


Abstract
Hamster EM9 cells, which lack Xrcc1 protein, have reduced levels of DNA ligase III and are defective in nuclear base excision repair. The Xrcc1 protein stabilizes DNA ligase III and may even play a direct role in catalyzing base excision repair. Since DNA ligase III is also thought to function in mitochondrial base excision repair, it seemed likely that mitochondrial DNA ligase III function would also be dependent upon Xrcc1. However, several lines of evidence indicate that this is not the case. First, western blot analysis failed to detect Xrcc1 protein in mitochondrial extracts. Second, DNA ligase III levels present in mitochondrial protein extracts from EM9 cells were indistinguishable from those seen in similar extracts from wild-type (AA8) cells. Third, the mitochondrial DNA content of both cell lines was identical. Fourth, EM9 cells displayed no defect in their ability to repair spontaneous mitochondrial DNA damage. Fifth, while EM9 cells were far more sensitive to the cytotoxic effects of ionizing radiation due to a defect in nuclear DNA repair, there was no apparent difference in the ability of EM9 and AA8 cells to restore their mitochondrial DNA to pre-irradiation levels. Thus, mitochondrial DNA ligase III function is independent of the Xrcc1 protein.
INTRODUCTION
Human DNA ligase III interacts directly with the 70 kDa Xrcc1 protein (1–3). Mutant hamster ovary cell lines EM7, EM9, EMC-11 and EM-C12 that lack Xrcc1 are hypersensitive to ionizing radiation and simple alkylating agents, and display a 10-fold increase in the frequency of spontaneous sister chromatid exchange (4). The amount of DNA ligase III protein in these mutant cells is reduced compared to control cells (1). Based on these observations, it is now known that heterodimerization of DNA ligase III with Xrcc1 is necessary for maintenance of normal cellular levels of active DNA ligase III (2). Analysis of the sequence of the Xrcc1 protein has not provided any insight into its function, and no enzymatic activity has been ascribed to this protein. However, a multiprotein complex formed by Xrcc1, poly(ADP-ribose) polymerase and DNA polymerase β is implicated in the removal of single strand breaks in nuclear DNA via the base excision repair (BER) pathway (5–8). Consistent with this view, the NMR solution structure of the N-terminal domain of Xrcc1 and a structural characterization of its interaction with DNA-polymerase-β bound to single-strand nicked or gapped DNA has recently been obtained (9).
DNA ligase III has been identified in mammalian mitochondria and is involved in mitochondrial BER (10,11). Since DNA ligase IIIβ is a testis-specific isoform and somatic cells were used for our studies we assume that the mitochondrial DNA ligase III is a DNA ligase IIIα isoform. More recently, we showed that depletion of DNA ligase III in the mitochondria by antisense DNA ligase III mRNA expression led to a decrease in cellular mitochondrial DNA copy number and increased levels of single-strand DNA breaks within the mitochondrial genome (U.Lakshmipathy and C.Campbell, submitted). We therefore hypothesized that the mitochondrial DNA ligase III protein would also require Xrcc1 for its normal functioning, and that cells lacking Xrcc1 should resemble cells deficient in DNA ligase III. To test this prediction, we analyzed mitochondrial protein extracts and mitochondrial DNA from Chinese hamster ovary (CHO) EM9 cells and their parental cell line AA8 (12,13). EM9 cells carry a frameshift mutation in the Xrcc1 gene that results in a truncated polypeptide lacking two-thirds of the normal sequence (14). There is no detectable Xrcc1 protein in these cells and the function of nuclear DNA ligase III is significantly reduced (2).
The results presented herein provide evidence that while Xrcc1 is necessary for nuclear DNA ligase III function, it is not required for the function of the mitochondrial form of DNA ligase III. We show that although DNA ligase III activity was reduced in nuclear extracts from EM9 cells compared to AA8 cells, there was no significant difference between the level of DNA ligase III activity detected in mitochondrial extracts of AA8 and EM9 cells. We also show that there was no reduction in either the copy number or integrity of mitochondrial DNA in EM9 cells, compared to AA8 cells. Furthermore, although EM9 cells were 1.6-fold more sensitive to the cytotoxic effects of 10 Gy of ionizing radiation compared to AA8 cells, both cell lines restored their mitochondrial DNA to pre-irradiation levels with equivalent kinetics. Finally, we show that the Xrcc1 protein cannot be detected in mitochondrial protein extracts from either AA8 or EM9 cells.
MATERIALS AND METHODS
Cells
The hamster Xrcc1 mutant cell line EM9 and its parental cell line AA8 were purchased from ATCC (Bethesda, MD). Cells were maintained as monolayers in α-MEM supplemented with 10% fetal bovine serum and 1 mM pyruvate.
Nuclear extracts
Nuclear extracts were prepared from AA8 or EM9 cells as described earlier (15). Briefly, cells were washed three times with phosphate buffered saline and scraped into a sterile tube. Following a wash with phosphate buffered saline, cells were homogenized in a buffer containing 10 mM Tris, pH 7.4, 10 mM KCl, 10 mM MgCl2 and 10 mM DTT. Nuclei were sedimented at 1500 g and nuclear protein extracts prepared by hypoosmotic shock. Protein concentration was measured by the Bradford method using bovine serum albumin (BSA) (Sigma Chemical Co., St Louis, MO) as the standard (16).
Mitochondrial extracts
Mitochondria were isolated from cells by differential centrifugation as described earlier (17). Cells were homogenized in a buffer containing 0.3 M sucrose, 1 mM EGTA, 5 mM MOPS, pH 7.4, 5 mM KH2PO4 and 0.1% BSA. Cell debris and nuclei were removed by centrifugation at 1500 g. The supernatant was subjected to centrifugation at 15 000 g to sediment the mitochondria. The crude mitochondrial preparation was washed several times and protein extracts prepared by hypoosmotic shock. The identity of the mitochondrial preparation was confirmed by cytochrome c oxidase assay (18).
Adenylation reaction
Five micrograms of protein was incubated with 0.5 µCi [α-32P]ATP in the presence of 60 mM Tris–HCl, pH 8.0, 10 mM MgCl2, 5 mM DTT and 50 mg/ml BSA at room temperature for 15 min, as described (19). The reaction was terminated by the addition of sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) loading dye and boiling for 5 min. The material was resolved on a 10% SDS–polyacrylamide gel and the adenylated product detected by exposure to phosphorimager screen (Molecular Dynamics, Foster City, CA).
Western blot
Five micrograms of nuclear or mitochondrial protein extract was separated on a 10% polyacrylamide gel and transferred electrophoretically to nitrocellulose. The membrane was incubated for 1 h in 5% BSA prior to probing with a 1:250 dilution of Xrcc1-specific mouse monoclonal antibody (LabVision Inc.). Following a 1 h incubation in the primary antibody, blots were treated with alkaline phosphatase-conjugated goat anti-mouse antibody. Color was developed with the alkaline phosphatase substrate, 5-bromo-4-chloro-indoyl phosphate/nitroblue tetrazolium (Sigma).
DNA ligase assay
DNA nick–seal assay was performed to measure DNA ligase activity as described earlier (20). Briefly, two oligonucleotides (15 and 17mer) were annealed to M13 single-strand DNA, creating a DNA duplex containing a single-strand nick. The 17mer oligonucleotide had been previously end-labeled with [γ-32P]ATP by polynucleotide kinase (New England Biolabs, Beverly, MA). Ligase-mediated sealing of this single-strand nick would create a radioactive 32mer oligonucleotide product that was distinguished from the labeled 17mer substrate by electrophoresis on a denaturing polyacrylamide gel containing 7 M urea. Five micrograms of protein extracts were used for the assay and 1 U of T4 DNA ligase (New England Biolabs) was used as the positive control
Isolation of total cellular DNA
Cells were lysed on the culture dish with 10 mM Tris–HCl, 1 mM EDTA and 0.5% SDS and incubated with proteinase K for 12 h at 37°C. DNA was extracted with phenol:chloroform (1:1) and precipitated with ethanol. DNA was washed with 70% ethanol, air dried and resuspended in 10 mM Tris, 1 mM EDTA, pH 8.0 buffer. DNA concentration was determined by the diphenylamine method using herring sperm DNA as a standard (21).
Southern blot
One microgram of total cellular DNA was digested with KpnI (New England Biolabs) to linearize the mitochondrial DNA. The sample was separated on a 0.5% agarose gel and transferred to a nylon membrane. For denaturing conditions, samples were treated with 0.1 N NaOH for 30 min at 37°C prior to electrophoresis. The blots were hybridized with an [α-32P]dATP labeled fragment of the mouse mitochondrial genome (nucleotides 13 551–15 329).
Gamma irradiation of cells
Cells were suspended in serum free media and exposed to 0, 2.5 or 10 Gy of radiation from a 137Cs source. For cytotoxicity assays, 500 irradiated cells were seeded in culture dishes in triplicate and allowed to grow in serum containing standard growth media. After 10 days, the cells were stained using crystal violet and the number of colonies counted. For analysis of the mitochondrial DNA, irradiated cells were lysed either immediately following irradiation, or at specific times as indicated, and total cellular DNA isolated. The DNA was subjected to Southern blot analysis as described.
RESULTS
DNA ligase III is the major form of DNA ligase in mammalian mitochondria (10,11). In human cells, reducing the amount of this protein by antisense DNA ligase III mRNA expression leads to a decrease in mitochondrial DNA copy number as well as the accumulation of mitochondrial DNA single-strand breaks (U.Lakshmipathy and C.Campbell, submitted). Nuclear DNA ligase III requires Xrcc1 protein for its function (22). Loss of this protein leads to a decrease in the nuclear DNA ligase III protein and activity. This is well documented in the hamster mutant cell line EM9 and allowed us to ask whether mitochondrial DNA ligase III requires Xrcc1 for its function.
To determine if the Xrcc1 protein is present in the mitochondria, western blot analysis was performed on nuclear and mitochondrial protein extracts from a number of mammalian cell lines. Figure 1A depicts the Coomassie blue stained image of these extracts resolved by PAGE, indicating that equal amounts of protein had been loaded. Figure 1B confirms earlier observations that while the Xrcc1 protein is present in nuclear extracts from AA8 (wild-type) cells (lane 1), this protein is absent from nuclear extracts made from the Xrcc1-deficient cell line, EM9 (lane 3). Next, mitochondrial extracts were made from both AA8 and EM9 cells. As Figure 1B shows, neither mitochondrial extract had detectable levels of Xrcc1 protein (lanes 2 and 4). An identical analysis was performed on nuclear and mitochondrial extracts from a second wild-type CHO-derived cell line, V79. We again observed that Xrcc1 protein was detected in the nuclear extract (Fig. 1B, lane 5) but was absent from the mitochondrial extract (Fig. 1B, lane 6). Similar experiments performed on a human fibrosarcoma-derived cell line, HT1080, also indicated that the Xrcc1 protein could be detected in a nuclear protein extract, but not in a mitochondrial extract (data not shown). The results described above suggest that Xrcc1 may not be present in mammalian mitochondria. However, it is possible that undetectable levels of Xrcc1 are present in the mitochondrial compartment, and that this low level of mitochondrial Xrcc1 protein may be essential for DNA ligase III function in this compartment.
Figure 1.
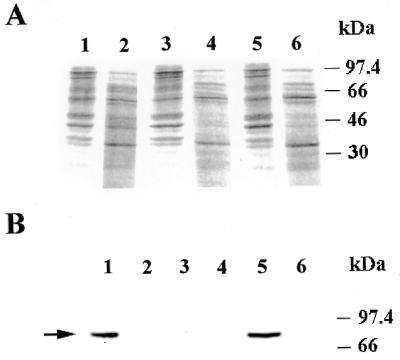
The Xrcc1 protein in not present in mitochondrial protein extracts. (A) Coomassie blue stained gel of 10 µg of nuclear (lanes 1, 3 and 5) or mitochondrial extracts (lanes 2, 4 and 6) from AA8 (lanes 1 and 2), EM9 (lanes 3 and 4) and V79 (lanes 5 and 6) cells. (B) Western blot analysis using an Xrcc1-specific monoclonal antibody on the same samples described in (A). The position of Xrcc1 is indicated with an arrow.
To test this hypothesis, we determined the level of DNA ligase III present in mitochondrial and nuclear extracts from AA8 and EM9 cells. Previously, Caldecott et al. (1) measured nuclear DNA ligase III levels in Xrcc1-deficient cells using adenylation assays. Nuclear extracts have multiple forms of DNA ligase. DNA ligase IV (125 kDa) is generally pre-adenylated and does not adenylate under the assay conditions used. Adenylated DNA ligase I (125 kDa) and DNA ligase III (100 kDa) can be clearly distinguished from each other based on their mobility (1). Mitochondrial extracts show a single adenylation band (∼75 kDa) that has been identified as the product of DNA ligase III gene (11). As Figure 2A indicates, there was substantially less adenylation activity seen in a nuclear extract (N) prepared from EM9 cells (E) than there was in a similar extract from AA8 (A) cells. In contrast, mitochondrial extracts (M) from both cells had similar levels of adenylation activity. To quantify this difference, adenylation assays of this type were performed on independently prepared nuclear and mitochondrial extracts (n = 3) from both cell lines. The results of this analysis are presented in Figure 2B. The amount of adenylation product was reduced by ~4-fold in nuclear extracts from EM9 cells compared to its parental cell line, AA8, as was seen in a previous study (1). In contrast, the adenylation activity seen in mitochondrial protein extracts from AA8 and EM9 cells (black bars, Fig. 2A) were identical, suggesting that the absence of Xrcc1 does not affect the mitochondrial DNA ligase III activity.
Figure 2.

Mitochondrial protein extracts from EM9 cells have DNA ligase activity but nuclear protein extracts do not. (A) Adenylation experiments were performed on 5 µg of nuclear (N) or mitochondrial (M) extracts from AA8 (A) and EM9 (E) cells. (B) Adenylation experiments were performed on multiple nuclear (Nuc.) and mitochondrial (Mito.) extracts from AA8 and EM9 cells. The amount of adenylation activity in extracts from EM9 cells (shaded bars) was normalized relative to that present in extracts from AA8 cells (open bars). Error bars represent the standard error of the mean (SEM, n = 3).
The total DNA ligase activity in crude extracts of EM9 cells is about half that of AA8 cells (3). Further, a 5-fold reduction in the rate of IR-induced single-strand breaks was observed in EM9 cells (4). To confirm that mitochondrial extracts from AA8 and EM9 cells have similar levels of DNA ligase activity, nuclear and mitochondrial extracts from these cells were used to perform nick-sealing assays (Fig. 3). This assay, described in detail in Materials and Methods, measures the ability of an extract to ligate together two synthetic oligonucleotides (one of which is radioactive) that are annealed side-by-side on a circular M13 virion molecule. The ligation product (indicated by the arrow in Fig. 3) migrates more slowly on a denaturing gel electrophoresis than does the un-ligated substrate. As a positive control, T4 DNA ligase was used. As Figure 3 indicates, there was far more DNA ligase activity in the nuclear extract prepared from AA8 cells than in the nuclear extract from EM9 cells. This lack of nick-sealing DNA ligase activity in the EM9 nuclear extract is consistent with earlier reports that demonstrate defective processing of nicked BER intermediate by cell free extracts of EM9 cells (6). In contrast, as Figure 3 also reveals, mitochondrial protein extracts prepared from both AA8 and EM9 cell lines have equivalent amounts of DNA ligase activity. To ensure that the kinetics of DNA ligase activity in the AA8 and EM9 mitochondrial extracts were identical, nick-sealing assay was carried out with protein extracts varying in concentration from 0.5 to 5 mg. In all cases, the amount of product formation was identical between the AA8 and EM9 mitochondrial extracts (data not shown). Since DNA single-strand nick sealing is equivalent to the final step of base excision repair, these results suggest that, in contrast to the nucleus, the mitochondria of EM9 cells are unlikely to have a defect in base excision repair.
Figure 3.
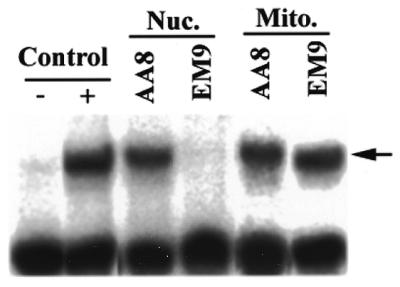
Nick-sealing activity of nuclear and mitochondrial extracts from EM9 and AA8 cells. A nick-seal assay was carried out on 5 µg samples of nuclear (Nuc.) and mitochondrial (Mito.) protein extracts as described in Materials and Methods. As a positive control, the substrate was incubated with 1 U of T4 DNA ligase. – Control, substrate alone; + control, substrate plus T4 DNA ligase; Nuc. AA8, substrate plus nuclear extract from AA8 cells; Nuc. EM9, substrate plus nuclear extract from EM9 cells; Mito. AA8, substrate plus mitochondrial extract from AA8 cells; Mito. EM9, substrate plus mitochondrial extract from EM9 cells. The arrow indicates the position of mobility of the sealed product.
If this hypothesis were correct, there would be no difference between the copy number and integrity of mitochondrial DNA in EM9 cells compared to that of AA8 cells. Alternatively, if mitochondrial BER were deficient in EM9 cells one would predict that these cells would have increased levels of damaged mitochondrial DNA, as was seen in mammalian cells deficient in DNA ligase III (U.Lakshmipathy and C.Campbell, submitted). To address this question, total cellular DNA was isolated from AA8 and EM9 cells and digested with KpnI to linearize the mitochondrial DNA. This material was resolved on an agarose gel, transferred to a membrane and probed with a mitochondrial DNA probe. Figure 4A reveals that 1 µg of AA8 (lane 1) and EM9 (lane 2) genomic DNA contained roughly equivalent amounts of mitochondrial DNA, indicating that the mitochondrial DNA copy number of both cell lines are similar. This analysis was repeated three times on separate genomic DNA preparations from each cell line. Quantitation of these mitochondrial DNA-specific hybridization signals from these Southern blots revealed that there was no significant difference between the mitochondrial DNA contents of these two cell lines (data not shown).
Figure 4.
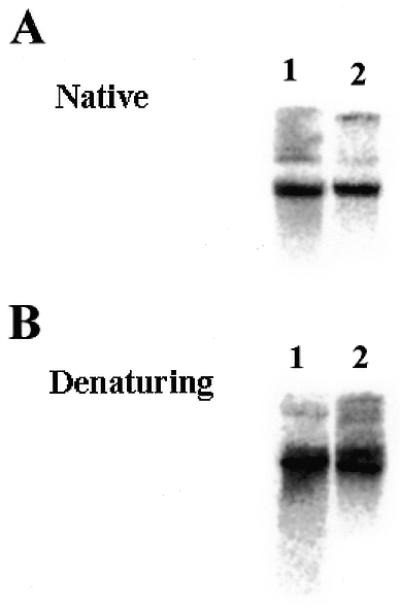
Mitochondrial DNA copy number and integrity is similar in AA8 and EM9 cells. (A) Total cellular DNA (1 µg) from AA8 (lane 1) or EM9 (lane 2) cells was digested with the restriction enzyme PvuII to linearize the mitochondrial DNA and resolved on a 0.5% agarose gel. The gel was transferred onto a nylon membrane and hybridized with a mitochondrial DNA probe. (B) Cellular DNA was isolated from AA8 and EM9 cells and treated as above. The DNA was treated with 0.1 N NaOH for 30 min at 37°C prior to electrophoresis. The gel was then transferred to nylon and hybridized with a mitochondrial DNA probe.
To determine whether the mitochondrial DNA in EM9 cells had accumulated damage in the form of DNA nicks due to defective mitochondrial ligase III activity, total cellular DNA was digested with KpnI and treated with 0.1 N NaOH for 10 min at 37°C prior to electrophoresis. While intact mitochondrial DNA migrates as a well-defined band, mitochondrial DNA with single-strand nicks migrates as a broad low-molecular weight smear (U.Lakshmipathy and C.Campbell, submitted). As Figure 4B reveals, there was no apparent difference in the integrity of the mitochondrial DNA in AA8 and EM9 cells (compare lanes 1 and 2). This analysis was repeated several times using separate DNA preparations; however, we did not detect any difference in the amount of DNA damage in mitochondrial DNA from EM9 cells. These results suggest that EM9 cells are fully capable of carrying out BER of spontaneous mitochondrial DNA damage.
It was of interest to determine whether there might be more subtle defects in mitochondrial BER in EM9 cells. We thus examined the kinetics of mitochondrial DNA recovery following treatment with ionizing radiation. We first performed a series of cytotoxicity experiments to confirm that these cells had a defect in their ability to repair ionizing radiation-induced DNA damage to the nuclear genome. EM9 and AA8 cells were suspended at a density of 10 000 cells/ml in serum free α-MEM and exposed to 0, 2.5, 5 or 10 Gy of radiation from a 137Cs source. Following irradiation, 500 cells were plated per 10 cm dish, in triplicate, and allowed to grow for 10 days in α-MEM at 37°C. At this time the total number of colonies formed per dish were counted by staining with crystal violet, and the percent survival at each dose was calculated. A total of three independent experiments were carried out, and the mean percent survival of cells was plotted against irradiation dose (Fig. 5). As this Figure indicates, AA8 cells (open circles) had an LD90 of 5 Gy, while EM9 cells (closed circles) had an LD90 of 3 Gy. This experiment revealed that EM9 cells are 1.66 times more sensitive to γ-irradiation compared to AA8 cells, consistent with earlier reports of a 1.7-fold greater sensitivity of EM9 cells to γ-irradiation compared to AA8 cells (18).
Figure 5.

EM9 cells are hypersensitive to ionizing radiation. EM9 (closed circles) and AA8 (open circles) cells were exposed to ionizing radiation, and the percent survival determined as described in Materials and Methods. Error bars indicate the SEM (n = 3).
To determine whether EM9 cellular mitochondrial DNA was more sensitive to damage due to irradiation the following experiment was performed. AA8 or EM9 cells (∼1 × 107 cells) were treated as above with either 5 or 10 Gy of radiation, and cellular DNA harvested immediately. This DNA was digested with KpnI, resolved on a 0.5% agarose gel, transferred to nylon membrane and hybridized with a mitochondrial DNA probe. Figure 6 depicts the results from a representative experiment, indicating that both cells sustained significant damage to their mitochondrial DNA immediately following irradiation. This experiment was repeated three additional times, and the relative amount of mitochondrial DNA present in the two cell lines was quantitated. As Figure 6B indicates, the mitochondrial DNA of both cells appeared to be similarly sensitive to ionizing radiation.
Figure 6.

Sensitivity of mitochondrial DNA from EM9 and AA8 cells to ionizing radiation. EM9 and AA8 cells were exposed to 0, 5 or 10 Gy of ionizing radiation. Total genomic DNA was immediately purified, cut with PvuII, resolved by agarose gel electrophoresis and hybridized to a mitochondrial DNA specific probe. (A) The results from a representative experiment. (B) Four experiments similar to that presented in (A) were performed and the relative amounts of mitochondrial DNA present were quantitated. Open bars, AA8 cells; shaded bars, EM9 cells. The error bars represent the SEM (n = 4).
We next examined mitochondrial DNA recovery in these two cell lines. Irradiation experiments similar to those described above were performed. Southern blot analysis was used to examine mitochondrial DNA either immediately following radiation (–), or after a 12 h recovery period (+). The major band represents KpnI linearized mitochondrial DNA genome (16.5 kb). A minor high molecular weight species seen in some lanes corresponds to uncut mitochondrial DNA. Figure 7A provides a representative experiment, and the results of a more detailed series of experiments (n = 3) are presented in Figure 7B. The data presented in Figure 7 indicate that both EM9 and AA8 have essentially restored their mitochondrial DNA content to pre-irradiation levels by 12 h. We also examined mitochondrial DNA in these cell lines at 8 h post irradiation. At this time, both cells had mitochondrial DNA content that was ~80% of the pre-irradiation level (data not shown). Taken together, these results indicate that EM9 cells are not deficient in their ability to restore mitochondrial DNA to pre-irradiation levels. DNA ligase III represents the major, if not the only, form of DNA ligase in the mitochondrial compartment (3,23). This latter result therefore strongly suggests that EM9 cells have no defect in mitochondrial DNA ligase III.
Figure 7.

Post-irradiation mitochondrial DNA recovery kinetics in AA8 and EM9 cells is similar. AA8 and EM9 cells were treated with ionizing radiation and their mitochondrial DNA was analyzed immediately, or following a 12 h recovery period, as described in the text. (A) Representative experiment. 0, pre-irradiation; –, immediately post-irradiation; +, 12 h post-irradiation. (B) Three experiments similar to that depicted in (A) were performed and mitochondrial DNA content measured by Southern blot hybridization. Opens bars, AA8 cells; shaded bars, EM9 cells. The error bars represent the SEM (n = 3).
DISCUSSION
It has been known for some time that Xrcc1 is required to stabilize nuclear DNA ligase III. We therefore expected to detect substantially reduced levels of mitochondrial DNA ligase III protein in mitochondrial protein extracts from EM9 cells, compared to mitochondrial extracts from AA8. The fact that no such decrease was seen indicates that the mitochondrial form of DNA ligase III, unlike the nuclear form, is not dependent upon Xrcc1 for its stability. In addition to being stable in the absence of Xrcc1, the data presented above suggest that mitochondrial DNA ligase III function is not dependent upon the Xrcc1 protein. The level of DNA ligase III activity seen in mitochondrial protein extracts from EM9 cells was equal to that seen in mitochondrial extracts from wild-type AA8 cells. We also observed that there was no detectable increase in the steady-state level of single-strand breaks in EM9 cell mitochondrial DNA. Likewise, the mitochondrial DNA copy number of EM9 cells was essentially identical to that seen in AA8 cells. Furthermore, EM9 cells, which are hypersensitive to the cytotoxic effects of gamma radiation, are wild-type in their ability to restore their mitochondrial DNA to pre-irradiation levels. We recently showed that reduced levels of mitochondrial DNA ligase III activity leads to increased mitochondrial single-strand breaks, and also reduces the mitochondrial DNA copy number in cultured mammalian cells (U.Lakshmipathy and C.Campbell, submitted). Thus, these findings indicate that in addition to having wild-type levels of mitochondrial DNA ligase III protein, EM9 cells have a fully functional mitochondrial ligase in vivo.
This conclusion was somewhat surprising, based on recent evidence suggesting that the Xrcc1 protein may play a more direct role in nuclear BER. It was previously determined that the Xrcc1 protein forms a multi-protein complex with DNA ligase III, DNA polymerase β and poly(ADP-ribose) polymerase (5). An NMR solution structure of a complex of the N-terminal domain of Xrcc1 protein, DNA polymerase β and single-strand nicked or gapped DNA showed that the Xrcc1 protein makes specific contact with the single-strand break of the DNA molecule (9). This finding suggests that the N-terminal domain of Xrcc1 binds to the DNA polymerase β–damaged DNA complex, while the C-terminal BRCT-containing domain of Xrcc1 is bound to the BRCT-containing domain of DNA ligase III (9). This model of interaction is consistent with Xrcc1 recruitment of DNA ligase III to the site of single-strand DNA nicks that are subsequently repaired by the DNA ligase III enzyme. It is therefore possible that for BER in the nucleus, DNA polymerase β and Xrcc1 interact at the DNA gap together with recruitment of DNA ligase III. For BER in the mitochondria, DNA polymerase γ performs gap-filling together with ligation by DNA ligase III in the absence of Xrcc1. Thus the differing polymerases may relate to the absence of Xrcc1 in mitochondrial BER.
If this model is correct, it raises the question of how DNA ligase III finds mitochondrial DNA single-strand nicks without the assistance of Xrcc1. By all accounts, there are greater relative numbers of DNA single-strand nicks within the mitochondrial genome, than the nuclear genome, presumably due to the proximity of the former to the oxygen-radical-generating oxidative phosphorylation cascade of the inner mitochondrial membrane. It is therefore tempting to speculate that the mammalian mitochondria may have an Xrcc1 homolog. In addition to targeting mitochondrial DNA ligase III to DNA single-strand nicks, such a protein could stabilize the mitochondrial DNA ligase III molecule. This is an attractive prospect, since it is known that in the nucleus, both DNA ligase III and DNA ligase IV have binding proteins (Xrcc1 and Xrcc4, respectively) that are essential for their stability (1, 2,23–25). Thus intracellular instability may be a common feature of DNA ligases, thereby requiring the existence of a mitochondrial DNA ligase III-binding protein. Alternatively, the possibility that the mitochondrial DNA ligase III is a β-isoform cannot be ruled out. The α-isoform is stabilized by interaction with Xrcc1 through its C-terminal BRCT while the testis-expressed β-isoform that lacks the C-terminal BRCT domain does not interact with Xrcc1 and is yet stable (26).
On the other hand, it is conceivable that DNA ligase III is inherently stable within the mammalian mitochondria, and thus does not require a stabilizing binding partner. If mitochondrial BER does not require a Xrcc1 homolog, it is conceivable that this organelle has developed an alternate mechanism to target the DNA ligase III molecule to sites of DNA damage.
Acknowledgments
ACKNOWLEDGEMENTS
Greg Coffey provided much-appreciated editorial advice. This work was supported by grants from the National Institute of Health (R29-CA61906, R01-AG16678), the American Heart Association (9601390) and the American Cancer Society (DHP-171). U.L. was supported by a fellowship from the American Heart Association.
REFERENCES
- 1.Caldecott K.W., McKeown,C.K., Tucker,J.D., Ljunquist,S. and Thompson,L.H. (1994) Mol. Cell. Biol., 14, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caldecott K.W., McKeown,C.K., Tucker,J.D., Stanker,L. and Thompson,L.H. (1995) Nucleic Acids Res., 23, 4836–4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ljungquist S., Kenne,K., Olsson,L. and Sandstrom,M. (1994) Mutat. Res., 314, 117–186. [DOI] [PubMed] [Google Scholar]
- 4.Thompson L.H., Brookman,K.W., Jones,N.J., Allen,S.A. and Carrano,A.V. (1990) Mol. Cell. Biol., 10, 6160–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caldecott K.W., Aoufouchi,S., Johnson,P. and Shall,S. (1996) Nucleic Acids Res., 24, 4387–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cappelli E., Taylor,R., Cevasco,M., Abbondandolo,A., Caldecott,K. and Frosina,G. (1997) J. Biol. Chem., 272, 23970–23975. [DOI] [PubMed] [Google Scholar]
- 7.Kubota Y., Nash,R.A., Klugland,A., Schar,P., Barnes,D.E. and Lindahl,T. (1996) EMBO J., 15, 6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 8.Masson M., Niedergang,C., Schreiber,V., Muller,S., Menissier-de Murcia,J. and de Murcia,G. (1998) Mol. Cell. Biol., 18, 3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marintchev A., Mullen,M.A., Maciejewski,M.W., Pan,B., Grykand,M.R. and Mullen,G.P. (1999) Nat. Struct. Biol., 6, 884–893. [DOI] [PubMed] [Google Scholar]
- 10.Lakshmipathy U. and Campbell,C. (1999) Mol. Cell. Biol., 19, 3869–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinz K.G. and Bogenhagen,D.F. (1998) Mol. Cell. Biol., 18, 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson L.H., Fong,S. and Brookman,K. (1980) Mutat. Res., 74, 21–36. [DOI] [PubMed] [Google Scholar]
- 13.Thompson L.H., Brookman,K.W., Dillehay,L.E., Carrano,A.V., Mazrimas,J.A., Mooney,C.L. and Minker,J.L. (1982) Mutat. Res., 95, 427–440. [DOI] [PubMed] [Google Scholar]
- 14.Shen M.R., Zdzienicka,M.Z., Mohrenweiser,H., Thompson,L.H. and Thelen,M.P. (1998) Nucleic Acids Res., 26, 1032–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jessberger R. and Berg,P. (1991) Mol. Cell. Biol., 11, 445–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradford M.M. (1976) Anal. Biochem., 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 17.Darley-Usmar V.M., Rickwood,D. and Wilson,M.T. (1987) Mitochondria: A Practical Approach. IRL Press, New York, NY.
- 18.Storrie B. and Madden,E.A. (1990) Methods Enzymol., 182, 203–225. [DOI] [PubMed] [Google Scholar]
- 19.Tomkinson A.E., Roberts,E., Daly,G., Totty,N.F. and Lindahl,T. (1991) J. Biol. Chem., 286, 21728–21735. [PubMed] [Google Scholar]
- 20.Barker D.G., Johnson,A.L. and. Johnston,L.H. (1985) Mol. Gen. Genet., 200, 458–462. [DOI] [PubMed] [Google Scholar]
- 21.Abraham G.N., Scaletta,C. and Vaughan,J.H. (1972) Anal. Biochem., 49, 547–549. [DOI] [PubMed] [Google Scholar]
- 22.Thompson L.H. and West,M.G. (2000) Mutat. Res., 459, 1–18. [DOI] [PubMed] [Google Scholar]
- 23.Grawunder U., Wilm,M., Wu,X., Kulesza,P., Wilson,T.E., Mann,M. and Lieber,M.R. (1997) Nature, 388, 492–495. [DOI] [PubMed] [Google Scholar]
- 24.Bryans M., Valenzano,M.C. and Stamato,T.D. (1999) Mutat. Res., 433, 53–58. [DOI] [PubMed] [Google Scholar]
- 25.Critchlow S.E., Bowater,R.P. and Jackson,S.P. (1997) Curr. Biol., 7, 588–598. [DOI] [PubMed] [Google Scholar]
- 26.Chew S.L., Baginsky,L. and Eperon,I.C. (2000) Nucleic Acids Res., 28, 402–410. [DOI] [PMC free article] [PubMed] [Google Scholar]