


Abstract
Transcription of class III genes is conducted by multi-protein complexes consisting of polymerase III itself and several transcription factors. We established a reconstituted in vitro transcription system from which the autoantigen La was removed by immunodepletion. This system showed no RNP formation, but was still fully active in transcription. Supplementing such La-free transcription reactions with recombinant La restored the formation of La complexes with the newly synthesised RNA, but did not lead to enhanced transcription efficiency. Furthermore, we developed a technique for the generation and isolation of transcription complexes, assembled from purified transcription factors and isolated by glycerol centrifugation. These complexes were fully competent to re-initiate RNA synthesis but they were not associated with La and their transcription rate could not be stimulated by addition of recombinant La. Therefore, we conclude that La does not act as a human polymerase III transcription factor.
INTRODUCTION
Human RNA polymerase III (pol III) catalyses the synthesis of several small RNA molecules, among others 5S rRNA, all types of tRNAs, U6 RNA and the adenovirus-associated RNAs VAI and VAII (1). The first step in pol III transcription is the sequential binding of transcription factors (TFs) to the promoter. These factors form a stable pre-initiation complex on the transcribed gene and recruit the polymerase to the initiator (2,3). Binding of the multi-subunit complex TFIIIC2 to the B-box is the initial step to establish the transcription complex on most genes with internal promoters, like the tRNA genes and the adenoviral VAI gene (4,5). The binding of TFIIIC2 is reinforced by TFIIIC1, which is an essential transcription factor of all pol III genes, but little is known about its structure (4,6,7). The third component required for transcription is the TBP–TAF complex TFIIIBβ, which is involved in polymerase recruitment (8,9).
Once the polymerase is assembled to the complex, it melts the DNA around the start point of transcription. This open complex is then transferred to a productive elongating complex by initiating RNA synthesis (10). One round of transcription ends when the polymerase reaches the terminator. It recognises the oligo(T) stretch at the end of the gene and then the ternary polymerase–DNA–RNA complex is dissociated. The RNA is released and the polymerase is ready for a second round of transcription (11,12). It could be shown in yeast that once a transcription complex is assembled, polymerase is re-initiated on the same gene in a facilitated pathway, implying that a second round of transcription is performed much faster than the initial one. This pathway is strictly dependent on the terminator (13). As shown for Xenopus, termination of transcription is a two step event in which recognition of the terminator is followed by release of the polymerase from the ternary complex. The latter process crucially requires displacement of the RNA (14).
After transcription the RNA is found to be complexed into ribonucleoprotein particles (RNPs) with a 50 kDa protein, the autoantigen La (15 and references therein). La protects the RNA from exonucleolytic degradation and renders it accessible to a regulated maturation pathway (16–18). Interestingly, La binds to the oligo(U) stretch at the 3′-end of the RNA which corresponds to the termination signal of pol III genes (19).
It has been reported that depletion of a human cellular extract of La leads to a dramatic decrease in pol III transcription (20). Thus a model was proposed according to which La is required for efficient RNA release and for displacement of the ternary complex at the terminator in mammals (16,21). Also, the polymerase itself was considered to be redirected to the initiator by La for efficient re-initiation, thus conceivably acting as a re-initiation and/or even as an initiation factor (17,22). Moreover, a regulatory role in human transcription was attributed to reversible phosphorylation of La (23).
However, in recent years conflicting results were presented for pol III transcription in Xenopus and yeast cells showing that RNP assembly occurs independently of transcription and that La is not required for the latter process (18,24).
Other functions have also been attributed to La, particularly regulation of RNA transport between different compartments of the cell (25–27) and regulation of translation of viral RNAs from poliovirus and human immunodeficiency virus (HIV) (28 and references therein). A role for La has been shown in the stabilisation of histone mRNAs (29). Furthermore, La is involved in regulation of the interferon-inducible protein kinase (PKR), thereby acting as an unwindase of double-stranded RNA (30,31).
In this report we show that human pol III transcription operates faithfully and efficiently without detectable La and that transcription and formation of La RNPs are not functionally coupled in vitro. Moreover, we show that TBP but not La is associated with functional human transcription complexes generated from purified transcription factors and isolated by glycerol gradient centrifugation.
MATERIALS AND METHODS
DNA templates
pUVAI contains the adenoviral VAI gene inserted into pUC18 (32). pBh5S contains the human genomic 5S gene inserted into pBluescript (33).
Buffers and additives
HEPES buffer (for phosphocellulose chromatography) consisted of 20 mM HEPES, pH 7.9, 10% (v/v) glycerol, 3 mM dithiothreitol (DTT), 0.2 mM phenylmethylsulfonylfluoride (PMSF). Transcription buffer consisted of 20 mM Tris–HCl, pH 7.9, 10% (v/v) glycerol, 5 mM MgCl2, 60 mM KCl, 3 mM DTT, 0.2 mM PMSF. All fractions employed were dialysed against transcription buffer before use, except for the polymerase fraction (see below).
BSA (Boehringer Mannheim) and RNase Block Ribonuclease Inhibitor (Stratagene) were negatively tested for La several times.
Purification of transcription factors
Cytoplasmic extracts (S100) from HEK (human embryonic kidney) cells were prepared as described previously (7,34), with a protein concentration of 17.5 mg/ml. The extract was fractionated by phosphocellulose chromatography (Whatman P11) as described (34,35), into fractions PCA (10 mg/ml), PCB (4 mg/ml), PCC (1 mg/ml) and PCD (0.5 mg/ml).
TFIIIC1 and TFIIIC2 were purified from the PCC fraction by MonoQ chromatography as described (7).
The PCB fraction was separated into fractions containing pol III, TFIIIBβ and TFIIIBα as described previously by EMD DEAE Fractogel (Merck) chromatography (8). TFIIIBβ was further purified by EMD SO3– Fractogel (Merck) chromatography as described (7), but was step eluted. TFIIIBβ eluted in the 250–400 mM KCl fraction and was free of TFIIIC1, TFIIIC2 and polymerase activity.
The polymerase fraction was also applied to EMD SO3– Fractogel and was step eluted. The polymerase activity eluted in the 400–650 mM KCl fraction and was free of TFIIIB, TFIIIC1 and TFIIIC2 activities. An aliquot of 0.1 mg/ml BSA was added and the polymerase fraction was diluted with glycerol to a final concentration of 50% (v/v) glycerol.
TFIIIA was purified from the PCA fraction by rechromatography as described (35).
Recombinant La was prepared as described (36).
In vitro transcription
The in vitro transcription mixtures contained the respective protein fractions, 1 µg plasmid DNA, 600 µM ATP, CTP and UTP, 30 µM GTP, 3 µCi [α-32P]GTP (Amersham), 20 U RNase Block Ribonuclease Inhibitor (Stratagene), 60 mM KCl, 20 mM Tris–HCl, pH 7.9, 10% glycerol and 5 mM MgCl2 in a final volume of 70 µl. After 90 min incubation at 30°C, the RNA was purified and loaded onto a denaturing 7 M urea–6% polyacrylamide gel. The gel was analysed by autoradiography and with a Fuji FLA-3000 phosphorimager.
The amount of RNA synthesised (in fmol) was quantitated from the specific radioactivity of the [α-32P]GTP employed, assuming that one molecule of VAI RNA contains 54 guanosine residues.
Purification of antibodies
Monoclonal antibodies SW5 and 3B9 (36) against human La and antibodies against human TBP (8) were purified from hybridoma cell supernatant by chromatography over a protein A–Sepharose column. The antibodies were eluted with acetate buffer, pH 2.75, and subsequently dialysed against transcription buffer. Purification of the IgG fraction from rabbit serum was conducted accordingly.
Immunodepletion of transcription factors
An aliquot of 4 mg of monoclonal antibodies against La (SW5) or purified IgGs from rabbit were each loaded onto a 1 ml HiTrap rProtein A column (Pharmacia). The antibodies were coupled with dimethylpimelimidate as described (8).
Fractions containing purified transcription factors and the polymerase were mixed at the stoichiometry optimally required for transcription of the pUVAI and pBh5S genes and were loaded onto either SW5 or purified IgG (mock) columns which had been previously blocked with 1 mg/ml BSA. Immunodepletion was conducted with a Bio-Rad Biologic mid pressure system.
Immunoprecipitation
SW5 antibodies (5 mg) were coupled to immobilised protein A as described above, except that 1 ml of protein A–Sepharose CL-4B (Sigma) was used. After coupling the beads were blocked with 1 mg/ml BSA. Control beads without antibodies were treated identically. Before immunoprecipitation the beads were aliquoted into 40 µl portions, consisting of 20 µl beads and 20 µl supernatant buffer. The mixtures were washed three times with transcription buffer. The supernatant was subsequently removed.
Transcription samples were prepared as described above and after incubation they were mixed with the prepared protein A–Sepharose beads. The samples were incubated for 30 min at room temperature by gentle mixing. Then the supernatant was removed. The beads were washed three times with transcription buffer. The supernatant and the wash steps, both containing free RNA, were subsequently treated with proteinase K/SDS. To elute the bound RNA, the remaining beads were likewise treated with proteinase K/SDS. The RNA was prepared from all fractions as described and analysed on a denaturing 6% polyacrylamide gel.
The supernatants were also tested for their La content after immunoprecipitation by western blotting as described below. The efficiency of immunoprecipitation was >85%, even in the samples with the highest La content.
Purification of transcription complexes by glycerol gradient centrifugation
Preparation of transcription complexes. Incubation was performed as described above except for the following modifications. The volume of each sample was 25 µl and only 350 ng plasmid DNA were used. TFIIIC1, TFIIIC2, TFIIIBβ and pol III were mixed in a ratio corresponding to the optimal stoichiometry required for transcription. Radioactively labelled GTP was omitted from the reaction and the incubation was conducted for 30 min either without nucleotides (see Discussion), with ATP, GTP and CTP but no UTP (Fig. 5) or with all four nucleotides (see Discussion) in order to assemble transcription complexes which were either not initiated, stalled at nucleotide +7 or already cycling.
Figure 5.


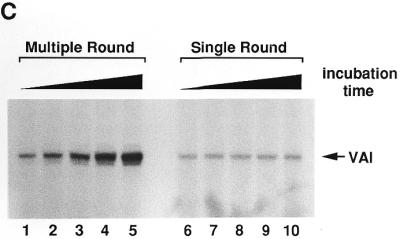
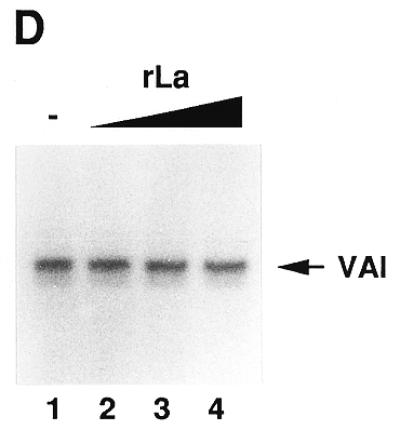
Analysis of functionally active pol III transcription complexes separated by a glycerol gradient. Several identical transcription reactions were performed in a volume of 25 µl each as described in Materials and Methods. After incubation they were pooled. Aliquots of 200 µl from this pool (corresponding to eight transcription reactions) were applied to a 4.2 ml glycerol gradient and centrifuged for 3 h at 50 000 r.p.m. in an SW 60 rotor. After centrifugation, fractions of 350 µl were collected. The corresponding fractions from six parallel gradients were pooled. (A) In vitro transcription. The fractions from the gradients and the load fraction were incubated with nucleotides, [α-32P]GTP and RNase Block as described above. No additional DNA was added. After incubation, the RNA in the samples was extracted as described above and loaded onto a 6% denaturing polyacrylamide gel. Lane 1, 25 µl load fraction; lanes 2–13, 50 µl glycerol gradient fractions. Fraction 1 is the top fraction (B) Western blot. The remaining pooled fractions were treated with Strataclean Resin (Stratagene) and loaded onto a 12.5% SDS gel. After blotting, the membrane was incubated with monoclonal anti-TBP antibodies and subsequently incubated with [125I]anti-mouse antibodies as secondary antibodies. The membrane was analysed with a phosphorimager. The membrane was not stripped, but incubated with a mixture of SW5 and 3B9 antibodies and subsequently incubated with 125I-labelled anti-mouse antibodies. The final analysis was performed with a phosphorimager and by autoradiography. Note that the band in fraction 8 at the height of the La signal was already visible after TBP incubation (data not shown). It is presumably an artefact due to the second antibody. Lane 1, 160 µl of load fraction; lanes 2–13, 1.85 ml glycerol gradient fractions; lane 14, 5 µl IIIBβ; lane 15, 5 µl of a La fraction, purified from PCA (20 ng La/µl). (C) In vitro transcription. The in vitro transcription was performed as described in (A) using a mixture of fractions 8 and 9 of a typical gradient. To ensure single round conditions in lanes 6–10, heparin was added at a final concentration of 600 µg/ml. Lanes 1–5, incubation without heparin for 2, 5, 10, 20 and 40 min; lanes 6–10, incubation with heparin for 2, 5, 10, 20 and 40 min. (D) Multiple round transcription of fractions 8 and 9 for 60 min. The gel was autoradiographed for a shorter time period as in (C). BSA (10 µg) was added to the reaction in order to avoid any protein effects. Lane 1, no additional La; lanes 2–4: addition of 15, 30 or 60 ng recombinant La.
Glycerol gradients were prepared in 4.2 ml SW 60 tubes with a gradient of 15–30% glycerol using a Biologic (Bio-Rad) gradient pump. The top 200 µl were removed. After the transcription incubation, identical transcription samples were pooled and 200 µl of this pool were loaded onto each gradient and centrifuged in a Beckmann SW 60 rotor at 50 000 r.p.m. for 3 h. The gradient was collected in 12 fractions (350 µl).
In vitro transcription of the separated transcription complexes. Aliquots of 50 µl of the gradient fractions were mixed with nucleotides, 3 µCi [α-32P]GTP (Amersham) and RNase Block and transcribed as described above in a total volume of 60 µl. No further template was added. To ensure single round conditions in one experiment, heparin was added at a final concentration of 600 µg/ml.
Western blotting of glycerol gradient fractions. Six identical gradients were run and the corresponding fractions were pooled. Before loading onto the SDS gel (see below) they were concentrated with Strataclean Resin (Stratagene).
Western blotting
After separation on SDS–12.5% polyacrylamide gels, proteins were transferred to a PVDF membrane (Immobilon-P; Millipore), stained with Ponceau S solution (Serva), destained and blocked with 10% skimmed milk in PBS/Tween. Primary antibodies were incubated at different concentrations depending on the expected signal strength. 3B9 and SW5 antibodies were used together in a molar ratio of 1:1. Both antibodies were comparably active and displayed no differences in detection (data not shown). 125I-labelled anti-mouse antibodies (Amersham) were used as secondary antibodies with 1 µCi/ml blocking solution. The membrane was analysed by autoradiography and with a Fuji FLA-3000 phosphorimager.
RESULTS
Partially purified fractions of the pol III transcription system contain La
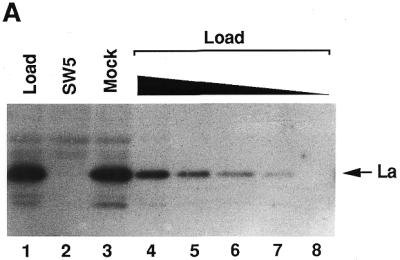
The role of La in transcription based on in vitro experiments using crude cell extracts is controversial. Although in one report a partially purified transcription system was used, it remained unclear how much La was still present in the fractions employed (37). Therefore, we initially determined the distribution of La in our conventional purification procedure. Interestingly, La showed an extremely diverse chromatographic behaviour (Fig. 1A). In the first step of purification, i.e. phosphocellulose chromatography, La could be found in all fractions PCA, PCB, PCC and PCD (lanes 2–5). Moreover, almost all purified factors required for pol III genes with internal promoters and also the polymerase fraction itself contained variable amounts of La (lanes 6–10) when further purified according to our conventional purification scheme, as outlined in Materials and Methods. Only the TFIIIBβ fraction was almost free of La (lane 6).
Figure 1.

Anti-La western blot of different chromatographic fractions containing human pol III and pol III transcription factors (A) Lane 1, S100 fraction (5 µl); lanes 2–5, phosphocellulose fractions, stemming from the S100 fraction, PCA (10 µl) and PCB–PCD (20 µl); lanes 6–10, further purified fractions according to the purification scheme described in Materials and Methods: TFIIIBβ (20 µl), pol III (20 µl), TFIIIC1 (80 µl), TFIIIC2 (40 µl) and TFIIIA (60 µl). (B) Comparison between a crude transcription system (lane 1, 5 µl S100 fraction), a system based on phosphocellulose fractions (lane 2, 10 µl PCB, 15 µl PCC and 7.5 µl TFIIIA) or the reconstituted system containing more purified fractions (lane 3, 2.5 µl TFIIIBβ, 2.5 µl pol III, 10 µl TFIIIC1, 5 µl TFIIIC2 and 7.5 µl TFIIIA). The amounts and compositions of all fractions correspond to the optimal stoichiometry of one sample used for in vitro transcription. PC, transcription system, based on phosphocellulose fractions; pur. frac., reconstituted system from more purified fractions.
However, conventional purification of the transcription factors led to a significant reduction in the La content of a typical transcription sample. In Figure 1B the La content was analysed in each of three transcription reactions containing either S100 fraction, phosphocellulose fractions or a system reconstituted from more purified transcription factors. The reconstituted transcription system was supplemented with TFIIIA and was hence also competent for transcription of the 5S gene.
In comparison to the S100 reaction (lane 1), it is evident that the PCB/PCC reaction still contained significant amounts of La (lane 2). However, the remaining La in the reconstituted system (lane 3) corresponded to <5% of the La in the S100 fraction, although it was equally active in transcription (data not shown).
A La depleted reconstitution system is still active in transcription
In order to further reduce the La concentration of the reconstituted transcription system, we prepared a batch of transcription factors and the polymerase having the same stoichiometry as presented in Figure 1B, lane 3. An aliquot of 40 µl of this mixture exactly corresponded to one transcription sample and contained ∼10 ng La, as quantitated in comparison to recombinant La in a western blot (data not shown). Subsequently, this mixture was applied to a protein A column coupled either with monoclonal anti-La antibodies (SW5) or with purified but non-specific IgG antibodies from rabbit (mock). The flow-throughs obtained from both these columns were tested for the presence of La (Fig. 2A) and for their transcriptional activity (Fig. 2B). Since only the central portion of the flow-through was collected from both columns, the protein content was not reduced compared to that of the loading fraction (data not shown).
Figure 2.
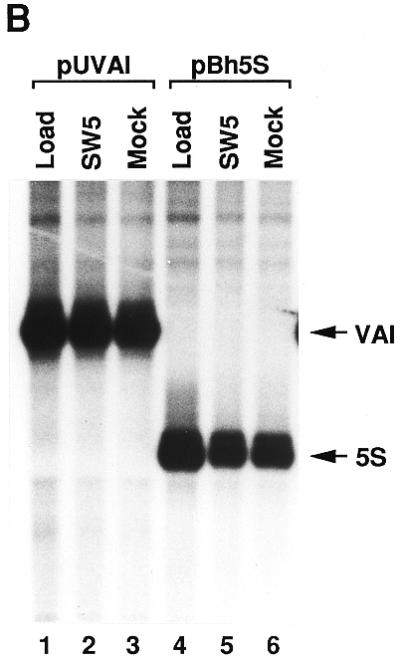
Immunodepletion of La from a reconstituted transcription system. Transcription factors IIIBβ, IIIC1, IIIC2 and IIIA together with pol III were mixed in optimal stoichiometry, corresponding to the reconstituted transcription system in Figure 1B, lane 3. For each transcription sample 12.5 µl of buffer were added. The mixture, with a final volume of 15 ml, was divided into three parts. One part was kept as the load fraction as a control. The other two parts (each 6.5 ml) were applied to a 1 ml HiTrap rProtein A–Sepharose column (Pharmacia), to which either 4 mg SW5 antibodies or 4 mg purified IgGs from rabbit had been coupled. The central portion of the flow-through was collected. (A) Western blot analysis. An aliquot of 80 µl of each fraction stemming from the immunodepletion was tested for their La content; the load fraction (lane 1), the flow-through from the SW5 column (lane 2) and the flow-through from the rabbit IgG column (Mock, lane 3). Lanes 4–8 show a titration of the load fraction for comparison, corresponding to 20, 10, 5, 2.5 and 1% of the amount of the load fraction. (B) An aliquot of 40 µl of the load fraction (lanes 1 and 4) or the flow-through fractions (lanes 2–3 and 5–6) was analysed by in vitro transcription of the VAI gene (pUVAI, lanes 1–3) or the 5S rRNA gene (pBh5S, lanes 4–6).
Because the exposure time of the autoradiogram in Figure 2A was extended to 10 days, the signal from the load fraction (lane 1) was much stronger than in Figure 1B, which was only exposed overnight. Additionally, the protein amount tested here was double in comparison to Figure 1B. It was found that the content of La was not reduced by mock chromatography (compare Fig. 2A, lanes 1 and 3). On the contrary, no La could be detected in the flow-through after SW5 chromatography (lane 2). The detection limit of our western blot was between 0.25 (5 fmol) and 0.1 ng (2 fmol) La as estimated from titration of the load fraction (lanes 4–8). Therefore, the residual amount of La in one transcription sample is certainly <5 fmol La.
In spite of this, transcription efficiency in the SW5 flow-through was not reduced compared to the mock depleted fraction (Fig. 2B). This is true for VAI (lanes 2 and 3) as well as for 5S transcription (lanes 5 and 6). However, it is apparent that the transcription capacities of both the SW5 and the mock flow-throughs is somewhat lower compared to the untreated loading mixture, presumably due to physical stress on the fractions during chromatography.
Using both the La depleted and the mock depleted transcription systems, ∼300 fmol VAI RNA were synthesised during transcription. Therefore, the amount of RNA synthesised in the La depleted transcription sample exceeds its conceivable La content by at least 60-fold.
Since, surprisingly, this transcription system, virtually devoid of La, still transcribes human pol III genes with high fidelity and efficiency, this finding contradicts the earlier assumption that La acts as an essential transcription factor in human cells.
Increasing quantities of La lead to an increasing amount of La–RNA complex but not to stimulation of transcription
Hitherto it was unclear whether the RNA synthesised in our reconstituted transcription system was assembled into La–RNP complexes. We attempted to detect La–RNA complexes by co-immunoprecipitation of the newly synthesised RNA with monoclonal anti-La antibodies. The SW5 antibodies were immobilised on protein A–Sepharose, while protein A–Sepharose without antibodies served as a control. Non-specific binding was minimised with BSA.
The transcription reactions with mock or La depleted mixtures were performed as described above. After incubation, coupled (or uncoupled) protein A–Sepharose was added to the transcription samples and was incubated for 30 min with gentle mixing. The supernatant was removed and the Sepharose was washed three times with transcription buffer. Bound components in the precipitate were released from the Sepharose by adding a proteinase K/SDS mixture.
Again, the control transcription reactions with La and mock depleted mixtures yielded equal RNA levels (Fig. 3, lanes 1 and 8). When both these reactions were treated with bare protein A–Sepharose after transcription, this resulted in hardly detectable traces of precipitated RNA (lanes 4 and 11), corresponding to the non-specific background. However, the situation was different when the mock depleted mixture was treated with immobilised SW5 antibodies. Although the major portion of the RNA was likewise not complexed (lane 12), ∼15% of the RNA could be found in the precipitate (lane 14), corresponding to specific RNA–La complexes.
Figure 3.

Immunoprecipitation of RNA–La complexes. In vitro transcription was conducted as described in Figure 2B, using either the SW5 depleted protein mixture (lanes 1–7) or the mock depleted mixture (lanes 8–14). After transcription, immunoprecipitation was conducted as described in Materials and Methods. Lanes 1 and 8 show the control reactions (C), which were stopped after transcription without further immunoprecipitation; lanes 2, 5, 9 and 12 show the supernatants (S); lanes 3, 6, 10 and 13 show the third wash steps (W); lanes 4, 7, 11 and 14 indicate the eluted precipitates (P). The synthesised RNA was quantitated with a phosphorimager.
When treating the La depleted mixture with the protein A-coupled SW5 antibodies (lane 7), no RNA could be precipitated. This result shows that the amount of La remaining after immunodepletion is insufficient to complex a significant proportion of the newly synthesised RNA. This observation underscores the quantitative measurements of the residual La concentration in the reaction (see Fig. 2 and the relevant discussion). Assuming that each La molecule binds to one RNA molecule, calculations have shown that <1.5% of the RNA could theoretically be complexed.
In the next experiment (Fig. 4) we added different quantities of recombinant La to the transcription mixtures prior to incubation. After transcription, the reactions were either stopped with proteinase K/SDS to analyse the accumulated RNA (lanes 1–8) or the La-complexed RNA was precipitated using immobilised SW5 antibodies (lanes 9–16).
Figure 4.

Effect of added recombinant La on transcription and on RNA–La complex assembly. Transcription reactions were conducted as before, either with SW5 (lanes 1–4) or with mock (lanes 5–8) depleted protein mixtures. Recombinant La was added in increasing amounts; no additional La (lanes 1 and 5) or 15 (lanes 2 and 6), 30 (lanes 3 and 7) or 60 ng (lanes 4 and 8). The transcription samples were incubated and prepared as described in Figure 2B. Lanes 9–16 show corresponding transcription samples which were immunoprecipitated after transcription with protein A–Sepharose-bearing SW5 antibodies identical to the previous experiment. Only the immunoprecipitates are shown. The synthesised RNA was quantitated with a phosphorimager.
The recombinant La showed no stimulating influence on transcription, neither in the case of the La depleted nor in that of the mock depleted reactions (lanes 1–4 and 5–8). In a control reaction without recombinant La again no RNA could be precipitated using the SW5 depleted mixture (lane 9), while with the mock depleted mixture 11.5% of the RNA was precipitated in this experiment (lane 13). The addition of 15 ng (300 fmol) recombinant La to the SW5 depleted mixture lead to precipitation of ∼8% of the RNA (lane 10). Thus the recombinant La was slightly less active in complex formation than the endogenous La (lane 13). Addition of 60 ng (1.2 pmol) recombinant La to the mock depleted mixture led to precipitation of ∼40% of the RNA synthesised (lane 16).
Taken together, these latter experiments show that the La depleted mixture of transcription factors and polymerase contained too little La for precipitation of RNA–La RNPs, but it was not reduced in its transcriptional capability. Moreover, addition of increasing amounts of recombinant La were able to enhance RNP assembly in mock depleted and La depleted mixtures without affecting transcription.
La is not associated with the human transcription complex
La was postulated to act as a transcription factor, which is necessary not only for termination but also for initiation and re-initiation of transcription (22). If this were the case, La should be detectable in purified transcription complexes by western blotting. Therefore, we used a technique first described by Wingender et al. (38) and Jahn et al. (34), who purified transcription complexes by glycerol gradients. We modified this method using purified factors instead of crude cell extracts for transcription complex formation. Additionally, we determined the composition of the complexes by western blotting analysis.
Using the purified factors described above, it was possible to stall transcription after synthesis of the first 6 nt. This was possible by deleting UTP from the reaction mixture, because the first T appears on the non-coding strand at position +7 (data not shown). We prepared these stalled complexes and applied them to the glycerol gradient. After centrifugation, the gradient was fractionated and corresponding fractions from six parallel gradients were pooled and tested for transcriptional activity by adding nucleotides and [α-32P]GTP but no template DNA. We found that transcription took place in fractions 4–9 (Fig. 5A, lanes 5–10). Several control experiments clearly showed that this transcriptional activity is mediated by isolated transcription complexes and is not due to a fortuitous co-sedimentation of the DNA and the required protein components (data not shown).
Aliquots of these fractions were then applied to SDS gel electrophoresis. The gel was blotted and the membrane probed with TBP and subsequently with La antibodies. Interestingly, TBP was found in two regions of the gradient (Fig. 5B). One part, corresponding to unincorporated TBP (TFIIIBβ), sedimented mainly in fractions 1 and 2 (lanes 2 and 3), while the other part, sedimenting in fractions 4–9 (lanes 5–10), corresponded to TFIIIBβ, which was incorporated into the transcription complex. Control experiments revealed that the slight shift observed in the peak of transcription (fraction 6, lane 7) and TBP content (fraction 5, lane 6) resulted from a minor portion of TBP (TFIIIBβ) bound non-specifically to cryptic TATA boxes in the plasmid.
Although La could also be detected in the gradient, its distribution was restricted to the first two or three fractions. La definitely did not co-sediment with the transcriptional activity, as was the case for TBP.
To investigate whether the isolated transcription complexes were able to re-initiate multiple rounds of transcription, we performed ‘single round’ versus ‘multiple round’ transcription assays. To exclude La contamination we used fractions 8 and 9 of a typical gradient. Lanes 1–5 in Figure 5C show a time course of transcription under multiple round conditions and lanes 6–10 show the corresponding single round transcription. Under single round conditions all transcription complexes had prolonged their RNA to the full length transcript after 2 min (lane 6) and longer incubation times did not lead to an increased accumulation of transcripts (lanes 7–10). In contrast, under multiple round conditions the transcription signal significantly increased with prolonged incubation. This was due to multiple re-initiation events.
Furthermore, we analysed whether such gradient fractions, capable of multiple rounds of transcription, but containing no detectable La, could be stimulated by addition of recombinant La. As shown in Figure 5D, there was no stimulating effect after supplementation of the isolated transcription complexes with increasing amounts of recombinant La.
DISCUSSION
Transcription of pol III genes in vivo is followed by formation of La–RNA complexes and most, if not all, transcripts are complexed and hence these processes are spatially and temporally adjoined in vivo. In the early 1980s it was shown that the kinetics of transcription and RNP assembly were similar, but it was nevertheless proposed that these two processes are not directly linked (15).
However, data were presented proposing that transcription of human pol III genes strictly depends on La. La was reported to be necessary for release of the RNA from the ternary polymerase–DNA–RNA complex at the terminator (16,20,21). It was, furthermore, postulated that La was also responsible for recycling of the polymerase to the initiator and even plays a role as an initiation factor in human cells (21,22).
In contrast, reports from other groups indicate that La does not exhibit any function as a transcription factor in yeast and Xenopus. Extracts from Xenopus cells immunodepleted of La transcribed a tRNA gene efficiently in vitro, although no RNA–La complex formation could be detected (24). Even more interestingly, haploid yeast strains with a deleted gene for Lhp1 were still viable, unless other genes were co-mutated (39,40). It could be shown that in these cells maturation of tRNAs was via an alternative pathway. However, transcription of neither tRNA nor U6 RNA was down-regulated. Also, addition of exogenous La to the mutant extract in vitro did not increase transcription (18,41).
These conflicting results regarding the role of La as a transcription factor did not necessarily mean that either model had to be wrong. It was conceivable that the spatial association between transcription and RNP assembly might have evolved to a functional coupling of these processes during the evolution of mammalian cells.
In this report we have used an efficient transcription system for the VAI and human 5S genes based on conventionally purified transcription factors and could show that the amount of residual La in this system was <5% of that observed in the S100 fraction.
This value approximates the value previously found in the Xenopus system, where immunodepletion of a cytoplasmic extract efficiently removed La without loss of transcriptional activity, but 1–3% of the La was retained (24).
By immunodepletion of our reconstituted transcription system we could further reduce the La concentration to levels that were below the detection limit of the western blot. However, this system was still fully active in transcription and could not be stimulated either by the addition of recombinant La (Fig. 4) or partly purified native La from different Q-Sepharose fractions prepared from the phosphocellulose flow-through (PCA) (data not shown).
We could also exclude that La–RNA complexes were formed using this transcription system. The number of RNA molecules synthesised in the transcription reaction was at least 60-fold higher than the possibly remaining La molecules. Moreover, in the immunoprecipitation experiments (Figs 3 and 4) no RNA could be precipitated unless exogenous La was added. Although a clear relation between La concentration and RNP complex formation in vitro was observed, the level of transcription was not influenced by varying La concentrations. These results strongly indicate that the formation of La-containing RNPs is not functionally coupled to transcription.
It was also previously postulated that La not only releases the RNA from the terminator, but is also associated with the transcription complex during RNA synthesis, thereby acting as a transcription factor for termination, re-initiation and even initiation of RNA synthesis. It was proposed that re-initiation of the polymerase from the terminator to the initiator would depend on La as a ‘recycling factor’ (21,22).
It has been demonstrated that functional human transcription complexes assembled from cytoplasmic extracts can be purified by glycerol gradient centrifugation (34,38). We improved this technique in two aspects. First, we were able to generate and isolate active transcription complexes using purified factors. Secondly, it was possible to detect one of the integrated transcription factors, TBP, by western blot analysis in such complexes.
It was thus interesting to investigate whether La would display co-sedimentation with the transcription complexes. This was, however, not the case. La clearly failed to co-sediment with the transcriptional activity. In the experiments shown here the initiated transcription complexes were stalled after 6 nt before centrifugation. It was conceivable, however, that La would enter the transcription complex in a later phase of transcription, e.g. during elongation of the nascent RNA. To examine this possibility, experiments were performed without or with all four nucleotides, corresponding to either assembled but not initiated or to cycling complexes. In both cases functional transcription complexes could be separated which sedimented almost identically to those described above. In both cases no La could be detected co-sedimenting with the complexes (data not shown). Thus we conclude that La is not associated with the transcription complex in the human system.
Moreover, transcription complexes sedimenting in fractions that were clearly devoid of La were active for multiple rounds of transcription. This finding is inconsistent with reports indicating that transcription complexes formed by nuclear extracts on immobilised templates are strictly dependent on La for termination and re-initiation of transcription. Interestingly, ∼200 ng to 1 µg of La was needed in those experiments for efficient RNA synthesis and the amount of added La and transcription efficiency clearly correlated positively, suggesting that one La molecule is required for synthesis and release of each RNA molecule (16,17,22). In our experiments faithful and efficient transcription was observed without any detectable La. Addition of recombinant La in amounts sufficient for efficient RNP assembly did not alter the transcription efficiency of our purified transcription complexes (Fig. 4D).
Taken together, the data presented here clearly show that human pol III transcription operates independently from the autoantigen La. We observed faithful and efficient transcription with La concentrations so low that no RNA–La complex formation could be observed. This result corresponds to the findings in yeast and Xenopus showing that RNP formation is not linked to transcription and that La does not act as a transcription factor in these organisms.
Moreover, we demonstrate that La is not associated with active human transcription complexes assembled from purified transcription factors and isolated by glycerol gradient centrifugation. Therefore, we conclude that La is neither necessary for dissociation of the ternary complex at the terminator nor is it responsible for recruitment of the polymerase for initiation or re-initiation in human cells. Although we cannot exclude from our experiments that transcription is somehow correlated with RNP assembly in vivo, we believe that La did not acquire an additional function as a transcription factor during evolution from amphibians to human.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Ulla Kopiniak, Frauke Seifart, Uta Drathen and Sarah Fehl for expert technical assistance. We also thank Prakash Dube for helpful discussions. This work was supported by grants from the Deutsche Forschungsgemeinschaft and the European Union.
REFERENCES
- 1.Willis I.M. (1993) Eur. J. Biochem., 212, 1–11. [DOI] [PubMed] [Google Scholar]
- 2.Lassar A.B., Martin,P.L. and Roeder,R.G. (1983) Science, 222, 740–748. [DOI] [PubMed] [Google Scholar]
- 3.Bieker J.J., Martin,P.L. and Roeder,R.G. (1985) Cell, 40, 119–127. [DOI] [PubMed] [Google Scholar]
- 4.Yoshinaga S.K., Boulanger,P.A. and Berk,A.J. (1987) Proc. Natl Acad. Sci. USA, 84, 3585–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshinaga S.K., L’Etoile,N.D. and Berk,A.J. (1989) J. Biol. Chem., 264, 10726–10731. [PubMed] [Google Scholar]
- 6.Yoon J.B., Murphy,S., Bai,L., Wang,Z. and Roeder,R.G. (1995) Mol. Cell. Biol., 15, 2019–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oettel S., Härtel,F., Kober,I., Iben,S. and Seifart,K.H. (1997) Nucleic Acids Res., 25, 2440–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teichmann M. and Seifart,K.H. (1995) EMBO J., 14, 5974–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z. and Roeder,R.G. (1997) Genes Dev., 11, 1315–1326. [DOI] [PubMed] [Google Scholar]
- 10.Kassavetis G.A., Blanco,J.A., Johnson,T.E. and Geiduschek,E.P. (1992) J. Mol. Biol., 226, 47–58. [DOI] [PubMed] [Google Scholar]
- 11.Bogenhagen D.F. and Brown,D.D. (1981) Cell, 24, 261–270. [DOI] [PubMed] [Google Scholar]
- 12.Kovelman R. and Roeder,R.G. (1990) Genes Dev., 4, 646–658. [DOI] [PubMed] [Google Scholar]
- 13.Dieci G. and Sentenac,A. (1996) Cell, 84, 245–252. [DOI] [PubMed] [Google Scholar]
- 14.Campbell F.E. Jr and Setzer,D.R. (1992) Mol. Cell. Biol., 12, 2260–2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Francoeur A.M. and Mathews,M.B. (1982) Proc. Natl Acad. Sci. USA, 79, 6772–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maraia R.J., Kenan,D.J. and Keene,J.D. (1994) Mol. Cell. Biol., 14, 2147–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodier J.L., Fan,H. and Maraia,R.J. (1997) Mol. Cell. Biol., 17, 5823–5832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoo C.J. and Wolin,S.L. (1997) Cell, 89, 393–402. [DOI] [PubMed] [Google Scholar]
- 19.Stefano J.E. (1984) Cell, 36, 145–154. [DOI] [PubMed] [Google Scholar]
- 20.Gottlieb E. and Steitz,J.A. (1989) EMBO J., 8, 841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gottlieb E. and Steitz,J.A. (1989) EMBO J., 8, 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maraia R.J. (1996) Proc. Natl Acad. Sci. USA, 93, 3383–3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan H., Sakulich,A.L., Goodier,J.L., Zhang,X., Qin,J. and Maraia,R.J. (1997) Cell, 88, 707–715. [DOI] [PubMed] [Google Scholar]
- 24.Lin-Marq N. and Clarkson,S.G. (1998) EMBO J., 17, 2033–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boelens W.C., Palacios,I. and Mattaj,I.W. (1995) RNA, 1, 273–283. [PMC free article] [PubMed] [Google Scholar]
- 26.Simons F.H.M., Rutjes,S.A., Van Venrooij,W.J. and Pruijn,G.J.M. (1996) RNA, 2, 264–273. [PMC free article] [PubMed] [Google Scholar]
- 27.Grimm C., Lund,E. and Dahlberg,J.E. (1997) Proc. Natl Acad. Sci. USA, 94, 10122–10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Craig A.W.B., Svitkin,Y.V., Lee,H.S., Belsham,G.J. and Sonenberg,N. (1997) Mol. Cell. Biol., 17, 163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLaren R.S., Caruccio,N. and Ross,J. (1997) Mol. Cell. Biol., 17, 3028–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao Q., Sharp,T.V., Jeffrey,I.W., James,M.C., Pruijn,G.J., Van Venrooij,W.J. and Clemens,M.J. (1994), Nucleic Acids Res., 22, 2512–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hühn P., Pruijn,G.J.M., Van Venrooij,W.J. and Bachmann,M. (1997) Nucleic Acids Res., 25, 410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneider H.R., Waldschmidt,R., Jahn,D. and Seifart,K.H. (1989) Nucleic Acids Res., 17, 5003–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sørensen P.D., Simonsen,H. and Frederiksen,S. (1990) Nucleic Acids Res., 18, 3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jahn D. and Wingender,E. and Seifart,K.H. (1987) J. Mol. Biol., 193, 303–313. [DOI] [PubMed] [Google Scholar]
- 35.Seifart K.H., Wang,L., Waldschmidt,R., Jahn,D. and Wingender,E. (1989) J. Biol. Chem., 264, 1702–1709. [PubMed] [Google Scholar]
- 36.Bachmann M., Tröster,H., Bartsch,H. and Grölz,D. (1996) J. Autoimmun., 9, 747–756. [DOI] [PubMed] [Google Scholar]
- 37.Fan H., Goodier,J.L., Chamberlain,J.R., Engelke,D.R. and Maraia,R.J. (1998) Mol. Cell. Biol., 18, 3201–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wingender E., Shi,X.P., Houpert,A. and Seifart,K.H. (1984) EMBO J., 3, 1761–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoo C.J. and Wolin,S.L. (1994) Mol. Cell. Biol., 14, 5412–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin-Marq N. and Clarkson,S.G. (1995) J. Mol. Biol., 245, 81–85. [DOI] [PubMed] [Google Scholar]
- 41.Pannone B.K., Xue,D. and Wolin,S.L. (1998) EMBO J., 17, 7442–7453. [DOI] [PMC free article] [PubMed] [Google Scholar]