholar]
- Hsieh P.-W.; Chang F.-R.; Lee K.-H.; Hwang T.-L.; Chang S.-M.; Wu Y.-C. A New Anti-HIV Alkaloid, Drymaritin, and a New C-Glycoside Flavonoid, Diandraflavone, from Drymaria diandra. J. Nat. Prod. 2004, 67, 1175–1177. 10.1021/np0400196. [DOI] [PubMed] [Google Scholar]
- Fang H. W.; Liao Y.-R.; Hwang T.-L.; Shieh P.-C.; Lee K.-H.; Hung H.-Y.; Wu T.-S. Total synthesis of cordatanine, structural reassignment of drymaritin, and anti-inflammatory activity of synthetic precursors. Bioorg. Med. Chem. Lett. 2015, 25, 3822–3824. 10.1016/j.bmcl.2015.07.074. [DOI] [PubMed] [Google Scholar]
- Tremmel T.; Bracher F. New approaches to the synthesis of canthin-4-one alkaloids and synthetic analogues. Tetrahedron 2015, 71, 4640–4646. 10.1016/j.tet.2015.05.002. [DOI] [Google Scholar]
- Ohashi M.; Nishida H.; Shudo T.. Compounds having cGMP-Pde inhibitory effect. U.S. Patent 6,476,021 B1, 2002.
- Dighe S. U.; Mahar R.; Shukla S. K.; Kant R.; Srivastava K.; Batra S. Synthesis of S-(−)-5,6-Dihydrocanthin-4-ones via a Triple Cooperative Catalysis-Mediated Domino Reaction. J. Org. Chem. 2016, 81, 4751–4761. 10.1021/acs.joc.6b00613. [DOI] [PubMed] [Google Scholar]
- Tremmel T.; Puzik A.; Gehring A. P.; Bracher F. Arch. Pharm. Chem. Life Sci. 2016, 349, 710–723. 10.1002/ardp.201600137. [DOI] [PubMed] [Google Scholar]
- Broumidis E.; Koutentis P. A. A one-pot, two-step synthesis of 3-deazacanthin-4-ones via sequential Pd-catalyzed Suzuki-Miyaura and Cu-catalyzed Buchwald-Hartwig reactions. Tetrahedron 2017, 58, 2661–2664. 10.1016/j.tetlet.2017.05.076. [DOI] [Google Scholar]
- a Ioannidou H. A.; Martin A.; Gollner A.; Koutentis P. A. Three-Step Synthesis of Ethyl Canthinone-3-carboxylates from Ethyl 4-Bromo-6-methoxy-1,5-naphthyridine-3-carboxylate via a Pd-Catalyzed Suzuki–Miyaura Coupling and a Cu-Catalyzed Amidation Reaction. J. Org. Chem. 2011, 76, 5113–5122. 10.1021/jo200824b. [DOI] [PubMed] [Google Scholar]; b Gollner A.; Koutentis P. A. Two-Step Total Syntheses of Canthin-6-one Alkaloids: New One-Pot Sequential Pd-Catalyzed Suzuki–Miyaura Coupling and Cu-Catalyzed Amidation Reaction. Org. Lett. 2010, 12, 1352–1355. 10.1021/ol100300s. [DOI] [PubMed] [Google Scholar]
- a Umadevi M.; Muthuraj V.; Vanajothi R. Structural, cytotoxicity and molecular docking studies of some quinoline schiff bases and their Pd(II), Mn(II) and Ru(II) complexes. J. Mol. Struct. 2020, 1221, 128778 10.1016/j.molstruc.2020.128778. [DOI] [Google Scholar]; b Al Zoubi W.; Al-Hamdani A. A. S.; Putu Widiantara I.; Hamoodah R. G.; Ko Y. G. Theoretical studies and antibacterial activity for Schiff base complexes. J. Phys. Org. Chem. 2017, 30, e3707 10.1002/poc.3707. [DOI] [Google Scholar]
- Wu H.; Lin J.; Li Y.; Wei C.; Chen S.; Long C.; Chen X.; Liu Z.; Chen L.. Quinoline derivatives as SMO Inhibitors. EP3124482 A1, 2017.
- Hardwood L. M. Dry-Column Flash Chromatography. Aldrichimica Acta 1985, 18, 25–25. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.