


Abstract
To assess whether removal of UV-induced cyclobutane pyrimidine dimers (CPDs) occurs with equal efficiency at different stages of the cell cycle in a cell cycle-regulated gene, we have analyzed repair of CPDs, following a single dose of UV, in normal human fibroblasts that were synchronized in either G0 or S phase. Based on a single nucleotide resolution analysis, we established a detailed map of DNA repair rates along the promoter region and the transcription initiation area of the human CDC2 gene. The promoter of this gene is covered by an array of sequence-specific transcription factors located between nt –280 and –9 relative to the major transcription start site. In both quiescent and S phase-synchronized fibroblasts the majority of these sequences were poorly repaired even after 24 h, probably as a result of the constitutive binding of transcription factors throughout the cell cycle. A domain of fast repair was found at sequences surrounding the transcription initiation site and continuing downstream for ∼80 nt. CPD removal from this domain was preferential in both quiescent and proliferating fibroblasts, despite lower levels of global genome repair and a lack of CDC2 transcription in quiescent cells. We suggest that sequences involved in transcription initiation may be book-marked for efficient repair throughout the cell cycle, even when the gene is temporarily not expressed.
INTRODUCTION
The eukaryotic cell cycle is an orchestrated series of events where transitions between successive phases are tightly regulated by feedback mechanisms called checkpoints (1–3). These surveillance mechanisms, governed by the sequential activation and inactivation of cyclin/cyclin-dependent kinase (cdk) complexes, are able to recognize genomic perturbations, such as DNA damage, and, in response, they delay cell cycle progression at a specific stage. This will prevent premature entry of the cells into the next phase of the cycle prior to correct completion of the macromolecular events of the previous phase. Only when DNA lesions are removed from the DNA by the repair machinery can the cell cycle progression be resumed. Cell cycle arrest and repair of DNA damage therefore play a major role in minimizing the propagation of errors into critical cell cycle phases, ensuring the integrity of the genetic information (4). Unrepaired DNA or inefficient removal of DNA lesions can result in genomic instability, mutations and, ultimately, cancer (5).
Exposure to UV irradiation leads to the formation of different types of DNA photoproducts (6–9). Cyclobutane pyrimidine dimers (CPDs), formed between the 5,6 bonds of two adjacent pyrimidines (mostly at 5′-TpT and 5′-PymC sequences) are thought to be the most harmful UV-induced lesions in mammalian cells (6–10). Because their removal is significantly slower than that of other UV photoproducts, CPDs persist much longer in the mammalian genome and may lead to mutagenesis (9). Induction and repair of CPDs can vary significantly along human sequences and different repair rates are often seen even between neighboring base positions (11–13). For instance, it has been shown that binding of transcription factors can modulate the frequency of lesions in different promoters (14). Within a certain time window, some domains in the genome can undergo extensive repair, while in other domains such repair is slow (15,16). This domain-specific and position-dependent heterogeneity in the rate of DNA repair is probably the result of significant variations in the intensity of repair as well as in the chromatin structure along a gene (17). UV damage is repaired more rapidly in transcriptionally active DNA than in the whole genome, largely due to a faster repair of damage in the transcribed strand than in the non-transcribed strand of genes (18–21). It has been suggested that the presence of an RNA polymerase stalled at the site of the lesion on the transcribed strand serves as a signal to attract repair-specific proteins (20,22,23). In Escherichia coli the Mfd protein, a transcription–repair coupling factor, has been shown to displace the stalled RNA polymerase, to bind the UvrA subunit of the excision nuclease and to stimulate repair of the transcribed strand (24). There is no evidence yet that repair occurs by a similar mechanism in eukaryotic cells. However, strand selectivity in both human and yeast cells has been shown to be dependent on active transcription by RNA polymerase II (25–28). Fast repair of sequences near the transcription start site of genes has been linked to increased local concentrations of DNA repair factors that are associated with general transcription factors (e.g. TFIIH) functioning in transcription initiation (15,16,29).
Because of the strict connection between the transcriptional status of a gene and the speed of DNA repair, we asked whether a cell cycle-regulated gene that displays significant variability in its rate of transcription also shows substantial heterogeneity in DNA repair during the cell cycle. Some cell cycle-dependent genes encode products that are essential for cell cycle progression and one may expect that repair efficiency is at least maximized at/or before the phase when these genes reach their maximum expression level. Alternatively, repair may operate with equally high efficiency in all stages of the cell cycle. This may guarantee that these genes are maintained lesion free at all times so that they can be promptly transcribed and be functional when cell cycle progression resumes.
To investigate if efficiency of DNA repair is regulated in a cell cycle-dependent manner, we studied the repair rates of UV-induced CPDs during the G0 and S phases in the human CDC2 gene. The CDC2 gene is regulated during the cell cycle with a maximum induction in S phase and this regulation is exerted at both a transcriptional and post-transcriptional level. The CDC2 gene encodes a protein kinase, p34cdc2 (a member of the cdk family), and appears to be crucial for initiation of DNA replication and entry into mitosis (30–32). p34cdc2 kinase activity is modulated by its cyclin partners, in particular cyclin B, whose association is essential for the G2/M phase transition.
In the present study we measured repair rates of UV-induced CPDs along the promoter region and the transcribed sequences of the human CDC2 gene (33). DNA repair was examined in UV-irradiated human fibroblasts at different stages of the cell cycle, in which transcription of the CDC2 gene occurs at different rates.
MATERIALS AND METHODS
Cell culture and UV irradiation
Normal human foreskin fibroblasts (HF55) were grown in 4% CO2 at 37°C in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS). Cells were brought to quiescence by serum withdrawal (between 2 and 14 days) and then re-stimulated to proliferate by the addition of fresh DMEM supplemented with 15% FCS. The medium was removed and the cells were washed in phosphate-buffered saline (PBS), followed by UV irradiation using a 254 nm germicidal lamp. UV doses were determined with a UVX radiometer (Ultraviolet Products, Upland, CA). For DNA repair experiments the original medium was returned to the cells and the cells were incubated for various periods of time to allow repair before either RNA or DNA analysis. Non-irradiated cells and cells collected immediately after irradiation served as negative controls (no damage) and positive controls (no repair), respectively.
RNA analysis
For the experiments described in Figure 1A, total RNA was isolated at different phases of the cell cycle after a starvation period of either 2 or 14 days and analyzed on northern blots as previously described (33). For the experiments described in Figure 1B, human foreskin fibroblasts were serum starved for 4 days and then re-stimulated to proliferate by serum addition. Synchronized fibroblasts were independently irradiated with increasing UV doses (2, 5, 10 and 20 J/m2, respectively) at either 6 or 12 h after serum stimulation. Total RNA was isolated 20 h after serum stimulation and a 10 µg aliquot of each sample was subjected to northern analysis (33). For repair studies (Fig. 1C), sub-confluent HF55 human fibroblasts were rendered quiescent by incubation in serum-free DMEM for 7 days, UV irradiated at a dose of 10 J/m2 and re-incubated in the original medium to allow repair. Fibroblasts that were synchronized at 20 h following serum stimulation were also UV irradiated and then allowed time for repair. Total cellular RNA from non-irradiated fibroblasts and from fibroblasts at various times after UV irradiation was isolated from quiescent and synchronized cells by standard procedures (RNAgents; Promega). A 10 µg aliquot of each RNA sample was separated on formaldehyde–agarose gels and transferred to a nylon membrane. Hybridization was performed as described (33). Single-stranded probes, specific for the first exon of the CDC2 gene, were generated by linear PCR amplification using appropriate oligonucleotides. Single-stranded probes specific for the human GAPDH gene were used as a control.
Figure 1.

CDC2 mRNA levels in untreated and UV-irradiated fibroblasts at different phases of the cell cycle. Northern blot analysis was performed as described in Materials and Methods. The two bands represent two transcripts of 2.0 and 1.4 kb. (A) Induction of the CDC2 gene during the cell cycle in human fibroblasts. Cells were serum starved for either 2 or 14 days and incubated after re-addition of serum for the indicated periods of time. Fib, asynchronous population of fibroblasts. (B) Expression of CDC2 is dependent on the dose and time of UV irradiation. The first two lanes show total RNA from undamaged fibroblasts at 0 and 20 h after serum addition. (C) Northern blot analysis of total RNA from G0 and S phase fibroblasts, irradiated with a UV dose of 10 J/m2 and allowed to repair for the indicated periods of time. In all experiments the nylon membranes were consecutively hybridized with probes specific for the human CDC2 and GAPDH genes.
DNA isolation and cleavage at CPDs
After incubation to allow DNA repair, cells were lysed and DNA was isolated from purified nuclei as described previously (15). The UV-irradiated DNA was cleaved to completion with T4 endonuclease V for 1 h at 37°C to generate single-strand breaks at CPDs, then digested with E.coli photolyase to produce ligatable ends (15,34). After enzyme treatment, the DNA was purified by phenol/chloroform extraction and ethanol precipitated. DNA was dissolved in TE buffer (10 mM Tris–HCl, pH 7.6, 1 mM EDTA) at a final concentration of 1 µg/µl. Damage and repair of CPDs in the total genome was determined by separating T4 endonuclease V cleavage products on 0.8% alkaline agarose gels following published procedures (35).
Ligation-mediated PCR (LM–PCR)
LM–PCR was performed as previously described (15,34). The following strand-specific oligonucleotide primer sets were used to map CPDs in the promoter and along transcribed sequences of the human CDC2 gene. For the lower (transcribed) strand: C-1 bis, 5′-ACTGGAGGAGAGCGCTTG; C-2 bis, 5′-CACTCAGTTGGCGCCCGCCCTC; C-3 bis, 5′-CTTTTTCTCTAGCCGCCCTTTCCTC; L-1, 5′-TCTTTCTTTCGCGCTCTAGC; L-2, 5′-GGAAGGCCTGCCCAGCGTAGCT; L-3, 5′-GCTGGGCTCTGATTGGCTGCTT. For the upper (non-transcribed) strand: H-1 bis, 5′-CCCGTTCCTCATACTCGC; H-2 bis, 5′-CGACGCCTCGGCCGTCCCCTA; H-3 bis, 5′-ACGACCCTGACCCCAGCCACT. Oligonucleotide primers -1 are the Sequenase primers used in the first primer extension step of LM–PCR, primers -2 are the PCR primers and primers -3 were used to make single-stranded hybridization probes. The Tm values were calculated with a computer program (36).
Nylon membranes were exposed to a PhosphorImager (Molecular Dynamics; Sunnyvale, CA) and radioactivity was determined in all CPD-specific bands of the sequencing gel that could be resolved from neighboring bands and showed a clear and consistent signal above background. Background values (no UV lanes) were subtracted. Potential loading differences were minimized using equal amounts of DNA as starting material for each time point. When minor loading differences occurred, they were corrected by normalization to reference bands in the upstream promoter region that showed no repair over a time course of 24 h. For example, if the intensity of such a reference band at 4 h was 20% greater than that at 2 and 8 h, then the intensities of all bands in the 4 h lane were divided by a factor of 1.2. Repair at all positions was measured at least twice giving similar results. A repair curve was established for each CPD position that gave a sufficient signal above background. The time at which 50% of the initial damage was removed was then determined from each of these curves. These values were incorporated into Figure 5.
Figure 5.


Summary of DNA repair rates along the human CDC2 gene. Data are for either G0 (A) or S phase (B). Sequences of the upstream promoter and the area surrounding the transcription initiation sites are shown. Transcription factor binding sites were identified by in vivo genomic footprinting (33) and are indicated by boxes. Two horizontal arrows show the major transcription start sites. Repair rates, measured as the time at which 50% of the CPD signal was removed, were calculated for each CPD position that showed a consistent signal above background and are represented by vertical columns of different length.
RESULTS
Cell synchrony
Prior to UV irradiation, normal human foreskin fibroblasts were synchronized at either G0 or S phase by standard serum deprivation. We used starvation periods longer than 2 days (up to 1–2 weeks), since they are effective in increasing the level of cell synchrony after serum stimulation without altering normal cell cycle progression. As confirmed by flow cytometry and propidium iodide staining (33,37), >95% of the cells were in G0 after serum starvation. Cells started to move into S phase between 15 and 20 h and ∼80% reached late S and G2 phase after 25–30 h (37). A good degree of cell synchrony is generally required in studies of cell cycle-related processes. In the experiments described below the analysis of CPD repair through the cell cycle can dramatically improve if uniform populations of cells are used.
Cell cycle and DNA damage dependence of CDC2 expression
Expression of the human CDC2 gene is strictly regulated during the cell cycle and its induction is serum dependent in normal human cells (30,38,39). We determined expression of the human CDC2 gene after serum stimulation in synchronized foreskin fibroblasts by northern blot analysis. CDC2 mRNA was almost undetectable in serum-starved fibroblasts after a starvation period of either 2 or 14 days (Fig. 1A). mRNA levels increased sharply just before the cells entered S phase (between 15 and 20 h after serum stimulation) and reached maximum levels after 24–30 h, when the majority of the cells were in S/G2. The two bands in the northern blots are documented in the literature and represent two CDC2 transcripts, 2.0 and 1.4 kb long, respectively (30). It is unknown if these two transcripts are the result of alternative splicing or other mechanisms. Low expression of the gene was observed in an asynchronous population of fibroblasts, where the majority of the cells are expected to be in G1 (Fig. 1A, last lane).
To elucidate the effects of UV irradiation on expression of the human CDC2 gene, we UV-irradiated human diploid fibroblasts that were serum starved and then stimulated to re-enter the cell cycle by serum addition. Four different UV doses (2, 5, 10 and 20 J/m2) and two irradiation times (6 and 12 h after serum stimulation) were selected for our experiments. The 6 and 12 h irradiation time points were chosen based on the assumption that in human fibroblasts the restriction point R might fall in this time window (40,41). A transition from mitogen dependence to mitogen independence occurs in mid to late G1 phase of the cell cycle and is called the restriction point. If UV irradiation leads to a suppression of CDC2 transcription initiation and/or elongation (either directly or indirectly), then irradiation before S phase (i.e. before and/or after the R point) should lead to diminished levels of CDC2 mRNA in S phase, when the gene is expressed.
Following UV irradiation synchronized fibroblasts were harvested at 20 h post-serum addition and total RNA from each sample was analyzed by northern blotting. Compared to an unirradiated control (Fig. 1B, lane 2), increasing doses of UV light increasingly impair expression of the human CDC2 gene in S phase if cells have been irradiated before the R point (6 h). This suggests that UV irradiation before the R point suppresses initiation of CDC2 transcription, presumably by creating a cell cycle block. However, when fibroblasts were irradiated in late G1 (12 h), the effect on CDC2 induction is much less drastic. Cells accumulate high levels of CDC2 mRNA even after a UV dose of 10 J/m2. Higher UV doses of 20 J/m2 abolish CDC2 transcription and this effect is independent of the time of irradiation (Fig. 1B, last two lanes). We conclude that UV irradiation at 10 J/m2 after the R point does not prevent transcription initiation of CDC2. Nuclear run-on assays of cells irradiated with this dose in S phase support this supposition (data not shown).
To study repair of CDC2 throughout the cell cycle, we then irradiated serum-starved and S phase-synchronized fibroblasts (at 20 h after release from G0) with a single UV dose of 10 J/m2. As described above (Fig. 1), a UV dose of 10 J/m2 given at a time when the cells have passed the R point does not appear to interfere significantly with transcription of CDC2 and is therefore suitable for our studies.
After incubation for various periods of time to allow damage removal, total RNA from each sample was isolated and analyzed on northern blots as shown in Figure 1C. Figure 1C shows the transcriptional status of the human CDC2 gene before and after repair of UV-induced DNA damage in G0 and S phase-synchronized fibroblasts. This is important to interpret the DNA repair data. In S phase irradiated cells that underwent repair in a time window of 24 h, the levels of CDC2 mRNA are comparable to those in unirradiated fibroblasts. Signal intensity remained constant at subsequent repair times. Only at 8 and 24 h incubation after UV irradiation, when the cells had presumably removed some of the damage, was an increase in CDC2 mRNA seen (Fig. 1C). At 24 h normal fibroblasts should be cycling into S phase of the next cell cycle. However, UV irradiation inhibits DNA replication and causes a delay in S phase progression (42–46). mRNA levels were very low in serum-starved fibroblasts at the time of irradiation (0 h) and at the following repair times (Fig. 1C, lanes 3–7), as expected for a gene that is transcriptionally silent in G0 and is not induced by UV irradiation.
Repair of CPDs within the CDC2 upstream promoter region and along the transcription start sites in the G0 and S phases
Before measuring repair in a specific domain of the genome at different stages of the cell cycle, it is important to determine the average repair efficiency in the genome as a whole. Figure 2 shows the overall repair of genomic DNA from G0 and S phase fibroblasts that were UV irradiated and allowed to repair for different periods of time. Repair of CPDs in the total genome of S phase-synchronized cells was >80% complete after 24 h, as analyzed by size separating the T4 endonuclease V-cleaved DNA fragments on an alkaline agarose gel. Quiescent fibroblasts showed a much slower overall repair compared to proliferating fibroblasts (Fig. 2).
Figure 2.

Global DNA repair in serum-starved and S phase-synchronized fibroblasts. Cells were irradiated with a UV dose of 10 J/m2 and allowed time for DNA repair. After various time periods DNA was isolated, digested with T4 endonuclease V and separated by size on a 0.8% alkaline agarose gel. The position of markers is indicated on the right (kb).
We then used LM–PCR to determine the repair rates of UV-induced CPDs at single nucleotide resolution along the promoter and the transcribed sequences of the human CDC2 gene. Human foreskin fibroblasts in either the quiescent (serum free) or proliferative state (20 h after serum stimulation) were UV irradiated at a dose of 10 J/m2 and then incubated for various periods of time to allow repair. DNA was isolated and cleaved with T4 endonuclease V to create single-strand breaks at CPDs and then digested with E.coli photolyase to generate ligatable ends. The resulting break positions were amplified by LM–PCR and displayed on sequencing gels as previously described (47).
Figure 3 shows repair of UV-induced CPDs along the promoter region and near the transcription start sites of the human CDC2 gene in both quiescent and S phase-synchronized fibroblasts (Fig. 3A and B, respectively). The initial distribution of CPDs at two adjacent pyrimidines can be observed in the 0 h lanes. Repair of CPDs at a particular site is indicated by a progressive decrease in the intensity of the band on the sequencing gels, compared with that of the corresponding band at 0 h. The gels in Figure 3A and B show the lower (transcribed) strand of the human CDC2 gene. We have previously characterized this region by detailed genomic footprinting analysis (33) and shown that the CDC2 promoter is protected by an array of sequence-specific transcription factors, essential for both its basal and cell cycle-dependent regulation. These include two inverted CCAAT boxes, two Sp1 sites, one ets-2 site and an E2F-like element at position –20. As shown in Figure 3, pyrimidine dimers are removed very inefficiently at almost all sequence positions upstream of the major transcription start site, with many sites remaining unrepaired even after 24 h repair time. This slow repair coincides with the binding of transcription factors along the upstream promoter. There are certain bands with weak signals that show no consistent decrease over the time course. For example, a band reappears at later times in quiescent fibroblasts at position –21 (Fig. 3A). This corresponds to the underlined C in the sequence 5′-CCGCGCTAAA. Since no CPDs can form at a single C, the signal is method-associated background noise and should be ignored. Repair is generally slow along the promoter region in both serum-starved and S phase-synchronized fibroblasts (Fig. 3A and B), probably as a result of constitutive binding of transcription factors throughout the cell cycle. In the proximity of the –20 repressor element repair becomes more efficient on the transcribed strand in S phase compared to quiescent cells. This increase in repair coincides with the release of a repressor complex in S phase, as shown by in vivo footprinting data (33).
Figure 3.
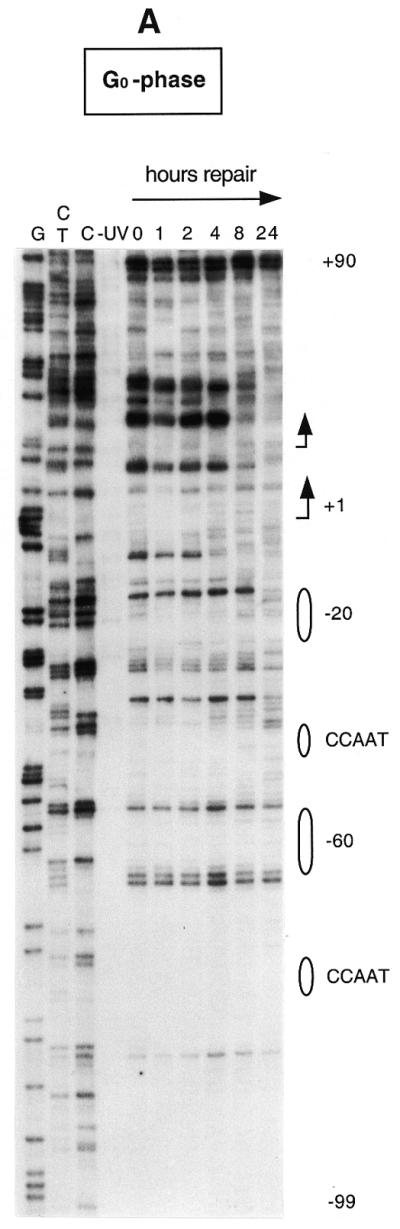
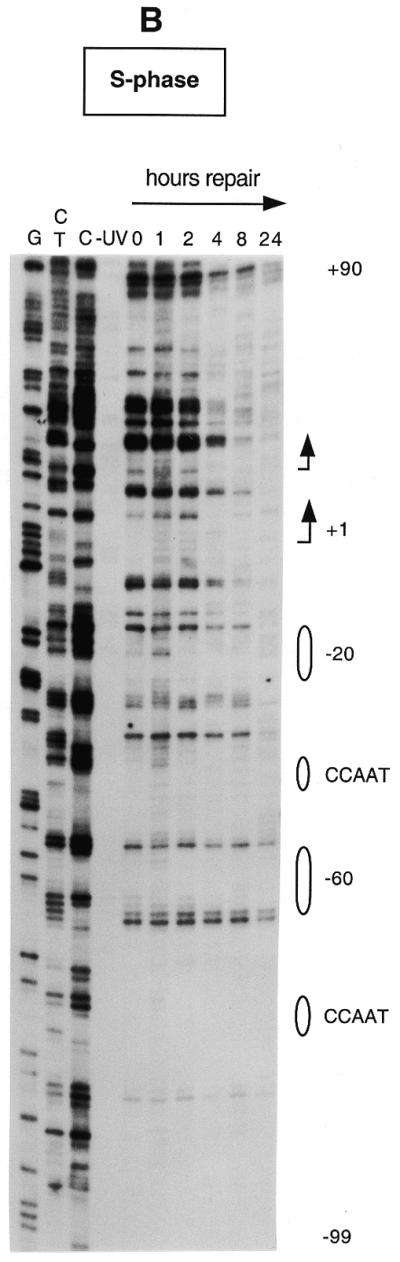
Repair of UV-induced CPDs along the promoter and the transcription initiation sites of the human CDC2 gene. Sub-confluent fibroblasts were either serum-starved (A) or synchronized in S phase by serum addition (B) and then UV irradiated at 10 J/m2. Repair was then allowed for the indicated periods of time. Data are for the lower (transcribed) strand and were obtained with primer set Cbis. Lanes G, C+T and C are Maxam–Gilbert sequencing reactions. Transcription factor binding sites are shown on the right. The arrows indicate the two major transcription start sites (at nt +1 and +19, respectively) and the direction of transcription.
The speed of CPD removal increases dramatically in the proximity of the major transcription start site and remains high for ∼80 bases downstream (Fig. 3A and B). In this domain (+1 to +80) most of the sequences are ∼90% repaired within 4 h in S phase cells, while repair is ∼2-fold slower in serum-starved fibroblasts.
We have also analyzed repair rates along the non-transcribed strand from nt +47 to +1 and along the same strand over the transcription start sites (arrows in Fig. 4A and B) into the upstream promoter region. The repair pattern of the non-transcribed strand is very similar to that of the transcribed strand and repair of the transcribed strand is not much faster in the region analyzed, except for the area immediately upstream of +1 in S phase. Only low signals were obtained from the few dipyrimidines that occur on the non-transcribed strand downstream of +30. All repair data are shown in Figure 5. Repair was quantitated for CPD-specific bands that showed a clear and consistent signal above background. There is a gradient for removal of DNA damage, where repair rate is faster in the proximity of the two major transcription start sites and sharply decreases towards the upstream promoter region. The only exception is represented by an E2F-like element located at nt –138. More than 50% of the lesions are repaired at a time of 5–8 h in S phase and quiescent fibroblasts. Increased repair rates are observed near the transcription start site up to nt +30 in both proliferating and quiescent fibroblasts (Figs 4A and B and 5). Repair of the transcribed strand downstream of the start sites is about twice as fast in S phase compared to G0.
Figure 4.
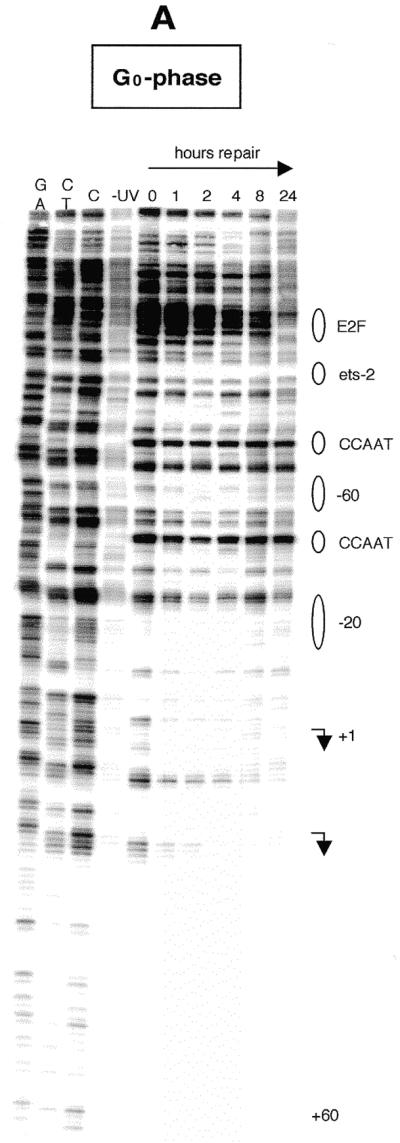
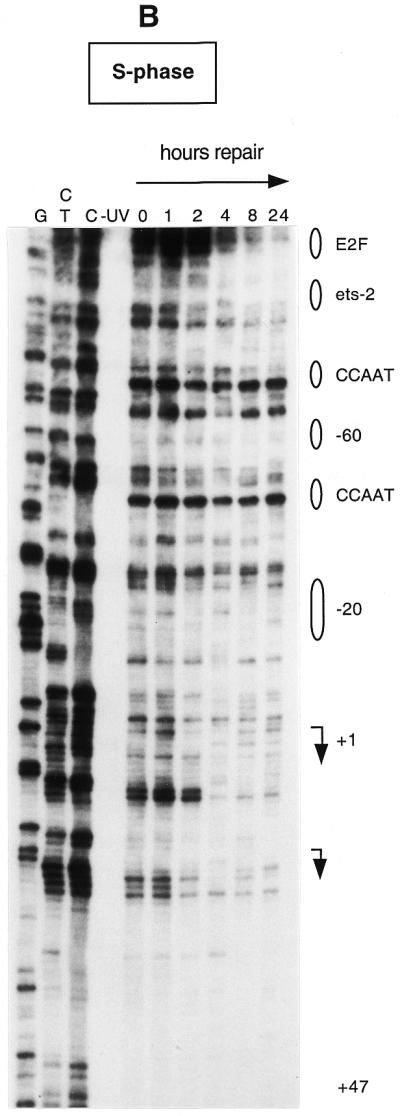
Repair of UV-induced CPDs along the transcription initiation sites and along the upstream promoter of the human CDC2 gene. Data are for the upper (non-transcribed) strand and were analyzed with primer set Hbis. (A) Human foreskin fibroblasts were brought to quiescence by serum withdrawal, UV irradiated at 10 J/m2 and then incubated for the times indicated to allow repair. Lanes G+A, C+T and C (and lane G in panel B) are Maxam–Gilbert sequencing reactions. Transcription factor binding sites are shown on the right. The arrows indicate the transcription start sites and the direction of transcription. (B) Data for the non-transcribed strand in S phase-synchronized fibroblasts.
DISCUSSION
Slow repair of the promoter region of the cell cycle-regulated CDC2 gene
We have previously characterized the promoter region of the human CDC2 gene by genomic footprinting. We found that the 5′-end sequences, spanning nt –280 to –9, are protected by at least 11 transcription factors, suggesting complex regulation of this gene. With the exception of the –20 element, and a less characterized –60 element, all the other factors bind constitutively throughout the cell cycle (33; see Fig. 5 for an overview of the binding sites).
We analyzed repair rates of UV-induced CPDs along both strands of the human CDC2 gene with the aid of the LM–PCR technique. In contrast to other techniques commonly used to study repair, this methodology provides a good level of sensitivity to study DNA lesions at single nucleotide resolution, even when they occur at very low frequency. Since UV irradiation is known to delay S phase (42–46), the disappearance of bands after a few hours cannot be explained by dilution of the signal following multiple rounds of replication. The evidence for a repair-associated signal decrease is also seen in quiescent fibroblasts, where DNA replication is absent (15,16), and in the persistence of several unrepaired sites in the promoter (Figs 3 and 4).
As shown in the repair maps (Fig. 5A and B), CPDs are removed inefficiently from the promoter region of the human CDC2 gene along sites that bind sequence-specific transcriptional regulatory proteins and also along most sequences between closely adjacent factors. This promoter-associated slow repair has previously been described for other human genes, such as the housekeeping gene PGK1 and the UV-induced JUN gene (12,15), and, thus, it may be a more general phenomenon. Most likely, a bulky complex resulting from the binding of sequence-specific transcription factors and other associated proteins makes the CPD lesions almost completely inaccessible to the DNA repair machinery. In addition, binding of sequence-specific transcription factors may strongly increase UV damage formation at some binding sites (14,47,48). The biological consequences of these phenomena are obvious. Inefficient repair of high frequency lesions, such as CPDs, within the promoter could impair important regulatory functions and, ultimately, may cause mutations if such damage is propagated through replication. We previously reported that the presence of CPDs strongly inhibits the binding of transcription factors to their binding sites in vitro (49). However, the slow repair across transcription factor binding sites suggests that the transcription factors do not become detached from the CDC2 promoter in vivo after lesion formation within a DNA-bound complex. Alternatively, detachment of one of the 11 factors still does not significantly increase repair protein accessibility to the damaged site.
A few appreciable differences in repair can be observed along the upstream sequences between the transcribed and non-transcribed strands and at different stages of the cell cycle. At position –52 on the upper strand repair becomes more efficient in S phase fibroblasts compared to G0 phase cells. We noticed that a dipyrimidine 5′-CT is near the –60 site, an element suspected of playing a role in cell cycle regulation of the gene. Indeed, in vivo genomic footprinting has shown that this site becomes hyperreactive to DMS modification at the beginning of S phase, coincident with the induction of CDC2 expression (33). This change in footprint patterns may be indicative of a switch in protein complex binding at the G1/S boundary and may therefore explain the faster repair observed after such a switch in S phase cells.
At the E2F element, located at position –138 relative to the major transcription start site, UV-induced CPDs on the non-transcribed strand are repaired faster than the rest of the promoter, although no distinction in repair rate was seen between G0 and S. This site, albeit a perfect E2F consensus binding site, remains unprotected in vivo at all stages of the cell cycle (33) and, therefore, it is hard to imagine its involvement in CDC2 cell cycle regulation as previously proposed (30). Faster removal of lesions at this sequence confirms that this E2F element is not associated with a stable transcription factor complex in both quiescent and S phase fibroblasts.
Fast repair of sequences surrounding the transcription start site(s)
The –20 element specifically interacts with a subset of E2F4/p130 complexes in G0 and early G1 phase and appears to be largely protein free in S phase (33). This site is more quickly repaired on the transcribed strand in S phase fibroblasts (2–3 h after UV irradiation), compared to quiescent fibroblasts. A general enhancement of the repair rate is also evident in the area surrounding the transcription start sites. This faster repair near the transcription start sites could reflect a concentration of DNA repair factors at this domain, which is different from transcription-coupled repair that acts at the level of mRNA elongation (15,16,50).
The repair patterns of the transcribed strand exhibited by cells in G0, when the gene is transcriptionally silent, is similar to that of cells in S phase that are actively transcribing CDC2, but repair is approximately twice as fast in S phase. Thus, in contrast to the upstream promoter area, the transcription initiation domain undergoes efficient DNA damage removal, regardless of the transcription level of the gene. It is generally accepted that faster repair correlates with transcription of a gene (18–20,51). Nevertheless, non-transcribed DNA may contain genes (such as cell cycle-dependent genes) that are activated under particular circumstances (poised) and therefore need to be repaired efficiently.
Despite several reports on the relationship between DNA repair and cell cycle phase in mammalian cells (44,52–56), the cell cycle-dependent nature of repair is still not well understood. In BB88 mouse leukemia cells non-transcribed genes were repaired faster in late S and G2 phase than in early S phase and the efficiency of repair became identical to that observed in transcribed genes (44). However, studies on CPD repair in the DHFR gene during the mitotic cycle of mammalian cells indicated no differences between the G1, S and G2 phases of the cell cycle (55,56). Repair endonuclease incision of UV lesions did not differ much in the G1, S and G2 phases of synchronized fibroblasts (53). Furthermore, the expression of NER proteins has not been reported to be cell cycle dependent. In this study we noticed that the overall repair of CPDs is low in cells that are driven to quiescence by serum withdrawal compared to S phase-synchronized fibroblasts (see Fig. 2). This is consistent with observations made by others (53; G.P.Holmquist, personal communication). However, some genes that are temporally not expressed but are essential for subsequent phases of the cell cycle, such as CDC2, may undergo quick repair at defined domains required for their activation, despite the low levels of global genome repair in quiescent cells. Book-marking for faster repair at the transcription start site(s) for these genes may be important for cell survival.
One might speculate on the mechanism(s) that allow(s) the persistence of fast repair in down-regulated genes before they are activated at the onset of S phase. Several mechanisms can be proposed. First, it is possible that transcription of CDC2 occurs at low levels in G0 cells and that this is sufficient for fast repair near the transcription start site. A faint signal is seen on northern blots of quiescent cells (Fig. 1). However, we believe that this signal comes from cells that have escaped synchronization by serum withdrawal. These represent <5% of the cell population (37) and should not contribute significantly to the overall repair signal. The most plausible mechanism is that accumulation of the nucleotide excision repair machinery at the transcription initiation domain is not a function of the cell cycle and acts independently of the transcriptional status of the gene. This model may suggest that repair proteins and possibly transcription initiation factors such as TFIIH are continuously engaged at the initiation site, even when the gene is in a temporally repressed state.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Steven Bates for assistance with cell culture, Aziz Sancar for kindly providing E.coli photolyase and Steven Lloyd for the gift of T4 endonuclease V. This work was supported by a grant from the National Institute of Environmental Health Sciences (ES06070) to G.P.P.
REFERENCES
- 1.Hartwell L.H. and Weinert,T.A. (1989) Science, 246, 629–634. [DOI] [PubMed] [Google Scholar]
- 2.Murray A.W. (1992) Nature, 359, 599–604. [DOI] [PubMed] [Google Scholar]
- 3.Nurse P., Masui,Y. and Hartwell,L. (1998) Nature Med., 4, 1103–1106. [DOI] [PubMed] [Google Scholar]
- 4.Nigg E.A. (1995) Bioessays, 17, 471–480. [DOI] [PubMed] [Google Scholar]
- 5.Hartwell L.H. and Kastan,M.B. (1994) Science, 266, 1821–1828. [DOI] [PubMed] [Google Scholar]
- 6.Brash D.E. (1988) Photochem. Photobiol., 48, 59–66. [DOI] [PubMed] [Google Scholar]
- 7.Mitchell D.L. and Nairn,R.S. (1989) Photochem. Photobiol., 49, 805–819. [DOI] [PubMed] [Google Scholar]
- 8.Sage E. (1993) Photochem. Photobiol., 57, 163–174. [DOI] [PubMed] [Google Scholar]
- 9.Pfeifer G.P. (1997) Photochem. Photobiol., 65, 270–283. [DOI] [PubMed] [Google Scholar]
- 10.Tommasi S., Denissenko,M.F. and Pfeifer,G.P. (1997) Cancer Res., 57, 4727–4730. [PubMed] [Google Scholar]
- 11.Hanawalt P.C. (1991) Mutat. Res., 247, 203–211. [DOI] [PubMed] [Google Scholar]
- 12.Gao S., Drouin,R. and Holmquist,G.P. (1994) Science, 263, 1438–1440. [DOI] [PubMed] [Google Scholar]
- 13.Tornaletti S. and Pfeifer,G.P. (1994) Science, 263, 1436–1438. [DOI] [PubMed] [Google Scholar]
- 14.Tornaletti S. and Pfeifer,G.P. (1995) J. Mol. Biol., 249, 714–728. [DOI] [PubMed] [Google Scholar]
- 15.Tu Y., Tornaletti,S. and Pfeifer,G.P. (1996) EMBO J., 14, 675–683. [PMC free article] [PubMed] [Google Scholar]
- 16.Tu Y., Bates,S. and Pfeifer,G.P. (1997) J. Biol. Chem., 272, 20747–20755. [DOI] [PubMed] [Google Scholar]
- 17.Meijer M. and Smerdon,M.J. (1999) Bioessays, 21, 596–603. [DOI] [PubMed] [Google Scholar]
- 18.Bohr V.A., Smith,C.A., Okumoto,D.S. and Hanawalt,P.C. (1985) Cell, 40, 359–369. [DOI] [PubMed] [Google Scholar]
- 19.Bohr V.A., Philipps,D.H. and Hanawalt,P.C. (1987) Cancer Res., 47, 6426–6436. [PubMed] [Google Scholar]
- 20.Mellon I., Spivak,G. and Hanawalt,P.C. (1987) Cell, 51, 241–249. [DOI] [PubMed] [Google Scholar]
- 21.van Hoffen A., Venema,J., Meschini,R., van Zeeland,A.A. and Mullenders,L.H.F. (1995) EMBO J., 14, 360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hanawalt P.C. (1994) Science, 266, 1957–1958. [DOI] [PubMed] [Google Scholar]
- 23.Leadon S.A. (1999) Am. J. Hum. Genet., 64, 1259–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Selby C.P. and Sancar,A. (1993) Science, 260, 53–58. [DOI] [PubMed] [Google Scholar]
- 25.Leadon S.A. and Lawrence,D.A. (1991) Mutat. Res., 255, 67–78. [DOI] [PubMed] [Google Scholar]
- 26.Leadon S.A. and Lawrence,D.A. (1992) J. Biol. Chem., 267, 23175–23182. [PubMed] [Google Scholar]
- 27.Christians F.C. and Hanawalt,P.C. (1992) Mutat. Res., 274, 93–101. [DOI] [PubMed] [Google Scholar]
- 28.Sweder K.S. and Hanawalt,P.C. (1992) Proc. Natl Acad. Sci. USA, 89, 10696–10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schaeffer L., Roy,R., Humbert,S., Moncollin,V., Vermeulen,W., Hoeijmakers,J.H.J., Chambon,P. and Egly,J.M. (1993) Science, 260, 58–63. [DOI] [PubMed] [Google Scholar]
- 30.Dalton S. (1992) EMBO J., 11, 1797–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ku D.H., Wen,S.-C., Engelhard,A., Nicolaides,C., Lipson,K.E., Marino,T. and Calabretta,B. (1993) J. Biol. Chem., 268, 2255–2259. [PubMed] [Google Scholar]
- 32.Nurse P. (1994) Cell, 79, 547–550. [DOI] [PubMed] [Google Scholar]
- 33.Tommasi S. and Pfeifer,G.P. (1995) Mol. Cell. Biol., 15, 6901–6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfeifer G.P. and Tommasi,S. (2000) In Tymms,M.J. (ed.), Methods in Molecular Biology. Humana Press, Totowa, NJ, Vol. 130, pp. 13–27. [DOI] [PubMed]
- 35.Drouin R., Gao,S. and Holmquist,G.P. (1996) In Pfeifer,G.P. (ed.), Technologies for Detection of DNA Damage and Mutations. Plenum Publishing, New York, NY, pp. 37–43.
- 36.Rychlik W. and Rhoads,R.E. (1989) Nucleic Acids Res., 17, 8543–8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tommasi S. and Pfeifer,G.P. (1997) J. Biol. Chem., 272, 30483–30490. [DOI] [PubMed] [Google Scholar]
- 38.Lee M.G. and Nurse,P. (1987) Nature, 327, 31–35. [DOI] [PubMed] [Google Scholar]
- 39.Furukawa Y., Piwnica-Worms,H., Ernst,T.J., Kanakura,Y. and Griffin,J.D. (1990) Science, 250, 805–808. [DOI] [PubMed] [Google Scholar]
- 40.Dou Q.P., Levin,A.H., Zhao,S. and Pardee,A.B. (1993) Cancer Res., 53, 1493–1497. [PubMed] [Google Scholar]
- 41.Di Leonardo A., Linke,S.P., Clarkin,K. and Wahl,G.M. (1994) Genes Dev., 8, 2540–2551. [DOI] [PubMed] [Google Scholar]
- 42.Kaufmann W.K. and Cleaver,J.E. (1981) J. Mol. Biol., 149, 171–187. [DOI] [PubMed] [Google Scholar]
- 43.Painter R.B. (1985) Mutat. Res., 145, 63–69. [DOI] [PubMed] [Google Scholar]
- 44.Russev G. and Boulikas,T. (1992) Eur. J. Biochem., 204, 267–272. [DOI] [PubMed] [Google Scholar]
- 45.Orren D.K., Petersen,L.N. and Bohr,V.A. (1997) Mol. Biol. Cell, 8, 1129–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cistulli C.A. and Kaufmann,W.K. (1998) Cancer Res., 58, 1993–2002. [PubMed] [Google Scholar]
- 47.Pfeifer G.P., Drouin,R., Riggs,A.D. and Holmquist,G.P. (1992) Mol. Cell. Biol., 12, 1798–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aboussekhra A. and Thoma,F. (1999) EMBO J., 18, 433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tommasi S., Swiderski,P.M., Tu,Y., Kaplan,B.E. and Pfeifer,G.P. (1996) Biochemistry, 35, 15693–15703. [DOI] [PubMed] [Google Scholar]
- 50.Tijsterman M., Verhage,R.A., van de Putte,P., Tasseron-de Jong,J.G. and Brouwer,J. (1997) Proc. Natl Acad. Sci. USA, 94, 8027–8032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Evans M.K., Chin,K.V., Gottesman,M.M. and Bohr,V.A. (1996) Oncogene, 12, 651–658. [PubMed] [Google Scholar]
- 52.Leadon S.A. and Hanawalt,P.C. (1986) Mutat. Res., 166, 71–77. [DOI] [PubMed] [Google Scholar]
- 53.Kaufmann W.K. and Wilson,S.J. (1990) Mutat. Res., 236, 107–117. [DOI] [PubMed] [Google Scholar]
- 54.Whisnant-Hurst N. and Leadon,S.A. (1999) Radiat. Res., 151, 257–262. [PubMed] [Google Scholar]
- 55.Petersen L.N., Orren,D.K. and Bohr,V.A. (1995) Mol. Cell. Biol., 15, 3731–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lommel L., Carswell-Crumpton,C. and Hanawalt,P.C. (1995) Mutat. Res., 336, 181–192. [DOI] [PubMed] [Google Scholar]