


Abstract
We have shown previously that CrhC is a unique member of the DEAD-box family of RNA helicases whose expression occurs specifically under conditions of cold stress. Here we show that recombinant His-tagged CrhC, purified from Escherichia coli, is an ATP-independent RNA binding protein possessing RNA-dependent ATPase activity which is stimulated most efficiently by rRNA and polysome preparations. RNA strand displacement assays indicate that CrhC possesses RNA unwinding activity that is adenosine nucleotide specific. Unwinding of partially duplexed RNA proceeds in the 5′→3′ but not the 3′→5′ direction using standard assay conditions. Immunoprecipitation and far-western analysis indicate that CrhC is a component of a multisubunit complex, interacting specifically with a 37 kDa polypeptide. We propose that CrhC unwinds cold-stabilized secondary structure in the 5′-UTR of RNA during cold stress.
INTRODUCTION
It is becoming increasingly evident that alteration of RNA secondary structure plays a crucial role in numerous intracellular processes, including translation initiation, ribosome biogenesis and RNA splicing, maturation and degradation (1,2). Modulation of RNA secondary structure is catalyzed by a large, ubiquitous protein family designated RNA helicases. RNA helicases are characterized by a ‘DEAD’ amino acid motif (Asp-Glu-Ala-Asp) as well as seven additional motifs whose sequences and spacing are highly conserved (1,3–5). Although several hundred DEAD-box RNA helicase-related sequences exist in the data banks, the majority are designated putative RNA helicases, as relatively few of their gene products have been characterized biochemically. Of those investigated, the majority exhibit RNA-dependent ATPase activity, with only a limited number shown to unwind RNA (2,6–8). In general, duplex RNA unwinding activity is not RNA substrate specific and can be either ATP dependent, as for bona fide RNA helicases, or ATP independent, as for RNA helix destabilizers (9,10). Unwinding of artificial double-stranded (ds)RNA duplexes requires a single-stranded (ss)RNA region and proceeds either unidirectionally, 3′→5′ (11–13), or bidirectionally (7,14).
We have recently reported the cloning and transcriptional analysis of a gene encoding a putative RNA helicase, crhC, from the cyanobacterium Anabaena sp. strain PCC 7120 (15,16). CrhC is a novel RNA helicase as it possesses a unique alteration of the highly conserved Ser-Ala-Thr (SAT) amino acid motif to a Phe-Ala-Thr (FAT) motif. This non-conservative alteration has potential implications for CrhC activity as crystal structure analysis of a viral RNA helicase indicates that the Ser residue (or its hydrogen bonding analog Thr) is required to link helicase and ATPase activities (17).
Of prime interest is the environmental regulation of CrhC expression at both the transcriptional and translational levels in response to cold stress (15,16). CrhC transcripts are not detectable in Anabaena cells grown at 30°C but accumulate rapidly and differentially upon exposure to temperatures <25°C. crhC transcripts remain elevated for the duration of the stress but return to undetectable levels upon incubation at 30°C. Furthermore, stabilization of crhC mRNA during cold stress plays an important role in crhC transcript accumulation. Consistent with specific cold shock-induced accumulation of the crhC transcript, CrhC protein is expressed only in cold-shocked cells (16). These results suggest that CrhC performs a function required for acclimation of cyanobacteria to cold stress.
Investigation of the role performed by CrhC in the response of cyanobacteria to cold stress necessitates biochemical characterization of CrhC activity. We report here the expression and purification of recombinant His-tagged CrhC from Escherichia coli. Biochemical characterization indicates that CrhC exhibits ATP-independent RNA binding and RNA-dependent ATPase and (d)ATP-dependent RNA unwinding activities, none of which are RNA substrate specific. The unwinding activity is unidirectional, as CrhC unwinds dsRNA duplexes in the 5′→3′ direction, suggesting that interaction with the 5′-end of target RNA(s) is required for CrhC function in vivo.
MATERIALS AND METHODS
CrhC expression and purification
Site-directed mutagenesis (18) was utilized to create a BamHI site immediately upstream of the ATG translation initiation codon and an EcoRI site 100 bp downstream of the stop codon of the crhC open reading frame (crhC accession no. AF040045). The mutagenic primers were 5′-TCGGAGAATTCTAAAGCGAT-3′ for the upstream sense strand and 5′-AGACATGGATCCCCAGA-3′ complementary to the coding strand for the downstream site. The resulting 1.4 kb BamHI–EcoRI fragment was cloned into the corresponding sites in pRSETA (Invitrogen), creating pRSET29, whose identity was confirmed by sequencing. Recombinant CrhC was expressed in JM109 (pRSET29) as a translational fusion with a 23 amino acid ORF containing a His6 tag domain at the N-terminus. An exponential JM109 (pRSET29) culture was induced by the addition of isopropyl-1-thio-β-d-galactopyranoside (IPTG) and M13/T7 DE3 phage (Invitrogen) to final concentrations of 0.5 mM and 1 × 109 p.f.u./ml, respectively. Cells were grown for a further 5 h at 20°C, harvested and stored as a pellet at –80°C. Frozen cell pellet, representing 5 ml of culture, was lysed by sonication in lysis buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl, 10 mM imidazole and 1 mM PMSF). Clarified lysate (4 ml) was added to 1 ml of 50% Ni–NTA slurry (Qiagen), as recommended by the manufacturer. The column was washed with increasing concentrations of imidazole in wash buffer (50 mM NaH2PO4, pH 8.0, 300 mM NaCl) and CrhC-containing fractions eluted with 250 mM imidazole.
ATPase assays
ATPase assays were performed by thin layer chromatography detection of γ-32P release from [γ-32P]ATP (19). The standard assay contained 200 ng recombinant CrhC in a reaction buffer containing 20 mM HEPES–KOH, pH 7.5, 5 mM magnesium acetate, 1 mM dithiothreitol (DTT), 40 µM ATP and 15 µCi/ml [γ-32P]ATP (4500 Ci/mmol; ICN) in a final volume of 20 µl. RNA (10 µg) was included as indicated. The mixture was incubated at 37°C for 30 min and quenched by addition of 0.1 M EDTA. Aliquots (2 µl) were spotted on PEI–cellulose TLC plates (Fisher Scientific) and developed with 0.15 M formic acid, 0.15 M LiCl, pH 3. 32P detected in inorganic phosphate (Pi) and ATP was quantified using a Molecular Dynamics PhosphorImager and ImageQuant software. Polysomes were isolated from Anabaena cells, cold shocked at 20°C for 3 h, by ultracentrifugation of lysates through a sucrose cushion (20).
RNA helicase substrates
RNA helicase assays and production of the artificial dsRNA substrate RNA II, containing only single-stranded 5′-ends, were performed as described (21). Transcriptions were performed using a Riboprobe in vitro Transcription System kit (Promega). RNA I was generated from BamHI-linearized pRNA I by transcription with SP6 RNA polymerase (Promega) in the presence of [α-32P]UTP (50 µCi, 3000 Ci/mmol; Amersham), yielding a 32P-labeled 92 nt transcript. The transcribed RNA spontaneously forms a 10 bp GC duplex region with 58 nt single-stranded 5′ tails and 24 nt 3′ tails. RNA II was generated from pGEM3 (Promega), linearized with BamHI and transcribed with SP6 RNA polymerase in the presence of [α-32P]UTP (50 µCi, 3000 Ci/mmol; Amersham), yielding a 32P-labeled 41 nt transcript. pGEM-MO1/2 was linearized with HincII and transcribed with T7 RNA polymerase (New England Biolabs), yielding an unlabeled 68 nt transcript. Transcripts were purified by separation on denaturing 8 M urea–6% polyacrylamide gels, visualized by autoradiography or UV shadowing, excised and eluted in 0.4 ml of RNA elution buffer (0.5 M ammonium acetate, 1 mM EDTA, 0.1% SDS) overnight at 4°C. RNAs were further purified by phenol/chloroform extraction and ethanol precipitation. RNA transcripts were hybridized in RNA annealing buffer (20 mM HEPES–KOH, pH 7.2, 250 mM NaCl, 1 mM EDTA) by heating to 95°C for 5 min, followed by cooling to 37°C and incubation at 37°C for 2 h. Duplex RNA was purified by electrophoresis on a native 8% polyacrylamide gel, excised and eluted as described above. The resulting partial RNA duplex substrate has 5′ single-stranded tails of 27 and 54 nt and a 14 bp duplex region with a Tm of 50°C. The artificial RNA strands required to produce a dsRNA duplex containing only free 3′ ssRNA tails was produced from pGEM-CS– (22). Briefly, 107 and 103 nt strands were prepared by transcription of HaeII-cleaved pGEM3CS– with SP6 RNA polymerase and T7 RNA polymerase transcription of pGEM-CS– cleaved with RsaI, respectively. Annealing and purification of the resulting strands, as described above, produced an RNA substrate containing 25 bp of duplex RNA and 3′ tails of 82 and 78 nt with a Tm of 65°C (23).
RNA helicase assays
RNA helicase assays were performed in 20 µl reaction mixtures containing 20 mM HEPES–KOH, pH 8.5, 3 mM MgCl2, 1 mM DTT, 3 mM ATP, 20 U RNasin (Promega), 200 µg/ml BSA, 50 fmol partially duplexed RNA substrate and 0.2 µg CrhC protein, unless otherwise stated. Reactions were quenched, after incubation at 37°C for 10 min, by addition of 5 µl of helicase stop solution (150 mM EDTA, 3% SDS, 0.25% Bromophenol blue and 15% Ficoll). Reaction products (10 µl) were separated by electrophoresis at 180 V for 40 min on 10% SDS–polyacrylamide gels. dsRNA and ssRNA species were visualized and quantified by phosphorimager analysis as described above. The percentage of the dsRNA substrate unwound to ssRNA is indicated below each lane.
RNA binding assays
RNA binding assays were performed using RNA II, essentially as described for the RNA helicase assays with minor modifications. SDS was removed from all buffers and the reaction mixtures were resolved on native 8% polyacrylamide gels. RNA–protein interaction was indicated by an RNA mobility shift, as detected and quantified by phosphorimager analysis as described above. The percentage of RNA substrate which is present in the complex is indicated below each lane.
Immunoprecipitation
Log phase (∼0.4 OD750) Anabaena cultures were grown and harvested at 30°C, resuspended in liquid BG-11 lacking sulfate, divided in half and incubation continued at either 20 or 30°C in the presence of [35S]methionine/cysteine (1000 Ci/mmol; Amersham Pro-Mix) at 1 µCi/ml for 1.5 h. Cells were harvested at the appropriate temperature, washed with sulfate-free BG-11 and lysed by sonication. Clarified lysate was diluted 5-fold with dilution buffer (10 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1% Triton X-100, 1 mg/ml BSA), mixed with anti-CrhC polyclonal antiserum (5 µl) and incubated on ice for 1.5 h. Protein A–Sepharose (12.5 mg/ml; Sigma) was added and incubation continued at 4°C with gentle shaking for 1.5 h. Sepharose beads were pelleted by centrifugation, washed twice each with dilution buffer (1× TBS and 10 mM Tris–HCl, pH 6.8), resuspended in Laemmli sample buffer, boiled and immunoprecipitated components separated on a 10% SDS–PAGE gel. Dried gels were analyzed by autoradiography using a Kodak BioMax TranScreen LE overnight at –80°C.
Far-western analysis
Far-western analysis was performed as described (24) with the exception that colorimetric detection of antibody–CrhC–protein complexes was performed. Briefly, soluble proteins (30 µg) were separated on 10% SDS–PAGE gels and electroblotted to nitrocellulose filters (Amersham Hybond-C). Immobilized proteins were denatured by incubation in 8 M urea in HBB buffer (25 mM HEPES–KOH, pH 7.5, 25 mM NaCl, 5 mM MgCl2, 1 mM DTT) for 10 min at room temperature. Proteins were renatured by incubation in 2-fold dilutions of urea in HBB for 10 min each. Filters were blocked with 5% skim milk powder (Difco) in HBB and incubated in hybridization buffer (20 mM HEPES–KOH, pH 7.5, 75 mM KCl, 2.5 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 0.1% Nonidet P-40, 1% skim milk powder) containing recombinant CrhC (10 µg/ml) at room temperature for 16 h. Filters were washed five times in 1× TBST for 10 min each and incubated with anti-CrhC polyclonal antibody (1:1000) in 1× BLOTTO (10 mM Tris–HCl, pH 7.5, 500 mM NaCl, 5% skim milk powder, 0.02% sodium azide) at room temperature for 16 h. CrhC–protein interactions were detected using goat anti-rabbit horseradish peroxidase conjugate (1:1000; Cappel) and 4-chloro-1-napthol as described previously (16).
RESULTS
Expression and purification of recombinant CrhC
To facilitate the biochemical characterization of CrhC, the crhC ORF was cloned as a translational fusion into pRSETA (Invitrogen), creating pRSET29. Soluble recombinant CrhC protein was obtained through controlled expression from the T7 promoter in JM109, accomplished by lowering the growth temperature to 20°C combined with IPTG induction for 5 h (Fig. 1). It was estimated that >50% of the CrhC was soluble under these conditions, while expression at higher temperatures (37 and 30°C) resulted in the production of insoluble protein present in inclusion bodies. We have also expressed CrhC using the pGEX (Pharmacia) system, however, the resulting protein was soluble only in 0.1% Sarkosyl or 8 M urea and did not bind to a glutathione–Sepharose column, presumably as a result of altered glutathione S-transferase conformation, and therefore could not be purified by affinity chromatography.
Figure 1.

Expression and purification of His-tagged CrhC. Histidine-tagged CrhC was overproduced in E.coli and purified by affinity chromatography on a nickel resin column. Lane 1, soluble proteins from uninduced E.coli strain JM109 (pRSET29); lane 2, soluble proteins from cells induced with IPTG (0.5 mM) in the presence of M13 phage/T7 at 20°C for 5 h; lane 3, flow-through collected after incubation of the lysate with Ni–NTA–agarose beads at 4°C for 1 h; lanes 4–6, proteins eluted with imidazole at 20, 20 and 40 mM; lanes 7–9, CrhC containing fractions eluted with 250 mM imidazole. Proteins were resolved by electrophoresis on a 10% SDS–polyacrylamide gel and stained with Coomassie Brilliant Blue. An arrow indicates the position of the 50 kDa His-tagged CrhC. The migration position of molecular weight makers is indicated on the left.
The 50 kDa His-tagged recombinant protein was affinity purified on a nickel–agarose column (Fig. 1, lane 8). Similar treatment of JM109 lacking pRSET29 indicated that the 28 kDa polypeptide observed in affinity-purified CrhC originated from JM109. Efforts to remove the His-tag portion of the fusion protein by cleavage with enterokinase (Invitrogen) were unsuccessful as the cleavage conditions resulted in CrhC protein aggregation and precipitation. Presence of the His-tag did not interfere with CrhC activity and, therefore, the affinity-purified His-tagged CrhC fusion protein was used for all biochemical analyses. Recombinant CrhC stored in 50% glycerol at –20°C remained active for at least 1 month.
RNA-dependent ATPase activity of CrhC
The ability of CrhC to hydrolyze ATP was assayed by incubating purified recombinant CrhC with [γ-32P]ATP and measuring the release of [32P]phosphate by thin layer chromatography. His-tagged CrhC exhibited a low level of ATPase activity in the absence of RNA, a level which was stimulated 7- to 10-fold by addition of exogenous RNA (Fig. 2A). RNA-dependent ATPase activity is also divalent cation dependent (Fig. 2B). Mg2+ could be replaced by Mn2+ but not by other divalent cations, including Ca2+, Cu2+ and Zn2+ (Table 1), whose presence inhibited the reaction. Examination of RNA substrate specificity indicated that CrhC does not exhibit RNA substrate specificity, as activity is observed with total RNA isolated from a number of prokaryotic and eukaryotic sources, tRNA and poly U (Table 1). The highest ATPase activity was observed with a mixture of E.coli 16S and 23S rRNA and crude polysome preparations isolated from cold-shocked Anabaena. Neither Anabaena 16S nor 23S rRNA enhanced ATPase activity significantly. In addition, ATPase activity was stimulated by an artificial DNA, poly(dI·dC). ATPase activity did not require NaCl nor was activity inhibited significantly by salt concentrations up to 200 mM.
Figure 2.

RNA-dependent ATPase activity of CrhC. (A) The effect of RNA concentration on the ATPase activity of CrhC. ATPase reactions were performed with 200 ng recombinant CrhC. The indicated amounts of total RNA isolated from cold-shocked Anabaena were added to the reactions. (B) CrhC ATPase activity is Mg2+ dependent. ATPase reactions were performed with 200 ng recombinant CrhC in the presence of 10 µg total RNA isolated from cold-shocked Anabaena. Mg2+ was added to the indicated final concentration.
Table 1. Substrate and cofactor stimulation of CrhC ATPase activity.
| Substrate addeda | Relative ATP hydrolysis (% of Anabaena total RNA) |
|---|---|
| None | 0 |
| Anabaena total RNA | 100 |
| Synechocystis total RNA | 155 |
| DH5α total RNA | 139 |
| Yeast total RNA | 33 |
| Yeast tRNA | 61 |
| E.coli 16S + 23S rRNA | 180 |
| Anabaena 16S rRNA | 127 |
| Anabaena 23S rRNA | 132 |
| Anabaena crude polysomes | 228 |
| Poly (U) | 150 |
| Poly (dI/dC) | 48 |
| Mg2+ | 100 |
| Ca2+ | 0 |
| Co2+ | 0 |
| Cu2+ | 0 |
| Mn2+ | 45 |
| Zn2+ | 0 |
| NaCl (0 mM) | 97 |
| NaCl (100 mM) | 93 |
| NaCl (200 mM) | 70 |
aAdditions replacing corresponding component in standard assay mixture.
Yeast refers to Saccharomyces cerevisiae.
RNA unwinding activity of CrhC
Recombinant purified CrhC unwinds artificial partially duplexed RNA substrates containing both 5′ and 3′ single-stranded regions (RNA I; data not shown) and only 5′ single-stranded regions (RNA II; Fig. 3). The reaction occurs in a pH-dependent manner with maximum activity occurring at pH 9.0, the highest pH tested. For this reason, all unwinding assays were performed at pH 8.5, since significantly lower activity was observed at the standard RNA helicase assay pH of 7.5. The reaction was also dependent on the presence of BSA in the reaction buffer, however, BSA itself did not unwind the dsRNA substrate as it was present in all assays, including CrhC minus controls (see for example Fig. 3B, lane 0 ng CrhC). RNA helicase activity was CrhC specific and not a result of contaminating protein, as unwinding was progressively inhibited by immunoprecipitation of CrhC with increasing concentrations of the anti-CrhC antibody (data not shown). A titration of CrhC protein indicated that RNA unwinding was complete (i.e. 100% unwinding) and saturated at 2.5 ng/µl CrhC at 37°C in 10 min (Fig. 3B). CrhC RNA unwinding activity was linear with respect to time for ∼30 min, using CrhC at 1 ng/µl (Fig. 3C). The reaction was also Mg2+ and ATP dependent with half-maximal unwinding activity at 36 and 71 µM, respectively (Fig. 3D and E). To determine the specificity of the energy source in RNA unwinding reactions, all four ribonucleotides (ATP, CTP, GTP and UTP), dATP and the non-hydrolyzable ATP analog ATP-γ-S (Sigma) were tested (Fig. 3F). CrhC exhibited a strong preference for adenosine nucleotides, with ATP and dATP being utilized with equal efficiency. CrhC displayed little, if any, activity with the other ribonucleoside triphosphates and limited but reproducible activity with ATP-γ-S (Fig. 3F).
Figure 3.

5′→3′ RNA unwinding activity of CrhC. RNA unwinding assays were performed under standard assay conditions (pH 8.5, 200 ng His-tagged CrhC, 50 fmol RNA II dsRNA substrate, 3 mM ATP and 3 mM Mg2+ for 10 min at 37°C) and modified as indicated. (A) Structure of the artificial duplex RNA substrate RNA II. (B) Influence of CrhC concentration on RNA unwinding activity. RNA helicase reactions were performed in the presence of the indicated amounts of CrhC. (C) A time course of RNA unwinding. RNA helicase reactions were performed with 20 ng His-tagged CrhC for the indicated times. (D) CrhC RNA unwinding activity is ATP dependent. ATP concentration in the reaction was varied as indicated. (E) CrhC unwinding activity is Mg2+ dependent. The Mg2+ concentration in the reaction was varied as indicated. (F) RNA unwinding activity of CrhC is ATP dependent. RNA helicase reactions were performed in the presence of the indicated nucleotide triphosphate provided at a final concentration of 3 mM. The percentage of the input duplex substrate RNA II unwound during the reaction is indicated below each lane.
As the ability of RNA helicases to unwind artificial partial RNA duplexes in both directions varies, we tested the ability of CrhC to unwind an artificial dsRNA containing only 3′ single-stranded regions. CrhC did not catalyze unwinding of this dsRNA substrate under the standard assay conditions described in Materials and Methods, even at CrhC concentrations 30-fold higher than those required for ∼50% unwinding of RNA II, which contains only 5′ single-stranded tails (Fig. 4). As a control, a second cyanobacterial RNA helicase, CrhR, unwound this substrate under identical conditions (S.L.Kujat and G.W.Owttrim, unpublished results).
Figure 4.

3′→5′ RNA unwinding is not catalyzed by CrhC. (A) Structure of the artificial 3′ tailed duplex RNA substrate. (B) RNA unwinding assays were performed as described for the standard assay in Figure 3 except that the duplex RNA substrate contains only 3′ ssRNA tails. His-tagged CrhC concentration was modified as indicated.
Characterization of RNA binding activity of CrhC
The RNA binding activity of recombinant CrhC was investigated by electrophoretic mobility shift assays (Fig. 5). On the basis of the non-specificity of the RNA substrate for ATPase activity, the target RNA for these studies was RNA II. The increasing appearance of a band with lower mobility than that of the RNA II substrate with increasing addition of CrhC indicates the formation of CrhC–RNA complexes (Fig. 5). Identical patterns of complex formation were observed in the presence or absence of ATP, indicating that RNA binding by CrhC is ATP independent (Fig. 5). CrhC at concentrations as low as 1 ng/µl altered migration of the RNA substrate. RNA binding by CrhC did not increase in a linear manner, with the percentage of the RNA substrate shifted increasing from 30 to >90% with an increase in CrhC concentration from 1 to 2.5 ng/µl.
Figure 5.

ATP-independent RNA binding activity of CrhC. CrhC binding to RNA II (50 fmol) was assayed in the presence of the indicated amounts of recombinant CrhC, in the presence and absence of ATP (3 mM) at 37°C for 30 min. Reaction products were separated by electrophoresis on native 8% polyacrylamide gels. The migration positions of duplex RNA substrate and RNA–CrhC complexes are indicated.
CrhC is a component of a multisubunit complex
To further investigate the biochemical characteristics of CrhC, co-immunoprecipitation experiments were performed to determine if CrhC is present in a protein complex in vivo. Sulfate-free medium supplemented with [35S]methionine/cysteine was utilized to label Anabaena cells subjected to cold stress (20°C) or normal growth temperature (30°C). CrhC will accumulate or be undetectable, respectively, under these conditions (16). A specific pattern of polypeptides was consistently immunoprecipitated with anti-CrhC antibodies from extracts prepared from cells grown at 20°C (Fig. 6, lane 1). These included polypeptides with apparent molecular weights of 80, 76, 72, 68, 62, 47, 38, 28, 24 and 22 kDa. Polypeptides were not observed to be immunoprecipitated from total lysates of cells labeled at 30°C (Fig. 6, lane 2). The 47 kDa polypeptide present in the immunoprecipitated products obtained from cells grown at 20°C was identified as CrhC by western analysis (Fig. 6, lane 3). As expected, CrhC was not observed in immunoprecipitates from Anabaena grown at 30°C (Fig. 6, lane 4).
Figure 6.

CrhC is a component of a multisubunit complex at 20°C. Anti-CrhC antiserum was used to immunoprecipitate 35S-labeled polypeptides from lysates prepared from Anabaena cells grown at 20 or 30°C (lanes 1 and 2). Western analysis of immunoprecipitated extracts probed with anti-CrhC antibodies was utilized to identify CrhC in the precipitates (lanes 3 and 4).
CrhC interacts with a 37 kDa polypeptide
While co-immunoprecipitation indicated that CrhC was a component of a multisubunit complex, it did not indicate which of the proteins interacts directly with CrhC. Far-western analysis using recombinant CrhC to probe soluble proteins isolated from Anabaena grown at 20 and 30°C was therefore performed (Fig. 7). This analysis resulted in the detection of 37 and 120 kDa polypeptides which were present in lysates isolated from both warm and cold grown cells. While we consistently observed a faint reaction with a 120 kDa polypeptide with the anti-CrhC antibody in western analyses, the 37 kDa polypeptide was unique and resulted from a specific interaction with CrhC. The 120 kDa polypeptide may correspond to a second RNA helicase in Anabaena. A faint signal, corresponding to CrhC, was also detected at 47 kDa only in lysates obtained from cells grown at 20°C. This signal was enhanced if the bound proteins were not denatured with urea before probing (data not shown).
Figure 7.
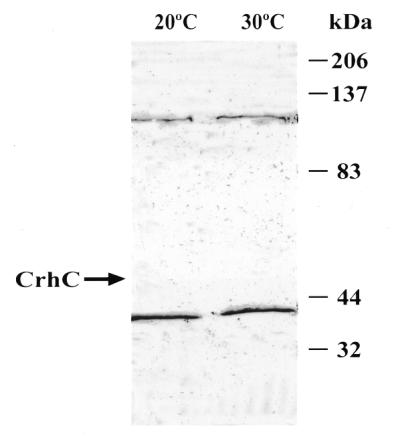
CrhC specifically interacts with a constitutively expressed 37 kDa polypeptide. Far-western analysis was utilized to detect polypeptides with which CrhC interacts directly. Immunoblots of soluble protein isolated from Anabaena grown at 20 or 30°C was probed with affinity-purified CrhC (10 µg/ml). Protein–protein interaction was indicated by anti-CrhC antibody binding and colorimetric detection using goat anti-rabbit horseradish peroxidase conjugate (1:1000; Cappel) and 4-chloro-1-napthol. An arrow indicates the presence of CrhC in the 20°C lane.
DISCUSSION
CrhC was identified as a putative RNA helicase on the basis of its amino acid sequence similarity to other DEAD-box RNA helicases (15,16). It is a unique DEAD-box RNA helicase specifically expressed during cold stress in the cyanobacterium Anabaena sp. strain PCC 7120. We show here that affinity-purified recombinant CrhC is a bona fide RNA helicase, possessing RNA-dependent ATPase activity, 5′→3′ (d)ATP-dependent RNA unwinding activity and ATP-independent RNA binding activity. Furthermore, immunoprecipitation and far-western analysis of cold-shocked Anabaena extracts indicates that CrhC is a component of a multisubunit complex, interacting specifically with a 37 kDa polypeptide.
CrhC exhibits RNA-dependent ATPase activity which is stimulated by a variety of natural and artificial RNA substrates. The highest activity was observed with rRNA and polysome preparations, while yeast total RNA or tRNA and the synthetic DNA polymer poly(dI·dC) were much less effective. RNA binding by CrhC was ATP independent and non-specific, characteristics similar to those observed for the majority of other RNA helicases (11,25) but in contrast to the ATP requirement for RNA binding by eIF-4A (21). Identical RNA gel shifts, irrespective of ATP addition, indicate that CrhC binds to both the partially dsRNA substrate and ssRNA resulting from ATP-stimulated unwinding activity. RNA binding by CrhC most likely occurs through the HRIGRXGR motif as a RGG repeat motif, known to be involved in non-specific RNA binding (26), is not present in the C-terminal region (15). The physiological RNA substrates stimulating both ATPase and RNA binding activities in vivo cannot be inferred from these results, a situation similar to that observed for the majority of DEAD-box proteins. In fact, only the E.coli DEAD-box protein DbpA exhibits significant RNA substrate specificity in these assays (27,28). Nevertheless, the fact that CrhC is stimulated to the greatest degree by rRNA and polysome preparations is consistent with CrhC being ribosome associated.
CrhC unwinds artificial partial RNA duplexes containing only 5′ or both 5′ and 3′ single-stranded regions in a pH-, (d)ATP- and Mg2+-dependent reaction. The adenosine nucleotide specificity for unwinding is similar to other ATPases (7,14,29,30), including an Arabidopsis RNA helicase (31) and a yeast DNA helicase (32). In contrast, the majority of RNA helicases utilize any of the eight deoxyribonucleotides and ribonucleotides to power unwinding (11,13,19,33). Kinetic analysis indicated that the Km for ATP for unwinding is similar to that determined for both RNA and DNA helicases (11,34,35). CrhC does not, however, unwind an artificial partial RNA duplex containing only 3′ overhangs under conditions identical to those utilized for assays containing duplex RNA substrates possessing only 5′ or both 5′ and 3′ single-stranded tails. This is also true at CrhC concentrations significantly higher than those required to unwind duplex RNA substrates containing 5′ single-stranded tails. While the Tm of the 5′ tailed substrate is lower than that of the 3′ substrate, inhibition of unwinding activity by substrate stability has only been observed with significantly more stable secondary structures (36). Although we cannot rule out the possibility that a cofactor is required to unwind in the 3′→5′ direction, it appears that CrhC catalyzes unidirectional RNA helicase activity, unwinding RNA duplexes exclusively in the 5′→3′ direction. The DEAD-box RNA helicase II from HeLa cells has also been reported to unwind only in the 5′→3′ direction (23). All other RNA helicases either unwind duplex RNA in both directions (7,14) or unidirectionally in the 3′→5′ direction (11,22,37).
The FAT box, a unique non-conservative alteration of the highly conserved SAT/TAT motif, does not adversely affect any biochemical activity performed by CrhC. The SAT/TAT motif has been reported to be essential for helicase activity, linking ATP hydrolysis and helicase activities (38). Mutation of the SAT box yields helicase proteins that are inactive in vitro (39) and in vivo (40). Crystallographic analysis of the hepatitis C virus RNA helicase suggests that the Ser residue of the SAT box is involved in hydrogen bond switching which links the ATPase and helicase activities (17). These considerations are consistent with a unique mechanism of CrhC catalyzed RNA unwinding. The unique FAT motif may be related to unidirectional unwinding of the RNA substrates with which CrhC interacts.
Relatively few DEAD-box proteins have been shown to possess RNA unwinding activity in vitro, including four of 39 in yeast (41), mouse eIF-4A (7), human nuclear protein p68 (6) and vasa from Drosophila (42). Of the prokaryotic DEAD-box proteins identified, only the E.coli proteins DbpA and DeaD (CsdA) have been analyzed biochemically. The latter proteins destabilize duplex RNA in the absence of ATP hydrolysis, suggesting that unwinding is not an enzymatic reaction but a result of binding to single-stranded regions created by thermodynamic breathing (9,10). Conversely, CrhC catalyzed unwinding is catalytic, as ATP-γ-S cannot substitute for ATP. Furthermore, the observed ATP hydrolysis is not required for RNA binding, as CrhC binds duplex RNA in the absence of ATP. In addition, the in vitro enzymatic analysis indicates that CrhC does not require an accessory protein(s) for biochemical activity. Although eIF-4A and Dbp5p require accessory proteins to promote unwinding, the majority of RNA helicases do not (1,40). Our demonstration that CrhC actively unwound RNA indicates that it possesses an intrinsic helicase activity. CrhC, therefore, is the first prokaryotic RNA helicase shown to actively unwind RNA.
Biochemically, CrhC functions as a non-specific RNA helicase in vitro, however, these assays may not reflect the situation in vivo. It is possible that CrhC interacts with an accessory protein(s) in vivo, an interaction which may optimize activity or provide RNA substrate specificity to the enzyme, as observed with the eIF-4A interaction with eIF-4B (43). Co-immunoprecipitation indicates that CrhC is a component of a multisubunit complex during cold stress. While the identity of the associating proteins is not known, the fact that they become labeled during cold shock indicates that their corresponding mRNAs continue to be translated during cold stress. These proteins are not necessarily cold shock proteins, as they may be constitutively expressed but are not immunoprecipitated from warm grown extracts due to the absence of CrhC at 30°C (16). Other RNA helicases are known to be present in multisubunit complexes in both prokaryotic (44) and eukaryotic (2) systems. Far-western analysis indicates that CrhC interacts specifically with a 37 kDa polypeptide within this complex, as a polypeptide having a similar molecular weight is immunoprecipitated by anti-CrhC antibodies from extracts prepared from Anabaena grown at 20°C. While the identity of this polypeptide is not known, we speculate that it is ribosome associated. This conjecture is supported by our observation that polysomes stimulate ATPase activity significantly.
What role does CrhC perform during adaptation to cold stress in Anabaena? Specificity of function is provided to a degree by the regulated expression of CrhC as a result of cold stress. In addition, the 5′→3′ RNA unwinding activity and polysome association indicate that CrhC could actively unwind secondary structures at the 5′-end of target RNAs which will be thermodynamically stabilized at low temperature. The resulting enhancement of the target mRNA translational efficiency or the functional activity of rRNA could contribute to the removal of the metabolic block in translation initiation induced by cold shock (45). In this scenario the selective translation of specific mRNAs observed during cold stress could also be regulated by CrhC recognition and unwinding of mRNAs with specific secondary structures in their 5′-UTRs. Cold-induced genes are characterized by possessing extensive, highly structured 5′-UTRs that are required for cold shock-specific expression (45). The crhC sequence is consistent with this model as it possesses an extended, highly structured 5′-UTR. This region is crucial for the cold stress regulation of CrhC expression, as we show here that replacing the crhC promoter and 5′-UTR sequences with heterologous sequences required for expression in E.coli abolishes temperature-regulated expression. This effect is not a result of the heterologous system as crhC transcript accumulation is regulated by temperature similarly in E.coli and Anabaena (D.Chamot and G.W.Owttrim, unpublished results). Thus, CrhC may also regulate its own translation.
In summary, a DEAD-box protein, CrhC, from the cyanobacterium Anabaena sp. strain PCC 7120 was expressed, purified and characterized biochemically as an RNA helicase. CrhC is an ATP-independent RNA binding protein which possesses RNA-dependent ATPase and (d)ATP-dependent RNA helicase activities. RNA unwinding in the 5′→3′ direction provides the potential for CrhC interaction and unwinding of the 5′-UTR of target mRNAs to overcome the inhibition of translation initiation induced by cold stress. In vivo CrhC is a component of a multisubunit complex whose members are translated during cold shock, interacting specifically with a 37 kDa polypeptide. To date, the majority of cold stress response studies in cyanobacteria have focused on alterations in membrane composition. Biochemical characterization of CrhC as an RNA helicase involved in cold shock acclimation provides a unique opportunity to further our understanding of the mechanism by which cyanobacteria respond to this environmental stress.
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank Drs Nahum Sonenberg, Chee-Gun Lee and Hans Stahl for providing plasmids and Dr Warren Gallin for providing the DE3 M13/T7 phage. Thanks are also due to Dr Danuta Chamot for critical reading of the manuscript. This work was supported by a grant from the Natural Sciences and Engineering Research Council (NSERC) of Canada.
REFERENCES
- 1.Fuller-Pace F.V. (1994) Trends Cell Biol., 4, 271–274. [DOI] [PubMed] [Google Scholar]
- 2.Luking A., Stahl,U. and Schmidt,U. (1998) Crit. Rev. Biochem. Mol. Biol., 33, 259–296. [DOI] [PubMed] [Google Scholar]
- 3.Linder P., Lasko,P.F., Ashburner,M., Leroy,P., Nielsen,P.J., Nishi,K., Schnier,J. and Slonimsky,P.P. (1989) Nature, 337, 121–122. [DOI] [PubMed] [Google Scholar]
- 4.Gorbalenya A.E. and Koonin,E.V. (1993). Curr. Opin. Struct. Biol., 3, 419–429. [Google Scholar]
- 5.Pause A. and Sonenberg,N. (1993) Curr. Opin. Struct. Biol., 3, 953–959. [Google Scholar]
- 6.Hirling H., Schemffner,M., Restle,T. and Stahl,H. (1989) Nature, 339, 562–564. [DOI] [PubMed] [Google Scholar]
- 7.Rozen F., Edery,I., Meerovitch,K., Dever,T.E., Merrick,W.C. and Sonenberg,N. (1990) Mol. Cell. Biol., 10, 1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kadare G. and Haenni,A.L. (1997) J. Virol., 71, 2583–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones P.G., Mitta,M., Kim,Y., Jiang,W. and Inouye,M. (1996) Proc. Natl Acad. Sci. USA, 93, 76–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Böddeker N., Stade,K. and Franceschi,F. (1997) Nucleic Acids Res., 25, 537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee C.-G. and Hurwitz,J. (1992) J. Biol. Chem., 267, 4398–4407. [PubMed] [Google Scholar]
- 12.Shuman S. (1993) J. Biol. Chem., 268, 11798–11802. [PubMed] [Google Scholar]
- 13.Lee C.-G., Chang,K.A., Kuroda,M.I. and Hurwitz,J. (1997) EMBO J., 16, 2671–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tseng S., Weaver,P.L., Liu,Y., Hitomi,M., Tartakoff,A.M. and Chang,T.-H. (1998) EMBO J., 17, 2651–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chamot D., Magee,W.C., Yu,E. and Owttrim,G.W. (1999) J. Bacteriol., 181, 1728–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chamot D. and Owttrim,G.W. (2000) J. Bacteriol., 182, 1251–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho H.S., Ha,N.C., Kang,L.W., Chung,K.M., Back,S.H., Jang,S.K. and Oh,B.H. (1998) J. Biol. Chem., 273, 15045–15052. [DOI] [PubMed] [Google Scholar]
- 18.Zoller M.J. and Smith,M. (1987) Methods Enzymol., 154, 329–350. [DOI] [PubMed] [Google Scholar]
- 19.Rodriguez P.L. and Carrasco,L. (1993) J. Biol. Chem., 268, 8105–8110. [PubMed] [Google Scholar]
- 20.Angeloni S.V. and Potts,M. (1986) J. Bacteriol., 168, 1036–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pause A., Méthot,N. and Sonenberg,N. (1993) Mol. Cell. Biol., 13, 6789–6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scheffner M., Knippers,R. and Stahl,H. (1989) Cell, 57, 955–963. [DOI] [PubMed] [Google Scholar]
- 23.Flores-Rozas H. and Hurwitz,J. (1993) J. Biol. Chem., 268, 21372–21383. [PubMed] [Google Scholar]
- 24.Methot N., Song,M.S. and Sonenberg,N. (1996) Mol. Cell. Biol., 16, 5328–5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishi K., Morel-Deville,F., Hershey,J.W.B., Leighton,T. and Schnier,J. (1988) Nature, 336, 496–498. [DOI] [PubMed] [Google Scholar]
- 26.Lorkovic A.J., Herrmann,R.G. and Oelmuller,R. (1997) Mol. Cell. Biol., 17, 2257–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuller-Pace F.V., Nicol,S.M., Reid,A.D. and Lane,D.P. (1993) EMBO J., 12, 3619–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsu C.A. and Uhlenbeck,O.C. (1998) Biochemistry, 37, 16989–16996. [DOI] [PubMed] [Google Scholar]
- 29.Bayliss C.D. and Condit,R.C. (1995) J. Biol. Chem., 270, 1550–1556. [DOI] [PubMed] [Google Scholar]
- 30.Bayliss C.D. and Smith,G.L. (1996) J. Virol., 70, 794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okanami M., Meshi,T. and Iwabuchi,M. (1998) Nucleic Acids Res., 26, 2638–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bennett R.J., Sharp,J.A. and Wang,J.C. (1998) J. Biol. Chem., 273, 9644–9650. [DOI] [PubMed] [Google Scholar]
- 33.Shuman S. (1992) Proc. Natl Acad. Sci. USA, 89, 10935–10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roman L.J. and Kowalczykowski,S.C. (1989) Biochemistry, 28, 2873–2881. [DOI] [PubMed] [Google Scholar]
- 35.Thommes P. and Hubscher,U. (1990) J. Biol. Chem., 265, 14347–14354. [PubMed] [Google Scholar]
- 36.Tuteja N., Huang,N.W., Skopac,D., Tuteja,R., Hrvatic,S., Zhang,J., Pongor,S., Joseph,G., Faucher,C., Amalric,F. and Falaschi,A. (1995) Gene, 160, 143–148. [DOI] [PubMed] [Google Scholar]
- 37.Wagner J.D.O., Jankowsky,E., Company,M., Pyle,A.M. and Abelson,J.N. (1998) EMBO J., 17, 2926–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pause A. and Sonenberg,N. (1992) EMBO J., 11, 2643–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plumpton M., McGarvey,M. and Beggs,J.D. (1994) EMBO J., 13, 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmid S.R. and Linder,P. (1991) Mol. Cell. Biol., 11, 3463–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iost I., Dreyfus,M. and Linder,P. (1999) J. Biol. Chem., 274, 17677–17683. [DOI] [PubMed] [Google Scholar]
- 42.Liang L., Diehl-Jones,W. and Lasko,P. (1994) Development, 120, 1201–1211. [DOI] [PubMed] [Google Scholar]
- 43.Jaramillo M., Dever,T.E., Merrick,W.C. and Sonenberg,N. (1991) Mol. Cell. Biol., 11, 5995–5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Py B., Higgins,C.F., Krisch,H.M. and Carpousis,A.J. (1996) Nature, 381, 169–172. [DOI] [PubMed] [Google Scholar]
- 45.Jones P.G. and Inouye,M. (1994) Mol. Microbiol., 11, 811–818. [DOI] [PubMed] [Google Scholar]