

Abstract
A genetic selection method, the P22 challenge-phage assay, was used to characterize DNA binding in vivo by the prokaryotic β class [N6-adenine] DNA methyltransferase M·RsrI. M·RsrI mutants with altered binding affinities in vivo were isolated. Unlike the wild-type enzyme, a catalytically compromised mutant, M·RsrI (L72P), demonstrated site-specific DNA binding in vivo. The L72P mutation is located near the highly conserved catalytic motif IV, DPPY (residues 65–68). A double mutant, M·RsrI (L72P/D173A), showed less binding in vivo than did M·RsrI (L72P). Thus, introduction of the D173A mutation deleteriously affected DNA binding. D173 is located in the putative target recognition domain (TRD) of the enzyme. Sequence alignment analyses of several β class MTases revealed a TRD sequence element that contains the D173 residue. Phylogenetic analysis suggested that divergence in the amino acid sequences of these methyltransferases correlated with differences in their DNA target recognition sequences. Furthermore, MTases of other classes (α and γ) having the same DNA recognition sequence as the β class MTases share related regions of amino acid sequences in their TRDs.
INTRODUCTION
Prokaryotic type II DNA methyltransferase (MTase) enzymes provide an attractive opportunity to study DNA–protein interactions. RsrI [N6-adenine] DNA methyltransferase (M·RsrI) from Rhodobacter sphaeroides recognizes the duplex DNA sequence GAATTC and catalyzes the transfer of a methyl group from S-adenosylmethionine (AdoMet) to the exocyclic amino group of the central adenine to form GAmATTC and S-adenosylhomocysteine (AdoHcy) (1). We used the highly sensitive Salmonella bacteriophage P22 challenge-phage assay (2,3), which monitors cell viability as a function of site-specific DNA binding, to study M·RsrI–DNA interactions in vivo. The challenge-phage system assays the ability of wild-type and mutant M·RsrI enzymes to serve as pseudo-repressors of transcription of the reporter gene ant encoded on an engineered, infective P22 bacteriophage genome. An EcoRI pseudo-operator site is placed downstream (3′) of the promoter region of the ant gene. Successful repression of ant expression by an M·RsrI protein bound to the EcoRI pseudo-operator site prevents production of the Ant protein, which if expressed leads ultimately to a phage lytic cycle and death of the Salmonella typhimurium host. Thus, successful, site-specific DNA binding by M·RsrI represses ant transcription and promotes the establishment of lysogeny, which is quantified as a kanamycin-resistant colony.
We isolated a weakly catalytically active mutant, M·RsrI (L72P), that, unlike wild-type M·RsrI, bound in vivo to the EcoRI pseudo-operator site with enough affinity to give a robust DNA binding response in the challenge-phage assay. L72 is located in a loop (4) following catalytic motif IV, DPPY (residues 65–68). A double mutant, M·RsrI (L72P/D173A), had less site-specific DNA binding affinity compared to the L72P single mutant in vivo, suggesting that D173 is involved in target DNA recognition. The single mutant M·RsrI (D173A) retained catalytic activity comparable to the wild-type enzyme but failed to bind in vivo. In the crystal structure of M·RsrI, D173 forms an ion pair with R176 and is located in a 310-helix (4) that is part of the putative target recognition domain (TRD) of the enzyme (5).
TRD sequence alignment analyses of M·RsrI and several β class MTases revealed that D173 and R176 are moderately and highly conserved (≥70 and ≥90%), respectively. Conservation of D173 and R176 in the TRD of the β class MTases recognizing different DNA sequences suggests a role in maintaining the structural integrity of this region of the enzymes. Analogously, the ‘methylase fold’ forms the catalytic domain of all DNA MTases (6). Phylogenetic analysis of the aligned β class enzymes showed that MTases having the same DNA recognition sequence were more related than members of the same class recognizing other DNA sequences. In agreement with the phylogenetic analysis, β class MTases having the same or similar target recognition sequences displayed increased conservation of particular arrays of amino acid residues in certain regions of the TRD, apart from those conserved over the whole class. These results imply a more direct role for these sequence arrays in DNA sequence recognition.
Furthermore, comparison of the amino acid sequences of the β class TRDs with MTases of other classes (α and γ) recognizing similar DNA sequences revealed common stretches of amino acid sequence similarity. Thus, amino acids conserved throughout the TRDs of the β class, which comprises enzymes recognizing a variety of target sequences, may be involved in forming the nucleus of a general recognition structure. Conversely, the amino acids in the TRDs that differ over the entire β class but are conserved in subgroups that recognize similar targets may be more directly involved in DNA sequence recognition.
MATERIALS AND METHODS
Materials
Unpurified or OPC-purified oligodeoxyribonucleotide primers for PCR mutagenesis were purchased from Gibco BRL. Cloned Pfu DNA polymerase was from Stratagene. All other restriction or DNA modification enzymes were from Promega or New England Biolabs. λ DNA and all restriction, modification and PCR enzymes were purchased from Gibco BRL, Promega, New England Biolabs or Stratagene.
Plasmids, bacteriophage and bacterial strains
The Escherichia coli strain DHI (F– supE44 hsdk17 recA1 endA1 gyrA96 thi-1 relA1) was routinely used for transformation and expression of plasmids. Salmonella typhimurium strains MS1868 [leuA414(Am) Fels– hsdSB (r-m+)] and MS1883 [leuA414(Am) Fels– hsdSB (r-m+)supE] were used for challenge-phage assays and propagation of P22 bacteriophage 10-C containing a GAATTC pseudo-operator and P22-1000 lacking this site, respectively. P22 bacteriophage 10-C and P22-1000 were a gift of J. F. Gardner (University of Illinois, Urbana-Champaign, IL) and were prepared using the protocols of Maloy et al. (3). A derivative of pTrc99A (Amersham-Pharmacia) containing the R·RsrI (E117Q) gene, pTrc#6, was prepared by Yonghong Huan (University of Illinois, Urbana-Champaign, IL). P22 10-C bacteriophage containing methylated DNA was generated by isolating it from an MS1868 host culture containing 10 µM isopropyl-β-d-thiogalactoside (IPTG)-induced M·RsrI. Bacteriophage DNA from the induced cultures was prepared as described (3) and found to be completely resistant to R·EcoRI digestion. A clone containing the Van91II restriction–modification system was generously provided by Dr Arvydas Janulaitis (Fermentas ABI).
Challenge-phage assays to assess DNA binding in vivo
Colonies of S.typhimurium MS1868 containing a plasmid (a pTrc99A derivative) for the IPTG-inducible expression of wild-type (7) and mutant M·RsrI proteins and R·RsrI (E117Q) were grown overnight in 5 ml of LB and ampicillin (Ap; 100 µg/ml) cultures. An aliquot (50 µl) of these overnight cultures was inoculated into fresh broth (5 ml) of the same composition. The cultures were grown in a shaking water bath at 37°C to an OD650 ≈ 0.5–0.6 as measured in 1 cm quartz cuvettes with a Hewlett Packard 8451 diode array spectrometer. Portions (500 µl) of this culture were added to several tubes containing 1.5 ml of LB and Ap (100 µg/ml) and 1, 10, 100 or 1000 µM IPTG. These subcultures were incubated with shaking at 37°C for ∼1 h to induce enzyme production. Challenge phage (10-C, methylated 10-C or P22-1000) were added to 100 µl of the induced subcultures at a multiplicity of infection (m.o.i.) of 25 and incubated at ambient temperature for 1 h. The uninfected and infected subcultures were then serially diluted 10-fold to 1 × 10–10 in LB broth. To determine the viable cell concentration, dilutions of the uninfected subcultures were spotted (5–10 µl) onto LB, agar and Ap (100 µg/ml) containing the same concentration of IPTG used for induction and grown at 37°C for 48–72 h to allow colony formation. Dilutions of infected subcultures were plated onto LB, agar, Ap (100 µg/ml) and Kn (50 µg/ml) with the proper concentration of IPTG to determine the lysogens/ml. The absence of a lysogen was defined as one colony and the lowest frequency (∼1 × 10–8–1 × 10–9) is given as the reciprocal of the viable cell count at each IPTG concentration. The frequency of lysogeny is expressed as the number of lysogens divided by the number of viable cells at each IPTG concentration and is plotted as the log of the lysogeny frequency as a function of the log of the IPTG concentration.
Random PCR mutagenesis
Forced nucleotide random PCR mutagenesis of the rsrIM gene using Taq DNA polymerase, one dNTP in excess of the other three and high MgCl2 concentrations in the presence of MnCl2 was used to generate a population of mutagenized PCR products (8). The forward and reverse PCR primers d(CCTGC-CATGGCAAACCGATCTCA) and d(AAGGATCCGAATTCTATGAAGCAACATCTCC) were used to amplify the rsrIM gene from template DNA. The NcoI, EcoRI and BamHI sites in each primer are underlined. Four 100 µl PCR reactions were performed, each containing 300 nM forward and reverse primer, 200 µM dNTPs with one dNTP added to a final concentration of 3.2 mM in each reaction, 1× PCR buffer supplied by the manufacturer (Gibco BRL), 9.5 mM MgCl2, 0.5 mM MnCl2, 5 U Taq DNA polymerase (Gibco BRL) and ∼0.5 µg DNA template. A three-step PCR thermocycling protocol was used to amplify the rsrIM gene: (i) 94°C for 5 min; (ii) 30 cycles of 94°C for 1 min, 25°C for 1 min and 72°C for 1 min; (iii) 72°C for 10 min. For each reaction, the PCR product of the correct size was confirmed by agarose gel electrophoresis. Each product was isolated using a Qiagen QIAquick PCR purification kit following the protocol of the manufacturer. The combined reactions were digested with R·NcoI and R·BamHI and concentrated by organic extraction and ethanol precipitation. After suspension in sterile water, the mutagenized PCR fragments were ligated into R·NcoI and R·BamHI digested dephosphorylated pTrc99A. To maximize the number of clones the ligation reactions were first transformed into E.coli DH1, because the transformation efficiency of ligated DNA in S.typhimurium MS1868 was at least 3-fold less efficient. Clones from the DH1 transformation plates were combined by washing the plates with 10 ml of LB and plasmid DNA was isolated using a Qiagen Qiaprep Spin Plasmid Kit following the protocol of the manufacturer. Plasmid DNA was then transformed into MS1868.
Challenge-phage assays and isolation of pTrc99A::rsrIM-(L72P/D173A)
Approximately 1000 colonies were washed from three MS1868 transformation plates by suspension in 10 ml of LB supplemented with 50 µg/ml Ap. IPTG was added to the 10 ml cell suspension to 0.5 mM and the bacteria grown at 37°C for 2 h with shaking. A portion of this culture was diluted to A650 = 0.6 (∼5 × 108 cells/ml) in LB supplemented with 50 µg/ml Ap and 0.5 mM IPTG. Two aliquots (500 µl) of these diluted cultures were infected with either 10-C or P22-1000 bacteriophage at an m.o.i. of 25 at ambient temperature for 1 h. Cells were collected by brief centrifugation and 400 µl of the supernatant removed. The pelleted cells were resuspended and the remaining 100 µl was plated onto LB, IPTG (0.5 mM), Ap (100 µg/ml) and Kn (40 µg/ml) plates and grown overnight at 37°C. Only two of several lysogens picked and inoculated into 5 ml of LB broth supplemented with IPTG (0.5 mM), Ap (100 µg/ml) and Kn (40 µg/ml) grew into cultures overnight at 37°C. Plasmids isolated from these two cultures were re-transformed into MS1868 for further challenge-phage assays. Plasmids were also transformed into DH1 for restriction digestion analysis with R·NcoI and R·BamHI to confirm the presence of the gene (7) and for sequence analysis. Sequence analysis revealed that both plasmids contained genes encoding the double mutation (L72P/D173A).
Site-directed PCR mutagenesis to generate M·RsrI single mutants L72P and D173A
We separated the L72P and D173A mutants by whole plasmid site-directed PCR mutagenesis (based on the Quick Change Site-Directed Mutagenesis Kit from Stratagene). A 50 µl reaction contained 850 nM each of the OPC-purified forward mutagenic primers LeutoProForward d(CCTCCATACAACATCATGCCGGCGGACTGGGATGATCAC) or AsptoAlaForward d(GTATTTCTTCGATCTTGCAGCTGTGCGGGAACCATATG) and their reverse complements, 50 ng template plasmid pTrc99A::rsrIM, Pfu DNA polymerase (Stratagene) and 200 µM dNTPs. A three-step PCR thermocycling protocol was used: (i) 95°C for 3 min; (ii) 18 cycles of 95°C for 30 s, 55°C for 1 min and 68°C for 12 min; (iii) 68°C for 20 min. Subsequently, 1 µl (10 U) of R·DpnI was added directly to the PCR reaction and allowed to incubate at 37°C for 1 h to digest any remaining parental DNA. Portions of each PCR reaction were then electroporated into DH1 and subsequently the plasmid DNA from several clones was isolated for restriction analysis. A single R·NgoMIV site was introduced into the plasmid as a result of incorporation of the proline mutation and a third R·PvuII site was introduced by the Ala173 mutation. The sequences of clones exhibiting the correct restriction digestion patterns were confirmed.
Preparation of cellular extracts of IPTG-induced MS1868 cells containing wild-type or mutant M·RsrI for SDS–PAGE analysis
A portion (200 µl) of an overnight culture of MS1868 harboring a plasmid for the IPTG-inducible expression of wild-type or mutant M·RsrI proteins was added to four 100 ml aliquots of LB broth supplemented with 200 µg/ml Ap and subsequently grown at 37°C to an optical density of 0.15–0.2 at 650 nm and induced by the addition of IPTG to final concentrations of 1, 10, 100 or 1000 µM. Cells were grown to an OD of 0.4–0.6 at 650 nm, which reflects the OD after approximately 1 h IPTG induction of M·RsrI proteins, before addition of challenge-phage. Subsequently, cells were harvested by centrifugation for 10 min at 4000 g at 4°C. The wet cell paste was resuspended in 500 µl of sonication buffer (20 mM Tris–HCl, pH 8.0, 10% glycerol, 20 mM β-mercaptoethanol, 1 mM EDTA, pH 8.0, 50 mM KCl, 0.4 mM phenylmethylsulfonyl fluoride) and sonicated using a Heat Systems Ultrasonic Processor W-385 microtip at 25% amplitude four times for 30 s interspersed with 1 min incubations on ice to prevent overheating. The resultant sonicates were centrifuged immediately at 10 000 g at 4°C for 50 min. The clarified extracts were collected, the protein quantified using the Bradford assay (9) and the solutions frozen on dry ice and stored at –80°C. Equivalent amounts (1–10 µg) of IPTG-induced MS1868 extracts were subsequently analyzed for M·RsrI induction using either 20% homogeneous Phastsystem minigels using the Phastsystem (Amersham Pharmacia Biotech) or 12.5% pre-cast gels from Bio-Rad. SDS–PAGE, Coomassie Blue staining and gel densitometric analysis was performed as described (7).
Sequence alignments and phylogenetic analysis
Searches within the non-redundant database containing 457 798 sequences were performed using PSI-BLAST (10), available at the National Center for Biological Information (NCBI). Amino acid sequences of 17 β MTases having significant similarity (PSI-BLAST scores > 40) with M·RsrI were aligned with Clustal W (11) without manual adjustment using BioEdit Sequence Alignment Editor v.8.4.5 (12). A phylogenetic tree created from the Clustal W alignment data was generated using TreeView 1.5.2 (13). Amino acid sequences contained in the TRDs of β MTases were also aligned with other MTase sequences using Clustal W. Chemical similarity analysis was performed using the GeneDoc program v.2.5 (developed by K. B. Nicholas at the Pittsburgh Supercomputing Center and available at http://www.cris.com/∼Ketchup/genedoc.shtml ). The following classification of chemically similar amino acids was used: 1, basic (K, R and H); 2, acidic (E and D); 3, polar (N, Q, S, T and C); 4, aromatic (F, Y and W); 5, non-polar aliphatic (L, I, M, V and A); 6, proline (P); 7, glycine (G).
RESULTS
Challenge-phage assays
The challenge-phage assay measures DNA binding by an IPTG-induced, plasmid-encoded protein expressed in S.typhimurium. Protein binding to an altered promoter region of the infecting P22 bacteriophage genome prevents expression of an anti-repressor (ant) gene (2). The Ant protein binds and inactivates the primary c2 repressor protein, which allows expression of phage lytic genes. Upon successful binding of the test protein, ant gene expression is repressed and the P22 phage genome instead integrates into the host chromosome by site-specific recombination to generate a lysogen. In the P22 challenge phage used in this study an M·RsrI (EcoRI) binding site (GAATTC) was engineered downstream (3′) of the Pant promoter. The bacteriophage carries a kanamycin resistance (kn9) gene, so that binding of M·RsrI to the EcoRI site gives rise to a colony on an agar plate containing kanamycin.
Prior to infection, growth of cells harboring the rsrIM gene with increasing IPTG concentrations produced increasing amounts of M·RsrI. A challenge phage lacking the EcoRI pseudo-operator sequence served as a negative control. The catalytically inactive RsrI endonuclease R·RsrI (E117Q), known to show a site-specific binding response in the P22 challenge-phage assay (14), was used as a positive control. DNA binding was quantified by counting the number of KnR lysogens formed as a function of the number of viable cells at a given IPTG concentration.
Methylated EcoRI pseudo-operator site-containing P22 bacteriophage was prepared to determine whether or not M·RsrI binding in vivo was sensitive to methylation of the GAATTC binding site. Methylated bacteriophage preparations were generated by cultivating bacteriophage in a strain containing IPTG-induced M·RsrI to provide site-specific methylation. DNA from unmethylated or methylated bacteriophage preparations was then purified and subjected to digestion with R·EcoRI. DNA isolated from a bacteriophage preparation derived from an M·RsrI-containing Salmonella host was completely resistant to cleavage by R·EcoRI (data not shown). On the other hand, bacteriophage DNA isolated from preparations from cells lacking M·RsrI was completely susceptible to R·EcoRI digestion.
DNA binding analysis in vivo
Wild-type M·RsrI failed to elicit a binding response with either site-specific or control challenge phage at any of the IPTG concentrations tested. Thus, low concentrations of wild-type M·RsrI in the S.typhimurium cells did not bind with enough affinity to act as a repressor of ant expression. At higher concentrations of IPTG, production of the wild-type enzyme was toxic (Fig. 1, line D). Using PCR random mutagenesis (8), we selected a double mutant, M·RsrI (L72P/D173A), that showed IPTG-inducible, sequence-specific binding (Fig. 1, line J). Subsequently, we used site-directed mutagenesis to separate the mutants. Only the mutants that contained the L72P substitution bound DNA in vivo, as illustrated in Figure 1 (line F for L72P and line J for L72P/D173A). Neither R·RsrI (E117Q) nor any of the M·RsrI proteins bound to phage lacking a target sequence, showing that lysogen production is not due to non-specific binding. The lysogenic frequencies are given in Table 1.
Figure 1.

Average viable cell count and frequency of lysogeny using unmethylated and methylated P22 phage with various RsrI proteins as a function of IPTG concentration. Lines A–E (viable cells/ml): (A) M·RsrI (D173A) (closed triangles); (B) M·RsrI (L72P/D173A) (open squares); (C) R·RsrI (E117Q) (open circles); (D) M·RsrI (crosses); (E) M·RsrI (L72P) (closed diamonds). Lines F–L (frequency of lysogeny): (F) M·RsrI (L72P) with unmethylated P22 10-C phage (closed diamonds); (G) M·RsrI (L72P) with methylated P22 10-C phage (open diamonds); (H) R·RsrI (E117Q) with unmethylated P22 10-C phage (closed circles); (I) R·RsrI (E117Q) with methylated P22 10-C phage (open circles); (J) M·RsrI (L72P/D173A) with unmethylated P22 10-C phage (closed squares); (K) M·RsrI (L72P/D173A) with methylated P22 10-C phage (open squares); (L) M·RsrI (D173A) (inverted open triangles).
Table 1. Frequency of lysogeny with unmethylated or methylated EcoRI site-containing P22 phage using M·RsrI variants.
| Enzyme | [IPTG] (µM) | Viable cell count (cells/ml) | Lysogeny frequency |
|
|---|---|---|---|---|
| Unmethylated phage | Methylated phage | |||
| M·RsrI (L72P) | 1 | 6.6 (3.5)a × 108 | 1.8 (1.3) × 10–6 | 1.5 × 10–9 |
| 10 | 2.6 (3.4) × 108 | 7.8 (0.4) × 10–6 | 3.8 × 10–6 | |
| 100 | 8.4 (10) × 107 | 5.6 (0.4) × 10–1 | 4.8 (0.4) × 10–1 | |
| 1000 | 2.7 (2.3) × 102 | 5.6 (2.8) × 10–1 | 1.9 (0.5) × 10–1 | |
| M·RsrI (L72P/D173A) | 1 | 7.2 (4.5) × 108 | 1.4 × 10–9 | 1.4 × 10–9 |
| 10 | 6.8 (3.4) × 108 | 1.5 × 10–9 | 1.5 × 10–9 | |
| 100 | 5.7 (4.3) × 108 | 3.3 (1.7) × 10–6 | 1.8 × 10–9 | |
| 1000 | 1.4 (1.8) × 108 | 1.1 (0.7) × 10–1 | 1.0 (0.4) × 10–2 | |
| R·RsrI (E117Q) | 1 | 9.5 (17) × 107 | 3.5 (1.6) × 10–6 | 1.0 × 10–8 |
| 10 | 5.9 (19) × 107 | 1.5 (0.2) × 10–5 | 1.7 × 10–8 | |
| 100 | 5.1 (10) × 107 | 4.8 (0.8) × 10–2 | 6.9 × 10–2 | |
| 1000 | 4.5 (2.5) × 107 | 1.5 (0.2) × 10–1 | 2.3 (1.1) × 10–2 | |
| M·RsrI | 1 | 3.3 (8.4) × 108 | NDb | ND |
| 10 | 3.4 (6.7) × 108 | ND | ND | |
| 100 | 2.5 (2.0) × 105 | ND | ND | |
| 1000 | 1.8 (1.1) × 104 | ND | ND | |
| M·RsrI (D173A) | 1 | 1.9 (6.6) × 108 | ND | ND |
| 10 | 7.6 (3.2) × 108 | ND | ND | |
| 100 | 9.1 (7.8) × 108 | ND | ND | |
| 1000 | 3.2 (4.2) × 108 | ND | ND | |
aValues in parentheses represent 1 SD from the mean of three individual experiments.
bND, no lysogenic response was detected.
M·RsrI (L72P/D173A), M·RsrI (L72P) and R·RsrI (E117Q) were also tested for their site specificity of binding with 10-C phage containing methylated GAATTC sites. The binding of all proteins that displayed a lysogenic response was impaired when tested with methylated 10-C P22 challenge phage. From 1 to 10 µM the positive control R·RsrI (E117Q) bound ∼300- to 900-fold less well to the methylated phage (Fig. 1, line I versus line H). At higher concentrations of IPTG the difference in binding became negligible, suggesting that the crippled endonuclease binds a methylated site when sufficiently concentrated. For M·RsrI (L72P), ∼1200-fold less binding occurred to phage containing methylated DNA at 1 µM, 2-fold less binding at 10 µM and equal binding occurred at higher concentrations (Fig. 1, lines F and G). At lower concentrations of IPTG (1 and 10 µM) little binding was detected with the double mutant (L72P/D173A) (Fig. 1, line J). However, at 100 µM IPTG the double mutant has ∼1800-fold more affinity for the unmethylated phage and at 1 mM IPTG a 5-fold binding difference was detected (Fig. 1, line K). The D173A mutation appears to interfere with site-specific binding, as indicated by the decreased binding of the double mutant with respect to L72P itself. The impaired binding of these proteins to a methylated EcoRI site further demonstrates the site specificity of the binding.
Production of the wild-type and M·RsrI (L72P) proteins resulted in decreased viability of the S.typhimurium host at higher IPTG concentrations (Fig. 1, lines D and E, respectively). The D173A-containing proteins showed little change in cell viability over the range of IPTG concentrations tested (Fig. 1, lines A and B). The reduced toxicity of this mutation may correlate with the weaker binding of M·RsrI (L72P/D173A) with respect to M·RsrI (L72P).
The differing lysogenic frequencies of the L72P-containing mutants could result from differential protein production. To test this possibility, cultures of Salmonella MS1868 containing wild-type, L72P/D173A, L72P or D173A were prepared, induced with varying IPTG concentrations and cellular extracts prepared. The induction conditions were identical to those used for challenge-phage assays immediately before addition of phage. Equivalent amounts of total protein from each extract were subjected to SDS–PAGE, Coomassie Blue staining and densitometric analysis. Only cultures induced with 0.1 and 1.0 M IPTG showed readily detectable overproduction of the M·RsrI proteins (Fig. 2). All the M·RsrI proteins were similarly expressed at these IPTG concentrations, exhibiting at most a 2-fold variation in amount as analyzed by densitometry. Thus, gross differences in protein concentration cannot explain the differences in the challenge phage binding responses. The 170 000-fold decrease in binding to unmethylated phage at 100 µM IPTG for the double mutant (L72P/D173A) compared to the single mutant (L72P) is therefore likely due to the D173A mutation.
Figure 2.
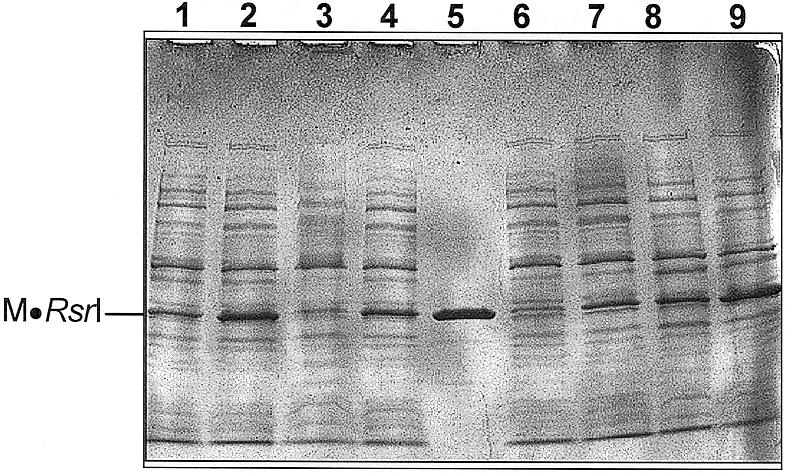
IPTG-induced production of wild-type and mutant M·RsrI proteins. Coomassie Blue stained SDS–PAGE gel of samples (∼10 µg) of sonicated extracts. Lanes 1 and 2, 0.1 and 1 mM IPTG-induced lysates, respectively, of wild-type M·RsrI; lanes 3 and 4, 0.1 and 1 mM IPTG-induced lysates, respectively, containing M·RsrI (D173A); lane 5, 2 µg purified M·RsrI; lanes 6 and 7, 0.1 and 1 mM IPTG-induced lysates, respectively, containing M·RsrI (L72P/D173A); lanes 8 and 9: 0.1 and 1 mM IPTG-induced lysates, respectively, containing M·RsrI (L72P).
Catalytic activity of mutant RsrI MTases
Plasmid DNA isolated from both uninduced and 1 h 1 mM IPTG-induced cultures containing the wild-type or D173A mutant proteins were completely resistant to digestion in vitro by R·EcoRI, indicating that the EcoRI sites contained at least one methyl group (data not shown). In contrast, plasmid DNA isolated from 1 mM IPTG-induced cultures of either the L72P or L72P/D173A mutants were ∼75% susceptible to digestion with R·EcoRI, indicating that unmethylated EcoRI sites remained. Whereas purified wild-type M·RsrI completely protected λ DNA from EcoRI digestion in λ DNA protection assays (7) at an enzyme concentration of 17 nM, purified M·RsrI (L72P) displayed only partial protection of λ DNA substrate at the highest concentration tested, 1.1 µM (data not shown). Therefore, the M·RsrI (L72P) protein is at least 65-fold less catalytically active than the wild-type protein. λ DNA protection assays with a cellular extract containing the D173A mutant, which was refractory to purification, showed ∼3- to 5-fold lower activity than extracts containing wild-type enzyme (data not shown). Whereas the L72P/D173A double mutant has less DNA binding affinity than the single mutant L72P as measured by the challenge-phage assay, the presence of the D173A mutation alone has little effect on the catalytic activity of the enzyme in vitro. These combined results suggest that the effects of the D173A mutation are limited to direct, canonical DNA binding rather than to an indirect effect due to hindered cofactor binding.
DNA binding analysis in vitro
M·RsrI (L72P) and M·RsrI (L72P/D173A) have been purified to >90% homogeneity, allowing DNA binding analysis in vitro with the electrophoretic gel retardation assay used to characterize wild-type M·RsrI (7). Table 2 lists the apparent DNA dissociation constants for wild-type and mutant M·RsrI proteins on unmethylated, hemimethylated and control DNA substrates in the presence of sinefungin, which promotes site-specific binding (7). The cofactor binding affinity of the L72P mutants has not been determined, excluding a direct comparison of the apparent DNA dissociation constants with the wild-type enzyme. However, both L72P-containing mutants displayed dissociation constants in the nanomolar range and, like the wild-type enzyme, preferred hemimethylated to unmethylated substrates. Generally, both wild-type and M·RsrI (L72P) bound DNA substrates with decreasing affinities: abasic > hemimethylated > unmethylated > dimethylated > control. Thus, M·RsrI (L72P) shares with the wild-type enzyme similar methylation-sensitive discrimination in canonical DNA binding. However, M·RsrI (L72P) showed markedly less discrimination for dimethylated DNA relative to the wild-type enzyme. Thus, Leu72 may help mediate discrimination between substrate and product DNA. The enhanced affinity of M·RsrI (L72P) for an abasic DNA substrate suggests that, like the wild-type enzyme, the mutant can flip the target adenine base.
Table 2. Apparent DNA dissociation constants for wild-type and mutant M·RsrI proteins in the presence of sinefungin.
| Enzyme | 5 nM unmethylated (TB)a | 5 nM hemimethylated (mTB) | 0.5 nM abasic hemimethylated (abTmB) | 5 nM dimethylated (mTmB) | 5 nM control (NS) |
|---|---|---|---|---|---|
| M·RsrI (L72P) | 33.8 (8.7)b | 9.7 (3.0) | 0.7 (0.1) | 44.5 (11.7) | >>250 |
| M·RsrI (L72P/D173A) | 374.9 (70.6) | 41.5 (12.1) | NDc | ND | ND |
| M·RsrI | 17 (1.5) | 6 (1.6) | 2 (0.8) | 156 (22.6) | >>800 |
aT, top strand; B, bottom strand; ab, abasic; m, methylated.
bApparent dissociation constants are in nM and values in parentheses represent ± 1 SD from the mean values from three experiments. Purification, DNA sequences and gel retardation assay conditions were performed as described (8).
cND, not done.
In agreement with the challenge-phage assays, the double mutant L72P/D173A bound less well than the single mutant L72P, having 11- and 4.3-fold less affinity for unmethylated and hemimethylated target DNA substrates, respectively. The double mutant M·RsrI (L72P/D173A) had less affinity for canonical DNA relative to M·RsrI (L72P) in vitro, providing evidence that the presence of the D173A mutation hinders DNA binding. However, the possibility exists that DNA binding by the D173A mutant may be affected by hampered cofactor binding. This explanation seems unlikely because D173 resides in the TRD and not the cofactor binding region of the protein. Additionally, the fungiform structure of the enzyme shows D173 lying in the stalk far removed from the cofactor binding site (4). Collectively, these results support those showing decreased DNA binding in vivo by the L72P/D173A double mutant relative to the L72P single mutant as determined by challenge-phage assays.
Sequence alignments
We performed an amino acid sequence alignment of M·RsrI with several β class N4mC and N6mA MTases to examine the possible conservation of amino acid residues in the regions containing the L72 and D173 mutations. Seventeen β class N6 and N4 MTases having significant similarity with M·RsrI as determined by a PSI-BLAST (10) search were then aligned using Clustal W sequence analysis (11). Leu72 is four amino acids C-terminal to the highly conserved catalytic motif IV, DPPY (residues 65–68). Both this Clustal W alignment and those of Malone et al. (5) and Bujnicki and Radlinska (15) reveal no preservation of an aliphatic amino acid at this position. In contrast, we found that the acidic D173 residue is moderately conserved in a part of the putative TRD of β MTases identified by sequence alignments by Malone et al. (5) as a region between amino acid sequence motifs involved in catalysis (IV–VIII) or cofactor binding (I–III and X) (Fig. 3, region 2). However, other sequence alignments by Gong et al. (6) and Bujnicki and Radlinska (15) have identified region 1 (containing β-strand 7) in our alignment (Fig. 3) as part of catalytic motif VIII. Thus, the function of residues contained in region I in catalysis or DNA recognition is unclear. The alignment in Figure 3 details the conservation of chemically similar residues. The β MTase TRD is centrally located between the N-terminal catalytic segment containing motifs IV–VIII and the C-terminal cofactor binding portion containing motifs I–III and X. Two other residues are highly conserved in TRD region 2: an aromatic and a basic residue (positions 170 and 176, respectively, in M·RsrI). Both D173 and R176 are contained within the 310-helix F1 in the structure of M·RsrI and form an ion pair (4). Other highly conserved residues throughout the β MTase TRD alignment correlate with particular structural features of M·RsrI (Fig. 3, regions 1 and 3–5). For instance, residues in TRD regions 1 and 4 are contained within β-strands 7 and 8, respectively, in M·RsrI. Sequence alignments by Bujnicki and Radlinska (15) identified a weakly conserved region in the TRD region of N4mC MTases (motif IX-N4, N/Q/D-V/I-W-N/E/D-I/V), forming a short β-strand, which corresponds to β-strand 8 (Fig. 3, region 4) of M·RsrI in the alignment reported here. The Clustal W alignment shows that the second and third amino acid residues of motif IX-N4 (V and W) are ≥90% conserved (Fig. 3, region 4), showing that this motif (comprising a β-strand) is present in N6mA MTases as well as N4mC MTases. Furthermore, TRD regions 3 and 5 correspond to β-turns. The retention of chemically similar amino acid residues in the β MTase TRD alignment among N4mC and N6mA MTases recognizing different DNA sequences suggests their roles in maintaining the structure of the TRDs. The N4mC β MTase M·PvuII has been crystallized in a complex with AdoMet (6). A large portion of the TRD of this enzyme is disordered (residues 179–216), precluding a direct comparison of structural features of the TRD of M·RsrI with M·PvuII. However, amino acids comprising structural elements β-strand 7 (in region 1) and β-strand 8 (in region 4) in M·RsrI and M·PvuII align moderately well in the Clustal W alignment (Fig. 3, nos 1 and 17). Furthermore, residues 172–176 of M·PvuII comprise a 310-helix that may be structurally equivalent to the 310-helix in region 2 of M·RsrI (Fig. 3). Thus, most of the TRDs of β MTases in the Clustal W alignment may have common structural features.
Figure 3.

Relationships of DNA TRD amino acid sequences of several β class N4mC and N6mA MTases. The position of the conserved acidic residue D173 in TRD region 2 of M·RsrI is indicated by the red triangle. The target A or C base in the target recognition sequence is indicated in bold. The methylatable base of M·Pac25I (residue 15) has not been identified. Chemically similar amino acid residues (see Materials and Methods) having a high degree of conservation (≥90%) are red; moderately conserved amino acid residues are blue (≥70%); less conserved amino acid residues are green (≥50%). The secondary structure in which each amino acid of the TRD of M·RsrI participates is indicated: B, β-strand; H, helix; t, β-turn; (–), loop; (∼), gaps generated as a result of alignment. The names of some structural elements are indicated along the top in blue (5). A segment of region 2, outlined within a stippled box, is expanded in Figure 4.
The lack of conservation of some parts of the β MTase TRDs suggests their possible participation in site-specific DNA recognition. We find that β MTases sharing similar target DNA sequences have linear conservations of chemically similar amino acid residues throughout their TRDs. Analysis of a 15 amino acid residue sequence of the TRD from region 2 (outlined by a dashed black box in Fig. 3) details this observation (Fig. 4). For instance, in TRD region 2 the adenine MTases DpnII B and LlaDCHI B recognize GATC (Fig. 4, nos 6 and 7) and contain the sequence RHYYNY(D/E)LMKE(L/F)NDG. BabI, SmeI and HinFI, which recognize GANTC (Fig. 4, nos 9–11), contain (F/Y)TFNY(E/K)(A/T)MK. N4mC MTases Cfr9I, XcyI and Pac25I (Fig. 4, nos 13–15) all recognize CCCGGG and contain the sequence KY(F/Y)YDW(E/Q)A. Structurally, TRD region 2 maps to a β-turn, 310-helix F1 and α-helix F in M·RsrI (Fig. 3). Thus, the less conserved portions of TRD region 2 (the β-turn and α-helix F in M·RsrI) may share similar structural elements composed of chemically dissimilar amino acid functionalities that mediate sequence-specific DNA recognition. Other parts of the β MTase TRD are conserved only within certain subgroups in the alignment shown in Figure 3, including segments between regions 2 and 3 and 4 and 5. The guide tree generated as a result of Clustal W analysis of the 17 β class MTases showed that MTases sharing the same DNA recognition sequence were more closely related than enzymes recognizing less similar targets (Fig. 5). Phylogenetic analyses of α class MTases (16) and N4mC MTases (15) have also revealed a similar correlation between DNA recognition sequence specificities and amino acid sequence similarities in their TRDs.
Figure 4.
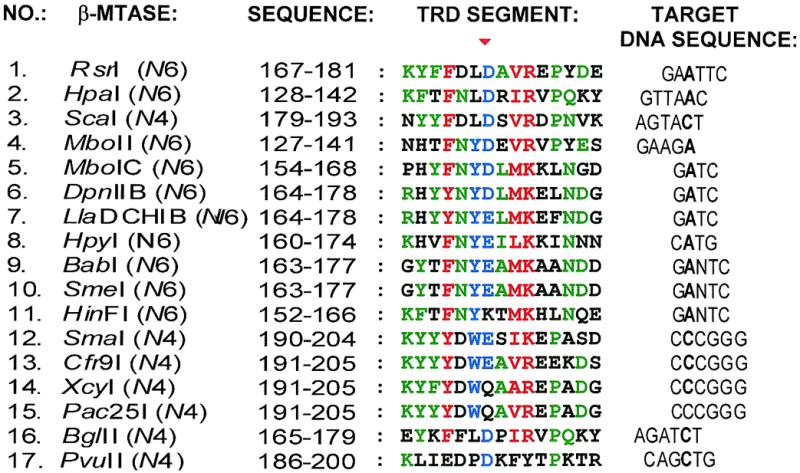
Amino acid sequence segment from β MTase TRD region 2 (Fig. 3). The classification scheme is the same as in Figure 3.
Figure 5.

Dendrogram of the 17 β class N4mC and N6mA MTases sequences generated by the Clustal W analysis in Figure 3. Branch lengths denote the percent divergence between the MTases. The recognition sequence of each MTase is shown.
Portions of amino acid sequences from various β MTase TRDs are also present in the TRDs of MTases of other classes having similar or identical DNA recognition specificities. For instance, similarity exists between the GATC- and GANTC-recognizing β MTases and the α and γ class, GATC- and GANTC-recognizing MTases (Fig. 6, A–C.). The TRDs are located in the central and C-terminal regions of α and γ enzymes, respectively. Alignment analyses of α MTases recognizing GATC and GANTC by several groups (16–18) have identified the segments of residues shown in Figure 6A and C as part of the TRD of these enzymes. The C5 MTase M·LlaR2I, which methylates the cytosine in the DNA sequence GATC, shares a linear array of chemically similar amino acids in its TRD with the β MTase M·LlaCHIB, which methylates an adenine (Fig. 6B). The GAATTC-recognizing TRD of the β MTase M·RsrI is also evident in the TRD of the γ MTases M·EcoRI and M·Van91II, which, like M·RsrI, recognize the DNA sequence GAATTC (Fig. 6D). Similarity also occurs in the M·RsrI TRD motif and the unclassified MTase XmnI, which recognizes the interrupted EcoRI DNA sequence GAA(N4)TTC (Fig. 6E). The conserved residues present in these alignments suggest that a specific linear arrangement of chemically similar functionalities contribute to forming a structure suitable for site-specific DNA recognition of similar sequences.
Figure 6.

Clustal W alignments of partial amino acid sequences of the β class TRD with other MTases. (A) Comparison of the β class GATC motif with GATC-recognizing α class MTases. Chemically similar amino acid residues (see Materials and Methods) having a high degree of conservation (≥80%) are red; moderately conserved amino acid residues are blue (≥60%). (B) Comparison of the β class LlaDCHIB GATC motif with GATC-recognizing C5 MTase LlaR2I. (C) Comparison of the β class HinFI GANTC motif with α class CviBI. (D) Comparison of M·RsrI with M·EcoRI and M·Van91II. (E) Comparison of M·RsrI TRD with M·XmnI.
DISCUSSION
Failure of wild-type M·RsrI to bind DNA in vivo
Wild-type M·RsrI and M·RsrI (D173A) failed to bind DNA site specifically in the challenge-phage assay at lower concentrations of IPTG; higher concentrations of the wild-type enzyme killed the cells. The absence of lysogens at low concentrations is presumably because the enzymes methylate the target recognition sequence and dissociate. In vitro DNA binding studies using gel retardation assays show that M·RsrI bound weakly to fully methylated GAATTC (KD = 117 nM; 7).
Higher levels of wild-type M·RsrI proteins could be toxic (Fig. 1, line D) because of ectopic methylation at functionally critical sites, as observed with M·EcoRI (19), or because of increased occupancy of important sites on the host genome. The L72P mutant also displays decreased cell viability, but requires higher concentrations of IPTG induction than the wild-type enzyme (Fig. 1, lines D and E). M·RsrI (L72P) is catalytically compromised, suggesting that excessive methylation may contribute to the decreased viability of wild-type enzyme (Fig. 1, lines D and E). M·RsrI (D173A) production was not toxic (Fig. 1, line A), although it displays activity in vitro comparable to the wild-type enzyme. Thus, the D173A mutation appears to disrupt DNA binding enough to decrease toxicity. M·RsrI (D173A) may also act like M·EcoRI (H235N), a bending-deficient mutant, which displays enhanced discrimination against non-canonical DNA relative to the wild-type enzyme (20).
DNA binding by M·RsrI mutants
M·RsrI (L72P) bound site-specifically in vivo. The binding is not likely due to grossly differential levels of protein expression. The L72P mutation lies four amino acids C-terminal to the highly conserved catalytic motif, DPPY (residues 65–68), that is found in all amino MTases. The crystal structure of DpnM in complex with AdoMet (16) as well as mutational studies with EcoDam (21) and T4 Dam (22) support the role of motif IV in both cofactor binding and catalysis. Studies with two DPPY mutants of M·EcoRV, D193N and D193A, showed that both lost catalytic activity but retained some degree of AdoMet and DNA binding (23). The residual catalytic activity of purified M·RsrI (L72P) in λ DNA protection assays demonstrates that the enzyme can bind both AdoMet and DNA functionally.
Both the DPPY motif and the L72P mutation lie in a loop in the structure of M·RsrI (4). The introduction of a Pro at position 72 in the loop may affect cofactor binding or catalytic activity without impairing site-specific DNA recognition. The mutant remains sensitive to methylation of the recognition site, suggesting that the mutation does not grossly perturb the protein–DNA recognition interface. Since the mutation is located near the active site of M·RsrI, its effects may include (i) interfering with base extrusion, (ii) aligning AdoMet and the flipped target base improperly so as to impair methyl transfer or (iii) trapping the target base in the enzyme to slow product release. The L72P mutation may result in the accumulation of a DNA-bound enzyme intermediate that is normally only transiently present, allowing the detection of site-specific DNA binding in vivo.
For DNA MTases DNA binding is tightly coupled to catalysis in a process likely involving conformational changes such as base extrusion and DNA bending. Solution studies on M·HhaI using electrophoretic gel retardation and 19F-NMR spectral analysis show that the enzyme can exist in a ‘closed’ state, with the target cytosine locked in the active site of the enzyme (24). Structural studies on the ternary complex of M·HhaI–AdoHcy–canonical DNA show that an active site pocket is formed only after the target base is extruded from the DNA. The formation of the pocket around the base results from large structural changes in the catalytic loop (25). The introduction of a Pro at position 72 in M·RsrI may reduce the conformational flexibility of the catalytic loop, resulting in altered ability to flip the target base into or release it from the active site of the enzyme.
Studies of DNA binding both in vivo and in vitro and sequence alignment analysis suggest that Asp173 is likely a part of the DNA TRD of the enzyme. D173 is located in the TRD of β class enzymes identified by Malone et al. (5). It is evident from Figure 1 that the double mutant M·RsrI (L72P/D173A) binds DNA less well than does the single mutant M·RsrI (L72P). Although weakened in binding, it retains the ability to discriminate between unmethylated and methylated GAATTC sites. Gel retardation assays in vitro also confirm that M·RsrI (L72P/D173A) binds target DNA substrates less well than M·RsrI (L72P). Furthermore, the single mutant D173A retains catalytic activity comparable to the wild-type enzyme in vivo and has ∼3-fold less activity in vitro. The relatively slight impairment of the catalytic activity suggests that the mutation must subtly affect the gross structure of the enzyme. Challenge-phage assays using the L72P mutant and selecting for weaker binding may identify other amino acids functionally involved in site-specific DNA recognition.
Structure-based alignment analysis
Clustal W alignment analysis of several β class MTases revealed that D173 resides within a cluster of conserved amino acids within the putative TRD (Fig. 3). The TRD, based on previous alignments (5,6,15), is defined as the region between motifs involved in cofactor binding and catalysis. These residues may participate directly in DNA recognition through interactions with base pairs or indirectly by supporting a structural framework of amino acid residues that themselves contact the bases. The crystal structure of M·RsrI reveals that D173 and R176 interact to form an ion pair (4). Disruption of this interaction may destabilize the helix in which it participates, thereby affecting DNA binding.
Sequence comparisons with other MTases
Clustal W sequence analysis revealed sequence similarities between the β MTase TRD and those of other MTases sharing the same or similar DNA recognition sequences (Fig. 6). Whereas conserved residues within all the β MTase TRD motifs imply a function in maintenance of the structure of the TRD, shared residues between different classes of MTases recognizing the same DNA sequence suggest a possible role in site-specific DNA recognition.
Four amino acids within the α MTase TRD sequence motif of enzymes recognizing GATC are missing in the β MTases (Fig. 6A). This missing stretch corresponds to residues 131–134 of M·DpnM, which has been crystallized in complex with AdoMet (16). The TRD of M·DpnM forms a highly structured loop. In the loop, residues 131–134 form part of a β-turn. Thus, this part of M·DpnM may serve to stabilize the structural framework of the TRD of α MTases. Residues in the TRD of other classes of MTases showing a lack of conservation when aligned with β MTase TRD motif sequences may serve to preserve a structural element not found in the β MTase TRD or contribute additional amino acid residues to mediate site-specific DNA recognition.
The Clustal W alignment in Figure 3 suggests that the DNA recognition specificity of β Mtases might be changed using site-directed mutagenesis. For example, the less conserved amino acid sequences in GATC-recognizing β MTases (Fig. 3, nos 5–8) in regions 2 or 4 could be mutagenized to residues contained in GANTC-recognizing sequences (Fig. 3, nos 9–11) and the mutant enzymes tested to see if the DNA specificity has been changed from GATC to GANTC. M·HpyI, a CATG-recognizing enzyme (Fig. 3, no. 8), might similarly be converted to recognize GATC by conservative changes in the TRD using M·LlaDCHIB (Fig. 3, no. 7) as a guide for mutagenesis.
Conclusions
We isolated a mutant, M·RsrI (L72P), that binds DNA in vivo site-specifically and in a methylation-sensitive manner as assessed by the challenge-phage assay. The introduction of a Pro at position 72 may deleteriously affect cofactor binding or alter the geometry of the active site during catalysis, leading to the observed impairment in catalytic activity. The mutation may also alter the flexibility of the loop in which it resides, thereby altering conformational changes associated with catalysis, such as DNA bending or base extrusion. The L72P mutation is near the highly conserved catalytic motif, DPPY, proposed to directly interact with the expelled target base. This mutant provides a starting point for an analysis in vivo of other amino acids involved in DNA recognition, as evidenced by the isolation of a double mutant M·RsrI (L72P/D173A). Addition of the D173A mutation weakened site-specific DNA binding in the challenge-phage assay, suggesting that D173, which is located in the target recognition domain, is involved in DNA binding. Residues that are conserved within the TRD of several β MTases are probably involved in maintaining their structures, as well as possibly contributing to site-specific DNA recognition. Residues conserved in the TRDs of different classes of MTases sharing the same DNA recognition sequence specificity may have a more direct role in site-specific DNA recognition.
Acknowledgments
ACKNOWLEDGEMENTS
The authors wish to thank Dr Arvydas Janulaitis for supplying a clone of the Van91II R-M system and Dr Mei Ge for sequencing of the M·Van91II gene. We are indebted to Dr Yonghong Huan for preparation of the R·RsrI (E117Q)-expressing-plasmid and Ms Nicole Collins for purification of M·RsrI (L72P). We also thank Drs Mair Churchill, Bernard Connolly, Sergei Degtyarev, Jeff Gardner, John Gerlt, Saulius Klimasauskas, Norbert Reich and Shuang-yong Xu for their helpful comments and discussions during preparation of the manuscript. This work was supported, in part, by NIH grant GM25621 to R.I.G., the Department of Biochemistry and a grant from the University of Illinois Research Board.
REFERENCES
- 1.Kaszubska W., Aiken,C., O’Connor,C.D. and Gumport,R.I. (1989) Nucleic Acids Res., 17, 10403–10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benson N., Adams,C. and Youderian,P. (1994) Mol. Microbiol., 11, 567–579. [DOI] [PubMed] [Google Scholar]
- 3.Maloy S. and Youderian,P. (1994) Methods Mol. Genet., 3, 205–233. [Google Scholar]
- 4.Scavetta R.D., Thomas,C.B., Walsh,M., Szegedi,S., Joachimiak,A., Gumport,R.I. and Churchill,M.E.A. (2000) Nucleic Acids Res., 28, 3950–3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Malone T., Blumenthal,R.M. and Cheng,X. (1995) J. Mol. Biol., 253, 618–632. [DOI] [PubMed] [Google Scholar]
- 6.Gong W., O’Gara,M., Blumenthal,R.M. and Cheng,X. (1997) Nucleic Acids Res., 25, 2702–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szegedi S.S., Reich,N.O. and Gumport,R.I. (2000) Nucleic Acids Res., 28, 3962–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fromant M., Blanquet,S. and Plateau,P. (1995) Anal. Biochem., 224, 347–353. [DOI] [PubMed] [Google Scholar]
- 9.Bradford M.M. (1976) Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 10.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thompson J.D., Higgins,D.G. and Gibson,T.J. (1994) Nucleic Acids Res., 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hall T.A. (1999) Nucleic Acids Symp. Ser., 41, 95–98. [Google Scholar]
- 13.Page R.D. (1996) Comput. Appl. Biosci., 12, 357–358. [DOI] [PubMed] [Google Scholar]
- 14.Fisher E.W., Yang,M.T., Jeng,S.T., Gardner,J.F. and Gumport,R.I. (1995) Gene, 157, 119–121. [DOI] [PubMed] [Google Scholar]
- 15.Bujnicki J.M. and Radlinska,M. (1999) Nucleic Acids Res., 27, 4501–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tran P.H., Korszun,Z.R., Cerritelli,S., Springhorn,S.S. and Lacks,S.A. (1998) Structure, 6, 1563–1575. [DOI] [PubMed] [Google Scholar]
- 17.Abdurashitov M.A., Netesova,N.A., Golikova,L.N., Gutorov,V.V., Belavin,P.A. and Degtyarev,S.K. (2000) Mol. Biol., 33, 87–94. [PubMed] [Google Scholar]
- 18.Guschlbauer W. (1988) Gene, 74, 211–4. [DOI] [PubMed] [Google Scholar]
- 19.Smith D.W., Crowder,S.W. and Reich,N.O. (1992) Nucleic Acids Res., 20, 6091–6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allan B.W., Garcia,R., Maegley,K., Mort,J., Wong,D., Lindstrom,W., Beechem,J.M. and Reich,N.O. (1999) J. Biol. Chem., 274, 19269–19275. [DOI] [PubMed] [Google Scholar]
- 21.Guyot J.B., Grassi,J., Hahn,U. and Guschlbauer,W. (1993) Nucleic Acids Res., 21, 3183–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kossykh V.G., Schlagman,S.L. and Hattman,S. (1993) Nucleic Acids Res., 21, 4659–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roth M., Helm-Kruse,S., Friedrich,T. and Jeltsch,A. (1998) J. Biol. Chem., 273, 17333–17342. [DOI] [PubMed] [Google Scholar]
- 24.Klimasauskas S., Szyperski,T., Serva,S. and Wuthrich,K. (1998) EMBO J., 17, 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Gara M., Horton,J.R., Roberts,R.J. and Cheng,X. (1998) Nature Struct. Biol., 5, 872–877. [DOI] [PubMed] [Google Scholar]