

Abstract
The aqueous uranyl dication has long been known to facilitate the UV light-induced decomposition of aqueous VOCs (volatile organic compounds), via the long-lived highly efficient, uranyl excited state. The lower-energy visible light excited uranyl ion is also able to cleave unactivated hydrocarbon C–H bonds, yet the development of this reactivity into controlled and catalytic C–H bond functionalization is still in its infancy, with almost all studies still focused on uranyl nitrate as the precatalyst. Here, hydrocarbon-soluble uranyl nitrate and chloride complexes supported by substituted phenanthroline (Ph2phen) ligands are compared to each other, and to the parent salts, as photocatalysts for the functionalization of cyclooctane by H atom abstraction. Analysis of the absorption and emission spectra, and emission lifetimes of Ph2phen-coordinated uranyl complexes demonstrate the utility of the ligand in light absorption in the photocatalysis, which is related to the energy and kinetic decay profile of the uranyl photoexcited state. Density functional theory computational analysis of the C–H activation steps in the reaction show how a set of dispersion forces between the hydrocarbon substrate and the Ph2phen ligand provide control over the H atom abstraction, and provide predictions of selectivity of H atom abstraction by the uranyl oxo of the ring C–H over the ethyl C–H in an ethylcyclohexane substrate.
We show how the effect of different ligands on the control of the functionalization of unactivated hydrocarbons by visible light excited uranyl catalysts.
Introduction
The controlled functionalisation of unactivated C(sp3)–H bonds remains a long-lasting challenge in synthetic chemistry. In recent years, visible light-induced catalytic C–H bond activation by the uranyl(vi) ion ([UO2]2+), the predominant form of uranium in the environment, has been rapidly gaining interest.1 The excitation of uranyl complexes by light of ca. 420 nm induces a ligand to metal charge transfer (LMCT) to give a highly reactive photoexcited uranyl ion which can react directly and efficiently with various organic substrates through hydrogen atom abstraction (HAA or hydrogen atom transfer, HAT) or single electron transfer (SET).1,2
Uranyl nitrate [UO2(NO3)2(OH2)2]·4H2O (UNO3) is by far the most widely studied photocatalyst. Early, groundbreaking studies were reported by Bacak on the aerobic oxidation of aqueous organics such as toluene under UV light excitation.3,4 Subsequent work started to focus on lower energy light-initiated reactions in organic solvents, with studies showing that uranyl nitrate is an excellent catalyst for alkane fluorination by an electrophilic fluorine source; simple cyclic and linear saturated alkanes such as cyclooctane could be fluorinated in 95% yield (95 turnovers) under illumination by a high intensity lamp.5 Uranyl acetate, [UO2(OAc)2·4H2O] was found to be an inferior photocatalyst to uranyl nitrate for this reaction. In 2019, our group reported the new uranyl–phenanthroline complex [UO2(NO3)2(Ph2phen)] 1-NO3 (Ph2phen = 4,7-diphenyl-1,10-phenanthroline), Fig. 1, as a selective catalyst for the oxidation of benzylic C–H bonds as well as C–C bond cleavage in 2-phenoxy-1-phenylethanol (2P1PE), a model for the parts of lignin that are most challenging to cleave.6,7 The oxidation of a range of benzylic substrates is notable since photoexcited uranyl nitrate is quenched by these substrates. The mechanism is proposed to involve a hydrogen atom abstraction (HAT) from the hydrocarbon by the oxo group of the photoexcited uranyl, which forms a carbon-centered radical that can react onwards with a variety of electrophiles.1 Uranyl complexes with phenanthroline (phen) bound in the equatorial plane instead of solvent molecules, were first reported fifty years ago.8 Phenanthroline is interesting as a neutral, L2-donor ligand since it is known for its derivatizability and light absorbing capacity, thus has the potential to act as an antenna ligand.9 We found that [UO2(NO3)2(Ph2phen)] gives higher conversions than uranyl nitrate for all substrates tested; two examples are shown in the table in Fig. 1.
Fig. 1. Uranyl nitrate and the phenylphenanthroline (Ph2phen) adduct that we reported previously (upper); a comparison of their capacity as precatalysts for hydrocarbon oxidation (center); and the Ph2phen adduct of uranyl chloride also studied as a precatalyst in this work (lower).

The Ph2phen ligand derives from phen, 1,10-phenanthroline, a weakly fluorescent molecule which has been incorporated into many luminescent coordination compounds; substitution at the 4 and 7 positions is not anticipated to significantly increase its fluorescence.9 A rapidly growing range of substrate oxidations is now being reported for uranyl nitrate hexahydrate, include the addition of (cyclo)alkanes to electron-poor olefins, ethers, acetals, and amides arising from HAT from the hydrocarbon substrate, ether, sulfone, aniline oxidations, and polymer degradation.10–20 Uranyl acetate, uranyl sulfate UO2SO4·4H2O, and uranyl triflate UO2(OTf)2·6H2O were all found to give lower yields than uranyl nitrate in those reactions.6,7,9,18
Uranyl chemistry is dominated by fast ligand exchange between the (normally) five ligands bound in the equatorial plane. In four instances a supporting (L-donor type) ligand has been used to control access to, or reactivity of the photocatalyst in solution. We have already shown that binding phenanthroline ligands equatorially, as described above, can improve product yield.6 A uranyl complex supported by a β-diketonatebis(catecholamide) ligand (CH2{COO(CH2)n(2,3-Ph(OH)2CONH)}2; n = 3, 4) showed good yields for photocatalytic degradation of rhodamine B.11 A chiral uranyl salen complex, [UO2(HOEt)(salen)] (salen = 2,2′-((1E,1′E)-((1R,2R)-cyclohexane-1,2-diylbis(azanylylidene))bis-(methanylylidene))diphenol), was used for α-cyanation of anilines.21 Again, uranyl acetate was found to only give trace amounts of cyanated product. Finally, the phosphine oxide – solvated cation [UO2(OPCyPh2)4]2+ catalyzes the oxygenation by molecular O2 of cyclohexene to four different oxygenated products (ketones, epoxide, alkenol) via a uranyl(vi) peroxo intermediate.22
Here we report an experimental, computational, and spectroscopic study on how the Ph2phen ligand helps control the reactivity of uranyl nitrate and chloride catalysts for alkane functionalization and explain why the choice of anionic co-ligand is also so important in this photocatalysis.
Results and discussion
Synthesis and structure
The syntheses of the photocatalysts under study here, the acetonitrile adduct of uranyl dichloride, three forms of the mono(ligand) adduct, and the bis(ligand) adduct, are shown in Scheme 1. We first targeted the mono(ligand) chloride complex [UO2Cl2(Ph2phen)(NCMe)] 1-Cl for comparison with the nitrate. We hypothesized that the uranyl catalysts could be more versatile than d-block metal oxo catalysts for hydrocarbon oxidation reactions in the absence of air.23 Oxidation using d-block metal oxo catalysts tend to yield oxygenated hydrocarbons arising from the transfer of the O atom to the product, whereas the strongly bound uranyl oxo groups should not participate in this ‘radical rebound mechanism’, enabling greater control of substrate functionalization.24
Scheme 1. The uranyl chloride complexes made for comparison with 1-NO3, containing 0, 1, and 2 Ph2phen ligands.

The complex 1-Cl can be made from the combination of equimolar Ph2phen and pale-yellow uranyl chloride hydrate (UO2Cl2·3H2O) in acetonitrile, which forms a dark yellow solution. The subsequent removal of volatiles yields 1-Cl as an orange solid which is very soluble in acetonitrile and acetone, Scheme 1. The 1H NMR spectra of CD3CN solutions of complex 1-Cl confirm the identity of the complex, with resonances shifted as far as 2 ppm higher in frequency compared to free ligand. For example, the two protons closest to the U(vi) center resonate at 11.27 ppm, compared to 9.17 in the unbound ligand, 9.25 ppm in Ph2phen, and 10.56 ppm in the related nitrate complex, [UO2(NO3)2(Ph2phen)] 1-NO3, that our group reported previously.6
The asymmetric U O stretch in the IR spectrum of the complex appears at 922 cm−1, which is lower than that of the uranyl starting material [UO2Cl2(THF)2]2 (947 cm−1). As expected, the O U O bond is thus weakened upon complexation of Ph2phen. The reported O U O stretch of a similar uranyl chloride complex with unsubstituted phenanthroline, [UO2Cl2(phen)], is higher at 932 cm−1.8 Similarly, the νasym(O U O) in uranyl nitrate complexes is slightly higher with phen vs. the Ph2phen ligand (942 cm−1 for [UO2(NO3)2(phen)] vs. 936 cm−1 for [UO2(NO3)2(Ph2phen)]).6
The cyclic voltammogram of 1-Cl was recorded in MeCN solution with 0.1 M TBA-BPh4 as the supporting electrolyte. The CV shows a relatively accessible, irreversible reduction at −1.163 V vs. Fc+/0 that we assign to the UVI/V couple.25,26
During the synthesis of 1-Cl, the formation of a small amount of a bright yellow solid, the less–soluble complex [UO2Cl2(Ph2phen)2] 2 (Scheme 1 and discussed below) is observed to form. Since further manipulations of 1-Cl result in further contamination with 2, samples for catalysis were prepared immediately prior to use. While we were unable to isolate single crystals of the acetonitrile adduct 1-Cl, vapor diffusion of hexanes into a THF solution of the crude solid yields crystals of [UO2Cl2(Ph2phen)(THF)], 1-Cl-THF, Fig. 2a and Table 1, while vapor diffusion of hexanes into a chlorobenzene solution affords the unsolvated dimer [UO2Cl2(Ph2phen)]2, [1-Cl]2Fig. 2b and Table 1.
Fig. 2. Solid-state structures of (a) [UO2Cl2(Ph2phen)(THF)], 1-Cl-THF, (b) unsolvated dimer [UO2Cl2(Ph2phen)]2, [1-Cl]2 (c) [UO2Cl2(Ph2phen)2], 2. Displacement ellipsoids set at 50%; H atoms omitted for clarity.

Key bond lengths (Å) and angles (°) for the solid-state crystal structures of 1-Cl-THF, [1-Cl]2, and 2 with uncertainties in parentheses compared with UO2(Ph2phen)2(NO3)2.
| Parameter | 1-Cl-THF | [1-Cl]2 | 2 | UO2(Ph2Phen)(NO3)2 |
|---|---|---|---|---|
| U O | 1.763(2), 1.766(2) | 1.746(3), 1.755(3) | 1.777(2), 1.777(3) | 1.747(3)–1.756(3) |
| U–Clterminal | 2.7023(7), 2.7057(6) | 2.644(1) | 2.684(1), 2.688(1) | N/A |
| U–Clbridging | N/A | 2.768(1), 2.793(1) | N/A | N/A |
| U–NPh2phen | 2.627(2), 2.639(2) | 2.562(3), 2.620(3) | 2.615(3), 2.662(4), 2.816(3), 2.782(3) | 2.589(3)–2.625(4) Å |
| ∠O U O | 177.41(8) | 178.1(1) | 163.4(1) | 177.28(14), 177.06(14) |
The asymmetric O U O stretch in the IR spectrum of 1-Cl appears at 922 cm−1, which is lower than that of the anhydrous uranyl chloride [UO2Cl2(MeCN)3] (953 cm−1), indicating that the O U O bond is weakened upon complexation of Ph2phen, and more so than upon complexation of the unsubstituted phenanthroline, which was already reported, [UO2Cl2(phen)] (932 cm−1).2 The band maximum in the electronic absorption spectrum of [UO2Cl2(Ph2phen)(MeCN)] in acetonitrile that corresponds to the U(5f) ← O(2p) LMCT transition appears at 449 nm (ε = 56 M−1 cm−1), which is red-shifted compared to uranyl chloride starting material (78 M−1 cm−1 at 434 nm) and the nitrate complex [UO2(NO3)2(Ph2phen)] (427 nm), indicating that [UO2Cl2(Ph2phen)(MeCN)] requires a lower energy to access the photoactive excited state. The solid-state structure of the THF solvate, 1-Cl-THF, is structurally similar with that of the previously reported mono Ph2phen complex 1-NO3, with the O U O angle essentially linear (177.41°) and U O bond lengths typical for uranyl complexes (1.764 Å ave.).
A complex with two unsubstituted phenanthrolines has also been reported; [UO2Cl2(phen)2] has bent and elongated U O bonds in the solid state (O–U–O angle = 161.88°) with distinct interactions between the uranyl oxo atoms and the H atoms in the phen that is most distorted away from the equatorial plane.27 Here, the addition of an acetonitrile solution of uranyl chloride hydrate (UO2Cl2·3H2O) to an acetonitrile solution of two equivalents of Ph2phen immediately results in the precipitation of a bright yellow powder characterized as [UO2Cl2(Ph2phen)2] 2, in 89% yield. We originally targeted the Ph-substituted ligand as it is so much more soluble than phen in organic solvents, but 2 is almost completely insoluble in acetonitrile or acetone, and sparingly soluble in methanol, chloroform, or dichloromethane. The 1H NMR spectrum of 2 in CD3CN solution shows that the Ph2phen resonances are again shifted to around 1 ppm higher frequency compared to unbound Ph2phen. In the solid state IR spectrum of 2, νasym(O U O) appears at 900 cm−1, which shows that the O U O bond is significantly weakened compared to the mono (Ph2phen) adduct 1-Cl (922 cm−1).3 As shown in Fig. 2c, the O U O angle in the solid-state structure of 2 is 163.4°, slightly less bent than in the analogue unsubstituted phen complex, while the U O distances are typical (mean 1.777 Å). The two Ph2phen ligands are nearly perpendicular to each other (74.5° between calculated planes of the phen backbones). The U–N bonds are also shorter for the Ph2phen parallel to the equatorial plane, and longer for the Ph2phen parallel to the uranyl group (2.615(3) and 2.662(4) vs. 2.816(3) and 2.782(3) Å), Table 1.
Absorption and emission spectroscopic study of the precatalysts 1-NO3 and 1-Cl
We have recorded absorbance and emission spectra for the precatalysts 1-NO3 and 2, recognising that in MeCN solution under catalytic conditions, 2 immediately loses one Ph2phen ligand to form 1-Cl. We also measured the photophysics of the uncomplexed ligand Ph2phen to be able to confirm that it does not interfere in the chemistry. We note that, recently, the absorption and emission properties of the unsubstituted analogue [UO2Cl2(phen)2] have been recorded.28
The absorption spectrum of 1-NO3, containing a single Ph2phen ligand bound to uranyl(vi) nitrate, recorded in MeCN solution, Fig. 3, consists of a single band peaking at 287 nm arising from the ligand, and a shoulder (350–390 nm) linked to metal–ligand interactions. The emission spectrum displays two convoluted bands which are observed at 445 nm and 520 nm; the lower energy band displays the well-known fine structure arising from vibronic coupling in the uranyl ion. This is confirmed through the use of time gated detection, where the short wavelength side of the emission band is significantly attenuated with a 50 μs gate, revealing more detail on the signal from the uranyl LMCT emission. Low temperature (77 K) studies, Fig. 3b, yield more resolution of the uranyl emission fine structure and a significant reduction in the relative intensity of the 445 nm feature which is indicative of the suppression of thermally activated back energy transfer processes that lead to population of a close-lying LMCT state. These observations, alongside previous work, lead to the assignment of the emission band centered at 445 nm, as phen ligand-to-uranium charge transfer, and at lower energy (520, 540 nm), as oxo ligand-to-uranium LMCT respectively.29 Indeed, the excitation spectra of these bands show some vibrational fine structure as commonly observed in uranyl-based charge transfer excitations which further confirms the assignments.30
Fig. 3. (a) Absorption, emission, and excitation spectra of 1-NO3 in MeCN at room temperature and (b) in frozen methyl-THF at 77 K.

A direct comparison of 1-NO3 with the new catalyst 1-Cl is of interest. Time resolved measurements of room temperature MeCN solutions of 1-NO3Fig. 4 (and Fig. S5† for 77 K spectra), show that the uranyl emission is much longer lived (42 μs at λem = 500 nm) than the equatorial LMCT emission (5 μs at λem = 430 nm) which has decayed to near background intensity after 30 microseconds.
Fig. 4. (a) Time resolved emission spectra map and (b) spectra recorded with increasing time delay (B) of 1-NO3 in MeCN at room temperature.

Samples of analytically pure 2 are essentially insoluble in MeCN, but release 1-Cl and Ph2phen into solution, enabling us to ensure that no uncomplexed uranyl chloride is present in solution, which we observe if we simply measure solutions of 1-Cl in MeCN. Background measurements were also made of a MeCN solution of the Ph2phen ligand alone. A solution of Ph2phen exhibits an absorption maximum at 273 nm, and when excited at this wavelength, exhibits both fluorescence (room temperature) and phosphorescence (77 K) at 380 nm and 511 nm respectively (see ESI†). However, these features are unlikely to contribute to the photocatalysis since the excitation wavelength of the light used in these experiments is much lower energy.
Emission spectra of 1-Cl in the presence of uncoordinated Ph2phen are shown in Fig. 5. The typical uranyl(vi) LMCT emission at ca. 520 nm is visible and long-lived (73 μs at λem = 500 nm) across the excitation region (280–360 nm). There is also a higher energy broad feature at 381 nm, which is much shorter lived and is assigned to Ph2phen fluorescence, from the singlet state. Thus, any contribution from Ph2phen to catalysis is unlikely.‡
Fig. 5. Left; Absorption, emission, and excitation spectra of 2 dissolved in MeCN (present as 1-Cl + Ph2phen) at room temperature. The blue trace was recorded using a 50 microsecond time delay and gate and shows the longer-lived uranyl emission can be resolved after the shorter lived emission centred at 380 and 444 nm has decayed.

Computational analysis of the differences between the catalysts and effect of the Ph2phen ligand on substrate binding
DFT calculations (ADF 2021 software, scalar-ZORA PBE0 and PBE levels, see ESI†) were carried out on models of the catalysts UNO3, 1-NO3, UCl, 1-Cl. Selected calculated geometry parameters and Mayer bond orders in the ground and excited states of each catalyst are listed in Tables 2 and S10† at PBE0 and PBE respectively.
Selected calculated geometry parameters and Mayer bond order for catalysts in the singlet ground state (S0) and lowest triplet excited state (T1) (bond lengths in Å, angles in °) at scalar-ZORA PBE0-D3BJ/TZP level.
| [U] catalyst | Parameters | S0 | T1 | ||
|---|---|---|---|---|---|
| Bond length/angle | Bond order | Bond length/angle | Bond order | ||
| [UO2(NO3)2(H2O)2] UNO3 | U–NO3 | 2.499 | 0.32 | 2.504 | 0.31 |
| 2.496 | 0.32 | 2.500 | 0.31 | ||
| 2.487 | 0.32 | 2.487 | 0.32 | ||
| 2.488 | 0.32 | 2.488 | 0.31 | ||
| U–H2O | 2.477 | — | 2.488 | — | |
| 2.514 | — | 2.512 | — | ||
| U O | 1.764 | 1.84 | 1.815 | 1.59 | |
| 1.764 | 1.84 | 1.814 | 1.60 | ||
| ∠O U O | 178.2 | 177.6 | |||
| [UO2(Ph2phen)(NO3)2] 1-NO3 | U–NO3 | 2.501 | 0.31 | 2.576 | 0.23 |
| 2.499 | 0.31 | 2.578 | 0.23 | ||
| 2.498 | 0.30 | 2.580 | 0.22 | ||
| 2.498 | 0.30 | 2.587 | 0.22 | ||
| U-Ph2phen | 2.593 | 0.31 | 2.705 | 0.20 | |
| (eq) | 2.589 | 0.31 | 2.693 | 0.21 | |
| U O | 1.768 | 1.83 | 1.842 | 1.70 | |
| 1.768 | 1.83 | 1.842 | 1.70 | ||
| ∠O U O | 177.9 | 179.6 | |||
| [UO2Cl2(MeCN)3] UCl | U–Cl | 2.697 | 0.74 | 2.728 | 0.72 |
| 2.697 | 0.74 | 2.717 | 0.72 | ||
| U–MeCN | 2.554 | 0.27 | 2.565 | 0.26 | |
| 2.539 | 0.28 | 2.598 | 0.26 | ||
| 2.552 | 0.27 | 2.567 | 0.26 | ||
| U O | 1.768 | 1.85 | 1.825 | 1.58 | |
| 1.768 | 1.85 | 1.825 | 1.59 | ||
| ∠O U O | 178.4 | 177.3 | |||
| ∠Cl U Cl | 147.1 | 146.9 | |||
| [UO2Cl2(Ph2phen)(MeCN)] 1-Cl | U–Cl | 2.705 | 0.70 | 2.837 | 0.45 |
| 2.706 | 0.70 | 2.842 | 0.45 | ||
| U-ph2phen | 2.616 | 0.29 | 2.706 | 0.20 | |
| (eq) | 2.624 | 0.29 | 2.715 | 0.24 | |
| U–MeCN | 2.554 | 0.28 | 2.625 | — | |
| U O | 1.769 | 1.85 | 1.827 | 1.73 | |
| 1.769 | 1.85 | 1.827 | 1.73 | ||
| ∠O U O | 179.8 | 177.9 | |||
| ∠Cl U Cl | 143.7 | 141.6 | |||
Uranyl chloride coordinates three acetonitrile solvent molecules to form [UO2Cl2(MeCN)3] UCl in solution. As Table 2 shows, the calculated bond lengths at the PBE0 level agree very well with the experimental data in Table 1. The complex has a similar coordination environment to compound 1-Cl, which has three nitrogen and two chlorine atoms bound. On the other hand, because of the repulsion from the chloride ions, the Ph2phen ligand in compound 1-Cl is tilted out of the equatorial plane with a dihedral angle of 26°. This weakens the U-Ph2phen bonding. Thus, UCl and 1-Cl have very similar U O bond lengths and bond orders with asymmetric U O stretching frequencies of ∼880 cm−1 and ∼960 cm−1 at PBE and PBE0 respectively. Again, PBE0 gives good agreement with experimental results mentioned above, notably in the U O bond lengths. The bending of the uranyl group in compound 2 weakens the U O bonds.
The U O bond length in the singlet S0 is elongated on excitation to the triplet T1 state. For example, the U O bond length for UCl lengthens from 1.768 Å to 1.825 Å at PBE0 level. On the other hand, the U O bond lengths for [UO2(ph2phen)(NO3)2] and 1-Cl lengthen by 0.074 Å and 0.058 Å. Along with this, the U O bond orders for [UO(NO3)2(H2O)2] and [UO2Cl2(MeCN)3] change dramatically by ∼0.25 and ∼0.27 respectively. By contrast, the U O bond order for [UO2(Ph2phen)(NO3)2] and 1-Cl show only ∼0.13 change at PBE0 level.
The U O bond length changes from S0 to T1 for [UO2(ph2phen)(NO3)2] 1-NO3 and 1-Cl are larger than in U0 and UCl. This indicates that the electronic structures are strongly affected by the ph2phen ligand. The spin density diagrams in Fig. 6 show that for the triplet excited state the spin density of UCl is localized on the uranium and oxygen atoms. This gives obvious oxyl-radical character which is similar to the results reported by Wu et al.31 However, in the presence of Ph2phen ligands, the system exhibits a distinct charge transfer mechanism, where the U O bond undergoes cleavage, leading to the formation of a U(v) complex.32 Concurrently, an electron is transferred to the Ph2phen ligand, instead of localizing on the uranyl oxygen atoms. This interaction is critical in defining the electronic structure and the reactive character of the complex under study. For complex 2, this electron localization, or spin density, is predominantly observed on the Ph2phen ligand situated axially (parallel to the uranyl axis) rather than the ligand in the equatorial plane, see Fig. 6 right.
Fig. 6. T1 state spin density of [UO2(NO3)2(H2O)2] UNO3, [UO2Cl2(MeCN)3] UCl, [UO2Cl2(Ph2phen)(MeCN)] 1-Cl, and [UO2Cl2(Ph2phen)2] 2 complexes calculated at the scalar-ZORA PBE0/TZP level of theory.

Looking at the energy levels of the excited states, after excitation to higher excited states, the system relaxes through intersystem crossing, ISC, to the lowest triplet state T1 which is lower in energy for compounds with the Ph2phen ligand, which stabilizes the radical. In the non-stabilized complexes, the lowest triplet state is much higher in energy and connected with the UV–O˙ electronic structure. The U O bond orders in compounds 1-NO3, 1-Cl and 2 agree with the state containing Uv character and a radical Ph2phen ligand with increased bond lengths for 1-NO3, 1-Cl and 2 due to lowering of the charge on uranyl from 2+ to 1+. This is also supported by TDDFT calculations as shown in Fig. 7 and S14 as well as Tables S11–13.†
Fig. 7. The orbital energy levels for the of [UO2Cl2(Ph2phen)(MeCN)] 1-Cl calculated at the scalar-ZORA PBE0/TZP level of theory.

The calculated reaction Gibbs free energies for the HAT reaction show that, upon excitation, the more distorted compound 2 is likely to lose one Ph2phen ligand to form compound 1-Cl. This explains the color changes observed experimentally and the similar reaction yield from compounds 1-Cl and 2.
The energy barriers for the HAT from cyclooctane by 1-Cl, 1-NO3, UCl and UNO3 are 8.2, 7.1, 1.0, and 0.0 kcal mol−1 respectively at the PBE level of theory. The energy profiles as well as spin density of reactant, transition state, and product are shown in Fig. 8 and S18.† As Fig. 8 right shows, the spin on the Ph2phen ligand moves to the uranyl to form a reactive oxyl-radical during the reaction. For [UO2(NO3)2(H2O)2] there is no reaction barrier for the HAT reaction at the scalar-ZORA PBE/TZP level of theory. Thus, single point energy correction at coupled cluster level (DLPNO-CCSD(T)) on the solution-optimized geometries from ADF PBE0 level were carried out, see Fig. 9 for the energy profiles for UNO3 and 1-Cl.
Fig. 8. Energy profiles for HAT from cyclooctane by the excited state of [UO2Cl2(MeCN)3] UCl (left) and [UO2Cl2(ph2phen)(MeCN)] 1-Cl (right) calculated at the scalar-ZORA PBE-D3BJ/TZP levels of theory. See Fig. S18† for the energy profile for 1-NO3. Spin densities of reactant, transition state, and product are shown (spins on different atoms are marked in different colours: Cl – azure, O – red, U – pink, and C – black).

Fig. 9. Energy profiles for HAT from cyclooctane by the excited state of [UO2(NO3)2(H2O)2] UNO3 (left) and [UO2Cl2(Ph2phen)(MeCN)] 1-Cl (right) calculated at the DLPNO-CCSD(T)//scalar-ZORA PBE0-D3BJ/TZP level of theory. Spin density of reactant, transition state, and product are shown (spins on different atoms are marked in different colours: Cl – azure, O – red, U – pink, and C – black).

The HAT reaction barrier for [UO2(NO3)2(H2O)2] UNO3 at the DLPNO-CCSD(T)//scalar-ZORA PBE0 D3BJ/TZP level is about 1.2 kcal mol−1, which is smaller than the results at the CASPT2 level reported by Wu et al.31 The profile for 1-NO3 shows very similar spin densities to 1-Cl, which has a barrier of 23.1 kcal mol−1. Thus, the predicted reaction barriers follow the order as UNO3 < UCl < 1-NO3 ≈ 1-Cl. The reason for compounds 1 having a higher barrier might be because the electron needs to transfer from the Ph2phen ligand to the uranyl oxygen, but this can be offset by the favorable weak interactions between Ph2phen ligand and reactants in the pre-reaction complex. This can explain why the observed trend in reactivity for fluorination and oxidation is rather UNO3 < 1-NO3 ≈ 1-Cl < UCl, and why it does not match what is predicted from calculated reaction barriers. The calculated energies of weak interactions between different reactants and Ph2phen are summarized in Table 3. The dispersion contribution has a positive correlation with the number of atoms in the reactants. The reactants should be larger than CH3CN, the solvent, to gain an advantage in the reaction, which might give higher selectivity for larger reactants. As Table S14† shows, the dispersion interactions of the S0 and T1 states of cyclooctane-UO2 compounds are the same. On the other hand, comparing to cyclooctane-UCl, the change of dispersion energy from T1 state to the transition state (TS) of cyclooctane 1-Cl compound is about 2 kcal mol−1. This contributes very little to the barrier associated with the HAT, as the barrier from the excited state is calculated to be +23.1 kcal mol−1.
Dispersion interactions between different reactants and Ph2phen ligand in kcal mol−1 calculated at the scalar-ZORA PBE-D3BJ/TZP level of theory.
| CH3CN | CH4 | C2H6 | C3H8 | C4H10 | C5H12 | C6H14 | Cyclohexane | Cyclooctane | |
|---|---|---|---|---|---|---|---|---|---|
| Dispersion interactions | −5.3 | −3.6 | −5.2 | −6.8 | −9.7 | −8.9 | −10.9 | −9.2 | −10.9 |
We were interested in the capacity of the Ph2phen ligand to afford some regioselectivity to the alkane substrate and have computed the energies of various orientations of ethylcyclohexane. We find that the conformer exhibiting the greatest contact area with the aromatic ligand is preferred. As illustrated in Fig. 10a, which focuses just on Ph2phen and ethylcyclohexane, when reactants present different faces the reaction will selectively favor functional groups on the most extensive surface. Apart from the dispersion forces attributed to the size of the reactant, the shape of Ph2phen also influences the orientation of the reactants. As depicted in Fig. 10b, which looks at the ethylcyclohexane with the whole molecule of 1-Cl, a C–H bond on the cyclohexyl ring is more likely to be cleaved than one on the ethyl group. The differences are very small for this small substrate, but we have sought experimental evidence of such a regioselectivity in the reaction to fluorinate ethylcyclohexane (see below for experimental details). Preliminary studies, see ESI,† show no selectivity differences between 1-Cl and UCl, as determined by NMR spectroscopy, although we note that the calculated energy differences are small with respect to room temperature, and NMR spectroscopy is a relatively insensitive technique. Future work will focus on larger hydrocarbons that can form a larger number of dispersion forces with the coordinated Ph2phen.
Fig. 10. (a) Face-dependent adsorption of ethylcyclohexane to Ph2phen with steric strain at scalar-ZORA PBE-D3BJ/TZP level of theory; (b) orientation-dependent adsorption of ethylcyclohexane onto 1-Cl at scalar-ZORA PBE-D3BJ/TZP level of theory. Orange = uranium, red = oxygen, turquoise = carbon, white = hydrogen, blue = nitrogen, green = chlorine.

Experimental photocatalytic alkane functionalization and the roles of the different equatorial ligands
This new family of uranyl complexes are all photocatalysts for alkane functionalization. The substrate cyclooctane (C–H BDFE 95.7 kcal mol−1)5 was chosen as a model substrate that contains a strong C–H bond (95.7 kcal mol−1)13 and yields and products (which are all literature compounds) are readily tracked by NMR spectroscopy and GC. The general reaction is anticipated to occur via the abstraction of a hydrogen atom from the alkane by the oxo group of the photoexcited uranyl (HAT), which leads to a carbon centered radical which can undergo reactive quenching for functionalization. Three reaction types were studied here; allowing the radical to react with an electrophilic F source under anaerobic conditions (Scheme 2), to form a CC bond through reaction with a Michael acceptor, also anaerobically (Scheme 3), and aerobic oxidation (Scheme 4).
Scheme 2. Photocatalytic fluorination of cyclooctane with uranyl complexes.
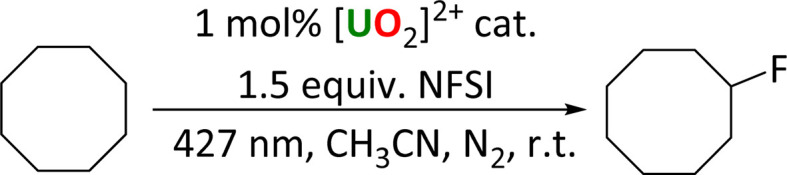
Scheme 3. Photocatalytic cyclooctane addition to benzylidene malononitrile.

Scheme 4. Photocatalytic oxidation of cyclooctane by uranyl complexes.
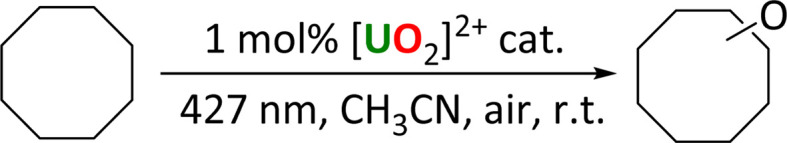
Fluorination reactions
The uranyl-catalyzed monofluorination of cyclooctane by N-fluorobenzenesulfonimide (NFSI), was chosen for study due to literature precedent set by West et al.5 Reactions were carried out under anaerobic conditions in order to avoid ketone and alcohol side products.9 Uranyl chloride acetonitrile adduct UCl, which can be made rigorously anhydrous, was chosen for a direct comparison with the Ph2phen ligated uranyl chloride, 1-Cl. A summary of the reactions is shown in Scheme 2.
The amount of NFSI and reaction volume were both optimized. Increasing the amount of NFSI was found to improve fluorocyclooctane yields with 1-Cl, but have essentially no effect on the reactivity of 1-Cl or 2, Table S8.† Even though two equivalents of NFSI gave the highest yields, e.g. 33% with UCl, 1.5 equiv. of NFSI was chosen for subsequent reactions as the option with better atom economy.
A catalyst loading of 1 mol% was found to be optimal as both higher and lower loadings of 2 gave lower fluorocyclooctane yields (Table S8† entries 1–3). The lower yield with 10 mol% of catalyst could possibly be due to the Ph2phen ligand quenching the excited state of the uranyl complex intermolecularly through exciplex decay since there will be more π–π interactions at higher concentration of the catalyst.3 The reaction conditions were also optimized for a single low-energy Kessil lamp with the same light output profile for each catalyst (Table S9†). Control reactions confirmed catalyst and light are essential for catalysis.
The reactivity of the different uranyl photocatalysts under optimized conditions is compared in Table 4. Both of the Ph2phen complexes show better yields of fluorocyclooctane than the simple uranyl chloride UCl. However, uranyl nitrate UNO3, the photocatalyst previously reported as efficient for cyclooctane fluorination by West et al.,3 was still found to give the highest yields under our conditions. The remaining mass balance in each case can be attributed to unreacted starting substrate, as observed by 1H NMR spectroscopy.
Cyclooctane fluorination reactions using different uranyl complexes.
| Entry | [U] catalyst | Fluorocyclooctane yield (%) |
|---|---|---|
| 1 | [UO2Cl2(MeCN)3] UCl | 26 |
| 2 | [UO2Cl2(Ph2phen)(MeCN)] 1-Cl | 35 |
| 3 | [UO2Cl2(Ph2phen)2] 2 | 40 |
| 4 | UO2(NO3)2·6H2O UNO3 | 46 |
Aside from 2, all the uranyl catalysts are soluble in the reaction mixtures. Under the reaction conditions, 2 dissolves fully within 1 minute of irradiation. The proton NMR spectrum of an aliquot taken after 5 and 10 minutes of the reaction shows the release of free Ph2phen as well as a small amount of another uranyl Ph2phen complex which is characterized as the fluoride-bridged dimer [UO2Cl(μ-F)(Ph2phen)]2[1-Cl-F]2. Storage of these aliquots in the dark overnight affords yellow crystals suitable for single crystal X-ray diffraction (Fig. 11). The isolation of a fluorinated uranyl complex is notable given the calculations performed by Wu et al. on cyclooctane fluorination by uranyl nitrate in which fluorine atom abstraction by uranium NFSI was calculated to have a higher activation barrier (15.5 kcal mol−1) than HAT from the cyclooctane substrate (10.8 kcal mol−1).31 However, the U–F bond is thermodynamically strong and both terminal and bridging fluorides are commonly observed.33
Fig. 11. Solid-state structure of [UO2ClF(Ph2phen)]2[1-Cl-F]2 with thermal ellipsoids at the 50% probability level. Hydrogen atoms are omitted for clarity.

Proton NMR spectra of cyclooctane fluorination with 1-Cl also shows the presence of some free Ph2phen ligand after five minutes, although no fluorinated complex could be observed in this case.
The changes in uranyl complexes during the first two hours of fluorination reactions were confirmed by UV-vis spectrophotometry, where the characteristic vibronic coupling is readily visible. The UV-vis electronic absorption spectra of both 1-Cl and UCl reactions (Fig. S10†) show small changes in the region associated with the uranyl LMCT absorption manifolds, so it is likely that these are due to some replacement of Cl by F.
C–C coupling reactions
The chlorides were also tested for C–C coupling reactions first reported by Capaldo et al.,10 using benzylidene malononitrile as a Michael acceptor. The reactions were carried out under an N2 atmosphere, and any products identified and quantified by proton NMR spectroscopy. In this case, UCl gave the highest yield of product (Table 5). In contrast to the fluorination reaction, complex 2 did not dissolve in the reaction mixture and yielded almost no product. The remaining mass balance in each case can be attributed to unreacted starting substrate, as observed by 1H NMR spectroscopy.
Cyclooctane addition to benzylidene malononitrile with different uranyl photocatalysts.
| Entry | [U] catalyst | Yield (%) |
|---|---|---|
| 1 | [UO2Cl2(MeCN)3] UCl | 21 |
| 2 | [UO2Cl2(Ph2phen)(MeCN)] 1-Cl | 18 |
Oxidation reactions
Uranyl complexes 1-Cl and 2 were also tested for cyclooctane oxidation, and their reactivity compared to our previously reported 1-NO3 as well as uranyl nitrate UNO3. The reactions were carried out in air so that the carbon-centered radical on the substrate created during the HAT step could be quenched with atmospheric oxygen. The reactions were analyzed by gas chromatography (GC). In all cases, the ketone product cyclooctanone was found to be the major product, Table 6, with smaller amounts of cyclooctanol produced, see ESI.† Similar to the fluorination reaction, uranyl nitrate was found to give the highest conversion of cyclooctane, while 1-NO3 gave the second highest yield. Of the three uranyl chloride complexes, both Ph2phen-ligated complexes produced slightly higher yields compared to uranyl chloride.
Cyclooctane oxidation reactions with different uranyl photocatalysts.
| Entry | [U] catalyst | Cyclooctane conversion (%) |
|---|---|---|
| 1 | [UO2Cl2(H2O)3] | 44 |
| 2 | [UO2Cl2(Ph2phen)(MeCN)] 1-Cl | 52 |
| 3 | [UO2Cl2(Ph2phen)2] 2 | 49 |
| 4 | [UO2(NO3)2(Ph2phen)] 1-NO3 | 69 |
| 5 | [UO2(NO3)2(OH2)2]·4H2O UNO3 | 80 |
Stern–Volmer analysis of the chloride catalysts in the presence of cyclooctane
The photoexcited uranyl(vi) moiety can undergo apparent quenching by both static and dynamic collisional quenching processes, and subsequently electron transfer processes, which would, to a first approximation, result in either productive or unproductive alkane H atom abstraction here.
The complexation between the uranyl molecule's excited state and a ground state conjugated molecule, exciplex formation, can provide a return to the ground state, i.e. a non-productive quenching, or “exciplex decay” competitive with HAT.34,35 Indeed, Bakac and co-workers previously observed that the vast majority of interactions between the aqueous uranyl excited state, [UO2]2+* and toluene return the molecules to the ground state rather than yielding the desired HAT and resulting oxidation of toluene.3 Here, the optical density of all catalyst solutions was maintained under 0.2 absorption units in order to avoid exciplex formation (and inner filter effects). Indeed, the emission intensity vs. concentration of Ph2phen behaves linearly as far as 0.35 absorption units (Fig. 12) as would be expected in the absence of self-absorption and exciplex formation.
Fig. 12. Luminescence spectra of 1-Cl in MeCN with increasing amounts of cyclooctane (λex = 427 nm) recorded at room temperature.

Stern–Volmer analyses of UCl and 1-Cl (made as described above) were carried out in the presence of the substrate cyclooctane, to study quenching of the uranyl moiety upon photoexcitation, and if the bound Cl reduces the effectiveness of the catalysis due to an energy transfer to the Cl rather than the oxo group.36 See Fig. 13 and 14.
Fig. 13. Stern–Volmer plot for samples of 1-Cl in MeCN, comparing the integrated luminescence intensity (λex = 427 nm) with increasing amounts of cyclooctane recorded at room temperature.

Fig. 14. Stern–Volmer plots for samples of 1-Cl in MeCN, comparing the luminescence lifetimes (λex = 427 nm) monitored at 515 nm (left) and 537 nm (right) with increasing amounts of cyclooctane.

Plots of emission intensity and luminescence lifetimes in the absence of substrate (I0, τ0) and in the presence of increasing amounts of substrate (I, τ) in acetonitrile at room temperature are presented in Fig. S11.† Any differences in the change in lifetime vs. change in emission intensity upon increasing substrate concentration (quencher) can indicate the predominant form of quenching. A linear decrease in luminescence intensity upon increasing quencher concentration indicates dynamic quenching processes are operative, whereas in the case of static quenching, the lifetime of the emitting species remains constant with increasing quencher concentration.
Monitoring the emission intensity lifetimes of the uranyl(vi) band for 1-Cl at 515 and 537 nm following equilibration after successive addition of microliter aliquots of cyclooctane (0–80 μM) affords a positive Stern–Volmer relationship in both the intensity and lifetime plots, with a quenching constant, KSV of ca. 6.8 M−1. Here, the slopes of the plots of I0/I vs. [cyclooctane] and τ0/τ vs. [cyclooctane] are within error of one another, suggesting that the cyclooctane substrate is dynamically quenching the excited state in 1-Cl.
The same global Stern–Volmer analysis of UCl with cyclooctane also shows a linear relationship with respect to emission intensity and lifetimes suggesting dynamic quenching predominates, but the KSV values (ca. 24 M−1 and 26 M−1) are an order of magnitude greater than 1-Cl (Table 7). However, KSV constants for UCl with cyclooctane display different behaviour on closer examination of the kinetic data, with plots of τ0/τ vs. [cyclooctane] being variable at 47, 19 and 14 M−1 respectively for the emission bands centred at 495, 516 and 540 nm (see ESI† for further discussion).
Averaged Stern–Volmer quenching constants calculated for 1-Cl and 1Cl in MeCN with cyclooctane.
Average of constants determined for lifetime data.
Measured as the THF adduct in MeCN.
Overall, these data suggest UCl may be a better photocatalyst than 1-Cl, which contradicts the experimental observations. However, it is important to consider the differences in the KSV values obtained through lifetime analysis, that indicate static quenching processes may also play a significant role in UCl (particularly at higher energy emissive transitions). In addition, the kinetic profile of UCl is biexponential, which complicates direct analysis with 1-Cl; other non-radiative competitive quenching processes that may be occurring, related to the emission quantum yield, such as photo-induced electron transfer processes with bound Cl, cannot be ruled out as contributing to the determined KSV values. Together, these data suggest that Stern–Volmer analysis for uranyl photochemistry may not be a suitable analysis procedure with which to determine the efficiency of these photocatalysts.
Conclusions
Uranyl nitrates and their Ph2phen adducts are better catalysts for the functionalization of cyclooctane by H atom abstraction than their chloride analogues because the chloride competes for charge transfer processes. Ligation of the Ph2phen confers advantages on both nitrate and chloride complexes; the ligand contributes strongly to the visible light energy absorption, via an LMCT excitation and the resulting excited states have significantly longer lifetimes than the simple uranyl salts. For example, both of the Ph2phen complexes 1 show better yields of fluorocyclooctane than the simple uranyl chloride UCl. The intra ligand π–π* excitations are involved in the energy migration pathway to afford uranyl LMCT emission. In the chloride complexes, competitive, and unwanted in this case, energy transfer to the Cl rather than to the oxo contribute to reducing the effectiveness of these precatalysts. The Ph2phen ligand affords some more protection to 1-Cl. There is more unwanted dynamic quenching of the excited state in the poorer catalyst UCl than in 1-Cl, but the photophysics measurements suggest that UCl should be better than 1-Cl, in contrast with experimental observations. The calculations agree with experiment.
Incorporation of the second Ph2phen to the uranyl ion (to form 2) bends and elongates the uranyl U Oyl bonds which could be considered as a useful way to increase the oxo group reactivity. However, the solid-state structure shows hydrogen bonding interactions between the ligand and oxo group which could prevent both substrate access, or quench the photoexcited state through non-reactive, radiative pathways. We were unable to determine which of these factors might dominate since the catalysis, spectroscopic, and computational results show that the second Ph2phen is lost readily to form the active catalyst.
Importantly, a molecular orbital analysis and TD-DFT calculations show two roles for the ligand contribute to the hydrocarbon C–H bond cleavage. The excited states of the complexes with one coordinated Ph2phen have radical character distributed away from the oxo group due to stabilization by the Ph2phen p system. The other advantage of the Ph2phen ligand is that it aligns the alkane substrate close to the oxo group through a set of dispersion forces. This will offset the barrier to the transition state in which the oxo takes on oxyl character and cleaves the strong alkane C–H bond.
Work is in progress to identify phenanthroline analogues that can enable regioselectivity in alkane functionalization reactions, and other functionalization chemistry that can be usefully achieved by hydrocarbon-soluble uranyl precatalysts.
Data availability
■■■■
Author contributions
RR, GH, YL made and characterized the compounds and reactions, AWW and LSN collected and interpreted the electronic spectra, XZ carried out and analyzed the computational parts, and with JP, SDW, MS, GS, and PLA, analyzed the data and wrote the manuscript. PLA raised the funding, conceptualized the project with contributions from LSN and GS, and all authors reviewed and edited the manuscript. RR and XZ contributed equally.
Conflicts of interest
The authors report no conflicts of interest.
Supplementary Material
Acknowledgments
The project was funded by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division, in the Heavy Element Program (parts of the spectroscopic efforts) and the Catalysis Program (RR) at the Lawrence Berkeley National Laboratory under Contract DE-AC02-05CH11231. Dr Cooper Citek and the Catalysis Laboratory in the DOE Catalysis Program also provided resource for instrumentation used in this work. We acknowledge Hasan Celik for NMR support and the NIH for funding part of the NMR facility under grant no. S10OD024998. We acknowledge access to the NNUF EPSRC CRR facilities at the University of Manchester, was supported by the National Nuclear User Facility (Call 6, Application 74 EP/T011289/1). PLA and RR thank the University of California, Berkeley for funding. GS and XZ acknowledge funding from the Natural Sciences and Engineering Research Council of Canada (NSERC Discovery Grant, GS) and the University of Manitoba (University of Manitoba Graduate Fellowship, XZ).
Electronic supplementary information (ESI) available: Additional experimental, computational, and crystallographic data (PDF). CCDC 2239853–2239856. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc01310g
Footnotes
This was verified by conducting the Stern–Volmer studies of both emission bands with and without a time-delay with cyclooctane. Negligible quenching and therefore participation of the fluorescence band was seen.
References
- Cowie B. E. Purkis J. M. Austin J. Love J. B. Arnold P. L. Chem. Rev. 2019;119:10595–10637. doi: 10.1021/acs.chemrev.9b00048. [DOI] [PubMed] [Google Scholar]
- Hu D. Jiang X. Synlett. 2021;32:1330–1342. doi: 10.1055/a-1493-3564. [DOI] [Google Scholar]
- Mao Y. Bakac A. J. Phys. Chem. 1996;100:4219–4223. doi: 10.1021/jp9529376. [DOI] [Google Scholar]
- Mao Y. Bakac A. Inorg. Chem. 1996;35:3925–3930. doi: 10.1021/ic960142n. [DOI] [PubMed] [Google Scholar]
- West J. G. Bedell T. A. Sorensen E. J. Angew. Chem., Int. Ed. 2016;55:8923–8927. doi: 10.1002/anie.201603149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold P. L. Purkis J. M. Rutkauskaite R. Kovacs D. Love J. B. Austin J. ChemCatChem. 2019;11:3786–3790. doi: 10.1002/cctc.201900037. [DOI] [Google Scholar]
- Ma R. Xu Y. Zhang X. ChemSusChem. 2015;8:24–51. doi: 10.1002/cssc.201402503. [DOI] [PubMed] [Google Scholar]
- Ahuja I. S. Singh R. J. Inorg. Nucl. Chem. 1973;35:2075–2078. doi: 10.1016/0022-1902(73)80149-6. [DOI] [Google Scholar]
- Accorsi G. Listorti A. Yoosaf K. Armaroli N. Chem. Soc. Rev. 2009;38:1690–1700. doi: 10.1039/B806408N. [DOI] [PubMed] [Google Scholar]
- Capaldo L. Merli D. Fagnoni M. Ravelli D. ACS Catal. 2019;9:3054–3058. doi: 10.1021/acscatal.9b00287. [DOI] [Google Scholar]
- Zhang Q. Jin B. Peng R. Wang X. Shi Z. Liu Q. Lei S. Liang H. Int. J. Photoenergy. 2017;2017:8041647. [Google Scholar]
- He W.-M. Lin Y.-W. Yu D.-H. Sci. China: Chem. 2020:291–293. doi: 10.1007/s11426-019-9675-5. [DOI] [Google Scholar]
- Li Y. Zhang G. Schwarz W. Li J. Inorg. Chem. 2020;59:6287–6300. doi: 10.1021/acs.inorgchem.0c00388. [DOI] [PubMed] [Google Scholar]
- Feng Z. J. Phys.: Conf. Ser. 2021;2079:012020. doi: 10.1088/1742-6596/2079/1/012020. [DOI] [Google Scholar]
- Yu J. Zhao C. Zhou R. Gao W. Wang S. Liu K. Chen S. Hu K. Mei L. Yuan L. Chai Z. Hu H. Shi W. Chem.–Eur. J. 2020;26:16521–16529. doi: 10.1002/chem.202003431. [DOI] [PubMed] [Google Scholar]
- Lv S. Li Q. Sang J.-W. Zhang Y. Wang J. Zhang W.-D. RSC Adv. 2023;13:11929–11937. doi: 10.1039/D3RA01037F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D. Jiang X. Green Chem. 2022;24:124–129. doi: 10.1039/D1GC04042A. [DOI] [Google Scholar]
- Zhou Y. Hu D. Li D. Jiang X. JACS Au. 2021;1:1141–1146. doi: 10.1021/jacsau.1c00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D. Zhou Y. Jiang X. Natl. Sci. Rev. 2022;9:nwab156. doi: 10.1093/nsr/nwab156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J. Zhou Y. Li D. Jiang X. Sci. Bull. 2023;68:1522–1530. doi: 10.1016/j.scib.2023.06.024. [DOI] [PubMed] [Google Scholar]
- Azam M. Al-Resayes S. I. Trzesowska-Kruszynska A. Kruszynski R. Kumar P. Jain S. L. Polyhedron. 2017;124:177–183. doi: 10.1016/j.poly.2016.12.033. [DOI] [Google Scholar]
- Mashita T. Tsushima S. Takao K. ACS Omega. 2019;4:7194–7199. doi: 10.1021/acsomega.9b00635. [DOI] [Google Scholar]
- Arnold P. L. McMullon M. W. Rieb J. Kuhn F. E. Angew. Chem., Int. Ed. 2015;54:82–100. doi: 10.1002/anie.201404613. [DOI] [PubMed] [Google Scholar]
- Rohe S. Morris A. O. McCallum T. Barriault L. Angew. Chem., Int. Ed. 2018;57:15664–15669. doi: 10.1002/anie.201810187. [DOI] [PubMed] [Google Scholar]
- Bell N. L. Shaw B. Arnold P. L. Love J. B. J. Am. Chem. Soc. 2018;140:3378–3384. doi: 10.1021/jacs.7b13474. [DOI] [PubMed] [Google Scholar]
- Arnold P. L. Love J. B. Patel D. Coord. Chem. Rev. 2009;253:1973–1978. doi: 10.1016/j.ccr.2009.03.014. [DOI] [Google Scholar]
- Schöne S. Radoske T. März J. Stumpf T. Patzschke M. Ikeda-Ohno A. Chem. - Eur. J. 2017;23:13574–13578. doi: 10.1002/chem.201703009. [DOI] [PubMed] [Google Scholar]
- Oher H. Gomes A. S. P. Wilson R. E. Schnaars D. D. Vallet V. Inorg. Chem. 2023;62:9273–9284. doi: 10.1021/acs.inorgchem.3c00847. [DOI] [PubMed] [Google Scholar]
- Redmond M. P. Cornet S. M. Woodall S. D. Whittaker D. Collison D. Helliwell M. Natrajan L. S. Dalton Trans. 2011;40:3914–3926. doi: 10.1039/C0DT01464H. [DOI] [PubMed] [Google Scholar]
- Lopez-Odriozola L., Walker L. and Natrajan L. S., in Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2022, 10.1016/B978-0-12-823144-9.00191-6 [DOI] [Google Scholar]
- Wu L. Cao X. Chen X. Fang W. Dolg M. Angew. Chem., Int. Ed. 2018;57:11812–11816. doi: 10.1002/anie.201806554. [DOI] [PubMed] [Google Scholar]
- Arnold P. L. Pécharman A.-F. Love J. B. Angew. Chem., Int. Ed. 2011;50:9456–9458. doi: 10.1002/anie.201104359. [DOI] [PubMed] [Google Scholar]
- Davidovich R. Goreshnik E. Struct. Chem. 2023;34:265–284. doi: 10.1007/s11224-022-02095-8. [DOI] [Google Scholar]
- Darmanyan A. P. Khudyakov I. V. Photochem. Photobiol. 1990;52:293–298. doi: 10.1111/j.1751-1097.1990.tb04184.x. [DOI] [Google Scholar]
- Natrajan L. S. Coord. Chem. Rev. 2012;256:1583–1603. doi: 10.1016/j.ccr.2012.03.029. [DOI] [Google Scholar]
- Parker D. Kanthi Senanayake P. Gareth Williams J. A. J. Chem. Soc., Perkin Trans. 2. 1998:2129–2140. doi: 10.1039/A801270I. doi: 10.1039/A801270I. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
■■■■