

SUMMARY
Reproduction is an energy-intensive process requiring systemic coordination. However, the inter-organ signaling mechanisms that relay nutrient status to modulate reproductive output are poorly understood. Here, we use Drosophila melanogaster as a model to establish the integrated stress response (ISR) transcription factor, Atf4, as a fat tissue metabolic sensor that instructs oogenesis. We demonstrate that Atf4 regulates lipase activity to mediate yolk lipoprotein synthesis in the fat body. Depletion of Atf4 in the fat body also blunts oogenesis recovery after amino acid deprivation and re-feeding, suggestive of a nutrient-sensing role for Atf4. We also discovered that Atf4 promotes secretion of a fat-body-derived neuropeptide, CNMamide, which modulates neural circuits that promote egg-laying behavior (ovulation). Thus, we posit that ISR signaling in fat tissue acts as a “metabolic sensor” that instructs female reproduction—directly by impacting yolk lipoprotein production and follicle maturation and systemically by regulating ovulation.
In brief
Across phyla, female reproduction is intimately tied to nutrient availability. Here, Grmai et al. demonstrate that the evolutionarily conserved integrated stress response acts a nutrient sensor in Drosophila fat tissues to regulate female reproduction via two ways: by regulating yolk production and by modulating neurons responsible for egg laying.
Graphical Abstract
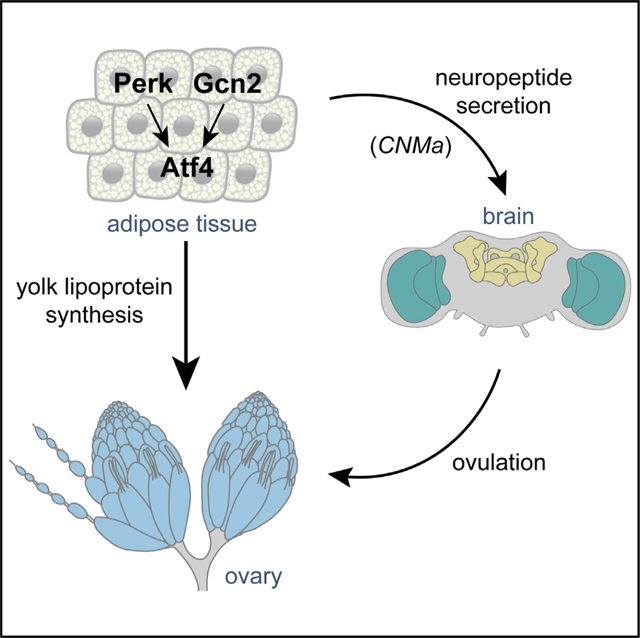
INTRODUCTION
The fundamental behaviors of an organism, such as feeding and reproduction, are informed by its metabolic status. Reproduction relies on nutrient availability to support the high energetic cost of gametogenesis and associated reproductive behaviors. Across multicellular organisms, the fat tissue serves as a lipid storage organ and acts as a signaling hub to peripheral organs to communicate nutrient status.1–3 Consequently, fat tissue homeostasis broadly influences reproductive capacity; defects in such homeostasis due to insufficientor excess dietary lipids result in decreased fertility, especially in women.4,5 There is thus tremendous interest in understanding the molecular mechanisms that maintain fat tissue homeostasis and inter-organ signals that relay the loss of such homeostasis to peripheral organs, such as the ovary.
Highly metabolic tissues, such as fat and liver, have been reported to rely on stress response pathways to maintain homeostasis.6–8 Specifically, constitutive activity of the integrated stress response (ISR), an evolutionarily conserved pathway that relies on stress-sensing kinases, has been observed in adipocytes and hepatocytes.7,9 There are four known ISR kinases—heme-regulated eIF2a kinase (HRI, protein kinase R [PKR]), general control nonderepressible 2 (GCN2), and protein kinase R-like ER kinase (PERK)—that all signal through stress-response transcription factors when activated by endogenous or external stressors.10 The best-studied transcription factor downstream of the ISR kinases is activating transcription factor 4 (ATF4).10 The importance of ISR signaling in metabolic tissues is highlighted by patient mutations in ISR effectors and loss-of-function studies in model organisms. PERK mutations in humans cause Wolcott-Rallison syndrome, characterized by neonatal diabetes,11,12 and loss of PERK signaling in mouse models results in dysregulated liver glycogen content and increased hepatocyte death in high-sugar dietary conditions.13,14 In mice and fruit flies (Drosophila melanogaster), Atf4-mutant animals show lower body fat content, and ATF4−/− mice show greater resistance to fatty liver under high-dietary-intake conditions.6
We and others have shown PERK and GCN2 to be constitutively active in fat tissues in mice and in Drosophila.9,15,16 Similar to loss of Atf4, loss of Perk or Gcn2 results in dysregulation of fatty acid homeostasis, which is further exacerbated by dietary restriction or excess lipid or sugar intake.13,15 While the primary effects of the loss of ISR factors on fat tissues have been well studied, our understanding of how ISR-mediated fat tissue homeostasis impacts peripheral tissue function has been limited. Here, we use the Drosophila model to investigate how ISR signaling in fat tissue (called the “fat body”) affects female reproduction. The fat body is a highly metabolic tissue comprised of multiple cell types that together execute vertebrate liver and adipose tissue functions, including fat storage, detoxification, and immune response.3 A number of recent studies have demonstrated multiple signaling pathways, including GCN2, in the fat body to be essential for non-autonomous regulation of germline stem cell numbers in the ovary (reviewed by Lin and Hsu2). Another prominent role of the female fat body is the synthesis and trafficking of yolk lipoprotein to maturing oocytes, which is critical for oocyte maturation.17
The Drosophila ovary is organized as long chains of developing follicles called ovarioles1 (Figure 1A). Germline and somatic stem cells reside at the anterior apex and undergo differentiation along the ovariole in individual follicles. The germline stem cells differentiate to give rise to 16-cell germ cell “cysts,” with one designated to be the oocyte and 15 supporting nurse cells. Concomitantly differentiating somatic cells envelop the germ cyst to form a follicle. Each follicle undergoes 14 stages of oogenesis, culminating in a mature oocyte (Figure 1A). From stages 8–14, the oocyte accumulates yolk lipoprotein, which is synthesized both by follicle cells within the ovary and fat body surrounding the ovary.18 We have reported previously that fat-body-specific depletion of Atf4, encoded by cryptocephal (crc) in Drosophila, results in a decreased rate of oogenesis (termed oogenesis arrest previously and here), accompanied by follicle death.19 In this study, we determine the underlying mechanisms of these oogenesis defects.
Figure 1. Atf4 is required in the fat body for proper oogenesis.

(A) Schematic of adult Drosophila ovaries, showing clusters of ovarioles containing maturing follicles. The diagram at the bottom shows an individual ovariole with various follicle stages, with germ cells depicted in blue and somatic cells in gray. Vitellogenesis begins in stage 8, during which time yolk lipoproteins (orange spots) are trafficked from somatic cells into maturing oocytes (dark blue). Border cells (purple) are shown migrating in a stage 9 follicle and complete their migration at the anterior end of the stage 10 follicle.
(B and C) Representative images of adult ovaries following control (lacZ, B) or Atf4 (C) depletion from the fat body using 3.1Lsp2-GAL4. Ovarioles marked with a dotted outline exhibit germ-cell nuclear fragmentation, indicative of dying follicles. DAPI marks nuclei in magenta.
(D) Quantification of follicle death from (B) and (C) based on the percentage of ovarioles with a dying follicle (based on Dcp1 staining).
(E) Quantification of follicle death from (B) and (C) as seen by cleaved caspase (Dcp1) staining. Death is quantified on the y axis as the number of ovarioles containing a dying follicle per ovary in each genotype.
(F) Quantification of the rate of oogenesis from (B) and (C), reported as the number of vitellogenic follicles (stages 8–10) per ovary.
(G and H) Quantification of rescue in follicle death (G) and oogenesis arrest (H) in crcGFSTF/+ heterozygous females with ectopic Atf4 expression in fat tissues using 3.1Lsp2-GAL4.
Here and in following figures, the data are pooled from at least 10 animals collected across two biologically independent crosses; statistical analyses were performed using a two-tailed unpaired Student’s t test with Welch’s correction for unequal standard deviations. Asterisks indicate statistical significance as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; ns, not significant. The specific transgenic lines used in each experiment here and in the following figures are listed in Table S1.
RESULTS
Atf4 in the fat body non-autonomously regulates oogenesis
We have shown previously that global Atf4-mutant animals exhibit reduced egg laying and increased death of mid-oogenesis follicles.19 To determine whether these oogenesis defects were ovary autonomous, we had examined Atf4 expression in the ovary. We observed that an Atf4-GFP fusion protein encoded by a protein trap allele (crcGFSTF) was not detected in adult ovaries.19 We had utilized the GAL4/upstream activating sequence (UAS) binary expression system20 to deplete Atf4 specifically in the fat body and observed that such loss was sufficient to recapitulate the reduced egg laying seen in Atf4 mutants (Figure S5).19 To further characterize this phenotype, we sought to test whether the observed reduction in egg laying was due to the increased follicle death seen in Atf4 mutants. We detected follicle death either by the presence of cleaved caspase staining (Dcp1 in Drosophila) or by observing germ cell nuclear fragmentation (discernible with DAPI staining), which is indicative of follicle death. We found that RNAi knockdown of Atf4 using the fat-body-specific driver 3.1Lsp2-GAL421 (hereafter referred to as 3.1Lsp2>Atf4RNAi) resulted in an increase in the number of Dcp1-positive follicles (Figures 1B and 1C). We observed a similar increase in follicle death upon Atf4 knockdown using another driver, Dcg-GAL4, which is active in the fat body and the ovary (Figure S1A). To eliminate the possibility that there were ovary-autonomous effects for Atf4, we also tested ovary germline (nos-GAL4) and somatic (tj-GAL4) drivers and found no significant increase in follicle death (Figures S1B and S1C). Together, these data convincingly implicate a non-autonomous role for fat-body Atf4 signaling in the oogenesis defect observed in crcGFSTF animals.
A prominent checkpoint in mid-oogenesis occurs at the onset of vitellogenesis (stage 8), during which maturing oocytes take up yolk proteins and lipids18 (Figure 1A). We observed that follicle death in 3.1Lsp2>Atf4RNAi was most frequent in stage 8–10 follicles, which are vitellogenic (Figures 1B and 1C). Such vitellogenic follicle death in 3.1Lsp2>Atf4RNAi is distinct from nurse cell death, which corresponds with nurse cell apoptosis, which is known to occur in stage 11–13 follicles22 (Figure S1D). The difference in vitellogenic follicle death between control and 3.1Lsp2>Atf4RNAi was statistically significant when quantified as the percentage of ovarioles containing at least one Dcp1-positive follicle (Figure 1D) or as the number of Dcp1-positive follicles per ovary (Figure 1E). In other words, we did not see a difference in the average number of ovarioles between control and 3.1Lsp2>Atf4RNAi animals (Figure S1E). Thus, for ease of data acquisition, we quantified further phenotypes on a per-ovary basis rather than on a per-ovariole basis.
In addition to increased follicle death, we also observed that 3.1Lsp2>Atf4RNAi animals had substantially fewer vitellogenic follicles (stages 8–10) per ovary, which is indicative of a defect in follicle maturation (Figures 1B, 1C, and 1F). To ensure that these defects were due to fat-body Atf4 signaling, we attempted rescuing the oogenesis defects in crcGFSTF mutants by restoring Atf4 expression only in the fat body using 3.1Lsp2-GAL4. In these experiments, we incidentally discovered that heterozygous crcGFSTF/+ animals also showed increased vitellogenic follicle death and fewer vitellogenic follicles (Figures S1F–S1H, 1G, and 1H). While we were unable to recover crcGFSTF homozygous animals with the UAS-Atf4 rescue transgene, we did recover a small number of crcGFSTF/+ heterozygous animals with the UAS-Atf4 rescue transgene. Analysis of these animals revealed that, indeed, restoring Atf4 expression in the fat tissues of Atf4 loss-of-function mutants was sufficient to rescue both vitellogenic follicle death and the number of vitellogenic follicles (Figures 1G and 1H).
Next, we asked whether the oogenesis defects in 3.1Lsp2>Atf4RNAi animals were the result of developmental contributions of Atf4. To do so, we utilized a GAL80TS transgene alongside 3.1Lsp2-GAL4 (hereafter called 3.1Lsp2TS). In the presence of GAL80TS, rearing animals at a restrictive temperature (18°C) prevents GAL4 activity during development, while shifting adults to a permissive temperature (24°C) permits GAL4 activity. We did not observe a statistically significant increase in follicle death or oogenesis arrest upon adult-only Atf4 depletion (Figures S1I and S1J), suggesting a developmental contribution for fat-body Atf4 in supporting oogenesis. Together, these oogenesis defects led us to pursue a non-autonomous role for fat-body Atf4 signaling in follicle maturation, which we further describe below.
Atf4 promotes yolk lipoprotein secretion from the fat body
Since we observed increased follicle death in vitellogenic stages, we examined whether loss of Atf4 signaling in the fat body affected yolk protein accumulation in maturing follicles. In Drosophila, yolk proteins are synthesized in both the somatic gonad and the abdominal fat body.17 We found that stage 14 oocytes from 3.1Lsp2>Atf4RNAi animals had substantially less yolk granule accumulation compared with controls (Figures 2A and 2B). Thus, we hypothesized that loss of ISR factors in the fat body compromises yolk protein trafficking to the ovary, which results in follicle death by disrupting vitellogenesis.
Figure 2. Atf4 activity in the fat body promotes vitellogenesis via yolk lipoprotein assembly in adipocytes.

(A and B) Representative confocal images of stage 14 oocytes from 3.1Lsp2>lacZRNAi (A) and >Atf4RNAi (B) females. Yolk granules were visualized by auto-fluorescence with a 405-nm laser; yolk granules appear as black/gray circles. Scale bars, 200 μm.
(C and D) Representative confocal images of Yp1::GFP (green) expression in adult fat bodies from 3.1Lsp2>lacZRNAi (C) and Atf4RNAi (D) females. Scale bars, 50 μm.
(E and F) Representative confocal images of neutral lipid staining (BODIPY, cyan) in adult fat bodies from 3.1Lsp2>lacZRNAi (E) and Atf4RNAi (F) females. Scale bars, 50 μm.
(G) Quantification of follicle death in control ovaries compared with loss of Atf4 (3.1Lsp2>Atf4RNAi; >GFP) and rescue by bmm co-expression (3.1Lsp2>Atf4RNAi, >bmm).
(H) Schematic depicting Atf4 binding sites (gray stars) within the first intron of the bmm gene. A 385-bp region from this intron (1p) containing three Atf4 binding sites was cloned upstream of the GFP gene to regenerate the bmm1p-GFP reporter, which was used for S2 cell expression analysis in (I).
(I) Relative GFP expression as determined by qPCR analysis in S2 cells transfected with wild type (WT) bmm1p-GFP and a mutant reporter lacking all three Atf4 binding sites (ΔAtf4, bmm1p-ΔAtf4-GFP in the text).
In confocal images, DAPI (magenta) labels DNA.
Yolk proteins are trafficked from the fat body to the ovary as lipoprotein vesicles, which contain a lipid core surrounded by the lipoproteins Yp1, Yp2, and Yp3.23 The lipid core of lipoproteins is derived from lipid droplets, which are extensions of the endoplasmic reticulum (ER) that bud off to form protein-coated vesicles.24 We tested whether there was an overall decrease in yolk lipoprotein production by visualizing expression of a yolk protein 1 (Yp1)-GFP fusion protein (Yp1::GFP)25 in the fat body. Knockdown of Atf4 in the fat body showed a dramatic reduction of Yp1::GFP in adipocytes (Figures 2C and 2D), suggesting that loss of Atf4 resulted in impaired yolk lipoprotein synthesis in the abdominal fat body.
Given the prominent role of Atf4 in lipid homeostasis,6,26 we next assayed for changes in lipid droplet formation in the fat body with loss of Atf4. Staining with the neutral lipid dye BODIPY showed that control fat bodies contained lipid droplets that characteristically organized into vesicles of varying sizes (Figures 2E, E′). In contrast, the BODIPY staining in Atf4-depleted fat bodies showed larger lipid droplets with a marked absence of discrete vesicular structures (Figures 2F and F′). We also observed similar large lipid droplets by BODIPY staining in fat bodies dissected from crcGFSTF mutants (Figures S2A and S2B). Consequently, we reasoned that the BODIPY staining in 3.1Lsp2>Atf4RNAi abdominal fat bodies (Figures 2E and 2F) is indicative of aberrant lipid droplet formation, which consequently impacts yolk lipoprotein synthesis.
We next sought to determine the underlying mechanism by which Atf4 affects lipoprotein synthesis in the fat body. Lipoprotein assembly relies on mobilization of triglyceride (TAG) reserves in lipid droplets by lipases, such as the adipose triglyceride lipase (ATGL)-like Drosophila lipase Brummer (encoded by the gene bmm).27 Interestingly, fat tissues from bmm mutants also contain large lipid droplets,27 similar to what we observed in 3.1Lsp2>Atf4RNAi and Atf4-mutant animals (Figures S2C and S2D). Based on this evidence, we pursued bmm as a potential Atf4 target in the context of lipoprotein assembly. We used the publicly available ENCODE chromatin immunoprecipitation sequencing (ChIP-seq) data28,29 to assess Atf4 occupancy on the bmm locus. As a positive control, this dataset showed enrichment of Atf4 binding at the Thor locus, a known transcriptional target of Atf49 (Figure S2E). A similar analysis showed several Atf4 binding events within the first intronic locus of bmm (Figure S2E). We next used the known position weight matrix for Atf4 to bioinformatically predict putative Atf4 binding sites, as described previously.30 Such analyses found several predicted Atf4 binding sites in the bmm intronic locus of varying strengths (Figure S2F). These observations prompted us to examine whether restoring bmm expression would be sufficient to rescue the follicle death seen with loss of Atf4 in the fat body (Figures 1B and 1C). Indeed, we found that co-expressing bmm in the fat body reduced both the number of dying follicles in 3.1Lsp2>Atf4RNAi animals (Figure 2G) and lipid droplet size in adipocytes (Figures S2G–S2I).
Based on our bioinformatics analysis and rescue experiments above (Figures S2E and S2F, 2G), we hypothesized that bmm is likely a direct transcriptional target of Atf4 in the fat body. To test this hypothesis, we generated a GFP-based enhancer reporter using the bmm intronic locus where we found the Atf4 binding sites (bmm1p-WT-GFP; Figure 2H; see STAR Methods for details). We also generated a second similar reporter where the predicted Atf4 binding sites had been deleted (bmm1p-ΔAtf4-GFP). We expressed these reporters in S2 cells to determine via qPCR whether deleting Atf4 binding sites led to loss in GFP expression. Contrary to our prediction, we found that deleting Atf4 binding sites resulted in derepression of the bmm1p element, as seen by elevated GFP expression in the bmm1p-ΔAtf4-GFP-expressing cells in comparison with the bmm1p-WT-GFP expressing cells (Figure 2I). Consistent with the reporter data, qPCR analysis of fat bodies isolated from 3.1Lsp2>Atf4RNAi animals also showed increased bmm in comparison with control animals (Figure S2J). These surprising data led us to conclude that Atf4-mediated regulation of yolk lipoprotein synthesis in the fat body is not enacted by Bmm but by an unidentified lipase whose activity can be substituted for by Bmm, as substantiated by our rescue experiments.
Atf4 in the fat body acts as a nutrient sensor to inform oogenesis
Nutrient deprivation has been demonstrated to induce oogenesis arrest and promote follicle death in a reversible manner.31–33 Nutrient deprivation, particularly amino acid deprivation, has also been documented extensively to induce Atf4.9,19,34–37 Thus, we asked whether oogenesis arrest during nutrient deprivation is mediated by Atf4. To do this, we performed a nutrient deprivation assay (Figure 3A, top), where mated females were raised for 4 days on 5% sucrose (starved) or nutrient-rich medium (fed). Notably, these dietary conditions have been independently shown to impact oogenesis38 and elevate Atf4 expression in the fat body.9 Since our data showed susceptibility to starvation in Atf4 mutants after the 4-day time point, we elected to starve the animals for a maximum of 4 days. We measured the rate of oogenesis by quantifying the number of vitellogenic follicles per ovary. As expected, at the end of the starvation period, ovaries from “starved” females contained significantly fewer vitellogenic follicles compared with ovaries from “fed” females in control animals (Figure 3B). In addition, we saw a significant increase in vitellogenic follicle death in “starved” versus “fed” ovaries, as determined by Dcp1 staining (Figure 3C). We observed a similar increase in follicle death and oogenesis arrest in ovaries from starved versus fed 3.1Lsp2>Atf4RNAi females (Figure 3B).
Figure 3. Atf4 is required for oogenesis recovery after amino acid deprivation.

(A) Diagram illustrating the starvation and re-feeding assay protocol. See STAR Methods and results for more details.
(B) Quantification of vitellogenic follicles per ovary from control and 3.1Lsp2>Atf4RNAi females under fed, starved, or re-fed conditions.
(C) Quantification of follicle death per ovary from control and 3.1Lsp2>Atf4RNAi females under fed, starved, or re-fed conditions.
(D–G) Representative confocal images of ovaries from control (D and E) and 3.1Lsp2>Atf4RNAi (F and G) females under fed (D and F) or starved and re-fed (E and G) conditions. DNA is labeled with DAPI (magenta). Scale bars, 100 μm.
The loss of maturing follicles induced by nutrient deprivation can typically be reversed by re-introduction of nutrients to the diet.32 Consistent with this, we found that re-feeding control females with nutrient-rich food following the 4-day starvation (Figure 3A, bottom) restored vitellogenic follicles to the ovary (Figure 3B). Additionally, we also found that the follicle death following starvation is substantially reduced after re-feeding in control animals (Figures 3C–3E). Analysis of ovaries from 3.1Lsp2>Atf4RNAi females showed resumption of oogenesis following starvation, as seen by the number of vitellogenic follicles (Figure 3B). However, we also saw a massive increase in the rate of death in both vitellogenic (stages 8–10) and pre-vitellogenic (stages 4–6) follicles in these ovaries after the re-feeding period (Figures 3C–3G), indicating impaired recovery of oogenesis after nutrient deprivation in 3.1Lsp2>Atf4RNAi females. Notably, there was no observable death in pre-vitellogenic follicles in control animals under these starvation conditions (Figure 3E). Such an increase in follicle death, particularly in pre-vitellogenic follicles, may be indicative of a sustained starvation response,31 suggesting that, in the absence of Atf4 in the fat body, female animals are unable to sense nutrient levels. Based on these data, we propose that Atf4 in the fat body is required for sensing changes in nutrient availability to regulate reproductive output.
Fat-body ISR signaling independently modulates ovulation
There are two known ISR kinases upstream of Atf4 in Drosophila: the ER-stress sensor Perk and amino acid sensor Gcn2. To assess which of these kinases are required for Atf4 signaling in the fat body, we used previously validated RNAi lines to deplete Perk or Gcn29 in the fat body using 3.1Lsp2-GAL4. As we observed in Atf4 knockdown animals, we found that knockdown of either Perk or Gcn2 in the fat body resulted in increased death of vitellogenic follicles and a decrease in the number of vitellogenic follicles (Figures 4A–4E). Examination of maturing eggs from these animals also revealed a decrease in yolk granules as seen with loss of Atf4 in the fat body (Figures S3A–S3C). These data suggest that both Perk and Gcn2 act upstream of Atf4 in the fat body.
Figure 4. ISR signaling in the fat body modulates egg laying behavior.

(A–C) Representative ovaries from 3.1Lsp2TS> lacZRNAi (A), >PerkRNAi (B), and >Gcn2RNAi (C) females. Dying follicles are encircled with a yellow dotted line, identified by nuclear breakdown. DAPI is shown in magenta.
(D) Quantification of follicle death depicted in (A)–(C).
(E) Quantification of vitellogenic follicles per ovary in (A)–(C).
(F and J) Representative bright-field images of ovaries from 3.1Lsp2TS>lacZRNAi (F), Atf4RNAi (G), PerkRNAi (H), Gcn2RNAi (I), and TnT (J) females. Opaque oocytes located at the base of each ovary represent eggs retained in each ovary at the time of dissection.
(K) Quantification of the egg retention phenotype shown in (F)–(J), reported as the number of stage 14 oocytes per ovary in the indicated genotypes.
Intriguingly, in addition to increased follicle death and mid-oogenesis arrest, we also observed a paradoxical accumulation of excess oocytes in Perk and Gcn2 knockdown animals. These effects were observed even when Perk or Gcn2 were knocked down only in adults, using 3.1Lsp2TS to suppress loss of ISR signaling in the fat body during development. Ovaries from mated females upon fat-body-specific depletion of Perk or Gcn2 (3.1Lsp2TS>PerkRNAi or >Gcn2RNAi) contained nearly twice as many mature oocytes per ovary (PerkRNAi: 55.4 ± 4.27, p < 0.0001; Gcn2RNAi: 43.56 ± 3.39, p < 0.001) than control ovaries (24.77 ± 2.78) (Figures 4F–4H, 4I, and 4K). Ovulation is exhibited in Drosophila by egg-laying behavior, and defects in this process result in retention of mature eggs in the ovary.39 While we did not observe this “egg retention” phenotype with 3.1Lsp2TS>Atf4RNAi (Figure 4G and 4K; 22.26 ± 2.00), we had previously reported a “swollen ovary” appearance in Atf4 hypomorphic mutants.19 We reasoned that, since both Perk and Gcn2 signal to induce Atf4, loss of either kinase may result in weaker effects than loss of Atf4. This interpretation is supported by our observation that loss of Perk or Gcn2 resulted in fewer Dcp1-positive follicles in comparison with loss of Atf4 (Figure 4D). We further tested this by simultaneous depletion of Perk and Gcn2 in the fat body. Indeed, 3.1Lsp2TS>PerkRNAi+Gcn2RNAi showed no egg retention phenotype (Figure S3D), similar to 3.1Lsp2TS>Atf4RNAi ovaries. Based on this, we conclude that substantial depletion of ISR signaling (by depleting the common downstream target Atf4) leads to severe oogenesis arrest. Such an arrest masks potential ovulation defects in these animals due to an overall decrease in maturing oocytes. Consequently, a role of ISR signaling in ovulation is only revealed with partial loss of function by depleting either upstream ISR kinase or by use of a hypomorphic Atf4 allele.
Since we observed that ISR signaling regulates yolk lipoprotein secretion from the fat body to support oogenesis, we sought to examine whether other factors are similarly secreted from the fat body to regulate ovulation. To test this, we expressed tetanus toxin (TnT) in the fat body to block secretion mediated by vesicular exocytosis.40 Expression of TnT using 3.1Lsp2TS-GAL4 recapitulated the “egg retention” phenotype we observed with 3.1Lsp2TS>PerkRNAi or >Gcn2RNAi compared with control animals (48.09 ± 4.94) (Figures 4J and 4K; 48.09 ± 4.94). Thus, in addition to regulating yolk lipoprotein synthesis, our genetic analysis revealed a new role of ISR signaling in ovulation, which we explore further below.
ISR signaling in the fat body promotes ovulation via CNMamide (CNMa)-dependent activation of sexually dimorphic neurons
Our results thus far suggest that ISR signaling in the fat body promotes ovulation via a secreted factor. In Drosophila, ovulation is stimulated in mated females via activation of sexually dimorphic neural circuits.41–45 Thus, we hypothesized that the secreted factor downstream of ISR signaling is a neuropeptide that signals to subsets of these neurons to regulate ovulation. To identify putative Atf4-regulated neuropeptides, we compiled a comprehensive list of the annotated neuropeptides in the D. melanogaster genome based on FlyBase categorizations (Table S2). We next examined Atf4 occupancy in the genomic loci of these neuropeptides using the ENCODE ChIP-seq dataset described above.28,29 Analysis of the overlap between these two datasets revealed four neuropeptides with evidence of Atf4 occupancy (Table S2, highlighted in green). We systematically depleted each of these neuropeptides in the fat body using 3.1Lsp2-GAL4 and quantified egg retention (Figures 5A and 5B). We found that depletion of all four individual neuropeptides predicted to be Atf4 targets led to increased egg retention: short neuropeptide F (sNPF), Ecdysis triggering hormone (Eth), Tachykinin (Tk), and CNMa (Figures 5A and 5B). Further qPCR analysis using Atf4 mutants showed a decrease in the mRNA abundance of two of these: CNMa and Eth (Figure S4A). Tk transcripts were undetectable by qPCR in both control and Atf4 mutants, and publicly available data via FlyAtlas and modENCODE sequencing projects report no detectable Tk expression in larval or adult fat bodies. Of these, we found that fat-body-specific depletion of CNMa had the largest effect on egg retention, to similar extents as seen with 3.1Lsp2TS>PerkRNAi or >Gcn2RNAi (Figures 5B and 5E; compare with Figures 4H, 4I, and 4K). Further, qPCR analysis of fat bodies from 3.1Lsp2TS>PerkRNAi or >Gcn2RNAi animals also showed a substantial decrease in CNMa mRNA levels (Figure 5C), which prompted us to characterize the role of this neuropeptide in ovulation.
Figure 5. Oviposition-descending neurons (oviDNs) respond to ISR-regulated CNMa to promote ovulation.

(A and B) Quantification of oocytes contained per ovary in a limited neuropeptide screen (see Table S2 for a list), where putative Atf4-regulated neuropeptides were individually depleted by RNAi in the fat body. Please note that RNAi lines were obtained from multiple sources, necessitating different negative controls.
(C) qPCR analysis of CNMa transcript abundance in fat bodies from control (3.1Lsp2>lacZRNAi) versus Perk-, Gcn2-, or Atf4-depleted fat bodies. Values were normalized to A-Tub84B as a housekeeping gene. Values are reported as the average of three biological replicates.
(D and E) Representative bright-field images of ovaries from 3.1Lsp2TS>control (D) and >CNMaRNAi (E) females.
(F) Quantification of oocytes per ovary in control versus 3.1Lsp2TS>CNMa females.
(G) Quantification of the egg retention phenotype in fat bodies lacking Atf4 (3.1Lsp2>Atf4RNAi; lacZ) and those rescued with simultaneous expression of ectopic CNMa (3.1Lsp2>Atf4RNAi; CNMa).
(H) Schematic depicting Atf4 binding sites (gray stars) within the promoter region and first intron of the CNMa gene. A 1,593-bp sequence from the promoter (1p) and a 2,486-bp sequence from the first intron (2p), each containing multiple Atf4 binding sites, were cloned upstream of the GFP gene to generate the CNMa1p-GFP and CNMa2p-GFP reporters, which were used for the S2 cell expression analysis in (I) and (J).
(I and J) Relative GFP expression, as determined by qPCR analysis, in S2 cells transfected with WT CNMa1p-GFP (I) and CNMa2p-GFP (J) as well as mutant reporters lacking the contained Atf4 binding sites (ΔAtf4, CNMa1p/2p-ΔAtf4-GFP in the text).
(K) Quantification of the egg retention phenotype in animals where the CNMa receptor (CNMaR) is depleted in sexually dimorphic neurons using a split oviDN-GAL4.
CNMa has been described previously to be induced in enterocytes of the gut in response to amino acid deprivation in an Atf4-dependent manner.37 Using a CNMa-GAL4 transgene,37 we found that the CNMa promoter is also active in both larval and adult female fat bodies (Figures S4B and S4C). Consistent with our prediction that CNMa is an Atf4 target, qPCR analysis showed lower levels of CNMa transcripts in Atf4 mutants and in 3.1Lsp2>Atf4RNAi animals (Figures S4A, 5C). While CNMa depletion in the fat body led to egg retention (Figures 5B, 5D, and 5E), we found that overexpression of CNMa in the fat body conversely caused a reduction in oocyte number per ovary (Figure 5F). Based on this, we performed a rescue experiment to determine whether ectopic expression of CNMa can rescue the ovulation defects in 3.1Lsp2TS>PerkRNAi or >Gcn2RNAi animals. However, we discovered that “diluting” GAL4 activity on the UAS-PerkRNAi/Gcn2RNAi transgenes with a second UAS element (such as UAS-lacZ or UAS-CNMa) resulted in no detectable decrease in ovulation between control and PerkRNAi/Gcn2RNAi animals (Fig. S4D). However, diluting GAL4 activity on the UAS-Atf4RNAi transgene with a second UAS element now resulted in an apparent ovulation defect that was rescued by restoring CNMa expression in the fat body (Figure 5G). Together, these data substantiate a dose-dependent role of ISR signaling in ovulation via regulation of CNMa.
We next wanted to test whether CNMa is a direct transcriptional target of Atf4. To do so, we employed the same strategy as we did with our analysis with bmm (Figures 2H, 2I, S2E, and S2F). modENCODE analyses showed Atf4 occupancy in the promoter and within the first intron of the CNMa locus (Figure S2E). Thus, we used the position weight matrix of Atf4 to determine whether there were putative Atf4 binding sites within the promoter and first intron of the CNMa locus (CNMa1p and CNMa2p, respectively). Our analysis revealed several predicted Atf4 binding sites in both CNMa elements (Figures S5A and S5B); from each of these elements, we generated GFP-based enhancer reporters (Figure 5H) similar to our construction of bmm1p-GFP reporters in Figure 2H. We transfected the resulting reporters, CNMa1p-WT-GFP, CNMa1p-ΔAtf4-GFP, CNMa2p-WT-GFP, and CNMa2p-ΔAtf4-GFP, into S2 cells and measured reporter activity by qPCR. These data revealed that, while deletion of the Atf4 binding sites in the CNMa1p element had no significant effect on reporter activity (Figure 5I), the activity of the intronic CNMa2p element was reduced by ~40% when the Atf4 binding sites were deleted (Figure 5J). Together, our qPCR analysis from fat bodies (Figure 5C) and enhancer element analysis (Figures S2E, S5A, S5B, and 5H–5J) submit that Atf4 likely directly targets the CNMa locus in the fat body.
Finally, we sought to identify which neurons respond to CNMa to promote egg-laying behavior. In Drosophila, egg-laying behavior is controlled by several groups of neurons, including two pairs of neurons descending from the mushroom body called oviposition descending neurons (oviDNs).41 The oviDNs are a subset of sexually dimorphic neurons in which the P1 promoter of fruitless (fruP1) is active.41 Synaptic silencing of all fruP1-expressing cells blocks ovulation, as does ablation of oviDNs.41 We tested whether the CNMa receptor, CNMaR, is broadly required in all fruP1-expressing neurons; however, RNAi depletion of CNMaR using fruP1-GAL4 resulted in an apparent block in oogenesis (Figures S5C–S5E), which likely would obscure an egg-retention phenotype. To circumvent this, we next depleted CNMaR specifically in the oviDNs using a split-GAL4 driver combination41 (oviDN-GAL4). Consistent with our hypothesis that CNMa from the fat body signals to neurons that promote ovulation, we saw that RNAi-mediated depletion of CNMaR using oviDN-GAL4 resulted in an increased number of eggs retained per ovary compared with control animals (Figure 5K; 29.19 ± 1.45 in oviDN>CNMaRRNAi vs. 24.17 ± 2.16 in control, p < 0.05). Taken together, these findings support a model where ISR signaling in the fat body induces CNMa production, which is secreted to activate ovulation-promoting neurons such as the oviDNs.
DISCUSSION
Single-celled organisms like yeast, where Atf4 (Gcn4 in yeast) was first discovered,34,46 rely on the ISR pathway under conditions of nutrient deprivation rather than for survival. In contrast, metabolically active tissues in higher organisms have evolved to rely on the ISR pathway for their homeostatic function.47 This is illustrated both by constitutive activity of Atf4 in fat tissues and by the metabolic phenotypes seen in Atf4-mutant Drosophila and mice.6,9 An increasing body of literature supports a role of fat tissues in modulating peripheral organ function,48 but whether and how this is regulated by ISR signaling is under active investigation. Our study makes a significant dent in this open problem by demonstrating multiple mechanisms by which homeostatic ISR signaling informs oogenesis in female flies.
Our findings show a requirement for both known ISR kinases, Perk and Gcn2, in both oogenesis and ovulation. Given that Perk is sensitive to changes in lipid content,49 and Gcn2 senses amino acid content,50 we favor a model where ISR signaling in the fat body acts as a conduit between nutrient status and reproductive capacity (Figure 6). In one branch of this model, oocyte maturation is compromised in the absence of Atf4 due to reduced yolk lipoprotein trafficking from the fat body to the ovary. In another seemingly parallel branch of this model, ISR signaling regulates ovulation, which is farther downstream of oogenesis, by neuromodulation of sexually dimorphic neurons. The sum effect of these two regulatory branches likely leads to the follicle death and oogenesis arrest observed in ISR-deficient animals and manifests as an overall decrease in fertility seen in global Atf4 mutants.19 However, the phenotypes reported here may only be part of the effects of ISR signaling on oogenesis since a previous study demonstrated that fat body Gcn2 signaling impacts germline stem cell maintenance under low-amino-acid conditions.51 Together, these findings implicate ISR signaling in parallel processes with synergistic effects on female reproductive output.
Figure 6. Model describing a role for ISR signaling in inter-organ regulation of reproduction.

ISR signaling in the fat body is required for follicles to pass the vitellogenesis checkpoint
During oogenesis, maturing follicles are subjected to at least two checkpoints: the first is a meiotic checkpoint in the germarium in stage 2A, and the second is a mid-oogenesis checkpoint around stage 8.52 We found that 3.1Lsp2>Atf4RNAi animals largely showed death in vitellogenic follicles (Figure 1D), suggesting that these follicles were failing the latter mid-oogenesis checkpoint. This checkpoint is also considered a “nutritional checkpoint,” and previous work has demonstrated that follicles fail to progress beyond mid-oogenesis when lipid and/or protein is scarce.31,53 The fat body is thought to contribute approximately half of the yolk lipoprotein that is accumulated in the oocyte between stages 6 and 10.17 Thus, our finding that 3.1Lsp2>Atf4RNAi follicles die or arrest at the mid-oogenesis checkpoint is consistent with our observation of decreased yolk lipoprotein abundance in both adipocytes and oocytes from 3.1Lsp2>Atf4RNAi animals (Figures 2A and 2B). The importance of yolk lipoprotein synthesized and trafficked from the fat body to the oocyte is further underscored by our preliminary data showing that depleting Yp1 in the fat body is sufficient to cause increased death in vitellogenic follicles (Figure S2K).
Yolk lipoprotein synthesis in the fat body is a multivariate process that relies on faithful formation of lipid droplets, which are then harvested by lipases to form lipoproteins. These particles are composed of a lipid core that is coated with yolk proteins, such as Yp1–3. Our data show that 3.1Lsp2>Atf4RNAi adipocytes have both aberrant lipid droplet formation (Figures 2E and 2F) and reduced levels of Yp1 protein (Figures 2C and 2D). Lipid droplets in 3.1Lsp2>Atf4RNAi animals appeared larger in size, which is consistent with what is seen in the absence of Bmm activity.27 However, our Atf4 binding site and qPCR analyses showed that bmm is likely not induced by Atf4 under homeostasis (Figures 2H, 2I, S2E, S2F, and S2J). Nonetheless, we found that ectopic bmm expression in 3.1Lsp2>Atf4RNAi animals rescued the vitellogenic follicle death phenotype and aberrant lipid droplet formation (Figures 2G and S2G–S2I), indicating that ectopic Bmm is likely substituting for another Atf4-regulated lipase. It also remains possible that Atf4 may regulate lipid droplet formation in additional ways other than lipase regulation. This possibility is supported by our preliminary data, where we see marked structural differences in the ER between control and 3.1Lsp2>Atf4RNAi animals (Figures S6A and S6B). That the ER is the primary site of lipid droplet biogenesis and that Atf4 is known to regulate fat content6,26,54 further lends credibility to the possibility that Atf4 impacts lipid droplet formation via multiple primary and secondary effects.
Our finding that 3.1Lsp2>Atf4RNAi and Atf4 mutant adipocytes have larger lipid droplets (Figures 2E and 2F) is somewhat contrary to previous studies demonstrating that Drosophila Atf4 mutants have lower overall TAG levels.6,26 However, consistent with these previous reports, our analysis also found that Atf4 mutants have lower overall TAG levels (Figure S6C) but nonetheless show an increase in lipid droplet size within adipocytes (Figure S2A). Together, these data lead us to consider that the lower TAG levels in Atf4 mutants may not be due to lipid storage defects in adipocytes but, rather, in another cell type. Fat bodies are comprised of two cell types: adipocytes, which are analogous to vertebrate adipose tissue, and oenocytes, which are analogous to vertebrate liver cells (hepatocytes). The decrease in TAG levels seen in Atf4 knockout mice has been demonstrated to be specific to the liver,26,54 suggesting the possibility that Atf4 has different roles in adipocytes and hepatocytes. Such cell-type-specific roles are also supported by our preliminary data showing that fat bodies from 3.1Lsp2>Atf4RNAi animals show similar TAG levels compared with control animals (Figure S6D). An important implication of these findings is that TAG levels as determined by biochemical assays do not correlate well with lipid droplet size. Additionally, the possibility that the change in lipid organization and usage in 3.1Lsp2>Atf4RNAi animals impacts other peripheral organs, like muscles, that have independent roles in reproductive behaviors involving motility remains to be tested.
The dramatic reduction in Yp1::GFP abundance in fat seen with loss of Atf4 (Figures 2C and 2D) is consistent with a defect in yolk lipoprotein synthesis but does not preclude the possibility that Atf4 may have effects on Yp1–3 transcription or protein synthesis due to its aforementioned impact on ER structure (Figures S6A and S6B). Analysis of the modENCODE project28,29 ChIP-seq data revealed no Atf4 occupancy at the Yp1, Yp2, or Yp3 locus (Figure S2B). Nonetheless, we observed a small but statistically significant decrease in Yp1–3 mRNA levels in 3.1Lsp2>Atf4RNAi animals (Figure S2J), though it remains unclear whether this occurs via direct transcriptional regulation of Yp1–3 by Atf4 or secondary effects of loss of Atf4 in the fat body. In either case, there remains the possibility that loss of Yp1–3 gene expression contributes to defects in lipoprotein assembly independent of the TAG mobilization defects seen in 3.1Lsp2>Atf4RNAi animals.
The role of ISR signaling in nutrient sensing
Atf4 is basally and constitutively active in the adult fat body,6,9 poising it to support reproduction under homeostatic conditions, as described above. However, Atf4 translation also increases in response to stress, notably amino acid deprivation, in the fat body and other tissues.9,55 Amino acid deprivation has a predictably negative impact on fertility, including a substantial increase in follicle death (paradoxically similar to loss of Atf4 in the fat) and eventual loss of germline stem cells (GSCs).31,56,57 How Atf4 signaling in the fat body differs between homeostatic vs. nutrient deprivation conditions remains an open question. Atf4 is a member of the ATF/CREB (activating transcription factor/cAMP response element binding protein) family of bZIP (basic leucine zipper) transcription factors with known interacting partners.58,59 Thus, an attractive possibility is that Atf4 regulates target gene expression in the fat body in concert with co-factors that change in abundance during starvation. It hence follows that, while Atf4 signaling is required for oogenesis during homeostasis,19 its homeostatic role would be mechanistically distinct from elevated Atf4 signaling under amino acid deprivation conditions. This distinction is supported by a previous study demonstrating that Gcn2 signaling in adipocytes mediates GSC loss under low-amino-acid conditions.51 However, the precise mechanism of how Gcn2 signaling implements GSC maintenance and the contribution of Perk signaling remain unknown.
Our data show that the increased vitellogenic follicle death and decreased rate of oogenesis seen with nutrient deprivation are not dependent on fat-body Atf4 signaling (Figure 3C). Strikingly, though, we saw increased death in not only vitellogenic but also pre-vitellogenic follicles even after re-feeding nutrient-deprived 3.1Lsp2>Atf4RNAi animals. We interpret these data as the 3.1Lsp2>Atf4RNAi animals failing to recognize that the organismal nutrient status had been restored, at least as it pertains to oogenesis. Thus, we posit that ISR signaling is a key metabolic sensor that passively permits or actively restricts oogenesis based on nutrient availability. In addition to Gcn2, amino acid levels are also sensed via the mTOR (mechanistic target of rapamycin) pathway,60 and similar to Gcn2, mTOR signaling in the adult fat body regulates GSC maintenance.51,61 Further, a vast body of literature demonstrates substantial interaction between Atf4 and mTOR signaling;6,62–64 Atf4 regulates expression of amino acid metabolism genes,35 and, consequently, Atf4 mutant mice have reduced mTOR activity.6 Hypomorphic Drosophila mTOR mutants also present with small ovaries and elevated vitellogenic follicle death,65 indicating some level of redundancy between ISR and mTOR signaling in the fat body. These observations introduce the possibility that the effects of fat body Atf4 signaling on oogenesis could be compounded by an accompanying decrease in mTOR activity. Our preliminary data show that ectopic expression of mTOR in the fat body results in no significant rescue of follicle death (Figure S7A). However, we did observe a partial rescue of the vitellogenic follicle number in 3.1Lsp2>Atf4RNAi animals (Figure S7B). Ongoing work aims to precisely identify the molecular mechanisms by which Atf4 interacts with mTOR signaling in the fat body to regulate oogenesis under homeostatic and nutrient deprivation conditions.
While there is a clear emerging role for Gcn2 as an amino acid sensor in the fat body,51 the molecular role of Perk is less obvious. Perk is best known as a sensor for ER stress, which can be induced by misfolded proteins and lipid imbalance, among others.10 Perk has been convincingly demonstrated to be sensitive to membrane lipid saturation (via its transmembrane domain) in mammalian cells.49 Since 3.1Lsp2>PerkRNAi adipocytes contained fewer yolk lipoprotein particles (Figure S3B), an attractive hypothesis is that Perk acts as a lipid sensor in the fat body, thus complementing the amino-acid-sensing role of Gcn2.
ISR signaling in the fat body promotes ovulation via CNMa secretion
Loss of either ISR kinase resulted in increased follicle death, decreased rate of oogenesis, and reduced yolk granules in maturing oocytes (Figures 4A–4E), demonstrating that both Perk and Gcn2 can act upstream of Atf4 in the adult fat body. Further, we observed that fat body knockdown of Perk or Gcn2 resulted in retention of mature oocytes in the ovary (Figures 4H, 4I, and 4K), which is indicative of a defect in ovulation. We reason that depletion of either ISR kinase results in milder loss of Atf4, resulting in only a partial loss of ISR signaling, as opposed to Atf4 knockdown, which results in greater loss of ISR signaling. Substantial loss of ISR signaling leads to a concomitant substantial oogenesis defect, thus obscuring any ovulation defects (Figures 4G and 4K). This notion is further supported by our data showing that joint knockdown of Perk and Gcn2, which leads to greater loss of ISR signaling in the fat body, does not display ovulation defects (Figure S3D). It is worth noting here that our data imply that proper ISR signaling in the developing fat body ensures yolk lipoprotein synthesis capacity in the adult (Figures S1I and S1J). However, ISR-mediated regulation of ovulation appears to be independent of a potential role of ISR signaling in fat body development (Figures 4F–4K).
Our data demonstrate that ISR mediates ovulation via multiple neuropeptides, including CNMa and Eth (Figures 5A and S4A), with CNMa being the largest contributor to ovulation. Based on our analysis, we propose that CNMa is likely directly transcriptionally regulated by Atf4 in the fat body (Figures 5C, 5H–5J, S2E, S5A, and S5B). The role of CNMa in the fat body is also supported by our finding that CNMa is constitutively expressed in larval and adult fat bodies under homeostatic conditions, similar to Atf4 (Figures S4B and S4C). Intriguingly, recent work has implicated CNMa in the amino acid deprivation response downstream of Gcn2 in enterocytes.37 However, this study utilized pan-neuronal drivers to demonstrate the role of the CNMa-CNMaR signaling axis in feeding behavior. In the context of ovulation, our data show that CNMa signaling via CNMaR appears to activate neurons, such as the oviDNs, that are known to promote egg-laying behavior.41 It is worth noting that CNMaR silencing specifically in oviDNs caused egg retention to a lesser degree than loss of CNMa from the fat body (Figure 5K). This leaves open the possibility that other cells (in addition to the oviDNs) may be responsive to CNMa in the context of egg-laying, including neurons in the central or peripheral nervous systems. For example, octopaminergic neurons in the brain and reproductive tract promote ovulation steps in response to mating-induced octopamine.42 Additionally, peripheral neurons that innervate the reproductive tract have been shown to regulate ovulation.44 High-throughput expression data generated by the modENCODE28,29 and FlyAtlas66,67 projects suggest that CNMaR expression is restricted to the head region, though it is possible that there are low levels of CNMaR in other cells. Further studies are needed to identify the entire cohort of cell types that are CNMa responsive in the regulation of ovulation downstream of ISR signaling in the fat.
Limitations of the study
Developmental contribution of Atf4
We observed that adult-only knockdown of Atf4 in the fat body by introducing a temperature-sensitive GAL80 transgene (GAL80TS) did not lead to increased follicle death. This suggests a possible developmental role of Atf4 in fat body development. An alternate possibility is that shifting animal rearing from 18°C to 24°C for 3 days did not allow sufficient Atf4 depletion to observe defects in lipoprotein synthesis and follicle death. The commonly used “permissive” temperature for GAL80TS is 29°C; we elected to upshift animals to 24°C instead of 29°C, as we observed severe fertility defects in females reared at 29°C.
Nutrient deprivation medium
It is worth noting that the dietary differences between our fed and starved animals extend beyond amino acids to carbohydrate composition, micronutrient availability, etc. Thus we remain open to the possibility that there may be additional nutrient-sensing mechanisms that may be at play for nutrient-deprivation-induced oogenesis arrest.
3.1Lsp2-GAL4 activity during starvation
It has been reported previously that nutrient deprivation can affect 3.1Lsp2-GAL4 activity.51 When performing the starvation assays described here, we also found such reduced GAL4 activity, which may impact the extent of RNAi knockdown. We acknowledge that this may impact the strength of phenotypes pertaining to the role of Atf4 in mediating oogenesis arrest during starvation. However, the key phenotypes observed in these experiments are during oogenesis recovery after starvation, at which time GAL4 activity should be restored. During this time, we definitively see that oogenesis rate is not restored in the absence of Atf4 and, more so, observe pre-vitellogenic follicle death in these animals that was not observed anywhere else in the study.
Cell-type contaminants in adult fat body preps
In this study, we report qPCR and TAG data on adult fat body isolates. Since adult fat bodies are tightly associated with the inner abdominal wall, they are exceptionally hard to remove from surrounding tissue without substantial tissue loss. Thus, we present these data with the caveat that these sample preparations include other contaminating tissues, such as abdominal muscle, trachea, and dorsal vessel. Nevertheless, these crude preparations appear to be sufficient to observe known differences in lipid content between males and females53,68 (Figure S6D). Further, our genetic manipulations in these samples are driven by 3.1Lsp2-GAL4, which is fat body specific and not active in any of the contaminating tissues, so we attribute the statistically significant changes reported here to loss of Atf4, Perk, or Gcn2 in the fat body.
GAL4 activity dilution with multiple UAS transgenes
The GAL4-UAS system is demonstrably sensitive to transgene dosage, with varying effects on the observed phenotypes depending on how many UAS binding sites are present per GAL4 transgene.69–71 This effect has somewhat limited our ability to perform certain rescue experiments in this study (e.g., Figure S4D), which we addressed using other complementary experiments that incidentally rely on the aforementioned dosage dependence (e.g., Figure 5G).
Effects of genetic background on number of vitellogenic follicles
In the course of compiling this study, the average number of vitellogenic follicles per ovary in control animals varied depending on the genetic background of the animals (e.g., control animals in Figure 1F vs. Figure 1H). Indeed, other studies have shown that there are substantial differences in stress response signaling across naturally occurring D. melanogaster genetic variants.72,73 Thus, though we controlled for genetic background whenever possible in this study, some of the differences we saw between control and loss-of-function animals may be due to unknown genetic background differences.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Deepika Vasudevan (deepika.vasudevan@pitt.edu).
Materials availability
Plasmids are available upon request, fly strains used in this study are publicly available from the Bloomington Drosophila Stock Center or have been sourced from indicated individual laboratories (Table S1).
Data and code availability
All data are available from the lead author upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Fly husbandry and stocks
The transgenic lines used in this study are publicly available via Bloomington Drosophila Stock Center or were sourced from other labs. See Table S1 for a complete list of lines used.
Animals were cultured in standard cornmeal agar media containing yeast and molasses (LabExpress, Inc). Fly stocks were maintained at either room temperature (RT) or 18°C and experimental crosses were maintained at 25°C. Adult females were collected at 0– 2 days following eclosion, mated with w1118 males for 3 days in standard media supplemented with yeast, and dissected after. For starvation assays, mated females were raised on amino acid-deficient medium containing 5% sucrose in 2% agarose for four days in the presence of w1118 males. To re-feed, females were transferred to fresh yeast-molasses medium vials for two more days. Conditional expression studies utilizing the GAL80TS transgene were maintained at 18°C to restrict GAL4 activity during development, and 0–2 day adult progeny were shifted to 25°C for 3 days (with w1118 males) to permit GAL4-mediated knockdown and/or overexpression.
METHOD DETAILS
Immunofluorescence
Adult ovaries and larval/adult fat body were dissected in PBS and fixed in 4% PFA for 15 min at RT. Samples were washed in PBS+0.1% detergent (Triton X-100 for ovary, Tween 20 for fat body) twice and incubated overnight at 4°C in primary antibody solution. Secondary antibodies along with DAPI (300nM final concentration) were incubated for 2 h at RT in the dark. Mouse anti-KDEL (Santa Cruz Biotech) was used at 1:100. Phalloidin-Rhodamine (Life Technologies) was used at 1:500. For yolk granule visualization, ovaries were fixed, washed, and mounted without DAPI. Yolk granules were visualized by auto-fluorescence upon excitation by 405nm laser.74 For neutral lipid staining in the adult fat body, samples were fixed and washed, followed by incubation in BODIPY solution (Thermo Scientific) for 20 min at a final concentration of 2 μg/mL. Confocal images were captured using a Nikon A1 confocal microscope through the Center for Biological Imaging at the University of Pittsburgh. Egg retention representative images were captured using a Nikon SMZ1270I with a ring light and DS-Ri2 camera attachment.
Fluorescent in situ hybridization (FISH)
FISH detection of Atf4 mRNA was performed using a commercially available kit (Molecular Instruments, Inc) and was conducted as per manufacturer’s instructions.
ChIP-seq data analysis via ENCODE
ChIP-seq was performed on Atf4-GFP by Dr. Kevin White (UChicago) and is publicly accessible via the ENCODE28,75 project website (www.encodeproject.org). ENCODE accession number for dataset used: ENCFF986LGA. Briefly, embryos (0–22h) were isolated from crc-MiMIC-GFP transgenic fly line and ChIP was performed using an anti-GFP antibody with sequencing on the Illumina HiSeq 2000 platform. Reads were aligned to the dm6 Drosophila melanogaster genome annotation. Read frequency plots shown in Figure S2E were generated by loading ENCODE bigWig file onto the Integrated Genome Viewer software (www.igv.org; Broad Institute).
Quantitative RT-PCR
Abdominal adult fat bodies were dissected and after removing the ovaries, gut and other reproductive tissues, it was left attached to the carcass alongside muscles, pericardiocytes and oenocytes. RNA was extracted from four female adults per replicate using TRIzol (Invitrogen) according to manufacturer’s protocol. Reverse transcription was performed using Maxima H minus reverse transcriptase (Thermo Scientific) according to manufacturer’s protocol. qPCR amplification reactions were prepared using SYBR Green Master Mix (MidSci) in the BioRad CFX96 real time system. Table S3 lists primers used for analysis. All data are from at least 3 biologically independent replicates.
Analysis of Atf4 binding sites in the bmm and CNMa locus
We used a previously published Python code30 (https://github.com/finnroach/transcription-factor-binding) to determine if the bmm1p, CNMa1p, and CNMa2p elements have Atf4 binding sites. The entire first intron of bmm and CNMa was used for bmm1p and CNMa2p respectively. CNMa1p utilized the intergenic sequence preceding the CNMa transcription start site. To generate the GFP-based enhancer reporters, short “enhancer fragments” from within each region that included the predicted Atf4 binding sites were cloned into a modified version of pHStinger76 lacking the Hsp70 promoter and containing attB recombination site. S2 cells were transfected with the reporters using Effectene (Qiagen) according to the DRSC protocol (https://fgr.hms.harvard.edu/stable-fly-cell-lines ) for 12-well format. After 48 h of transfection, RNA was extracted from cells using Trizol as described above.
QUANTIFICATIONS AND STATISTICAL ANALYSIS
All quantifications represent data pooled from at least 10 animals collected across at least two biologically independent crosses. All graphs show data points from every animal examined. For egg retention quantifications, ovaries were dissected with minimal perturbance, fixed, washed, and mounted. The number of mature oocytes per ovary was then scored manually. The appropriate statistical analyses for each assay (indicated in the accompanying legend) were performed using Microsoft Excel or GraphPad Prism. All statistical analyses except qPCRs used Student’s t-test with Welch’s correction, which is appropriate for datasets with unequal variances and unequal sample sizes.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Mouse anti-KDEL (10C3) | Santa Cruz Biotech | Cat# sc-58774 |
| Goat anti-mouse IgG (H + L) secondary, Alexa 488 | ThermoFisher Scientific | Cat# A-11029 |
| Phalloidin-Rhodamine | ThermoFisher Scientific | Cat# R415 |
| BODIPY 493/503 | ThermoFisher Scientific | Cat# D3922 |
| Rabbit anti-Dcp1 | Cell Signaling | Cat# 9578S |
| Chicken anti-GFP | Fisher scientific | Cat# 501962090 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| DH5Alpha | NEB | Cat #C2987H |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Paraformaldehyde 16% | Electron Microscopy sciences | 15710 |
| Vectashield mounting medium | Vector Labs | H100010 |
| Maxima H reverse transcriptase | Thermo Fisher | EP0753 |
| PR1MA qMAX Green qPCR Mix | MidSci | PR2000-H-5000 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Fluorescent in situ hybridization (FISH) kit | Molecular Instruments, Inc. | |
| Infinity Triglycerides Liquid Stable Reagent | ThermoFisher Scientific | Cat# TR22421 |
| Effectene transfection reagent | Qiagen | Cat# 301425 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| S2 cell line (Drosophila melanogaster) | Drosophila Genomics Resource Center | RRID:CVCL_TZ72 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| See Table S1 for list of transgenic and mutant fly lines used. | This paper | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S3 for list of oligonucleotides used for qPCR analysis. | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| See File S1 for sequence of recombinant DNA | This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| Integrated Genome Viewer | Broad Institute | https://igv.org/doc/desktop/ |
| Fiji-ImageJ | NIH | https://ImageJ.nih.gov/ij/ |
Highlights.
Loss of ISR signaling components in fat tissues leads to premature follicle death
ISR signaling regulates yolk lipoprotein production, which in turn impacts oocyte maturation
The ISR transcription factor Atf4 directly regulates transcription of the neuropeptide CNMa
CNMa regulates oviposition neurons; thus, loss of ISR signaling leads to ovulation defects
ACKNOWLEDGMENTS
We are deeply grateful to the Center for Biological Imaging at the University of Pittsburgh for imaging assistance and access to equipment. Additionally, we thank the Bloomington Drosophila Stock Center (BDSC; Bloomington, IN, USA), the Developmental Studies Hybridoma Bank (DSHB; Iowa City, IA, USA), the Vienna Drosophila Resource Center (VDRC; Vienna, Austria), and the Transgenic RNAi Project (Harvard Medical School, Cambridge, MA, USA) for making available reagents needed for this study. We are grateful to Drs. Marc Amoyel, Elizabeth Ables, and Shyama Nandakumar for providing project and manuscript feedback. We also thank Drs. Won-Jae Lee and Yusuke Hara for generously sharing transgenic fly lines. D.V. is funded by NIH R00EY029013 and R35GM150516, and L.G. is funded by NIH T32DK063922 and NIH K99GM149982.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.113863.
REFERENCES
- 1.Armstrong AR (2020). Drosophila melanogaster as a model for nutrient regulation of ovarian function. Reproduction 159, R69–R82. [DOI] [PubMed] [Google Scholar]
- 2.Lin K-Y, and Hsu H-J (2020). Regulation of adult female germline stem cells by nutrient-responsive signaling. Curr. Opin. Insect Sci 37, 16–22. [DOI] [PubMed] [Google Scholar]
- 3.Skowronek P, Wójcik q., and Strachecka A. (2021). Fat Body—Multifunctional Insect Tissue. Insects 12, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nikanfar S, Oghbaei H, Rastgar Rezaei Y, Zarezadeh R, Jafari-Gharabaghlou D, Nejabati HR, Bahrami Z, Bleisinger N, Samadi N, Fattahi A, et al. (2021). Role of adipokines in the ovarian function: Oogenesis and steroidogenesis. J. Steroid Biochem. Mol. Biol 209, 105852. [DOI] [PubMed] [Google Scholar]
- 5.Armstrong A, Berger M, and Al-Safi Z. (2022). Obesity and reproduction. Curr. Opin. Obstet. Gynecol 34, 184–189. [DOI] [PubMed] [Google Scholar]
- 6.Seo J, Fortuno ES 3rd, Suh JM, Stenesen D, Tang W, Parks EJ, Adams CM, Townes T, and Graff JM (2009). Atf4 Regulates Obesity, Glucose Homeostasis, and Energy Expenditure. Diabetes 58, 2565–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han J, and Kaufman RJ (2016). The role of ER stress in lipid metabolism and lipotoxicity. J. Lipid Res 57, 1329–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Volmer R, and Ron D. (2015). Lipid-dependent regulation of the unfolded protein response. Curr. Opin. Cell Biol 33, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang M-J, Vasudevan D, Kang K, Kim K, Park JE, Zhang N, Zeng X, Neubert TA, Marr MT 2nd, and Ryoo HD. (2017). 4E-BP is a target of the GCN2–ATF4 pathway during Drosophila development and aging. J. Cell Biol 216, 115–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Donnelly N, Gorman AM, Gupta S, and Samali A. (2013). The eIF2α kinases: their structures and functions. Cell. Mol. Life Sci 70, 3493–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH, and Hotamisligil GS (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461. [DOI] [PubMed] [Google Scholar]
- 12.Julier C, and Nicolino M. (2010). Wolcott-Rallison syndrome. Orphanet J. Rare Dis 5, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi W-G, Han J, Kim JH, Kim MJ, Park JW, Song B, Cha HJ, Choi HS, Chung HT, Lee IK, et al. (2017). eIF2a phosphorylation is required to prevent hepatocyte death and liver fibrosis in mice challenged with a high fructose diet. Nutr. Metab 14, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, and Cavener DR (2002). The PERK Eukaryotic Initiation Factor 2a Kinase Is Required for the Development of the Skeletal System, Postnatal Growth, and the Function and Viability of the Pancreas. Mol. Cell Biol 22, 3864–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo F, and Cavener DR (2007). The GCN2 eIF2alpha kinase regulates fatty-acid homeostasis in the liver during deprivation of an essential amino acid. Cell Metabol. 5, 103–114. [DOI] [PubMed] [Google Scholar]
- 16.Bobrovnikova-Marjon E, Hatzivassiliou G, Grigoriadou C, Romero M, Cavener DR, Thompson CB, and Diehl JA (2008). PERK-dependent regulation of lipogenesis during mouse mammary gland development and adipocyte differentiation. Proc. Natl. Acad. Sci. USA 105, 16314–16319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bownes M. (1990). The yolk proteins and their genes in Drosophila. Prog. Clin. Biol. Res 342, 336–342. [PubMed] [Google Scholar]
- 18.Bownes M, and Hames BD (1977). Accumulation and degradation of three major yolk proteins in Drosophila melanogaster. J. Exp. Zool 200, 149–156. [DOI] [PubMed] [Google Scholar]
- 19.Vasudevan D, Katow H, Huang H-W, Tang G, and Ryoo HD (2022). A protein-trap allele reveals roles for Drosophila ATF4 in photoreceptor degeneration, oogenesis and wing development. Dis. Model. Mech 15, dmm049119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brand AH, and Perrimon N. (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415. [DOI] [PubMed] [Google Scholar]
- 21.Lazareva AA, Roman G, Mattox W, Hardin PE, and Dauwalder B. (2007). A role for the adult fat body in Drosophila male courtship behavior. PLoS Genet. 3, e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buszczak M, and Cooley L. (2000). Eggs to die for: cell death during Drosophila oogenesis. Cell Death Differ. 7, 1071–1074. [DOI] [PubMed] [Google Scholar]
- 23.Palm W, Sampaio JL, Brankatschk M, Carvalho M, Mahmoud A, Shevchenko A, and Eaton S. (2012). Lipoproteins in Drosophila melanogaster—Assembly, Function, and Influence on Tissue Lipid Composition. PLoS Genet. 8, e1002828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olzmann JA, and Carvalho P. (2019). Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol 20, 137–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hara Y, and Yamamoto D. (2021). Effects of Food and Temperature on Drosophila melanogaster Reproductive Dormancy as Revealed by Quantification of a GFP-Tagged Yolk Protein in the Ovary. Front. Physiol 12, 803144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang C, Huang Z, Du Y, Cheng Y, Chen S, and Guo F. (2010). ATF4 regulates lipid metabolism and thermogenesis. Cell Res. 20, 174–184. [DOI] [PubMed] [Google Scholar]
- 27.Grönke S, Mildner A, Fellert S, Tennagels N, Petry S, Müller G, Jäckle H, and Kühnlein RP (2005). Brummer lipase is an evolutionary conserved fat storage regulator in Drosophila. Cell Metabol. 1, 323–330. [DOI] [PubMed] [Google Scholar]
- 28.ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.modENCODE Consortium; Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, et al. (2010). Identification of Functional Elements and Regulatory Circuits by Drosophila modENCODE. Science 330, 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown B, Mitra S, Roach FD, Vasudevan D, and Ryoo HD (2021). The transcription factor Xrp1 is required for PERK-mediated antioxidant gene induction in Drosophila. Elife 10, e74047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drummond-Barbosa D, and Spradling AC (2001). Stem cells and their progeny respond to nutritional changes during Drosophila oogenesis. Dev. Biol 231, 265–278. [DOI] [PubMed] [Google Scholar]
- 32.Terashima J, Takaki K, Sakurai S, and Bownes M. (2005). Nutritional status affects 20-hydroxyecdysone concentration and progression of oogenesis in Drosophila melanogaster. J. Endocrinol 187, 69–79. [DOI] [PubMed] [Google Scholar]
- 33.Pritchett TL, and McCall K. (2012). Role of the insulin/Tor signaling network in starvation-induced programmed cell death in Drosophila oogenesis. Cell Death Differ. 19, 1069–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hinnebusch AG, and Fink GR (1983). Positive regulation in the general amino acid control of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 80, 5374–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. (2003). An Integrated Stress Response Regulates Amino Acid Metabolism and Resistance to Oxidative Stress. Mol. Cell 11, 619–633. [DOI] [PubMed] [Google Scholar]
- 36.Kim K, Park JE, Yeom J, Park N, Trần TXT, and Kang MJ (2020). Tissue-specific roles of GCN2 in aging and autosomal dominant retinitis pigmentosa. Biochem. Biophys. Res. Commun 533, 1054–1060. [DOI] [PubMed] [Google Scholar]
- 37.Kim B, Kanai MI, Oh Y, Kyung M, Kim EK, Jang IH, Lee JH, Kim SG, Suh GSB, and Lee WJ (2021). Response of the microbiome-gut-brain axis in Drosophila to amino acid deficit. Nature 593, 570–574. [DOI] [PubMed] [Google Scholar]
- 38.Terashima J, and Bownes M. (2004). Translating available food into the number of eggs laid by Drosophila melanogaster. Genetics 167, 1711–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horváth B, and Kalinka AT (2018). The genetics of egg retention and fertilization success in Drosophila: One step closer to understanding the transition from facultative to obligate viviparity. Evolution 72, 318–336. [DOI] [PubMed] [Google Scholar]
- 40.Martin J-R, Keller A, and Sweeney ST (2002). 1 - Targeted Expression of Tetanus Toxin: A New Tool to Study the Neurobiology of Behavior**Dedicated to the memory of Heiner Niemann (1946–1999), without whose generosity the work here described would not have been possible. In Adv. Genet, Hall JC, Friedmann T, Dunlap JC, and Giannelli F, eds. (Academic Press; ), pp. 1–48e. [DOI] [PubMed] [Google Scholar]
- 41.Wang F, Wang K, Forknall N, Patrick C, Yang T, Parekh R, Bock D, and Dickson BJ (2020). Neural circuitry linking mating and egg-laying in Drosophila females. Nature 579, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee H-G, Seong C-S, Kim Y-C, Davis RL, and Han K-A (2003). Octopamine receptor OAMB is required for ovulation in Drosophila melanogaster. Dev. Biol 264, 179–190. [DOI] [PubMed] [Google Scholar]
- 43.Monastirioti M. (2003). Distinct octopamine cell population residing in the CNS abdominal ganglion controls ovulation in Drosophila melanogaster. Dev. Biol 264, 38–49. [DOI] [PubMed] [Google Scholar]
- 44.Häsemeyer M, Yapici N, Heberlein U, and Dickson BJ (2009). Sensory Neurons in the Drosophila Genital Tract Regulate Female Reproductive Behavior. Neuron 61, 511–518. [DOI] [PubMed] [Google Scholar]
- 45.Yang C-H, Rumpf S, Xiang Y, Gordon MD, Song W, Jan LY, and Jan YN (2009). Control of the postmating behavioral switch in Drosophila females by internal sensory neurons. Neuron 61, 519–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hinnebusch AG (1984). Evidence for translational regulation of the activator of general amino acid control in yeast. Proc. Natl. Acad. Sci. USA 81, 6442–6446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitra S, and Ryoo HD (2019). The unfolded protein response in metazoan development. J. Cell Sci 132, jcs217216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Funcke J-B, and Scherer PE (2019). Beyond adiponectin and leptin: adipose tissue-derived mediators of inter-organ communication. J. Lipid Res 60, 1648–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Volmer R, van der Ploeg K, and Ron D. (2013). Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA 110, 4628–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dong J, Qiu H, Garcia-Barrio M, Anderson J, and Hinnebusch AG (2000). Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell 6, 269–279. [DOI] [PubMed] [Google Scholar]
- 51.Armstrong AR, Laws KM, and Drummond-Barbosa D. (2014). Adipocyte amino acid sensing controls adult germline stem cell number via the amino acid response pathway and independently of Target of Rapamycin signaling in Drosophila. Development 141, 4479–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCall K. (2004). Eggs over easy: cell death in the Drosophila ovary. Dev. Biol 274, 3–14. [DOI] [PubMed] [Google Scholar]
- 53.Sieber MH, and Spradling AC (2015). Steroid Signaling Establishes a Female Metabolic State and Regulates SREBP to Control Oocyte Lipid Accumulation. Curr. Biol 25, 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H, Meng Q, Xiao F, Chen S, Du Y, Yu J, Wang C, and Guo F. (2011). ATF4 deficiency protects mice from high-carbohydrate-diet-induced liver steatosis. Biochem. J 438, 283–289. [DOI] [PubMed] [Google Scholar]
- 55.Hinnebusch AG, Sonenberg N, Ivanov IP, and Sonenberg N. (2016). Translational control by 5’-untranslated regions of eukaryotic mRNAs. Science 352, 1413–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nezis IP, Lamark T, Velentzas AD, Rusten TE, Bjørkøy G, Johansen T, Papassideri IS, Stravopodis DJ, Margaritis LH, Stenmark H, and Brech A. (2009). Cell death during Drosophila melanogaster early oogenesis is mediated through autophagy. Autophagy 5, 298–302. [DOI] [PubMed] [Google Scholar]
- 57.Barth JMI, Szabad J, Hafen E, and Köhler K. (2011). Autophagy in Drosophila ovaries is induced by starvation and is required for oogenesis. Cell Death Differ. 18, 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cohen DM, Won KJ, Nguyen N, Lazar MA, Chen CS, and Steger DJ (2015). ATF4 licenses C/EBPb activity in human mesenchymal stem cells primed for adipogenesis. Elife 4, e06821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huggins CJ, Mayekar MK, Martin N, Saylor KL, Gonit M, Jailwala P, Kasoji M, Haines DC, Quiñones OA, and Johnson PF (2015). C/EBPg Is a Critical Regulator of Cellular Stress Response Networks through Heterodimerization with ATF4. Mol. Cell Biol 36, 693–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shimobayashi M, and Hall MN (2016). Multiple amino acid sensing inputs to mTORC1. Cell Res. 26, 7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Armstrong AR, and Drummond-Barbosa D. (2018). Insulin signaling acts in adult adipocytes via GSK-3β and independently of FOXO to control Drosophila female germline stem cell numbers. Dev. Biol 440, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park Y, Reyna-Neyra A, Philippe L, and Thoreen CC (2017). mTORC1 Balances Cellular Amino Acid Supply with Demand for Protein Synthesis through Post-transcriptional Control of ATF4. Cell Rep. 19, 1083–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Selvarajah B, Azuelos I, Platé M, Guillotin D, Forty EJ, Contento G, Woodcock HV, Redding M, Taylor A, Brunori G, et al. (2019). mTORC1 amplifies the ATF4-dependent de novo serine-glycine pathway to supply glycine during TGF-β1–induced collagen biosynthesis. Sci. Signal 12, eaav3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Torrence ME, MacArthur MR, Hosios AM, Valvezan AJ, Asara JM, Mitchell JR, and Manning BD (2021). The mTORC1-mediated activation of ATF4 promotes protein and glutathione synthesis downstream of growth signals. Elife 10, e63326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y, Billington CJ, Pan D, and Neufeld TP (2006). Drosophila target of rapamycin kinase functions as a multimer. Genetics 172, 355–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chintapalli VR, Wang J, and Dow JAT (2007). Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet 39, 715–720. [DOI] [PubMed] [Google Scholar]
- 67.Leader DP, Krause SA, Pandit A, Davies SA, and Dow JAT (2018). FlyAtlas 2: a new version of the Drosophila melanogaster expression atlas with RNA-Seq, miRNA-Seq and sex-specific data. Nucleic Acids Res. 46, D809–D815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jehrke L, Stewart FA, Droste A, and Beller M. (2018). The impact of genome variation and diet on the metabolic phenotype and microbiome composition of Drosophila melanogaster. Sci. Rep 8, 6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Staller MV, Yan D, Randklev S, Bragdon MD, Wunderlich ZB, Tao R, Perkins LA, Depace AH, and Perrimon N. (2013). Depleting gene activities in early Drosophila embryos with the ‘maternal-Gal4-shRNA’ system. Genetics 193, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Barwell T, DeVeale B, Poirier L, Zheng J, Seroude F, and Seroude L. (2017). Regulating the UAS/GAL4 system in adult Drosophila with Tet-off GAL80 transgenes. PeerJ 5, e4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Singh M, and Kim JH (2022). Generation and validation of pX-UASTattB for dose-dependent misexpression studies in Drosophila. MicroPubl. Biol 2022. [DOI] [PMC free article] [PubMed]
- 72.Chow CY, Wolfner MF, and Clark AG (2013). Using natural variation in Drosophila to discover previously unknown endoplasmic reticulum stress genes. Proc. Natl. Acad. Sci. USA 110, 9013–9018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Palu RAS, Ong E, Stevens K, Chung S, Owings KG, Goodman AG, and Chow CY (2019). Natural Genetic Variation Screen in Drosophila Identifies Wnt Signaling, Mitochondrial Metabolism, and Redox Homeostasis Genes as Modifiers of Apoptosis. G3 (Bethesda). 9, 3995–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Andersen D, and Horne-Badovinac S. (2016). Influence of ovarian muscle contraction and oocyte growth on egg chamber elongation in Drosophila. Development 143, 1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Luo Y, Hitz BC, Gabdank I, Hilton JA, Kagda MS, Lam B, Myers Z, Sud P, Jou J, Lin K, et al. (2020). New developments on the Encyclopedia of DNA Elements (ENCODE) data portal. Nucleic Acids Res. 48, D882–D889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barolo S, Carver LA, and Posakony JW (2000). GFP and beta-galactosidase transformation vectors for promoter/enhancer analysis in Drosophila. Biotechniques 29, 726–732. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the lead author upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.